
Abstract
The term metastasis is widely used to describe the endpoint of the process by which tumour cells spread from the primary location to an anatomically distant site. Achieving successful dissemination is dependent not only on the molecular alterations of the cancer cells themselves, but also on the microenvironment through which they encounter. Here, we reviewed the molecular alterations of metastatic gastric cancer (GC) as it reflects a large proportion of GC patients currently seen in clinic. We hope that further exploration and understanding of the multistep metastatic cascade will yield novel therapeutic targets that will lead to better patient outcomes.
Similar content being viewed by others


Introduction
Gastric cancer (GC) is the fourth most common cancer and second leading cause of cancer-related deaths worldwide [1]. Over 70% of GC cases (~677,000 per annum) occur in the developing regions, mainly in Asia, Central and Eastern Europe and Latin America [2,3,4]. Despite improvements in GC incidence and mortality over the last decade, the disease burden still remains high. The majority of patients present with clinically advanced disease such that curative surgical resection is no longer possible and current therapeutics are poor at controlling the progression of metastatic disease. More worryingly, there are suggestions that advancements in GC treatment are likely to be surpassed by other diseases, consequently some foresee that GC is on a rising trend as a leading cause of death worldwide [5].
Metastasis is the main cause of cancer mortality (>90%) and a critical step that hampers the development of anti-cancer therapy due to its systemic nature and resistance to existing therapeutic drugs [6, 7]. Metastasis of gastric adenocarcinoma is no exception. It represents a multistep biological cascade that ultimately leads to widespread dissemination of carcinoma cells in various tissue sites [6, 8, 9]. In this review, we take you step-by-step through the GC metastatic cascade and the current understanding of the spectrum of molecular alterations involved. We look forward to this update being a guide for future research, and at the same time, highlighting its potential for translation into therapeutic strategies.
GC metastasis cascade
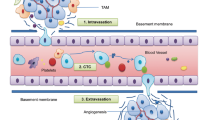
GC most commonly metastasises to the liver, peritoneum, lung, bone and lymph nodes [10] either through direct invasion or more distant seeding via the blood, lymphatic system and intraperitoneal spread. Notwithstanding these differences, they share the following series of sequential and interrelated events: (1) local invasion into the surrounding tumour-associated stroma, (2) intravasation into the haematopoietic or lymphatic systems, or intraperitoneal spread, (3) survival in vasculature transition or intraperitoneal fluid circulation, (4) extravasation into 'fertile soil' at distant organs with pre-metastatic niches and (5) colonisation and proliferation to form detectable metastases (Fig. 1) [7, 11,12,13]. These cellular events are normally kept in check under the orchestration of both intrinsic and extrinsic molecular pathways; however, aberrant molecular alterations allow the transformation of nascent tumour cells to highly invasive malignancies, which further lead to incurable metastatic disease with systemic spread and therapeutic resistance [6].

Metastatic routes and sites in gastric cancer. Major routes of distant metastasis in gastric cancer: intraperitoneal, lymphatic and haematogenous spread, and direct invasion into neighbouring organs. Common sites of metastases: spleen, pancreas, colon, liver, peritoneum, ovary, lymph nodes, lung and bone
Local invasion into surrounding tumour-associated stromal microenvironment
Local invasion occurs when tumour cells no longer obey the delineation of the basement membrane (BM), and the invasive front infiltrates the neighbouring tumour-associated stroma and surrounding normal tissues. Three major players facilitate this process: epithelial–mesenchymal transition (EMT), matrix metalloproteinases (MMPs) and the stromal environment, within which alterations and interactions amongst various molecular processes determine the tumour cells’ invasive propensity [14].
EMT
EMT describes the dissociation of tightly knitted epithelial cells and subsequent transdifferentiation into motile and invasive mesenchymal cells [15]. In the mesenchymal cell state, these cancer cells possess novel ability to invade into the surrounding microenvironment. Thus, EMT is considered to be the crucial step in the initiation of local invasion, and hence subsequent dissemination [14]. The transition involves relocalisation, dissolution and degradation of adherens junctions, subapical tight junctions, desmosomes and gap junctions between epithelial cells, ultimately leading to the loss of cell polarity and cytoskeleton changes [14,15,16]. As EMT becomes more established, mesenchymal phenotypes become more prominent, and the cells start to possess the ability to degrade extracellular matrix (ECM) proteins (Fig. 2) [15].

Epithelial–mesenchymal transition and tumour–stromal interactions in gastric cancer. Main phenotypic changes of EMT in gastric cancer include loss of cell polarity, degradation of cell-anchoring junctions, cytoskeleton changes, acquisition of invasiveness and ultimately degradation of basement membrane. Interactions within between key components of the stromal environment. EMT epithelial–mesenchymal transition, ECM extracellular matrix, CAF cancer-associated fibroblast, MSC mesenchymal stem cell, MMP matrix metalloproteinase
In GC, a number of signalling pathways have been found to regulate EMT, with the PI3K/AKT, MEK/ERK and WNT/β-Catenin pathways taking leading roles (Fig. 3). Transcription factors (TFs) and microRNAs, as described below, are the primary modulators. Although either can act independently, there is often some cross-modulation and interdependence that provides further complexity to their role in the regulation of signalling pathways.

Molecular mechanisms of EMT in gastric cancer. Major signalling pathways that regulate EMT in gastric cancer. PI3K/AKT, WNT/β-Catenin, ERK, TGF-β/SMAD and Snail signalling pathways promote EMT; Notch1/2 inhibits EMT in gastric cancer
Transcription factors
EMT is tightly regulated by TFs. Apart from the prominent TFs, such as Slug [17], Snail [18], Twist1/2 [19, 20], FOXQ1 [21] and ZEB1/2 [22,23,24,25] in GC, there are emerging novel TFs that have also been found to regulate EMT. For example, runt-related TF 3 (RUNX3), which has a role in suppressing EMT through the TGF-β-activated SMAD pathway, has been observed to be frequently downregulated [26, 27]. Similarly, there is loss of RUNX3-dependent miR-30a activation which normally inhibits vimentin expression and EMT [28]. In addition, a study based on array profiling identified significant upregulation of serum response factor (SRF) in metastatic GC cells. SRF functions to promote EMT through miR-199a-5p-mediated decrease in E-cadherin expression [29]. Also of interest, HOXB9 has been shown to halt GC progression. Studies have shown that restoration of HOXB9 expression in GC cells led to inhibited invasion and migration, at the same time stimulated the reversal of EMT process [30].
microRNAs
EMT can also be modulated by microRNAs [28]. For example, miR-544a induces EMT, as shown by the decreased expression of E-cadherin, APC2 and AXIN2, which stabilises the nuclear import of β-catenin and activates Wnt signalling to promote cell invasiveness in GC cell lines [31]. miR-2392 inhibits EMT through downregulating TFs, such as Slug and Twist1, in GC cells [32]. miR-223 promotes GC cell invasion and resistance to cisplatin by targeting FBXW7 [33, 34]. Conversely, miR-338-3p suppresses EMT through downregulating ZEB2, a TF that plays a vital role in promoting EMT in GC [25]. miR-506 suppresses EMT directly and its low expression is correlated with poor prognosis which indicates that it can serve as an independent prognosis fact in GC patients [27, 35]. Therefore, microRNAs can also be utilised as a potential therapeutic target for blocking EMT progression. For example, AC1MMYR2, a specific small-molecular inhibitor that can block the maturation of pre-miR-21 to miR-21, has been shown to reverse EMT and eventually lead to the suppression of GC cell metastasis [36].
Other molecules
In addition to TFs and microRNAs, EMT can be regulated by many other endogenous molecules. For example, Jumonji domain-containing protein 2B (JMJD2B) and erythropoietin-producing hepatocellular A2 (EphA2) which belongs to hydroxylase superfamily and protein-tyrosine kinase family, respectively, have both been suggested to induce EMT via the Wnt/β-catenin signalling pathway and further stimulate GC development and metastasis [37, 38]. Another example is melatonin, which a recent in vitro study revealed suppression of EMT in GC cells via the induction of endoplasmic reticulum stress and inhibition of β-catenin activity. Melatonin therapy decreased peritoneal dissemination in mice [39]. A more comprehensive list of the genes and microRNAs involved in regulating EMT are listed in Tables 1 and 2.
MMP
The BM is an important regulator of cellular behaviour in addition to its passive role in supporting surrounding tissues [40]. In cancer, BM functions as a mechanical barrier that prohibits cancer cells from penetrating the neighbouring stroma [41]. MMPs are proteolytic enzymes with a physiological role in degrading ECM proteins. However, dysregulation of MMPs, as seen in cancer cells, lead to uncontrolled proteolytic activity, tissue remodelling and disproportionate degradation of BM, thereby granting tumour cells stromal access [42, 43]. MMPs are upregulated in nearly all cancers, and their increased expressivity is generally associated with a poorer prognosis. Accumulating evidence has highlighted the role of MMPs in lymph node metastasis, peritoneal metastasis and distant metastasis [44,45,46,47,48,49].
MMP-1
MMP-1 is an interstitial collagenase that plays a role in the degradation of type I collagen (a major ECM component of stomach mucosa) [50,51,52]. One study reported that Helicobacter pylori infection can stimulate the upregulation of MMP-1, which could further enhance the potential of GC metastasis [51].
MMP-2 and MMP-9
MMP-2 and MMP-9 belong to the family of type IV collagenases or gelatinases. Both have been reported to contribute to vessel invasion and lymph node metastasis in intra-mucosal GC by degrading type IV collagen, which enabled infiltration of lymph capillaries [44, 53]. Certain oncogenic proteins play a role in regulating expression of MMP-2/9 in promoting cell invasion. For example, Bcl-w, which belongs to Bcl-2 protein family, has been demonstrated to induce MMP-2 expression via a sequential activation of PI3K, Akt and Sp1, thereby enhancing cell invasiveness and GC metastasis [54]. Similarly, androgen receptor, which is believed to play vital roles in various types of cancers, has been reported to bind directly to the promoter region of MMP-9, which upregulates MMP-9 expression and in turn promotes GC cell migration and invasion [55]. Furthermore, lipocalin-2, which is regarded as neutrophil gelatinase-associated lipocalin, has recently been shown to prevent auto-degradation of MMP-9. Collectively, both MMP-2 and MMP-9 are highly expressed in GC and their expression is positively associated with the poor survival of GC patients [56].
MMP-7
MMP-7 (matrilysin) is another MMP that is highly expressed in GC [57, 58]. It is the smallest (molecular weight) member of MMP family but with most efficient ECM-degrading activity on a wide spectrum of matrix substrates, such as proteoglycans, elastin, caseins, laminins, fibronectins, collagens, gelatins, entactins, vitronectins [59,60,61]. The expression level of MMP-7 at the invasive front of the tumour is relatively higher than the core, which indicates that the upregulation of MMP-7 is associated with cancer aggressiveness in GC [60]. Interestingly, several studies indicate that psychological stress-related changes may be involved in promoting cancer metastasis through stimulating the expression of MMPs [62,63,64]. For instance, it has been reported that catecholamines, which are stress-inducible hormones responsive to stress, depression or panic, can upregulate MMP-7 expression through AP-1 and STAT3 stimulation [59].
