
Abstract
Positron emission tomography (PET) enables non-invasive estimation of neurotransmitter fluctuations in the living human brain. While these methods have been applied to dopamine and some other transmitters, estimation of 5-hydroxytryptamine (5-HT; Serotonin) release has proved to be challenging. Here we demonstrate the utility of the novel 5-HT2A receptor agonist radioligand, [11C]CIMBI-36, and a d-amphetamine challenge to evaluate synaptic 5-HT changes in the living human brain. Seventeen healthy male volunteers received [11C]CIMBI-36 PET scans before and 3 h after an oral dose of d-amphetamine (0.5 mg/kg). Dynamic PET data were acquired over 90 min, and the total volume of distribution (VT) in the frontal cortex and the cerebellum derived from a kinetic analysis using MA1. The frontal cortex binding potential (BPNDfrontal) was calculated as (VTfrontal/VTcerebellum) − 1. ∆BPNDfrontal = 1 − (BPNDfrontal post-dose/BPNDfrontal baseline) was used as an index of 5-HT release. Statistical inference was tested by means of a paired Students t-test evaluating a reduction in post-amphetamine [11C]CIMBI-36 BPNDfrontal. Following d-amphetamine administration, [11C]CIMBI-36 BPNDfrontal was reduced by 14 ± 13% (p = 0.002). Similar effects were observed in other cortical regions examined in an exploratory analysis. [11C]CIMBI-36 binding is sensitive to synaptic serotonin release in the human brain, and when combined with a d-amphetamine challenge, the evaluation of the human brain serotonin system in neuropsychiatric disorders, such as major depression and Parkinson’s disease is enabled.
Similar content being viewed by others

Introduction
Positron emission tomography (PET) and single photon emission computed tomography (SPECT) enable the non-invasive measurement of neurotransmitter fluctuations in the living human brain. The measurement of extracellular dopamine release using PET/SPECT in combination with dopamine releasing pharmacological challenges has been well established over the past 20 years [1], but similar assessment of synaptic serotonin has proven to be difficult. Attempts to identify and characterize experimental paradigms and serotoninergic radioligands sufficiently sensitive to detect changes in endogenous serotonin levels following an acute pharmacological challenge, have had limited success [2]. Studies using serotonin 2A receptor antagonist radioligands have generally failed to demonstrate adequate sensitivity to challenges with fenfluramine in rodents [3] or non-human primates [4], or SSRIs or ketamine in humans [5,6,7]. Of the 19 human studies using radioligands targeting the serotonin 1A, 1B, 2A, and 4 receptors, and the serotonin transporter (SERT), three studies have detected a change in signal in the expected direction following an acute pharmacological challenge (for review, see [8, 9]). These three positive studies all have limitations. Specifically, the decrease in the cortical binding of the serotonin 2A receptor ligand, [18F]altanserin, observed in two of the studies [10, 11] are difficult to interpret, because the pharmacological challenges used, clomipramine and nor-dexfenfluramine (the major metabolite of dexfenfluramine used as a 5-HT releasing agent), have significant affinity for the 5-HT2A receptor, and direct occupancy of the target by these agents may have contributed to the observed reductions in [18F]altanserin binding [2]. The third study found displacement of the serotonin 1A receptor antagonist ligand, [18F]MPPF, after fluoxetine challenge in raphe—but not in any other investigated brain regions [12]. In contrast to these three positive studies, a paradoxical effect was seen in two studies with serotonin 1A [13] and 1B [14] receptor radioligands, where the binding of the ligands increased rather than decreased after a pharmacological challenge with a selective serotonin reuptake inhibiting compound.
The reasons for the limited success in the development of a robust method to measure acute serotonin release in humans may be related to the magnitude of the 5-HT release (a function of the pharmacological paradigm used), the affinity of 5-HT for specific molecular targets tested, the magnitude of the displaceable signal for the radioligands tested, or a combination of the factors above. Selective serotonin reuptake inhibitors (SSRIs)—the pharmacological challenge used in the vast majority of such studies—may not produce sufficiently large 5-HT release in the serotonergic projection areas following a single dose administration [15]. Using a combination of microdialysis and a novel serotonin 2A receptor agonist radioligand, [11C]CIMBI-36, in pig, Joergensen and colleagues recently showed that the SSRI, citalopram (2 mg/kg), was five times less potent in its ability to increase extracellular serotonin levels compared with the serotonin-releasing agent, fenfluramine (0.5 mg/kg) [16]. Until the introduction of [11C]CIMBI-36, the radioligands examined have been antagonists, the binding of which may be less sensitive to fluctuations of an endogenous agonist such as serotonin, than that of agonist radioligands. While an agonist has minimal affinity for the low-affinity state of a G-protein couple receptor (GPCR) an antagonist will bind with equal facility to both high and low-affinity states of the receptor. Thus a significant proportion of the total binding of an antagonist radioligand will be insensitive to the fluctuations of an endogenous agonist [2].
Recently, [11C]CIMBI-36 was found to be sensitive to serotonin-releasing challenges in both pigs [16] and rhesus monkeys [17]. A pharmacologically induced 8-fold increase in interstitial 5-HT led to 46% decrease in [11C]CIMBI-36 binding. When compared with similar data on dopamine release examined with with microdialysis and PET, these results indicated that [11C]CIMBI-36 was three times more sensitive to changes in serotonin than [11C]raclopride (widely used in measuring brain dopamine fluctuations), is to changes in extracellular dopamine [16]. In line with the proposition mentioned above, however, a recent study in humans showed that the combination of citalopram and pindolol was insufficient to elicit acute unidirectional changes in serotonin levels that could be detected with [11C]CIMBI-36 PET in neocortex, although a decline was seen in hippocampus [18]. To date, the effects of a more potent serotonin-releasing agent on cerebral [11C]CIMBI-36 binding has not been tested in humans.
D-amphetamine is a safe and widely used pharmacological challenge agent for human studies. Although typically viewed as a releaser of dopamine and noradrenaline, it has also been shown to increase extracellular serotonin levels in pre-clinical experiments [19,20,21,22]. Based on good serotonin-releasing potency, combined with a favourable selectivity profiles of d-amphetamine (negligible binding to serotonin 2A receptors [23]) and [11C]CIMBI-36 (>1000-fold selectivity for the serotonin 2A receptor over dopaminergic and noradrenergic targets [24]), we evaluated the utility of this combination as a robust method to detect endogenous 5-HT release in the living human brain.
Methods
The study was sponsored by Imanova Ltd (now Invicro), London, UK, and approved by local Ethics Committees (reference numbers: 14/EE/0103 and 12/LO/1117) and the Administration of Radioactive Substances Advisory Committee (ARSAC, reference numbers: RPC 630/3764/-31344 & -28855), and all participants provided informed, written consent. PET and Magnetic resonance (MR) scans of the brain were carried out at the Invicro Imaging Centre, Hammersmith Hospital, London, UK.
Participants
Seventeen healthy male volunteers were included in this study. They were recruited by advertisements and word of mouth, and received general medical and psychiatric screening. Historical or current neurologic or psychiatric diseases, including substance abuse or dependence, or significant traumatic brain injury (>2 min loss of consciousness and/or requiring hospital admission) were exclusion criteria, and none of the volunteers had history of present or past use of psychoactive medications including SSRI’s.
Subjective measures
State anxiety levels were assessed using Spielberger’s State Anxiety Inventory (SSAI, scores ranging from 20 to 80) prior to d-amphetamine administration (baseline) as well as at the beginning and end of the post-challenge PET scan (SSAI was only administered to 11 of the 17 participants). D-amphetamine-induced anxiety was calculated as the difference between the baseline and an average of the two post-challenge SSAI measures, capturing state anxiety during the second PET scan. Self-rating of d-amphetamine-induced “drug effect” was also acquired immediately before and after the second PET scan. An average of ratings just before and after the post-d-amphetamine PET scan was used. For 11 subjects ratings were acquired using a visual analogue scale (VAS) of “I feel a drug effect” ranging from “not at all” to “extremely”, for the remaining 6 subjects a VAS score was estimated from an average of a Likert rating of least (=1) to most (=5) high, measured before and after the post-d-amphetamine PET scan.
Plasma d-amphetamine measures
Blood samples for d-amphetamine levels were acquired immediately before and after the second PET scan, i.e. at 3 and 4.5 h post-d-amphetamine administration. An average of the 3 and 4.5 h plasma level concentrations (i.e. mid-scan) was used to examine the relationship between d-amphetamine dose and PET outcome parameters.
PET acquisition
An intravenous cannula was inserted into a cubital or forearm vein for radioligand administration, and a second cannula was inserted into the radial artery prior to the scan to enable the collection of arterial blood samples. All volunteers received [11C]CIMBI-36 PET scans before and 3 h after a single oral dose of d-amphetamine (0.5 mg/kg) (see Fig. 1 for study design). A low-dose computed tomography (CT) scan (30 mAs, 130 KeV, 0.55 pitch) was performed immediately before each PET scan in order to estimate attenuation. The radioligand was administered as a bolus (over 20 s) in a volume of 20 mL at the start of the PET scans, all acquired on a Biograph 6 TruePoint PET/CT scanner (Siemens Healthcare, Erlangen, Germany). Dynamic emission PET data were acquired over 90 min and were reconstructed into 26 frames (frame durations: 8 × 15, 3 × 60, 5 × 120, 5 × 300, 5 × 600 s) using discrete inverse Fourier transform (DIFT) reconstruction. Corrections were applied for attenuation, randoms and scatter, and subject motion.