MMP-11
MMP-11, also known as Stromelysin-3, is distinct from other MMPs as it can only weakly degrade the main components of ECM. Additionally, it needs to be proteolytically cleaved and activated intracellularly by Furin-like serine proteinases prior to its relocalisation to the cell membrane [65, 66]. MMP-11 was first identified as a breast cancer-related gene which was later found to be highly expressed in most metastatic primary tumours as well as in some of their metastases when compared to matched normal tissues. Not only was this evident in GC, it could also be seen in renal, colon and lung cancers [67]. Moreover, one study found that MMP-11 levels were markedly elevated in the serum of GC patients compared with those from healthy subjects, and the enhanced expression of MMP-11 was well associated with metastases in these GC patients [66].
MMP-14
MMP-14 belongs to one of the six membrane-anchored MMPs, unlike the majority which are secreted proteins [68]. MMP-14 is normally located at the leading edge or invadopodia of a cell, which facilitates the degradation of ECM and guides cells to invade in a specific direction [68]. Additionally, MMP-14 promotes the secretion and activation of pro-MMP-2 and pro-MMP-9 [68, 69]. A recent meta-analysis showed that MMP-14 levels were significantly higher in GC tissues, and the increased MMP-14 expression correlated to higher clinical stage and metastases [70].
While most of the studies thus far focus on cancer cells-derived MMPs, emerging evidence indicate that MMPs (including MMP-2 and MMP-9) can also be secreted by the surrounding stromal cells, such as endothelial cells, fibroblasts, myofibroblasts and inflammatory cells [65]. These findings imply the importance of the tumour microenvironment in GC metastatic progression, as discussed below (Table 3).
Stromal environment
When cancer cells reach the surrounding stroma following EMT and BM penetration, the next step involves overcoming the barriers to allow further infiltration. Recent studies revealed that tumours function as a complex multicellular organ composed of both cancer cells and tumour stroma with significant interactive cross-talks [71]. It is thus unsurprising that tumour progression may be driven by molecular alterations in cancer cells as well as the tumour-associated stromal microenvironment [71,72,73,74]. GC cells invading into surrounding stroma will thus be confronted with neutrophils [75], carcinoma-associated fibroblasts (CAFs) [72, 74, 76], and a range of bone marrow-derived cells such as mesenchymal stem cells (MSCs) [72, 77, 78] and tumour-associated macrophages (TAMs) [79]. In response, it has been shown that cancer cells generate a variety of growth factors, chemokines and proteases that modulate surrounding stroma to establish a tolerant and contributory stromal environment for tumour progression [71].
Stromal cells
Stromal cells can heighten the aggressiveness and invasiveness of cancer cells through different molecular signalling pathways. For instance, interactions between neutrophils and MSCs via an IL-6–STAT3 axis lead to neutrophil activation and MSCs differentiation into CAFs, which provide a pro-inflammatory habitat. These stromal cells in turn collaboratively induce angiogenesis and invasiveness of GC cells to stimulate metastatic dissemination [75]. Furthermore, CAFs, derived from bone marrow, and MSCs are frequently enriched during progression into dysplasia. They express cytokine IL-6, glycoprotein Wnt5α, bone morphogenetic protein BMP4, as well as exhibit DNA hypomethylation and induce invasive growth [72]. Additionally, enhanced IL-17B expression in GC tissues leads to MSCs activation and increased migration and stemness, which further accelerates GC cell migration [77]. Interestingly, a recent study showed that MSCs are recruited and reprogrammed in tumour-specific manner. For example, lung cancer cell characteristics are independent of their MSC counterparts while GC cell proliferation, migration and invasion are dependent on the activation of hepatocyte growth factor (HGF)/c-MET signalling pathway specifically induced by HGF from GC-MSCs [78]. Moreover, gene-expression profiling of GC patients has identified a 'stromal-response' expression signature, which is highly enriched in inflammation-, ECM-, cytokine- and growth factor-related proteins. Most of these genes are specifically expressed in the surrounding stroma, but not cancer cells themselves, indicating the important role of stromal cells in promoting GC cell migration and metastasis [79].
Angiogenesis
Angiogenesis represents a tumour response to the hypoxic and nutrient-deficient environment driven by uncontrolled cellular proliferation and consequent explosive enlargement of tumour bulk [80, 81]. This process is fine-tuned by multiple signalling molecules and pathways in the tumour microenvironment. For example, miR-130a and miR-495 mediated downregulation of RUNX3, a suppressor of tumour angiogenesis, induces the metastatic ability of GC cells [82]. Based on the hypothesis that neovasculature can be formed through sprouting new vessels from existing blood vessels, emerging evidence indicate that tumour-associated angiogenesis can be initiated by cells recruited from the bone marrow or differentiated from putative cancer stem-like cells [81, 83]. Tumour-induced neovascularisation serves to supply sufficient oxygen and nutrients to meet the metabolic needs of uncontrolled tumour growth. Furthermore, studies show that tumour-associated angiogenesis are usually leaky and tortuous with high permeability, which could increase the chance of surrounding tumour cells intravasating into the blood circulation and disseminating to distant sites [12, 81, 84].
Lymphangiogenesis
Recent studies have shown that the growth of lymphatic vasculature, also known as lymphangiogenesis, either around the tumour or in the sentinel lymph nodes, is associated with increased incidence of lymphatic metastasis [85, 86]. In GC patients, lymph nodes are among top metastatic destinations, and accumulating evidence has shown that LN metastasis predicts GC prognosis [87, 88]. A study reported that the lymphatic vessel density (LVD) within lymph nodes is closely associated with nodal metastasis and malignancy of GC. Concomitantly, GC patients with high LVD showed notably poorer prognosis compared to low-LVD group, suggesting that intranodal lymphangiogenesis is tightly correlated with lymph node metastasis and poor prognosis in GC patients [86]. Mechanistic studies have highlighted the molecular mechanisms underlying the regulation of lymphangiogenesis. For instance, it has been shown that VEGF-C, VEGF-D and VEGFR-3 have an inducive role in promoting lymphangiogenesis in various cancers [85, 89,90,91], including GC [86, 92, 93]. Using human lymphatic endothelial cells co-cultured with VEGF-C-induced high-lymphangiogenesis GC cell line MKN45 and SGC-7901, the researchers identified several lymphangiogenesis-associated microRNAs such as upregulation of miR-648, miR-5002-3p and downregulation of miR-3178, miR-593-5p, miR-4485 [92]. Rosiglitazone [87], a peroxisome proliferator-activated receptor γ (PPARγ) agonist, has shown promising suppressive effect on lymphangiogenesis by concurrently downregulating the expression of VEGF-C and VEGFR-3 in GC xenograft mice models [93].
Collectively, these findings provide evidence that interactions between cancer cells and the tumour-associated stromal microenvironment could establish a potential positive-feedback loop, which provides substantial contributions to GC progression and metastasis. Accordingly, it is reasonable to hypothesise that tumour malignancy may be suppressed or even reversed by normalising the stromal environment.
Intravasation into the circulation
During the path of local invasion, cancer cells may encounter blood vessels or lymphatics to facilitate movement towards distant pre-metastatic niches. Alternatively, they may reach and penetrate beyond the serosa to initiate intraperitoneal seeding or direct invasion into neighbouring organs. Here we focus on intravasation which describes the process in which cancer cells gain access into the tumour-associated vasculatures located in the gastric submucosa [10, 43, 93].
Intravasation can be accelerated by molecular alterations that improve the potency of cancer cells in transendothelial invasion. Accumulating evidence has shown the positive correlation among vascular invasion, intratumoral angiogenesis and distant metastasis [94, 95]. For example, the first cloned member of CCN family, Cysteine-rich 61 (Cyr61), was shown to enhance the IL-8-dependent chemotactic migration of GC cells through inducing CXCR1/CXCR2 function, which promotes transendothelial invasion and intravasation [96].
Apart from its role as passive channels for tumour cell dissemination, emerging evidence also illustrated that lymphatic vessels actively stimulate recruitment of tumour cells to lymph nodes, immune regulation and cancer cell survival [85, 89]. The quantity of lymphatic vessels in the vicinity of primary tumours correlates with the rate of lymph node metastasis, and lymphatic metastasis is a key factor for prognosis and tumour staging in majority of cancers [85, 90, 97].
Intraperitoneal spread after serosal penetration
In addition to distant metastasis, ~10–20% of GC patients were found to harbour peritoneal metastasis that have likely arisen from exfoliated cancer cells through penetration of the gastric serosa [13, 98, 99]. However, this is likely to be an underestimation as intraperitoneal seeding was subsequently found in some who had undergone radical gastrectomy [98]. These microscopic metastases can initially be difficult to identify by imaging or even during surgery, and is only realised when patients present with progressive disease despite curative surgery. The field has yet to identify any molecular alterations that facilitate this pathway.
Survival within vasculature transition and intraperitoneal environment
Following successful intravasation into the circulation, the disseminating cancer cells, now termed circulating tumour cells (CTCs), must survive the precarious microenvironment en route to new sites of dissemination. The exposure to blood introduces stressors such as haemodynamic shear forces and recognition by the innate immune system. Furthermore, CTCs must also gain the ability to survive in the absence of substratum [100,101,102].
Anoikis resistance
The concept of 'anoikis' represents a form of programmed cell death triggered by loss of ECM attachment in epithelial cells [103]. Anoikis is crucial for maintaining epithelial architecture by prohibiting abnormal proliferation in unwanted locations after detachment. Cancer cells are frequently resistant to anoikis, which enable them to survive and thrive even after detachment from its substratum. Anoikis resistance is mechanistically facilitated by cell adhesion molecules, integrins and apoptosis modulators [103, 104], which promotes cell survival and dissemination in the periphery, thereby increasing the possibility of metastatic spread. For example, the peritoneal dissemination of GC cells can be inhibited by Caspase-8-augmented anoikis, which reduced cell survival in vitro and in vivo [105]. Meanwhile, tight junction protein Claudin-1 can induce anoikis resistance through β-catenin-modulated cell–cell adhesion and survival signals [106]. Of interest, RhoA, which belongs to Rho family GTPases, is upregulated in primary GC and its activation has been suggested to be essential for anoikis resistance by eliciting pro-survival responses [4, 107]. Hypoxia-induced ANGPTL4A in GC cells also induces increased resistance to anoikis by activating ANGPTL4A-dependent FAK/Src/PI3K-Akt/ERK pathway, leading to elevated peritoneal metastasis in scirrhous GC cells [108].
Platelets
Auxiliary pro-metastatic signals exist during intravascular transition in the circulation to aid cancer metastasis. Emerging evidence has shown that the interaction between platelets and cancer cells, more specifically the formation of emboli, are constructive in priming CTCs for intravascular survival [100, 109,110,111]. On the one hand, the platelet-coated tumour cells can protect them from blood flow shear forces, substratum absence and direct lysis by natural killer cells [112]; on the other hand, their association could also induce EMT in cancer cells [109], enhance adhesion to endothelial cells [111], or even disrupt the function of endothelial barrier, making it more porous for extravasation of cancer cells [101]. One recent study postulated an association between microRNAs and platelets using microRNA microarray analysis of MKN45 cell line. The group identified miR-4670-5p as the most significantly upregulated microRNA that promoted GC cell proliferation and that its proliferation-promoting effects are inhibited by aspirin in vivo [113]. This finding is consistent with five large randomised clinical trials showing that platelet inhibition by low-dose aspirin is beneficial in reducing the incidence of cancer metastasis [114]. Platelet micro-particles (PMP), submicroscopic vesicles shed by activated platelets membrane, are significantly upregulated in GC patients as compared to healthy subjects. Plasma PMP can be used as a platelet activation marker for GC diagnosis and to screen GC patients with increased potential for metastasis [115]. These results indicate that interaction between platelets and CTCs function as intrinsic determinants for distant metastasis through promoting cancer cell survival during intravascular transition, thereby raising the prospect of developing platelet inhibition drugs to aid anti-metastasis therapy.