Chart showing main study procedures carried out on the imaging visit (visit 2).
Arterial blood acquisition
Whole blood activity was measured using a continuous automatic blood sampling system (Allogg AB, Marlefred, Sweden) acquired at a rate of 5 mL/min. Discrete blood samples were taken at 2, 5, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80 and 90 min after scan start and total radioactivity concentration was evaluated in both blood and plasma in a Perkin Elmer 1470 10-well gamma counter. Discrete blood samples were used to determine the fraction of plasma radioactivity constituted by unchanged parent radioligand (Ppf) using high-performance liquid chromatography (HPLC) analysis. For each ligand, the plasma free fraction fp was measured by ultrafiltration in triplicate using an arterial blood sample taken prior to tracer injection. Individual measurements of fp are given in Table 1.
MR acquisition
Structural magnetic resonance imaging (MRI) data were acquired on the same day as the PET scans, using 3T (Magnetom Trio and Verio, Siemens Healthcare Sector, Erlangen, Germany) with a 32-receiver channel head matrix coil, in the sagittal plane, utilising a 3D magnetization prepared rapid gradient echo (MP-RAGE) scan with the following parameters: repetition time = 2300 milliseconds, echo delay time = 2.98 ms, flip angle = 9°, isotropic voxels = 1.0 × 1.0 × 1.0 mm, 160 slices, total scanning time = 5 min, 3 s.
Image analysis
All image data were analysed using Invicro London’s in-house PET data quantification tool, MIAKATTM (version 4.3.7, http://www.miakat.org). MIAKATTM is implemented using MATLAB (version R2016a; Mathworks Inc., Natick, MA, USA), and makes use of SPM12 (Wellcome Trust Centre for Neuroimaging, http://www.fil.ion.ucl.ac.uk/spm) functions for image segmentation and registration.
Frame-by-frame motion correction was applied to the dynamic PET data, and the CIC neuroanatomical atlas [25] (grey matter masked using the subject’s own MRI) was applied to the PET data by non-linear deformation parameters derived from the transformation of the structural MRI into standard space. The region-of-interest (ROI) time-activity data were sampled from the frontal cortex, a large region with high 5-HT2A receptor density that was determined a priori as the main ROI. Exploratory analysis was conducted for the parietal, temporal and occipital cortices. The whole cerebellum was used as a reference region to estimate the [11C]CIMBI-36 non-displaceable binding [26].
Regional total volumes of distribution (VT) were derived from kinetic analysis using the multilinear analysis-1 (MA1) method [27]. Regional binding potential (BPNDROI) was calculated as (VTROI/VTcerebellum) − 1. Serotonin release was quantified as ΔBPND = 1 – (BPNDROI post-dose/BPNDROI baseline).
Statistical inference was tested by means of a paired Students t-test evaluating a reduction in post-d-amphetamine [11C]CIMBI-36 BPND. The relationship between serotonin release (ΔBPND) and subjective drug effects and anxiety was tested using linear regression.
Results
Study conduct
Seventeen male volunteers were included in the analysis (see Table 1 for demographic information). There was no difference in average CIMBI-36 mass injected between the baseline and post-d-amphetamine conditions (1.33 ± 0.14 μg versus 1.31 ± 0.17 μg, p = 0.383).
[11C]CIMBI-36 PET data
Robust decreases in [11C]CIMBI-36 VT were observed post-d-amphetamine administration across all cortical regions (13–15%, p-values < 0.01). We also observed a post-d-amphetamine reduction in the cerebellar VT (7%, p = 0.004).
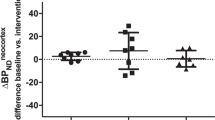
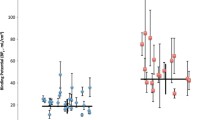
At baseline, the regional [11C]CIMBI-36 BPND distribution was consistent with previous reports [28, 29], with high binding across cortical areas (highest in temporal cortex mean BPND: 1.07 ± 0.3). Following d-amphetamine administration, BPND was significantly reduced in the frontal cortex (14 ± 13%, p = 0.002) (see Table 2 and Fig. 2), as well as across all neocortical regions examined (ranging from 9 to 14%). There was a significant relationship between the baseline BPND and the magnitude of ΔBPND (Pearson’s r = 0.67 for the frontal cortex, Fig. 3).

Individual (and average) [11C]CIMBI-36 non-displaceable binding potentials (BPND), before (PET1) and 3 h after (PET2) an oral d-amphetamine challenge.

Relationship between frontal cortex BPND and ∆BPND post-amphetamine administration.
D-amphetamine effects on PET and subjective measures
Consistent with previous imaging studies conducted by our team with d-amphetamine as the pharmacological challenge [30, 31], an oral dose of 0.5 mg/kg of d-amphetamine was well tolerated, and no significant adverse effects were reported; average plasma concentration mid-scan was 63 ± 20 µg/L, consistent with previous studies using oral d-amphetamine challenge [32, 33]. Average reported post-d-amphetamine VAS “drug effect” during the second PET scan was 33 ± 18% (range: 0–65%), and change in SSAI anxiety rating between baseline and post-d-amphetamine was on average + 1 ± 7 (range: −7 to +18, one-sample t-test: p = 0.628). No significant associations were identified between ΔBPND and plasma d-amphetamine concentration, or between plasma d-amphetamine concentration and ratings of anxiety or subjective drug effects.
Discussion
We provide the first demonstration of a practical and robust method to evaluate serotonin release in the human cortex. [11C]CIMBI-36 PET combined with an oral d-amphetamine challenge, provides a useful method to investigate acute serotonin release in the living human brain, similar to the methods used to investigate dopamine release [1, 33]. Reductions of the order of ~14% in [11C]CIMBI-36 BPND in the frontal cortex of healthy volunteers are similar in magnitude and variability to the signal change seen in the striatum with the d-amphetamine/[11C]raclopride combination. A signal change of this magnitude allows the investigation of both increased and decreased 5-HT release in patient populations, as was done for the dopamine system (for review see [34, 35]).
A robust change in [11C]CIMBI-36 binding, in the hypothesized direction, following a serotonin-releasing challenge, seen in our data, is consistent with combined PET/microdialysis study in the pig brain, where a 11-fold increase in extracellular 5-HT following the administration of d-fenfluramine, resulted in a 44% reduction in [11C]CIMBI-36 BPND [16]. D-amphetamine is a potent serotonin releaser [19, 20, 22, 36] although usually seen as an agent for dopamine and noradrenaline release. Intra-peritoneal administration of 4 mg/kg d-amphetamine led to increases in extracellular serotonin similar to those following 3 mg/kg fenfluramine, and exceeding those seen following 10 mg/kg administration of the SSRI fluoxetine [19]. Furthermore, pre-treatment with d-amphetamine was recently shown to reduce binding of the 5-HT1b antagonist, [11C]AZ10419369, to the same degree as the potent 5-HT releaser [37], MDMA, when administered in same dose (1 mg/kg) in non-human primates [36]. In contrast to fenfluramine which was withdrawn from the market two decades ago [38], d-amphetamine is readily available for human research and is considered to be safe as an acute pharmacological challenge. In multiple studies conducted in our centre, d-amphetamine demonstrated very good tolerability and low level of adverse events [30, 31, 33]. Importantly, d-amphetamine has negligible affinity for the serotonin 2A receptor (Ki > 10,000 nM in [23], whereas [11C]CIMBI-36 has >1000-fold selectivity for the serotonin 2A receptor over dopaminergic and noradrenergic targets [24]. Therefore, competition between (1) d-amphetamine and [11C]CIMBI-36 on the 2A receptor, or (2) between non-serotonin mono-amines and [11C]CIMBI-36 at dopaminergic or noradrenergic targets, cannot explain the decrease in [11C]CIMBI-36 BPND after a d-amphetamine challenge. Overall, available data strongly supports the conclusion that the observed d-amphetamine-induced reduction of [11C]CIMBI-36 BPND can be interpreted as a consequence of increased extracellular serotonin. Of note, past studies evaluating other 5-HT PET ligands found some relationship between sleep/wake cycles and 5-HT receptor density [39, 40]. Our subjects may have had some changes in arousal following amphetamine administration, potentially introducing a small additional contribution the signal change we observed.
We saw no correlation between plasma levels of d-amphetamine and changes in [11C]CIMBI-36 BPND, or between [11C]CIMBI-36 BPND changes and either anxiety scores or subjective “drug effects”. In contrast, the magnitude of subjective/euphoric effects of amphetamine has previously been observed to correlate with both endogenous dopamine and opioid release as assessed with [11C]raclopride-PET [41,42,43] and [11C]carfentanil-PET [30], respectively. Importantly, these studies not only focused on release of two transmitters known to be directly involved in reward processing (for reviews, please see [44] and [45] for opioid and dopamine systems, respectively), but also reported specifically from the ventral striatum, a key region for rewarding effects of substances [46]. Due to the relatively low density of serotonin 2A receptors in most subcortical brains regions, including the striatum (BPND < 0.3 [26]), [11C]CIMBI-36 does not provide sufficient signal to examine serotonin release in this region.
We observed a small but significant reduction in the cerebellar [11C]CIMBI-36 VT post-d-amphetamine (7%). As the cerebellum serves as the reference region used in the calculation of the BPND, changes in the cerebellar VT should be assessed carefully to estimate their impact on the study findings. In our case, the magnitude of the reduction is small, and cannot explain a reduction in cortical BPND, as it would lead to an increase rather than a decrease in the cortical BPND, moderating any reductions due to serotonin release.