Extravasation into 'fertile soil' at distant pre-metastatic niches
Despite the theoretical possibility that CTCs can be deposited at any metastatic niche within or surrounding both circulation systems, clinical observations have shown that certain cancer types have a higher probability of giving rise to metastasis in certain target organ(s) because of exosome-initiated pre-metastatic niches formation. For example, GC tends to form distant metastasis in the liver, peritoneum, lung, bone and lymph nodes [10]. Two hypotheses have postulated the pattern of metastasis tropism: (1) passive transfer, whereby the site of dissemination is dependent on vessel diameter as circulating cancer cells are arrested as they reach the microvasculature, which suggests that the metastatic pattern could be influenced by the layout of circulation systems [116]; (2) active homing, whereby the CTCs have genetically programmed receptor–ligand signalling that have predetermined predilections to target specific organs [6].
Extravasation
Extravasation represents the exiting of circulating cancer cells out of the vessel lumen to establish new sites of metastasis. There are two recognised forms of extravasation dependent on vessel diameter. Firstly, CTCs with adhesive molecules on the surface can attach to and penetrate the endothelium of the vessel walls irrespective of vessel size [6]. Alternatively, CTCs may be arrested and trapped at the microvasculature due to their relatively larger diameters of 20–30 μm compared to that of around 8 μm [6]. Once trapped, CTCs tend to grow into microcolonies which disrupt the luminal wall and invade into the surrounding tissue environment. Emerging evidence shows that the latter choice is the prevalent pathway by which CTCs grow into a distant metastasis, as single extravasated cancer cells may easily be eliminated by the surrounding microenvironment [117].
Naturally, factors that promote vasculature permeability are associated with increased extravasation. Calponin h1, an actin-binding protein which is mainly expressed in smooth muscle cells, plays a role in stabilising the actin filament system. Calponin h1 deficiency can induce the fragility of blood vessels and peritoneum, leading to the increased incidence of extravasation and tumour metastasis [118]. Accumulating evidence has shown that ANGPTL-4 plays a role in promoting metastasis by inducing the permeability of vasculatures in cancers that metastasise to the lungs [119, 120], and that ANGPTL-4 can increase the frequency of venous invasion. The potential role of ANGPTL-4 in disrupting vascular permeability in promoting GC metastasis requires further investigation [121].
Furthermore, studies focusing on targeting extravasation has led to the discovery of a double anti-angiogenic decoy receptor, double anti-angiogenic protein (DAAP), which simultaneously targets VEGF-A and angiopoietins to block tumour-associated angiogenesis and vascular leakage [122]. Hence this suggests that there is potential for analogues to be developed that can limit primary tumour growth as well as inhibit distant spread.
Exosome and pre-metastatic niche formation
Exosomes are membranous nanoparticles 40–50 nm in diameter and they can be released by both tumour cells and surrounding stromal cells, which will interact reciprocally to modulate immune responses, remodel tumour microenvironments and facilitate cancer metastases [123,124,125]. The role of GC-derived exosomes in metastasis has been extensively studied over the years.
GC-derived exosomes can modulate immune responses. For example, GC-derived exosomes can stimulate macrophages to generate a pro-inflammatory microenvironment via activation of nuclear factor κB (NFκB) signalling pathway, resulting in increased cell proliferation and migration [126]. Similarly, GC-derived exosomes enveloped with miR-451 can be translocated to infiltrating T cells and induces mTOR signalling pathway activation, which in turn leads to T-helper 17 (Th17) cells differentiation [127]. Hence, GC-derived exosomes may play important roles in mediating immune surveillance escape.
In terms of tumour microenvironment remodelling and cancer metastasis, mounting evidence indicates that GC-derived exosomes can initiate or accelerate pre-metastatic formation [124, 128, 129]. For example, EGFR-containing exosomes secreted by GC cells can be transported to liver and activate hepatocyte growth factor (HGF), which interacts with c-MET on disseminated GC cells in a paracrine fashion, thereby further promotes their colonisation and proliferation [130]. In addition, another study demonstrated that GC-derived exosomes can bolster pre-metastatic niche formation in peritoneum by inducing fibrosis and the disruption of mesothelium, which originally functions as a protective barrier to restrain peritoneal metastasis [131]. Similarly, GC-derived exosomes can promote expression of adhesion-related molecules, such as fibronectin 1 (FN 1) and laminin gamma 1 (LAMC 1), in mesothelial cells, which result in a favourable microenvironment for disseminating cancer cells to colonise and initiate metastasis [132].
Nevertheless, researchers are utilising the unique features of exosomes for drug delivery. Exosomes are loaded with drugs or siRNA to target the tumour regions [133, 134]. Exosomes loaded with HGF siRNAs have shown promising efficacy in inhibiting tumour growth, migration and angiogenesis in vitro and in vivo [133]. Exosomes isolated from heat stress-treated malignant ascites of GC patients showed elevated immunogenicity and might be employed as a cancer vaccine. Such exosomes can induce dendritic cell maturation and stimulate a tumour-specific cytotoxic T lymphocyte response [135].
Despite intensive efforts, limitations still exist in the study of exosomes as they are mainly restricted to in vitro co-culture or in vivo injection using labelled-exosomes, which is markedly different from their physiological location and concentration [123]. These concerns accentuate the necessity of developing novel models to overcome the limitations in exosome studies.
Colonisation and proliferation reactivation into clinical detectable metastases
Given the divergent microenvironment of the metastasised sites from that of the stomach, successfully extravasated cells need to adapt to the foreign microenvironments in order to survive and colonise. Currently, there are two universally acknowledged mechanisms by which cancer cells adapt to their new microenvironment: (1) cell autonomous programmes and (2) non-autonomous programmes [6].
For cell autonomous programmes, disseminating tumour cells (DTCs) acquire molecular alterations to increase their colonising ability. Colonising abilities are normally evaluated by detecting pulmonary metastases after intravenous (IV) injection in immune-deficient mice. For example, ectopic expression of RUNX3 repressed lung colonisation of GC cells in nude mice [28]. Similarly, the silencing of IL-32 in GC cells inhibited cell motility, invasion and lung colonisation in severe combined immunodeficiency (SCID) mice [136]. In the case of peritoneal metastasis after serosa penetration, increased expression of connexin 43 (Cx43) in GC cells exfoliated into peritoneal cavity was found to enhance their heterocellular gap-junctional intercellular communication (GJIC) with peritoneal mesothelial cells, which in turn mediated heterocellular gap junction and accelerated the infiltration of GC cells into peritoneal mesothelium for further colonisation [137]. This finding provides implications for further studies on GC cells seeded onto the lining mesothelial layer. Another mechanism is based on ligand-receptor interaction. For example, the expression of stromal cell-derived factor-1, together with its sole interactive receptor CXCR4, correlated with increased probability of lymph node and liver metastases [138]. For non-autonomous programmes, certain organ sites provide supportive niches which better facilitate the survival of DTCs. A recent retrospective study found that patients with STAT3 activation in cancer cell-free lymph nodes demonstrated higher rate of metastasis and poorer prognosis, which implicated the possibility of p-STAT3-induced pre-metastatic niches in lymph nodes [139]. Indeed, STAT3 blockade in myeloid cells abrogated the formation of pre-metastatic niches [140]. Moreover, the inflammatory cytokine tumour necrosis factor-α (TNFα) has been reported to induce morphological changes of mesothelial cells and regulate interactions between peritoneal mesothelial cells and DTCs, which in turn promotes peritoneal metastasis of GC cells in the intraperitoneal (IP) injected mouse model [141]. Hence, cytokines and chemokines also play a role in the shaping of pre-metastatic niches for GC peritoneal metastasis. Collectively, both autonomous and non-autonomous programmes promote cancer cells’ colonisation in pre-metastatic niches.
However, clinical observations showed that relapses are often detected long after removal of the primary tumour, spanning from months to years even when there was no previous evidence of metastasis [142]. This implies that these patients already carry DTCs in the body, where those DTCs remain dormant in two modes: (1) cellular dormancy and (2) tumour mass dormancy [143]. For example, in some GC patients, dormant DTCs can be detected harbouring inside bone marrow and they eventually develop into detectable metastasis in brain after 10 years, illustrating that dormant DTCs derived from GC retained both metastatic and growth ability for long periods of time [143, 144].
Latency represents a state in which metastatic cancer cells undergo proliferative quiescent in order to escape from immune clearance, attack from the new microenvironment and the surrounding growth inhibitory signals. They remain latent until certain, currently unknown factors re-activate their proliferative potency. At present, our knowledge of the underlying mechanisms of latent metastasis is limited due to the lack of mouse models that faithfully recapitulates the metastatic process and microenvironment.
Conclusion and perspective
Over the past decades, research progress on GC metastasis-related molecular alterations has provided valuable knowledge for deciphering this complex biological phenomenon. Although by no means comprehensive, we have rapidly gained an appreciation for the importance of stromal cells and the microenvironment. Nevertheless, due to the complexity and systemic nature of metastasis, a number of fundamental questions concerning the mechanisms of GC metastasis remain unanswered.
The major hurdle in the study of tumour metastasis is the lack of a mouse model with a competent immune system that can perfectly mimic the entire metastatic cascade. Therefore, this bottleneck imposes restrictions on in-depth study of the latter stages in the GC metastatic cascade. Attempts to establish a better metastatic mouse model have recently achieved intriguing progress, such as the implementing genome-wide or high-throughput screening approaches into immune-competent mice for identification of novel regulators of metastases [145, 146]. In this way, researchers can evaluate both tumour-cell-intrinsic (molecular manipulation of cancer cells) and tumour-cell extrinsic factors (tumour microenvironment of genetically engineered mice or drug treated mice) that modulates the metastasis cascade. Interestingly, a recent study used vascular endothelial growth factor receptor (VEGFR3) as an 'lymphorecporter' and established a novel mouse model that allows whole-body imaging of lymphovascular niches, which shed new lights on pre-metastatic niches [147]. These innovative technologies can all be considered as tools for future GC metastasis study.
Metastatic cancer cells that have successfully intravasated into the circulation system can survive and extravasate efficiently (>80%) [145]. This phenomenon suggests that effective mechanisms exist to protect CTCs from being eliminated during the transition. Mounting evidence has shown the correlations between neutrophil/lymphocyte ratio (NLR) and GC patients outcome that high NLR predicts poor prognosis and survival status [148,149,150]. However, few research has done on the mechanism by which neutrophils interact with CTCs in the circulation system to promote metastasis progression. Meanwhile, the role of other tumour-infiltrating immune cells, such as TAMs, natural killer (NK) cells, CAFs, also deserves further investigation.
Mechanistically, apart from the widely reported function of MMPs in the degradation of ECM, recent studies have revealed alternative roles of MMPs in metastasis, such as regulation of growth signals, apoptosis, tumour vasculature, inflammation and non-proteolytic functions [42]. Moreover, long noncoding RNAs (lncRNAs) are also gaining attention, since emerging data indicate that the deregulation of lncRNAs might contribute to tumour metastasis [151,152,153,154]. Apart from cell intrinsic alterations that contribute to GC metastasis, tumour-derived exosomes also showed great influences on inducing pre-metastatic niches [128]. These potential targets also deserve further exploration.