We have chosen the MA1 model to quantify our data, rather than the more standard two tissue compartment model (2TCM) as MA1 does not make a priori assumptions about the number of compartments in the model, an advantage for a ligand such as [11C]CIMBI-36, where for some ROI, a one tissue compartment model (1TCM) appears to be preferable to 2TCM in some subjects. The 2TCM produced very similar results (a reduction in cortical BPND of 9–14%, see Fig. 4 and Table 2). An analysis not requiring the use of an arterial plasma input function (such as a simplified reference tissue model—SRTM) would provide significant advantages with regard to study logistics, costs and subject comfort. We have also evaluated our data using SRTM with a cerebellar input function, but while the SRTM based results provide generally similar conclusions to the 2TCM and MA1 data, there was only moderate correspondence between SRTM and 2TCM/MA1 results (see Fig. 4 and Table 2). The relationship between SRTM and 2TCM quantification, seen in our study is consistent with that reported previously [26]. We have not explored extensively the reasons for the less than optimal correspondence between 2TCM/MA1 and SRTM data, but assumption of one tissue compartment kinetics and the lack of correction for blood volume contribution inherent in the SRTM model may be relevant factors here. The latter factor may be particularly relevant in a study using a highly vasoactive challenge such as d-amphetamine challenge, that may result in changes in vascular parameters that would be particularly notable in a region with very low binding, such as the cerebellum.

Comparison of regional BPND values derived by 2TCM method to those derived by MA1 and SRTM.
We observed a significant relationship between the baseline [11C]CIMBI-36 BPND and the magnitude of d-amphetamine induced change. We have no robust explanation, but may speculate about the potential mechanisms underlying such an effect. If significant differences in synaptic 5-HT concentration are present across individuals, and the absolute magnitude of 5-HT released by a given dose of d-amphetamine were similar across individuals (rather than being a function of the baseline 5-HT concentration)—then a relationship between baseline [11C]CIMBI-36 BPND and the magnitude of d-amphetamine induced change, similar to the one we observed, would be expected.
Similarly to the unsuccessful previous attempts to develop a neuroimaging method to assess acute serotonin release using 5-HT2A radioligands [8, 9], studies using 5-HT1A selective radioligands such as [11C]WAY100-635 and [11C]CUMI-101 have also produced mixed results. A paradoxical effect (increases in radioligand binding following the administration of the selective serotonin reuptake inhibitor citalopram) has been reported in two other studies using serotonin 5-HT1A [13] and 5-HT1B [14] antagonist radioligands. The authors explained their results by hypothesizing a reduction in 5-HT release in the terminal fields, due to the activity of increased 5-HT on the inhibitory somatodendritic 5-HT1A autoreceptors in the raphe nuclei, consistent with some microdialysis data in rodents [47, 48]. Such an explanation would be consistent with a reduction in radioligand binding in the raphe region in these studies, but this has not been conclusively demonstrated. Studies with 5-HT1B antagonists ([11C]P943, [11C]AZ10419369) in non-human primates have demonstrated significant changes in radioligand binding in the hypothesized direction, following the administration of the 5-HT releasing agents fenfluramine (REFS), amphetamine and high doses of SSRIs [9].
The inconsistent results above may therefore reflect the deficiency of many of the pharmacological paradigms used to date, to elicit large magnitude release of 5-HT. Recent data also suggest that the pharmacological response in terms of serotonin release may be highly interindividually different [18]. The limited sensitivity of the antagonist radioligands used to date, to detect the binding of the agonist serotonin, is a contributory factor. Serotonin releasers such as fenfluramine and amphetamine are known to produce significantly higher change in extracellular serotonin levels compared with the challenges above. Microdialysis evaluation in the pig brain found a 11-fold increase in extracellular serotonin concentration following a 0.5 mg/kg challenge with the serotonin releaser fenfluramine, compared with a twofold increase after 2 mg/kg of citalopram [16]. Such small increases in cortical serotonin following a single dose of citalopram are likely to explain the lack of effect on [11C]CIMBI-36 BPND seen in a recent human PET study using a citalopram challenge [49].
A limitation of our study was the fixed order of the two scans; with post-d-amphetamine scans conducted 6 h after the baseline scans. This fixed order raises the possibility of an order effect—due to either a systemic variation serotonin 2A receptor binding, or serotonin levels (due to diurnal variability or a non-specific effects of the experimental procedures—such as a difference in anxiety levels between baseline- and post-d-amphetamine scans) or due to a direct occupancy effect of the unlabelled CIMBI-36 carried over from the baseline d-amphetamine scan. We do not believe these considerations are relevant for the explanation of our data. [11C]CIMBI-36 test-retest data in eight healthy human volunteers, obtained with test and retest scans conducted at different times of the day (with duration of between 3 h and several weeks between the two scans) do not suggest any diurnal effect. In that data set, of the 8 “late scans” 4 were 3–6% higher whereas 4 others were 1–6% lower as compared with the 8 “early scans” ([29] and additional communication with NeuroPharm, Copenhagen). With regard to a potential carry-over effect from early to late scans, CIMBI-36 in the 0–5 µg range did not affect cortical binding in the pig brain (Møller, personal communication). In our study, the maximal administered dose of CIMBI-36 was 1.5 µg, indicating that a carry-over effects are unlikely to explain the outcome.
In conclusion, we find that cerebral [11C]CIMBI-36 binding is sensitive to acute synaptic serotonin release in the human brain, and when combined with a d-amphetamine challenge the human brain serotonin system can be evaluated in neuropsychiatric disorders, such as major depression.
Funding and disclosure
There were no conflicts of interests to declare for this study. The funding came from an Imanova (now Invicro) IMPETUS pilot PET grant for Erritzoe, a grant to establish the Center for Experimental Medicine NeuroPharmacology (www.neuropharm.eu) from Innovation Fund Denmark (grant number 4108-00004B) for Knudsen, and an MRC Project grant for Howes.
References
Laruelle M. Imaging synaptic neurotransmission with in vivo binding competition techniques: a critical review. J Cereb Blood Flow Metab. 2000;20:423–51.
Paterson LM, Tyacke RJ, Nutt DJ, Knudsen GM. Measuring endogenous 5-HT release by emission tomography: promises and pitfalls. J Cereb Blood Flow Metab. 2010;30:1682–706.
Hirani E, Sharp T, Sprakes M, Grasby P, Hume S. Fenfluramine evokes 5-HT2A receptor-mediated responses but does not displace [11C]MDL 100907: small animal PET and gene expression studies. Synapse. 2003;50:251–60.
Staley JK, Van Dyck CH, Tan PZ, Al Tikriti M, Ramsby Q, Klump H, et al. Comparison of [18F]altanserin and [18F]deuteroaltanserin for PET imaging of serotonin2A receptors in baboon brain: pharmacological studies. Nucl Med Biol. 2001;28:271–9.
Matusch A, Hurlemann R, Rota Kops E, Winz OH, Elmenhorst D, Herzog H, et al. Acute S-ketamine application does not alter cerebral [18F]altanserin binding: a pilot PET study in humans. J Neural Transm (Vienna). 2007;114:1433–42.
Meyer JH, Cho R, Kennedy S, Kapur S. The effects of single dose nefazodone and paroxetine upon 5-HT2A binding potential in humans using [18F]-setoperone PET. Psychopharmacology. 1999;144:279–81.
Pinborg LH, Adams KH, Yndgaard S, Hasselbalch SG, Holm S, Kristiansen H, et al. [18F]altanserin binding to human 5HT2A receptors is unaltered after citalopram and pindolol challenge. J Cereb Blood Flow Metab. 2004;24:1037–45.
Paterson LM, Kornum BR, Nutt DJ, Pike VW, Knudsen GM. 5-HT radioligands for human brain imaging with PET and SPECT. Med Res Rev. 2013;33:54–111.
Finnema SJ, Scheinin M, Shahid M, Lehto J, Borroni E, Bang-Andersen B, et al. Application of cross-species PET imaging to assess neurotransmitter release in brain. Psychopharmacology. 2015;232:4129–57.
Larisch R, Klimke A, Hamacher K, Henning U, Estalji S, Hohlfeld T, et al. Influence of synaptic serotonin level on [18F]altanserin binding to 5HT2 receptors in man. Behav Brain Res. 2003;139:21–9.
Quednow BB, Treyer V, Hasler F, Dorig N, Wyss MT, Burger C, et al. Assessment of serotonin release capacity in the human brain using dexfenfluramine challenge and [18F]altanserin positron emission tomography. Neuroimage. 2012;59:3922–32.
Sibon I, Benkelfat C, Gravel P, Aznavour N, Costes N, Mzengeza S, et al. Decreased [18F]MPPF binding potential in the dorsal raphe nucleus after a single oral dose of fluoxetine: a positron-emission tomography study in healthy volunteers. Biol Psychiatry. 2008;63:1135–40.
Selvaraj S, Turkheimer F, Rosso L, Faulkner P, Mouchlianitis E, Roiser JP, et al. Measuring endogenous changes in serotonergic neurotransmission in humans: a [11C]CUMI-101 PET challenge study. Mol Psychiatry. 2012;17:1254–60.
Nord M, Finnema SJ, Halldin C, Farde L. Effect of a single dose of escitalopram on serotonin concentration in the non-human and human primate brain. Int J Neuropsychopharmacol. 2013;16:1577–86.
Tyacke RJ, Nutt DJ. Optimising PET approaches to measuring 5-HT release in human brain. Synapse. 2015;69:505–11.