We hope that our understanding of the evolution of cancer metastasis continues to excel at this impressive pace, and that some of these findings will be translated into clinical use, especially in light of the current proportion of patients with metastatic GC.
Disclaimer
Neither the submitted paper nor any similar paper, in whole or in part, other than an abstract or preliminary communication, has been submitted to or published in any other primary scientific journal. All the authors are aware of and agree to the content of the paper, copyright assignment and authorship responsibility.
References
Van Cutsem E, Sagaert X, Topal B, Haustermans K, Prenen H. Gastric cancer. Lancet. 2016;388:2654–64.
Ferro A, Peleteiro B, Malvezzi M, Bosetti C, Bertuccio P, Levi F, et al. Worldwide trends in gastric cancer mortality (1980-2011), with predictions to 2015, and incidence by subtype. Eur J Cancer. 2014;50:1330–44.
Ferlay J,Soerjomataram I,Dikshit R,Eser S,Mathers C,Rebelo M, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136:86–E359.
Cai J, Niu X, Chen Y, Hu Q, Shi G, Wu H, et al. Emodin-induced generation of reactive oxygen species inhibits RhoA activation to sensitize gastric carcinoma cells to anoikis. Neoplasia. 2008;10:41–51.
Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006;3:2011–30.
Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell. 2011;147:275–92.
Gupta GP, Massague J. Cancer metastasis: building a framework. Cell. 2006;127:679–95.
Sun YT, Ma L. The emerging molecular machinery and therapeutic targets of metastasis. Trends Pharmacol Sci. 2015;36:349–59.
Turajlic S, Swanton C. Metastasis as an evolutionary process. Science. 2016;352:169–75.
Riihimaki M, Hemminki A, Sundquist K, Sundquist J, Hemminki K. Metastatic spread in patients with gastric cancer. Oncotarget. 2016;7:52307–16.
Fidler IJ. Timeline–the pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–8.
Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–72.
Iida T, Iwahashi M, Katsuda M, Ishida K, Nakamori M, Nakamura M, et al. Prognostic significance of IL-17 mRNA expression in peritoneal lavage in gastric cancer patients who underwent curative resection. Oncol Rep. 2014;31:605–12.
Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009;139:871–90.
Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Bio. 2014;15:178–96.
Scheel C, Onder T, Karnoub A, Weinberg RA. Adaptation versus selection: the origins of metastatic behavior. Cancer Res. 2007;67:11476–9. discussion11479–11480
Chen D, Zhou H, Liu G, Zhao Y, Cao G, Liu Q. SPOCK1 promotes the invasion and metastasis of gastric cancer through Slug-induced epithelial-mesenchymal transition. J Cell Mol Med. 2017;22:797–807.
Cho HJ, Park SM, Kim IK, Nam IK, Baek KE, Im MJ, et al. RhoGDI2 promotes epithelial-mesenchymal transition via induction of Snail in gastric cancer cells. Oncotarget. 2014;5:1554–64.
Guo WW, You X, Xu DQ, Zhang YX, Wang Z, Man KY, et al. PAQR3 enhances Twist1 degradation to suppress epithelial-mesenchymal transition and metastasis of gastric cancer cells. Carcinogenesis. 2016;37:397–407.
Wang L, Lin L, Chen X, Sun L, Liao YL, Huang N, et al. Metastasis-associated in colon cancer-1 promotes vasculogenic mimicry in gastric cancer by upregulating TWIST1/2. Oncotarget. 2015;6:11492–506.
Xiang XJ, Deng J, Liu YW, Wan LY, Feng M, Chen J, et al. MiR-1271 inhibits cell proliferation, invasion and EMT in gastric cancer by targeting FOXQ1. Cell Physiol Biochem. 2015;36:1382–94.
Zhao L,Li X,Song N,Li A,Hou K,Qu X, et al. Src promotes EGF-induced epithelial-to-mesenchymal transition and migration in gastric cancer cells by upregulating ZEB1 and ZEB2 through AKT. Cell Biol Int. 2017;42:294–302.
Kurashige J, Kamohara H, Watanabe M, Hiyoshi Y, Iwatsuki M, Tanaka Y, et al. MicroRNA-200b regulates cell proliferation, invasion, and migration by directly targeting ZEB2 in gastric carcinoma. Ann Surg Oncol. 2012;19:S656–64.
Li H, Wang ZW, Zhang W, Qian K, Xu W, Zhang SR. Fbxw7 regulates tumor apoptosis, growth arrest and the epithelial-to-mesenchymal transition in part through the RhoA signaling pathway in gastric cancer. Cancer Lett. 2016;370:39–55.
Huang N, Wu ZZ, Lin L, Zhou MY, Wang L, Ma HR, et al. MiR-338-3p inhibits epithelial-mesenchymal transition in gastric cancer cells by targeting ZEB2 and MACC1/Met/Akt signaling. Oncotarget. 2015;6:15222–34.
Peng ZH, Wei DY, Wang LW, Tang HM, Zhang J, Le XD, et al. RUNX3 inhibits the expression of vascular endothelial growth factor and reduces the angiogenesis, growth, and metastasis of human gastric cancer. Clin Cancer Res. 2006;12:6386–94.
Chen Y, Wei XF, Guo CC, Jin HF, Han ZY, Han Y, et al. Runx3 suppresses gastric cancer metastasis through inactivation of MMP9 by upregulation of TIMP-1. Int J Cancer. 2011;129:1586–98.
Liu ZF, Chen L, Zhang XC, Xu X, Xing HX, Zhang YJ, et al. RUNX3 regulates vimentin expression via miR-30a during epithelial- mesenchymal transition in gastric cancer cells. J Cell Mol Med. 2014;18:610–23.
Zhao X, He L, Li T, Lu Y, Miao Y, Liang S, et al. SRF expedites metastasis and modulates the epithelial to mesenchymal transition by regulating miR-199a-5p expression in human gastric cancer. Cell Death Differ. 2014;21:1900–13.
Chang Q, Zhang L, He CY, Zhang BG, Zhang J, Liu BY, et al. HOXB9 induction of mesenchymal-to-epithelial transition in gastric carcinoma is negatively regulated by its hexapeptide motif. Oncotarget. 2015;6:42838–53.
Yanaka Y, Muramatsu T, Uetake H, Kozaki K, Inazawa J. miR-544a induces epithelial-mesenchymal transition through the activation of WNT signaling pathway in gastric cancer. Carcinogenesis. 2015;36:1363–71.
Li J, Li T, Lu Y, Shen G, Guo H, Wu J, et al. MiR-2392 suppresses metastasis and epithelial-mesenchymal transition by targeting MAML3 and WHSC1 in gastric cancer. FASEB J. 2017;31:3774–86.
Li JH, Guo YY, Liang XD, Sun M, Wang GL, De W, et al. MicroRNA-223 functions as an oncogene in human gastric cancer by targeting FBXW7/hCdc4. J Cancer Res Clin. 2012;138:763–74.
Zhou XY, Jin WJ, Jia HY, Yan J, Zhang GX. MiR-223 promotes the cisplatin resistance of human gastric cancer cells via regulating cell cycle by targeting FBXW7. J Exp Clin Cancer Res. 2015;34:28.
Sakimura S, Sugimachi K, Kurashige J, Ueda M, Hirata H, Nambara S, et al. The miR-506-induced epithelial-mesenchymal transition is Involved in poor prognosis for patients with gastric cancer. Ann Surg Oncol. 2015;22:S1436–43.
Shi ZD, Zhang JX, Qian XM, Han L, Zhang KL, Chen LY, et al. AC1MMYR2, an inhibitor of Dicer-mediated biogenesis of Oncomir miR-21, reverses epithelial-mesenchymal transition and suppresses tumor growth and progression. Cancer Res. 2013;73:5519–31.
Zhao L, Li WJ, Zang W, Liu ZF, Xu X, Yu H, et al. JMJD2B promotes epithelial-mesenchymal transition by cooperating with beta-catenin and enhances gastric cancer metastasis. Clin Cancer Res. 2013;19:6419–29.
Huang J, Xiao D, Li G, Ma J, Chen P, Yuan W, et al. EphA2 promotes epithelial-mesenchymal transition through the Wnt/beta-catenin pathway in gastric cancer cells. Oncogene. 2014;33:2737–47.
Wu SM, Lin WY, Shen CC, Pan HC, Keh-Bin W, Chen YC, et al. Melatonin set out to ER stress signaling thwarts epithelial mesenchymal transition and peritoneal dissemination via calpain-mediated C/EBP and NFB cleavage. J Pineal Res. 2016;60:142–54.
Kalluri R. Basement membranes: structure, assembly and role in tumour angiogenesis. Nat Rev Cancer. 2003;3:422–33.
Liotta LA, Tryggvason K, Garbisa S, Hart I, Foltz CM, Shafie S. Metastatic potential correlates with enzymatic degradation of basement-membrane collagen. Nature. 1980;284:67–8.
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141:52–67.
Zhong J, Chen Y, Wang LJ. Emerging molecular basis of hematogenous metastasis in gastric cancer. World J Gastroenterol. 2016;22:2434–40.
Kabashima A, Maehara Y, Kakeji Y, Baba H, Koga T, Sugimachi K. Clinicopathological features and overexpression of matrix metalloproteinases in intramucosal gastric carcinoma with lymph node metastasis. Clin Cancer Res. 2000;6:3581–4.
Al-Batran SE, Pauligk C, Wirtz R, Werner D, Steinmetz K, Homann N, et al. The validation of matrix metalloproteinase-9 mRNA gene expression as a predictor of outcome in patients with metastatic gastric cancer. Ann Oncol. 2012;23:1699–705.
Yonemura Y, Fujimura T, Ninomiya I, Kim BS, Bandou E, Sawa T, et al. Prediction of peritoneal micrometastasis by peritoneal lavaged cytology and reverse transcriptase-polymerase chain reaction for matrix metalloproteinase-7 mRNA. Clin Cancer Res. 2001;7:1647–53.
Chu DK, Zhang ZX, Li YM, Zheng JY, Dong GL, Wang WZ, et al. Matrix metalloproteinase-9 is associated with disease-free survival and overall survival in patients with gastric cancer. Int J Cancer. 2011;129:887–95.
Wu CY, Wu MS, Chiang EP, Chen YJ, Chen CJ, Chi NH, et al. Plasma matrix metalloproteinase-9 level is better than serum matrix metalloproteinase-9 level to predict gastric cancer evolution. Clin Cancer Res. 2007;13:2054–60.
Tang Y, Zhu JW, Chen L, Chen LY, Zhang S, Lin JY. Associations of matrix metalloproteinase-9 protein polymorphisms with lymph node metastasis but not invasion of gastric cancer. Clin Cancer Res. 2008;14:2870–7.
Ghilardi G, Biondi ML, Mangoni J, Leviti S, DeMonti M, Guagnellini E, et al. Matrix metalloproteinase-1 promoter polymorphism 1G/2G is correlated with colorectal cancer invasiveness. Clin Cancer Res. 2001;7:2344–6.
Wu JY, Lu H, Sun Y, Graham DY, Cheung HS, Yamaoka Y. Balance between polyoma enhancing activator 3 and activator protein 1 regulates Helicobacter pylori-stimulated matrix metalloproteinase 1 expression. Cancer Res. 2006;66:5111–20.
Poola I, DeWitty RL, Marshalleck JJ, Bhatnagar R, Abraham J, Leffall LD. Identification of MMP-1 as a putative breast cancer predictive marker by global gene expression analysis. Nat Med. 2005;11:481–3.
Wu CY, Wu MS, Chen YJ, Chen CJ, Chen HP, Shun CT, et al. Clinicopathological significance of MMP-2 and TIMP-2 genotypes in gastric cancer. Eur J Cancer. 2007;43:799–808.
Bae IH, Park MJ, Yoon SH, Kang SW, Lee SS, Choi KM, et al. Bcl-w promotes gastric cancer cell invasion by inducing matrix metalloproteinase-2 expression via phosphoinositide 3-kinase, Akt, and Sp1. Cancer Res. 2006;66:4991–5.
Zhang BG, Du T, Zang MD, Chang Q, Fan ZY, Li JF, et al. Androgen receptor promotes gastric cancer cell migration and invasion via AKT-phosphorylation dependent upregulation of matrix metalloproteinase 9. Oncotarget. 2014;5:10584–95.
Kubben FJGM, Sier CFM, Hawinkels LJAC, Tschesche H, van Duijn W, Zuidwijk K, et al. Clinical evidence for a protective role of lipocalin-2 against MMP-9 autodegradation and the impact for gastric cancer. Eur J Cancer. 2007;43:1869–76.
Aihara R, Mochiki E, Nakabayashi T, Akazawa K, Asao T, Kuwano H. Clinical significance of mucin phenotype, beta-catenin and matrix metalloproteinase 7 in early undifferentiated gastric carcinoma. Br J Surg. 2005;92:454–62.
Honda M, Mori M, Ueo H, Sugimachi K, Akiyoshi T. Matrix metalloproteinase-7 expression in gastric carcinoma. Gut. 1996;39:444–8.
Shi M, Liu D, Duan HJ, Han CL, Wei B, Qian L, et al. Catecholamine up-regulates MMP-7 expression by activating AP-1 and STAT3 in gastric cancer. Mol Cancer. 2010;9:269.
Sentani K, Matsuda M, Oue N, Uraoka N, Naito Y, Sakamoto N, et al. Clinicopathological significance of MMP-7, laminin gamma 2 and EGFR expression at the invasive front of gastric carcinoma. Gastric Cancer. 2014;17:412–22.
Bebb JR, Letley DP, Thomas RJ, Aviles F, Collins HM, Watson SA, et al. Helicobacter pylori upregulates matrilysin (MMP-7) in epithelial cells in vivo and in vitro in a Cag dependent manner. Gut. 2003;52:1408–13.
Yang EV, Sood AK, Chen M, Li Y, Eubank TD, Marsh CB, et al. Norepinephrine up-regulates the expression of vascular endothelial growth factor, matrix metalloproteinase (MMP)-2, and MMP-9 in nasopharyngeal carcinoma tumor cells. Cancer Res. 2006;66:10357–64.
Sood AK, Bhatty R, Kamat AA, Landen CN, Han L, Thaker PH, et al. Stress hormone-mediated invasion of ovarian cancer cells. Clin Cancer Res. 2006;12:369–75.
Thaker PH, Han LY, Kamat AA, Arevalo JM, Takahashi R, Lu C, et al. Chronic stress promotes tumor growth and angiogenesis in a mouse model of ovarian carcinoma. Nat Med. 2006;12:939–44.
Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:161–74.
Yang YH, Deng H, Li WM, Zhang QY, Hu XT, Xiao B, et al. Identification of matrix metalloproteinase 11 as a predictive tumor marker in serum based on gene expression profiling. Clin Cancer Res. 2008;14:74–81.
Peruzzi D, Mori F, Conforti A, Lazzaro D, De Rinaldis E, Ciliberto G, et al. MMP11: a novel target antigen for cancer immunotherapy. Clin Cancer Res. 2009;15:4104–13.
Zarrabi K, Dufour A, Li J, Kuscu C, Pulkoski-Gross A, Zhi JZ, et al. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J Biol Chem. 2011;286:33167–77.
Zuo QF, Cao LY, Yu T, Gong L, Wang LN, Zhao YL, et al. MicroRNA-22 inhibits tumor growth and metastasis in gastric cancer by directly targeting MMP14 and Snail. Cell Death Dis. 2015;6:e2000.
Dong YC, Chen GH, Gao MM, Tian X. Increased expression of MMP14 correlates with the poor prognosis of Chinese patients with gastric cancer. Gene. 2015;563:29–34.
Mueller MM, Fusenig NE. Friends or foes–bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–49.
Quante M, Tu SP, Tomita H, Gonda T, Wang SSW, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19:257–72.
Coussens LM, Werb Z. Inflammation and cancer. Nature. 2002;420:860–7.
Shibata W, Ariyama H, Westphalen CB, Worthley DL, Muthupalani S, Asfaha S, et al. Stromal cell-derived factor-1 overexpression induces gastric dysplasia through expansion of stromal myofibroblasts and epithelial progenitors. Gut. 2013;62:192–200.
Zhu Q, Zhang X, Zhang L, Li W, Wu H, Yuan X, et al. The IL-6-STAT3 axis mediates a reciprocal crosstalk between cancer-derived mesenchymal stem cells and neutrophils to synergistically prompt gastric cancer progression. Cell Death Dis. 2014;5:e1295.
Abe A, Nagatsuma AK, Higuchi Y, Nakamura Y, Yanagihara K, Ochiai A. Site-specific fibroblasts regulate site-specific inflammatory niche formation in gastric cancer. Gastric Cancer. 2017;20:92–103.
Bie QL, Zhang B, Sun CX, Ji XY, Barnie PA, Qi C, et al. IL-17B activated mesenchymal stem cells enhance proliferation and migration of gastric cancer cells. Oncotarget. 2017;8:18914–23.
Berger L, Shamai Y, Skorecki KL, Tzukerman M. Tumor specific recruitment and reprogramming of mesenchymal stem cells in tumorigenesis. Stem Cells. 2016;34:1011–26.
Busuttil RA, George J, Tothill RW, Ioculano K, Kowalczyk A, Mitchell C, et al. A signature predicting poor prognosis in gastric and ovarian cancer represents a coordinated macrophage and stromal response. Clin Cancer Res. 2014;20:2761–72.
Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol. 2002;29:15–8.
Weis SM, Cheresh DA. Tumor angiogenesis: molecular pathways and therapeutic targets. Nat Med. 2011;17:1359–70.
Lee SH, Jung YD, Choi YS, Lee YM. Targeting of RUNX3 by miR-130a and miR-495 cooperatively increases cell proliferation and tumor angiogenesis in gastric cancer cells. Oncotarget. 2015;6:33269–78.
Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307.
Weis SM, Cheresh DA. Pathophysiological consequences of VEGF-induced vascular permeability. Nature. 2005;437:497–504.
Achen MG, McColl BK, Stacker SA. Focus on lymphangiogenesis in tumor metastasis. Cancer Cell. 2005;7:121–7.
Watanabe M, Tanaka H, Ohira M, Yoshii M, Sakurai K, Toyokawa T, et al. Intranodal lymphangiogenesis precedes development of lymph node metastasis and accelerates progression of gastric cancer. J Gastrointest Surg. 2014;18:481–90.
Marchet A, Mocellin S, Ambrosi A, Morgagni P, Garcea D, Marrelli D, et al. The ratio between metastatic and examined lymph nodes (N ratio) is an independent prognostic factor in gastric cancer regardless of the type of lymphadenectomy–results from an Italian multicentric study in 1853 patients. Ann Surg. 2007;245:543–52.
Arigami T, Natsugoe S, Uenosono Y, Yanagita S, Arima H, Hirata M, et al. CCR7 and CXCR4 expression predicts lymph node status including micrometastasis in gastric cancer. Int J Oncol. 2009;35:19–24.
Karaman S, Detmar M. Mechanisms of lymphatic metastasis. J Clin Invest. 2014;124:922–8.
Tammela T, Alitalo K. Lymphangiogenesis: molecular mechanisms and future promise. Cell. 2010;140:460–76.
Alitalo K, Tammela T, Petrova TV. Lymphangiogenesis in development and human disease. Nature. 2005;438:946–53.
Yang B, Jing C, Wang J, Guo X, Chen Y, Xu R, et al. Identification of microRNAs associated with lymphangiogenesis in human gastric cancer. Clin Transl Oncol. 2014;16:374–9.
Chen FZ, Mo XM, Wang QP, Li J, Zhang L. Effects of rosiglitazone on the growth and lymphangiogenesis of human gastric cancer transplanted in nude mice. Oncol Rep. 2013;30:2705–12.
Maehara Y, Kabashima A, Koga T, Tokunaga E, Takeuchi H, Kakeji Y, et al. Vascular invasion and potential for tumor angiogenesis and metastasis in gastric carcinoma. Surgery. 2000;128:408–16.
Wang YD, Wu P, Mao JD, Huang H, Zhang F. Relationship between vascular invasion and microvessel density and micrometastasis. World J Gastroenterol. 2007;13:6269–73.
Lin BR, Chang CC, Chen LR, Wu MH, Wang MY, Kuo IH, et al. Cysteine-rich 61 (CCN1) enhances chemotactic migration, transendothelial cell migration, and intravasation by concomitantly up-regulating chemokine receptor 1 and 2. Mol Cancer Res. 2007;5:1111–23.
Karpanen T, Alitalo K. Molecular biology and pathology of lymphangiogenesis. Annu Rev Pathol. 2008;3:367–97.
Gianni Mura BV. The features of peritoneal metastases from gastric cancer. J Cancer Metastasis Treat. 2016;2:365–74.
Sadeghi B, Arvieux C, Glehen O, Beaujard AC, Rivoire M, Baulieux J, et al. Peritoneal carcinomatosis from non-gynecologic malignancies–results of the EVOCAPE 1 multicentric prospective study. Cancer. 2000;88:358–63.
Palumbo JS, Talmage KE, Massari JV, La Jeunesse CM, Flick MJ, Kombrinck KW, et al. Platelets and fibrin(ogen) increase metastatic potential by impeding natural killer cell-mediated elimination of tumor cells. Blood. 2005;105:178–85.
Schumacher D, Strilic B, Sivaraj KK, Wettschureck N, Offermanns S. Platelet-derived nucleotides promote tumor-cell transendothelial migration and metastasis via P2Y2 receptor. Purinergic Signal. 2014;10:730–730.
Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895–904.
Frisch SM, Ruoslahti E. Integrins and anoikis. Curr Opin Cell Biol. 1997;9:701–6.
Liotta LA, Kohn E. Anoikis–cancer and the homeless cell. Nature. 2004;430:973–4.
Nishimura S, Adachi M, Ishida T, Matsunaga T, Uchida H, Hamada H, et al. Adenovirus-mediated transfection of caspase-8 augments anoikis and inhibits peritoneal dissemination of human gastric carcinoma cells. Cancer Res. 2001;61:7009–14.
Huang J, Zhang L, He C, Qu Y, Li J, Zhang J, et al. Claudin-1 enhances tumor proliferation and metastasis by regulating cell anoikis in gastric cancer. Oncotarget. 2015;6:1652–65.