Jorgensen LM, Weikop P, Villadsen J, Visnapuu T, Ettrup A, Hansen HD, et al. Cerebral 5-HT release correlates with [11C]Cimbi36 PET measures of 5-HT2A receptor occupancy in the pig brain. J Cereb Blood Flow Metab. 2016;37:425–34.
Yang KC, Stepanov V, Martinsson S, Ettrup A, Takano A, Knudsen GM, et al. Fenfluramine reduces [11C]Cimbi-36 binding to the 5-HT2A receptor in the nonhuman primate brain. Int J Neuropsychopharmacol. 2017;20:683–91.
da Cunha-Bang S, Ettrup A, Mc Mahon B, Skibsted AP, Schain M, Lehel S, et al. Measuring endogenous changes in serotonergic neurotransmission with [(11)C]Cimbi-36 positron emission tomography in humans. Transl Psychiatry. 2019;9:134.
Heal DJ, Cheetham SC, Prow MR, Martin KF, Buckett WR. A comparison of the effects on central 5-HT function of sibutramine hydrochloride and other weight-modifying agents. Br J Pharm. 1998;125:301–8.
Kuczenski R, Segal DS, Cho AK, Melega W. Hippocampus norepinephrine, caudate dopamine and serotonin, and behavioral responses to the stereoisomers of amphetamine and methamphetamine. J Neurosci. 1995;15:1308–17.
Ridler K, Plisson C, Rabiner EA, Gunn RN, Easwaramoorthy B, Abi-Dargham A, et al. Characterization of in vivo pharmacological properties and sensitivity to endogenous serotonin of [11C] P943: a positron emission tomography study in Papio anubis. Synapse. 2011;65:1119–27.
Kehr J, Ichinose F, Yoshitake S, Goiny M, Sievertsson T, Nyberg F, et al. Mephedrone, compared with MDMA (ecstasy) and amphetamine, rapidly increases both dopamine and 5-HT levels in nucleus accumbens of awake rats. Br J Pharm. 2011;164:1949–58.
Toll L, Berzetei-Gurske IP, Polgar WE, Brandt SR, Adapa ID, Rodriguez L, et al. Standard binding and functional assays related to medications development division testing for potential cocaine and opiate narcotic treatment medications. NIDA Res Monogr. 1998;178:440–66.
Ettrup A, Hansen M, Santini MA, Paine J, Gillings N, Palner M, et al. Radiosynthesis and in vivo evaluation of a series of substituted 11C-phenethylamines as 5-HT2A agonist PET tracers. Eur J Nucl Med Mol Imaging. 2011;38:681–93.
Tziortzi AC, Searle GE, Tzimopoulou S, Salinas C, Beaver JD, Jenkinson M, et al. Imaging dopamine receptors in humans with [11C]-(+)-PHNO: dissection of D3 signal and anatomy. Neuroimage. 2011;54:264–77.
Ettrup A, da Cunha-Bang S, McMahon B, Lehel S, Dyssegaard A, Skibsted AW, et al. Serotonin 2A receptor agonist binding in the human brain with [11C]Cimbi-36. J Cereb Blood Flow Metab. 2014;34:1188–96.
Ichise M, Toyama H, Innis RB, Carson RE. Strategies to improve neuroreceptor parameter estimation by linear regression analysis. J Cereb Blood Flow Metab. 2002;22:1271–81.
Beliveau V, Ganz M, Feng L, Ozenne B, Hojgaard L, Fisher PM, et al. A high-resolution in vivo atlas of the human brain’s serotonin system. J Neurosci. 2017;37:120–8.
Ettrup A, da Cunha-Bang S, McMahon B, Lehel S, Dyssegaard A, Skibsted AW, et al. Serotonin 2A receptor agonist binding in the human brain with [11C]Cimbi-36: test-retest reproducibility and head-to-head comparison with the antagonist [18F]altanserin. Neuroimage. 2016;130:167–74.
Colasanti A, Searle GE, Long CJ, Hill SP, Reiley RR, Quelch D, et al. Endogenous opioid release in the human brain reward system induced by acute amphetamine administration. Biol Psychiatry. 2012;72:371–7.
Mick I, Myers J, Stokes PR, Erritzoe D, Colasanti A, Bowden-Jones H, et al. Amphetamine induced endogenous opioid release in the human brain detected with [11C]carfentanil PET: replication in an independent cohort. Int J Neuropsychopharmacol. 2014;17:2069–74.
Narendran R, Mason NS, Paris J, Himes ML, Douaihy AB, Frankle WG. Decreased prefrontal cortical dopamine transmission in alcoholism. Am J Psychiatry. 2014;171:881–8.