Hayakawa Y, Ariyama H, Stancikova J, Sakitani K, Asfaha S, Renz BW, et al. Mist1 expressing gastric stem cells maintain the normal and neoplastic gastric epithelium and are supported by a perivascular stem cell niche. Cancer Cell. 2015;28:800–14.
Baba K, Kitajima Y, Miyake S, Nakamura J, Wakiyama K, Sato H, et al. Hypoxia-induced ANGPTL4 sustains tumour growth and anoikis resistance through different mechanisms in scirrhous gastric cancer cell lines. Sci Rep UK. 2017;7(1):11127.
Labelle M, Begum S, Hynes RO. Direct signaling between platelets and cancer cells induces an epithelial-mesenchymal-like transition and promotes metastasis. Cancer Cell. 2011;20:576–90.
Camerer E, Qazi AA, Duong DN, Cornelissen I, Advincula R, Coughlin SR. Platelets, protease-activated receptors, and fibrinogen in hematogenous metastasis. Blood. 2004;104:397–401.
Stanger BZ, Kahn ML. Platelets and tumor cells: a new form of border control. Cancer Cell. 2013;24:9–11.
Nieswandt B, Hafner M, Echtenacher B, Mannel DN. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999;59:1295–300.
Mikami J, Kurokawa Y, Takahashi T, Miyazaki Y, Yamasaki M, Miyata H, et al. Antitumor effect of antiplatelet agents in gastric cancer cells: an in vivo and in vitro study. Gastric Cancer. 2016;19:817–26.
Rothwell PM, Wilson M, Price JF, Belch JFF, Meade TW, Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012;379:1591–601.
Kim HK, Song KS, Park YS, Kang YH, Lee YJ, Lee KR, et al. Elevated levels of circulating platelet microparticles, VEGF, IL-6 and RANTES in patients with gastric cancer: possible role of a metastasis predictor. Eur J Cancer. 2003;39:184–91.
Friedl P, Alexander S. Cancer invasion and the microenvironment: plasticity and reciprocity. Cell. 2011;147:992–1009.
Al-Mehdi AB, Tozawa K, Fisher AB, Shientag L, Lee A, Muschel RJ. Intravascular origin of metastasis from the proliferation of endothelium-attached tumor cells: a new model for metastasis. Nat Med. 2000;6:100–2.
Taniguchi S, Takeoka M, Ehara T, Hashimoto S, Shibuki H, Yoshimura N, et al. Structural fragility of blood vessels and peritoneum in calponin h1-deficient mice, resulting in an increase in hematogenous metastasis and peritoneal dissemination of malignant tumor cells. Cancer Res. 2001;61:7627–34.
Galaup A, Cazes A, Le Jan S, Philippe J, Connault E, Le Coz E, et al. Angiopoietin-like 4 prevents metastasis through inhibition of vascular permeability and tumor cell motility and invasiveness. Proc Natl Acad Sci USA. 2006;103:18721–6.
Padua D, Zhang XHF, Wang QQ, Nadal C, Gerald WL, Gomis RR, et al. TGF beta primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell. 2008;133:66–77.
Nakayama T, Hirakawa H, Shibata K, Nazneen A, Abe K, Nagayasu T, et al. Expression of angiopoietin-like 4 (ANGPTL4) in human colorectal cancer: ANGPTL4 promotes venous invasion and distant metastasis. Oncol Rep. 2011;25:929–35.
Koh YJ, Kim HZ, Hwang SI, Lee JE, Oh N, Jung K, et al. Double antiangiogenic protein, DAAP, targeting VEGF-A and angiopoietins in tumor angiogenesis, metastasis, and vascular leakage. Cancer Cell. 2010;18:171–84.
Ruivo CF, Adem B, Silva M, Melo SA. The biology of cancer exosomes: insights and new perspectives. Cancer Res. 2017;77:6480–8.
Fong MY, Zhou WY, Liu L, Alontaga AY, Chandra M, Ashby J, et al. Breast-cancer-secreted miR-122 reprograms glucose metabolism in premetastatic niche to promote metastasis. Nat Cell Biol. 2015;17(2):183–94.
Thery C, Ostrowski M, Segura E. Membrane vesicles as conveyors of immune responses. Nat Rev Immunol. 2009;9:581–93.
Wu L, Zhang X, Zhang B, Shi H, Yuan X, Sun Y, et al. Exosomes derived from gastric cancer cells activate NF-kappaB pathway in macrophages to promote cancer progression. Tumour Biol. 2016;37:12169–80.
Liu F, Bu Z, Zhao F, Xiao D. Increased T-helper 17 cell differentiation mediated by exosome-mediated microRNA-451 redistribution in gastric cancer infiltrated T cells. Cancer Sci. 2018;109:65–73.
Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Mark MT, et al. Tumour exosome integrins determine organotropic metastasis. Nature. 2015;527:329–35.
Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816–26.
Zhang H, Deng T, Liu R, Bai M, Zhou L, Wang X, et al. Exosome-delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat Commun. 2017;8:15016.
Deng G, Qu J, Zhang Y, Che X, Cheng Y, Fan Y, et al. Gastric cancer-derived exosomes promote peritoneal metastasis by destroying the mesothelial barrier. FEBS Lett. 2017;591:2167–79.
Arita T, Ichikawa D, Konishi H, Komatsu S, Shiozaki A, Ogino S, et al. Tumor exosome-mediated promotion of adhesion to mesothelial cells in gastric cancer cells. Oncotarget. 2016;7:56855–63.
Zhang HY, Wang Y, Bai M, Wang JY, Zhu KG, Liu R, et al. Exosomes serve as nanoparticles to suppress tumor growth and angiogenesis in gastric cancer by delivering hepatocyte growth factor siRNA. Cancer Sci. 2018;109:629–41.
Wang JJ, Wang ZY, Chen R, Xiong J, Yao YL, Wu JH, et al. Macrophage-secreted exosomes delivering miRNA-21 inhibitor can regulate BGC-823 cell proliferation. Asian Pac J Cancer Prev. 2015;16:4203–9.
Zhong H, Yang Y, Ma S, Xiu F, Cai Z, Zhao H, et al. Induction of a tumour-specific CTL response by exosomes isolated from heat-treated malignant ascites of gastric cancer patients. Int J Hyperthermia. 2011;27:604–11.
Tsai CY, Wang CS, Tsai MM, Chi HC, Cheng WL, Tseng YH, et al. Interleukin-32 increases human gastric cancer cell invasion associated with tumor progression and metastasis. Clin Cancer Res. 2014;20:2276–88.
Tang B, Peng ZH, Yu PW, Yu G, Qian F, Zeng DZ, et al. Aberrant expression of Cx43 is associated with the peritoneal metastasis of gastric cancer and Cx43-mediated gap junction enhances gastric cancer cell diapedesis from peritoneal mesothelium. PLoS ONE. 2013;8:e74527.
Iwasa S, Yanagawa T, Fan JL, Katoh R. Expression of CXCR4 and its ligand SDF-1 in intestinal-type gastric cancer is associated with lymph node and liver metastasis. Anticancer Res. 2009;29:4751–8.
Wu LJ, Li HX, Luo XT, Lu RZ, Ma YF, Wang R, et al. STAT3 activation in tumor cell-free lymph nodes predicts a poor prognosis for gastric cancer. Int J Clin Exp Pathol. 2014;7:1140–6.
Deng JH, Liu Y, Lee H, Herrmann A, Zhang W, Zhang CY, et al. S1PR1-STAT3 signaling is crucial for myeloid cell colonization at future metastatic sites. Cancer Cell. 2012;21:642–54.
Mochizuki Y, Nakanishi H, Kodera Y, Ito S, Yamamura Y, Kato T, et al. TNF-alpha promotes progression of peritoneal metastasis as demonstrated using a green fluorescence protein (GFP)-tagged human gastric cancer cell line. Clin Exp Metastasis. 2004;21:39–47.
Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou YL, et al. Metastatic latency and immune evasion through autocrine inhibition of WNT. Cell. 2016;165:45–60.
Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nat Rev Cancer. 2004;4:448–56.
Fukushima M, Katayama Y, Shigemori Y, Miyake H, Hirayama T, Kotani A. Clivus metastasis from gastric signet ring cell carcinoma after a 10-year disease-free interval—case report. Neurol Med Chir (Tokyo). 2012;52:751–3.
Speak AO, Swiatkowska A, Karp NA, Arends MJ, Adams DJ, van der Weyden L. A high-throughput in vivo screening method in the mouse for identifying regulators of metastatic colonization. Nat Protoc. 2017;12:2465–77.
van der Weyden L, Arends MJ, Campbell AD, Bald T, Wardle-Jones H, Griggs N, et al. Genome-wide in vivo screen identifies novel host regulators of metastatic colonization. Nature. 2017;541:233–6.
Olmeda D, Cerezo-Wallis D, Riveiro-Falkenbach E, Pennacchi PC, Contreras-Alcalde M, Ibarz N, et al. Whole-body imaging of lymphovascular niches identifies pre-metastatic roles of midkine. Nature. 2017;546:676–80.
Lian L, Xia YY, Zhou C, Shen XM, Li XL, Han SG, et al. Application of platelet/lymphocyte and neutrophil/lymphocyte ratios in early diagnosis and prognostic prediction in patients with resectable gastric cancer. Cancer Biomark. 2015;15:899–907.
Ock CY, Nam AR, Lee J, Bang JH, Lee KH, Han SW, et al. Prognostic implication of antitumor immunity measured by the neutrophil-lymphocyte ratio and serum cytokines and angiogenic factors in gastric cancer. Gastric Cancer. 2017;20:254–62.
Zhang H, Liu H, Shen Z, Lin C, Wang X, Qin J, et al. Tumor-infiltrating neutrophils is prognostic and predictive for postoperative adjuvant chemotherapy benefit in patients with gastric cancer. Ann Surg. 2016;267(2):311–8.
Kung JTY, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013;193:651–69.
Wilusz JE, Sunwoo H, Spector DL. Long noncoding RNAs: functional surprises from the RNA world. Gene Dev. 2009;23:1494–504.
Lian D, Amin B, Du D, Yan W. Enhanced expression of the long non-coding RNA SNHG16 contributes to gastric cancer progression and metastasis. Cancer Biomark. 2017;21(1):151–60.
Sun TT, He J, Liang Q, Ren LL, Yan TT, Yu TC, et al. LncRNA GClnc1 promotes gastric carcinogenesis and may act as a modular scaffold of WDR5 and KAT2A complexes to specify the histone modification pattern. Cancer Discov. 2016;6:784–801.
Yang T, He W, Cui F, Xia J, Zhou R, Wu Z, et al. MACC1 mediates acetylcholine-induced invasion and migration by human gastric cancer cells. Oncotarget. 2016;7:18085–94.
Xu SH, Huang JZ, Xu ML, Yu GC, Yin XF, Chen D, et al. ACK1 promotes gastric cancer epithelial-mesenchymal transition and metastasis through AKT-POU2F1-ECD signalling. J Pathol. 2015;236:175–85.
Xu SH, Huang JZ, Chen M, Zeng M, Zou FY, Chen D, et al. Amplification of ACK1 promotes gastric tumorigenesis via ECD-dependent p53 ubiquitination degradation. Oncotarget. 2017;8:12705–16.
Cui Y, Wang Y, Li H, Li Q, Yu Y, Xu X, et al. Asparaginyl endopeptidase promotes the invasion and metastasis of gastric cancer through modulating epithelial-to-mesenchymal transition and analysis of their phosphorylation signaling pathways. Oncotarget. 2016;7:34356–70.
Ma HY, Gao LL, Li SC, Qin J, Chen L, Liu XZ, et al. CCR7 enhances TGF-beta 1-induced epithelial-mesenchymal transition and is associated with lymph node metastasis and poor overall survival in gastric cancer. Oncotarget. 2015;6:24348–60.
Mashino K, Sadanaga N, Yamaguchi H, Tanaka F, Ohta M, Shibuta K, et al. Expression of chemokine receptor CCR7 is associated with lymph node metastasis of gastric carcinoma. Cancer Res. 2002;62:2937–41.
Zhang JP, Zhou YZ, Yang YG. CCR7 pathway induces epithelial-mesenchymal transition through up-regulation of Snail signaling in gastric cancer. Med Oncol. 2015;32(2):467.
Deng J, Lei W, Xiang X, Zhang L, Lei J, Gong Y, et al. Cullin 4A (CUL4A), a direct target of miR-9 and miR-137, promotes gastric cancer proliferation and invasion by regulating the Hippo signaling pathway. Oncotarget. 2016;7:10037–50.
Qi M, Jiao M, Li X, Hu J, Wang L, Zou Y, et al. CUL4B promotes gastric cancer invasion and metastasis-involvement of upregulation of HER2. Oncogene. 2017;37(8):1075–85.
Gan L,Meng J,Xu M,Liu M,Qi Y,Tan C, et al. Extracellular matrix protein 1 promotes cell metastasis and glucose metabolism by inducing integrin beta4/FAK/SOX2/HIF-1alpha signaling pathway in gastric cancer. Oncogene. 2017;37:744–55.
Zhang YJ, Du J, Zheng JC, Liu JJ, Xu R, Shen T, et al. EGF-reduced Wnt5a transcription induces epithelial-mesenchymal transition via Arf6-ERK signaling in gastric cancer cells. Oncotarget. 2015;6:7244–61.
Han JY, Xie YC, Lan FH, Yu YH, Liu W, Chen JH, et al. Additive effects of EGF and IL-1 beta regulate tumor cell migration and invasion in gastric adenocarcinoma via activation of ERK1/2. Int J Oncol. 2014;45:291–301.
Zhu HY, Wu YL, Zheng W, Lu SL. CO-029 is overexpressed in gastric cancer and mediates the effects of EGF on gastric cancer cell proliferation and invasion. Int J Mol Med. 2015;35:798–802.
Hou FT, Yuan WJ, Huang J, Qian LY, Chen ZK, Ge J, et al. Overexpression of EphA2 correlates with epithelial-mesenchymal transition-related proteins in gastric cancer and their prognostic importance for postoperative patients. Med Oncol. 2012;29:2691–700.
Li RX, Yuan WJ, Mei WJ, Yang KD, Chen ZH. MicroRNA 520d-3p inhibits gastric cancer cell proliferation, migration, and invasion by downregulating EphA2 expression. Mol Cell Biochem. 2014;396:295–305.
Peng Y, Zhang P, Huang X, Yan Q, Wu M, Xie R, et al. Direct regulation of FOXK1 by C-jun promotes proliferation, invasion and metastasis in gastric cancer cells. Cell Death Dis. 2016;7:e2480.
Yang L, Cui M, Zhang L, Song L. FOXM1 facilitates gastric cancer cell migration and invasion by inducing Cathepsin D. Oncotarget. 2017;8:68180–90.
Yu S, Yu Y, Zhang W, Yuan W, Zhao N, Li Q, et al. FOXO3a promotes gastric cancer cell migration and invasion through the induction of cathepsin L. Oncotarget. 2016;7:34773–84.
Yang SW, Zhang ZG, Hao YX, Zhao YL, Qian F, Shi Y, et al. HIF-1 alpha induces the epithelial-mesenchymal transition in gastric cancer stem cells through the Snail pathway. Oncotarget. 2017;8:9535–45.
Liu YW, Sun M, Xia R, Zhang EB, Liu XH, Zhang ZH, et al. LincHOTAIR epigenetically silences miR34a by binding to PRC2 to promote the epithelial-to-mesenchymal transition in human gastric cancer. Cell Death Dis. 2015;6:e1802.
Xu L, Zhou R, Yuan L, Wang S, Li X, Ma H, et al. IGF1/IGF1R/STAT3 signaling-inducible IFITM2 promotes gastric cancer growth and metastasis. Cancer Lett. 2017;393:76–85.
Wu X, Tao P, Zhou Q, Li J, Yu Z, Wang X, et al. IL-6 secreted by cancer-associated fibroblasts promotes epithelial-mesenchymal transition and metastasis of gastric cancer via JAK2/STAT3 signaling pathway. Oncotarget. 2017;8:20741–50.
Jiang YX, Yang SW, Li PA, Luo X, Li ZY, Hao YX, et al. The promotion of the transformation of quiescent gastric cancer stem cells by IL-17 and the underlying mechanisms. Oncogene. 2017;36:1256–64.
Han FJ, Ren JC, Zhang JJ, Sun YD, Ma F, Liu ZF, et al. JMJD2B is required for Helicobacter pylori-induced gastric carcinogenesis via regulating COX-2 expression. Oncotarget. 2016;7:38626–37.
Li WJ, Zhao L, Zang W, Liu ZF, Chen L, Liu TT, et al. Histone demethylase JMJD2B is required for tumor cell proliferation and survival and is overexpressed in gastric cancer. Biochem Biophys Res Commun. 2011;416:372–8.
Mariotti S, Barravecchia I, Vindigni C, Pucci A, Balsamo M, Libro R, et al. MICAL2 is a novel human cancer gene controlling mesenchymal to epithelial transition involved in cancer growth and invasion. Oncotarget. 2016;7:1808–25.
Xia J, Wang H, Huang H, Sun L, Dong S, Huang N, et al. Elevated Orai1 and STIM1 expressions upregulate MACC1 expression to promote tumor cell proliferation, metabolism, migration, and invasion in human gastric cancer. Cancer Lett. 2016;381:31–40.
Dong W, Qin G, Shen R. Rab11-FIP2 promotes the metastasis of gastric cancer cells. Int J Cancer. 2016;138:1680–8.
Liang X, Zeng J, Wang L, Shen L, Ma X, Li S, et al. Histone demethylase RBP2 promotes malignant progression of gastric cancer through TGF-beta1-(p-Smad3)-RBP2-E-cadherin-Smad3 feedback circuit. Oncotarget. 2015;6:17661–74.
Cho HJ, Baek KE, Park SM, Kim IK, Choi YL, Cho HJ, et al. RhoGDI2 expression is associated with tumor growth and malignant progression of gastric cancer. Clin Cancer Res. 2009;15:2612–9.
Cho HJ, Kim IK, Park SM, Baek KE, Nam IK, Park SH, et al. VEGF-C mediates RhoGDI2-induced gastric cancer cell metastasis and cisplatin resistance. Int J Cancer. 2014;135:1553–63.
Zhang L, Xu Z, Xu X, Zhang B, Wu H, Wang M, et al. SALL4, a novel marker for human gastric carcinogenesis and metastasis. Oncogene. 2014;33:5491–500.
Ren YH, Liu KJ, Wang M, Yu YN, Yang K, Chen Q, et al. De-SUMOylation of FOXC2 by SENP3 promotes the epithelial-mesenchymal transition in gastric cancer cells. Oncotarget. 2014;5:7093–104.
Yoo YA, Kang MH, Lee HJ, Kim BH, Park JK, Kim HK, et al. Sonic hedgehog pathway promotes metastasis and lymphangiogenesis via activation of Akt, EMT, and MMP-9 pathway in gastric cancer. Cancer Res. 2011;71:7061–70.
Kim HP, Han SW, Song SH, Jeong EG, Lee MY, Hwang D, et al. Testican-1-mediated epithelial-mesenchymal transition signaling confers acquired resistance to lapatinib in HER2-positive gastric cancer. Oncogene. 2014;33:3334–41.
Yan L, Sun K, Liu Y, Liang J, Cai KR, Gui JQ. MiR-129-5p influences the progression of gastric cancer cells through interacting with SPOCK1. Tumor Biol. 2017;39.1010428317706916.
Qiao JL, Liu ZJ, Yang C, Gu LK, Deng DJ. SRF promotes gastric cancer metastasis through stromal fibroblasts in an SDF1-CXCR4-dependent manner. Oncotarget. 2016;7:46088–99.
Zhou Q, Wang X, Yu Z, Wu X, Chen X, Li J, et al. Transducin (beta)-like 1 X-linked receptor 1 promotes gastric cancer progression via the ERK1/2 pathway. Oncogene. 2017;36:1873–86.
Jung H, Lee KP, Park SJ, Park JH, Jang YS, Choi SY, et al. TMPRSS4 promotes invasion, migration and metastasis of human tumor cells by facilitating an epithelial-mesenchymal transition. Oncogene. 2008;27:2635–47.
Li J, Deng ZJ, Wang Z, Wang D, Zhang LJ, Su Q, et al. Zipper-interacting protein kinase promotes epithelial-mesenchymal transition, invasion and metastasis through AKT and NF-kB signaling and is associated with metastasis and poor prognosis in gastric cancer patients. Oncotarget. 2015;6:8323–38.
Duan J, Zhang H, Qu Y, Deng T, Huang D, Liu R, et al. Onco-miR-130 promotes cell proliferation and migration by targeting TGFbetaR2 in gastric cancer. Oncotarget. 2016;7:44522–33.
Mi Y, Zhang D, Jiang W, Weng J, Zhou C, Huang K, et al. miR-181a-5p promotes the progression of gastric cancer via RASSF6-mediated MAPK signalling activation. Cancer Lett. 2017;389:11–22.
Zhou Q, Zheng X, Chen L, Xu B, Yang X, Jiang J, et al. Smad2/3/4 pathway contributes to TGF-beta-induced MiRNA-181b expression to promote gastric cancer metastasis by targeting Timp3. Cell Physiol Biochem. 2016;39:453–66.
Hsu KW, Wang AM, Ping YH, Huang KH, Huang TT, Lee HC, et al. Downregulation of tumor suppressor MBP-1 by microRNA-363 in gastric carcinogenesis. Carcinogenesis. 2014;35:208–17.
Ge X, Liu X, Lin F, Li P, Liu K, Geng R, et al. MicroRNA-421 regulated by HIF-1alpha promotes metastasis, inhibits apoptosis, and induces cisplatin resistance by targeting E-cadherin and caspase-3 in gastric cancer. Oncotarget. 2016;7:24466–82.
Liu X, Ge X, Zhang Z, Zhang X, Chang J, Wu Z, et al. MicroRNA-940 promotes tumor cell invasion and metastasis by downregulating ZNF24 in gastric cancer. Oncotarget. 2015;6:25418–28.
Yan HB, Wang XF, Zhang Q, Tang ZQ, Jiang YH, Fan HZ, et al. Reduced expression of the chromatin remodeling gene ARID1A enhances gastric cancer cell migration and invasion via downregulation of E-cadherin transcription. Carcinogenesis. 2014;35:867–76.