Shotbolt P, Tziortzi AC, Searle GE, Colasanti A, van der Aart J, Abanades S, et al. Within-subject comparison of [11C]-(+)-PHNO and [11C]raclopride sensitivity to acute amphetamine challenge in healthy humans. J Cereb Blood Flow Metab. 2012;32:127–36.
Erritzoe D, Talbot P, Frankle WG, Abi-Dargham A. Positron emission tomography and single photon emission CT molecular imaging in schizophrenia. Neuroimaging Clin N Am. 2003;13:817–32.
Martinez D, Narendran R. Imaging neurotransmitter release by drugs of abuse. Curr Top. Behav Neurosci. 2010;3:219–45.
Yang KC, Takano A, Halldin C, Farde L, Finnema SJ. Serotonin concentration enhancers at clinically relevant doses reduce [11C]AZ10419369 binding to the 5-HT1B receptors in the nonhuman primate brain. Transl Psychiatry. 2018;8:132.
Baumann MH, Wang X, Rothman RB. 3,4-Methylenedioxymethamphetamine (MDMA) neurotoxicity in rats: a reappraisal of past and present findings. Psychopharmacol (Berl). 2007;189:407–24.
Connolly HM, Crary JL, McGoon MD, Hensrud DD, Edwards BS, Edwards WD, et al. Valvular heart disease associated with fenfluramine-phentermine. N Engl J Med. 1997;337:581–8.
Elmenhorst D, Kroll T, Matusch A, Bauer A. Sleep deprivation increases cerebral serotonin 2A receptor binding in humans. Sleep. 2012;35:1615–23.
Derry C, Benjamin C, Bladin P, le Bars D, Tochon-Danguy H, Berkovic SF. et al. Increased serotonin receptor availability in human sleep: evidence from an [18F]MPPF PET study in narcolepsy. Neuroimage. 2006;30:341–8.
Abi-Dargham A, Kegeles LS, Martinez D, Innis RB, Laruelle M. Dopamine mediation of positive reinforcing effects of amphetamine in stimulant naive healthy volunteers: results from a large cohort. Eur Neuropsychopharmacol. 2003;13:459–68.
Martinez D, Slifstein M, Broft A, Mawlawi O, Hwang DR, Huang Y, et al. Imaging human mesolimbic dopamine transmission with positron emission tomography. Part II: amphetamine-induced dopamine release in the functional subdivisions of the striatum. J Cereb Blood Flow Metab. 2003;23:285–300.
Drevets WC, Gautier C, Price JC, Kupfer DJ, Kinahan PE, Grace AA, et al. Amphetamine-induced dopamine release in human ventral striatum correlates with euphoria. Biol Psychiatry. 2001;49:81–96.
Le Merrer J, Becker JA, Befort K, Kieffer BL. Reward processing by the opioid system in the brain. Physiol Rev. 2009;89:1379–412.
Schultz W. Dopamine reward prediction error coding. Dialogues Clin Neurosci. 2016;18:23–32.
Di Chiara G, Imperato A. Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proc Natl Acad Sci USA. 1988;85:5274–8.
Hervas I, Artigas F. Effect of fluoxetine on extracellular 5-hydroxytryptamine in rat brain. Role of 5-HT autoreceptors. Eur J Pharm. 1998;358:9–18.
Romero L, Bel N, Artigas F, de Montigny C, Blier P. Effect of pindolol on the function of pre- and postsynaptic 5-HT1A receptors: in vivo microdialysis and electrophysiological studies in the rat brain. Neuropsychopharmacology. 1996;15:349–60.
Ettrup A, McMahon B, Skibsted A, da Cunha-Bang S, Lehel S, Dyssegaard A, et al. Serotonin 2A receptor agonist binding with [11C]Cimbi-36 in the human brain is unaltered by citalopram/pindolol and acute tryptophan depletion, in 29th ECNP Congress 2016: Vienna. S307-8; 2016.
Acknowledgements
We would like to thank all the volunteers for their participation as well as the invaluable contributions of the Imanova/Invicro clinical staff for their excellent technical support. EAR wishes to acknowledge the support and funding by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Erritzoe, D., Ashok, A.H., Searle, G.E. et al. Serotonin release measured in the human brain: a PET study with [11C]CIMBI-36 and d-amphetamine challenge. Neuropsychopharmacol. 45, 804–810 (2020). https://doi.org/10.1038/s41386-019-0567-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41386-019-0567-5
- Springer Nature Switzerland AG
This article is cited by
-
Exploring mechanisms of psychedelic action using neuroimaging
Nature Mental Health (2024)
-
Neuroimaging in psychedelic drug development: past, present, and future
Molecular Psychiatry (2023)
-
Endogenous dopamine release in the human brain as a pharmacodynamic biomarker: evaluation of the new GPR139 agonist TAK-041 with [11C]PHNO PET
Neuropsychopharmacology (2022)
-
Application of positron emission tomography in psychiatry—methodological developments and future directions
Translational Psychiatry (2022)