Li H, Xu L, Li C, Zhao L, Ma Y, Zheng H, et al. Ubiquitin ligase Cbl-b represses IGF-I-induced epithelial mesenchymal transition via ZEB2 and microRNA-200c regulation in gastric cancer cells. Mol Cancer. 2014;13:136.
Yuan W, Li T, Mo X, Wang X, Liu B, Wang W, et al. Knockdown of CMTM3 promotes metastasis of gastric cancer via the STAT3/Twist1/EMT signaling pathway. Oncotarget. 2016;7:29507–19.
Ye J, Xu J, Li Y, Huang Q, Huang J, Wang J, et al. DDAH1 mediates gastric cancer cell invasion and metastasis via Wnt/beta-catenin signaling pathway. Mol Oncol. 2017;11:1208–24.
Wu WD, Ding HH, Cao J, Zhang WH. FBXL5 inhibits metastasis of gastric cancer through suppressing Snail1. Cell Physiol Biochem. 2015;35:1764–72.
Liu JK, Shi H, Li XD, Chen G, Larsson C, Lui WO. miR-223-3p regulates cell growth and apoptosis via FBXW7 suggesting an oncogenic role in human testicular germ cell tumors. Int J Oncol. 2017;50:356–64.
Yokobori T, Mimori K, Iwatsuki M, Ishii H, Onoyama I, Fukagawa T, et al. p53-altered FBXW7 expression determines poor prognosis in gastric cancer cases. Cancer Res. 2009;69:3788–94.
Calcagno DQ, Freitas VM, Leal MF, De Souza CRT, Demachki S, Montenegro R, et al. MYC, FBXW7 and TP53 copy number variation and expression in gastric cancer. BMC Gastroenterol. 2013;13:141.
Eto K, Iwatsuki M, Watanabe M, Ishimoto T, Ida S, Imamura Y, et al. The sensitivity of gastric cancer to trastuzumab is regulated by the miR-223/FBXW7 pathway. Int J Cancer. 2015;136:1537–45.
Gong JH, Cui Z, Li L, Ma Q, Wang QF, Gao YH, et al. MicroRNA-25 promotes gastric cancer proliferation, invasion, and migration by directly targeting F-box and WD-40 Domain Protein 7, FBXW7. Tumor Biol. 2015;36:7831–40.
Li MR, Zhu CC, Ling TL, Zhang YQ, Xu J, Zhao EH, et al. FBXW7 expression is associated with prognosis and chemotherapeutic outcome in Chinese patients with gastric adenocarcinoma. BMC Gastroenterol. 2017;17(1):60.
He CY, Wang ZQ, Zhang L, Yang LY, Li JF, Chen XH, et al. A hydrophobic residue in the TALE homeodomain of PBX1 promotes epithelial-to-mesenchymal transition of gastric carcinoma. Oncotarget. 2017;8:46818–33.
Sha SM, Gu Y, Xu B, Hu H, Yang Y, Kong XY, et al. Decreased expression of HOXB9 is related to poor overall survival in patients with gastric carcinoma. Dig Liver Dis. 2013;45:422–9.
Hou JJ, Wang T, Xie QQ, Deng WX, Yang JY, Zhang SQ, et al. N-Myc-interacting protein (NMI) negatively regulates epithelial-mesenchymal transition by inhibiting the acetylation of NF-kappa B/p65. Cancer Lett. 2016;376:22–33.
Ling ZQ, Guo W, Lu XX, Zhu X, Hong LL, Wang Z, et al. A Golgi-specific protein PAQR3 is closely associated with the progression, metastasis and prognosis of human gastric cancers. Ann Oncol. 2014;25:1363–72.
Qiao SS, Guo WW, Liao LJ, Wang L, Wang Z, Zhang R, et al. DDB2 is involved in ubiquitination and degradation of PAQR3 and regulates tumorigenesis of gastric cancer cells. Biochem J. 2015;469:469–80.
Du C, Zhang CY, Hassan S, Biswas MHU, Balaji KC. Protein kinase D1 suppresses epithelial-to-mesenchymal transition through phosphorylation of Snail. Cancer Res. 2010;70:7810–9.
Cho SJ, Kook MC, Lee JH, Shin JY, Park J, Bae YK, et al. Peroxisome proliferator-activated receptor gamma upregulates galectin-9 and predicts prognosis in intestinal-type gastric cancer. Int J Cancer. 2015;136:810–20.
Yang Y, Zhang J, Yan Y, Cai H, Li M, Sun K, et al. Low expression of Rap1GAP is associated with epithelial-mesenchymal transition (EMT) and poor prognosis in gastric cancer. Oncotarget. 2017;8:8057–68.
Noh HS, Hah YS, Ha JH, Kang MY, Zada S, Rha SY, et al. Regulation of the epithelial to mesenchymal transition and metastasis by Raf kinase inhibitory protein-dependent Notch1 activity. Oncotarget. 2016;7:4632–46.
Martinho O, Simoes K, Longatto A, Jacob CE, Zilberstein B, Bresciani C, et al. Absence of RKIP expression is an independent prognostic biomarker for gastric cancer patients. Oncol Rep. 2013;29:690–6.
Jia BQ, Liu HY, Kong QL, Li B. RKIP expression associated with gastric cancer cell invasion and metastasis. Tumor Biol. 2012;33:919–25.
Wang HQ, Zhou R, Sun L, Xia JL, Yang XC, Pan CQ, et al. TOP1MT deficiency promotes GC invasion and migration via the enhancements of LDHA expression and aerobic glycolysis. Endocr-Relat Cancer. 2017;24:565–78.
He B, Li W, Wu Y, Wei F, Gong Z, Bo H, et al. Epstein-Barr virus-encoded miR-BART6-3p inhibits cancer cell metastasis and invasion by targeting long non-coding RNA LOC553103. Cell Death Dis. 2016;7:e2353.
Zhao X, Dou W, He L, Liang S, Tie J, Liu C, et al. MicroRNA-7 functions as an anti-metastatic microRNA in gastric cancer by targeting insulin-like growth factor-1 receptor. Oncogene. 2013;32:1363–72.
Huang TT, Ping YH, Wang AM, Ke CC, Fang WL, Huang KH, et al. The reciprocal regulation loop of Notch2 pathway and miR-23b in controlling gastric carcinogenesis. Oncotarget. 2015;6:18012–26.
Yu WW, Jiang H, Zhang CT, Peng Y. The SNAIL/miR-128 axis regulated growth, invasion, metastasis, and epithelial-to-mesenchymal transition of gastric cancer. Oncotarget. 2017;8:39280–95.
Lei C, Du F, Sun L, Li T, Li T, Min Y, et al. miR-143 and miR-145 inhibit gastric cancer cell migration and metastasis by suppressing MYO6. Cell Death Dis. 2017;8:e3101.
Kurashige J, Mima K, Sawada G, Takahashi Y, Eguchi H, Sugimachi K, et al. Epigenetic modulation and repression of miR-200b by cancer-associated fibroblasts contribute to cancer invasion and peritoneal dissemination in gastric cancer. Carcinogenesis. 2015;36:133–41.
Tao YM, Yang SB, Wu YY, Fang XD, Wang YN, Song Y, et al. MicroRNA-216a inhibits the metastasis of gastric cancer cells by targeting JAK2/STAT3-mediated EMT process. Oncotarget. 2017;8:88870–81.
Li P, Chen XH, Su LP, Li CL, Zhi QM, Yu BQ, et al. Epigenetic silencing of miR-338-3p contributes to tumorigenicity in gastric cancer by targeting SSX2IP. PLoS ONE. 2013;8(6):e66782.
Chen JT, Yao KH, Hua L, Zhang LP, Wang CY, Zhang JJ. miR-338-3p inhibits the proliferation and migration of gastric cancer cells by targeting ADAM17. Int J Clin Exp Pathol. 2015;8:10922–8.
Guo B, Liu LY, Yao JY, Ma RL, Chang DM, Li ZF, et al. miR-338-3p suppresses gastric cancer progression through a PTEN-AKT axis by targeting P-REX2a. Mol Cancer Res. 2014;12:313–21.
Song G, Zhang H, Chen C, Gong L, Chen B, Zhao S, et al. miR-551b regulates epithelial-mesenchymal transition and metastasis of gastric cancer by inhibiting ERBB4 expression. Oncotarget. 2017;8:45725–35.
Zheng HC, Takahashi H, Murai Y, Cui ZG, Nomoto K, Niwa H, et al. Expressions of MMP-2, MMP-9 and VEGF are closely linked to growth, invasion, metastasis and angiogenesis of gastric carcinoma. Anticancer Res. 2006;26:3579–83.
Chen YS, Huang YF, Huang YL, Xia XW, Zhang JB, Zhou Y, et al. JWA suppresses tumor angiogenesis via Sp1-activated matrix metalloproteinase-2 and its prognostic significance in human gastric cancer. Carcinogenesis. 2014;35:442–51.
Zhao ZS, Wang YY, Chu YQ, Ye ZY, Tao HQ. SPARC is associated with gastric cancer progression and poor survival of patients. Clin Cancer Res. 2010;16:260–8.
Ren G, Tian QF, An YX, Feng B, Lu YY, Liang J, et al. Coronin 3 promotes gastric cancer metastasis via the up-regulation of MMP-9 and cathepsin K. Mol Cancer. 2012;11:67.
Boulay A, Masson R, Chenard MP, El Fahime M, Cassard L, Bellocq JP, et al. High cancer cell death in syngeneic tumors developed in host mice deficient for the stromelysin-3 matrix metalloproteinase. Cancer Res. 2001;61:2189–93.
Kou YB, Zhang SY, Zhao BL, Ding R, Liu H, Li S. Knockdown of Mmp11 inhibits proliferation and invasion of gastric cancer cells. Int J Immunopathol Pharmacol. 2013;26:361–70.
Lowy AM, Clements WM, Bishop J, Kong L, Bonney T, Sisco K, et al. beta-catenin/Wnt signaling regulates expression of the membrane type 3 matrix metalloproteinase in gastric cancer. Cancer Res. 2006;66:4734–41.
Funding
This project was supported by research funds from RGC/GRF Hong Kong (14114615, 14101917, 14111216, 14106145); National Natural Science Foundation of China (NSFC) (81772501, 81502064); HMRF Hong Kong (03140856); Vice-Chancellor’s Discretionary Fund CUHK; Shenzhen Virtual University Park Support Scheme to CUHK Shenzhen Research Institute; CUHK direct grant.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, W., Ng, J.MK., Wong, C.C. et al. Molecular alterations of cancer cell and tumour microenvironment in metastatic gastric cancer. Oncogene 37, 4903–4920 (2018). https://doi.org/10.1038/s41388-018-0341-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-018-0341-x
- Springer Nature Limited
This article is cited by
-
CircGLIS3 promotes gastric cancer progression by regulating the miR-1343-3p/PGK1 pathway and inhibiting vimentin phosphorylation
Journal of Translational Medicine (2024)
-
HIF1A protein expression is correlated with clinical features in gastric cancer: an updated systematic review and meta-analysis
Scientific Reports (2024)
-
Microbiota and metabolites alterations in proximal and distal gastric cancer patients
Journal of Translational Medicine (2022)
-
Sec62 promotes gastric cancer metastasis through mediating UPR-induced autophagy activation
Cellular and Molecular Life Sciences (2022)
-
Inhibition of LPS-mediated TLR4 activation abrogates gastric adenocarcinoma-associated peritoneal metastasis
Clinical & Experimental Metastasis (2022)