
Abstract
We report a mechanism that underlies stress-induced cognitive inflexibility at the molecular level. In a mouse model under subacute cellular stress in which deficits in rule shifting tasks were elicited, the nuclear glyceraldehyde dehydrogenase (N-GAPDH) cascade was activated specifically in microglia in the prelimbic cortex. The cognitive deficits were normalized with a pharmacological intervention with a compound (the RR compound) that selectively blocked the initiation of N-GAPDH cascade without affecting glycolytic activity. The normalization was also observed with a microglia-specific genetic intervention targeting the N-GAPDH cascade. At the mechanistic levels, the microglial secretion of High-Mobility Group Box (HMGB), which is known to bind with and regulate the NMDA-type glutamate receptors, was elevated. Consequently, the hyperactivation of the prelimbic layer 5 excitatory neurons, a neural substrate for cognitive inflexibility, was also observed. The upregulation of the microglial HMGB signaling and neuronal hyperactivation were normalized by the pharmacological and microglia-specific genetic interventions. Taken together, we show a pivotal role of cortical microglia and microglia-neuron interaction in stress-induced cognitive inflexibility. We underscore the N-GAPDH cascade in microglia, which causally mediates stress-induced cognitive alteration.
Similar content being viewed by others


Introduction
Cognitive flexibility is a construct of executive function that adapts our decision-making to new experiences [1]. This behavioral construct is closely related to creativity and empathy, enabling us to not only adapt, but also to think outside of the box, taking other perspectives into consideration [1]. Cognitive flexibility is predominantly regulated by the prefrontal cortex [2,3,4]. This function is sensitive to, and negatively affected by conditions of stress [5,6,7]. Accordingly, cognitive inflexibility is observed in an array of brain disorders, including schizophrenia (SZ) and Alzheimer’s disease (AD) [8,9,10]. In these conditions, impairment of cognitive flexibility directly leads to functional deficits and worsens clinical outcomes for patients [11, 12].
At the molecular level, inflammation and redox imbalance have been frequently associated with poor cognitive performance [13,14,15,16,17,18,19,20,21,22]. Biofluids from patients with cognitive deficits have shown molecular signatures of inflammatory processes and excess oxidative stress [17, 20, 23,24,25]; however, it is unknown how such molecular changes are related to specific symptoms at the mechanistic level. Meanwhile, basic neurobiology has shown that neuroinflammatory molecules can modulate synaptic structures and functions in rodent models [18, 19]. An array of rodent models with cognitive deficits have been used to connect the gaps between basic neurobiology and clinical observations [26,27,28,29,30]. Some models are generated by administering lipopolysaccharide (LPS), polyinosinic:polycytidylic acid (poly I:C), ethanol, or cuprizone [31,32,33,34,35]. Repeated administration of low-dose LPS consistently elicits excess oxidative stress and prolonged changes in microglia [36,37,38,39,40,41,42]. The changes in cortical microglia reportedly include an enlargement of the cell body and processes with a lesser number of branches at the morphological levels as well as epigenetic and expression changes such as an increase in the histone 3 methylation at lysine 4 [37,38,39,40,41,42]. Cognitive performance has also been precisely addressed in this model after resolution of the transient sickness behavior that occurs upon LPS injection [43].
Among representative stress-triggered intracellular signaling, we have studied a cascade in which glyceraldehyde-3-phosphate dehydrogenase (GAPDH) acts as a sensor of cellular stress [44, 45] (Supplementary Fig. 1a). Beyond its classic role in glycolysis [46, 47], during stress, a pool of this multi-functional protein [45, 48] is post-translationally modified at specific residues, such as cysteine 150 (Cys-150) and lysine 160 (Lys-160), which converts this glycolytic enzyme into a signaling molecule [49]. A wide range of stressors that ultimately lead to oxidative/nitrosative stress in the cells can trigger this cascade, which include dexamethasone for thymocytes and LPS for macrophages [49, 50]. At the molecular levels such stressors lead to S-nitrosylation (or possibly oxidation) at Cys-150, which allows post-translationally modified GAPDH to interact with a chaperon protein Siah1 creating a protein complex of GAPDH-Siah1. Through this process, GAPDH loses its glycolytic activity and is translocated to the nucleus as a GAPDH-Siah1 complex [49]. The nuclear translocation of a pool of GAPDH implies a gain-of function that has a significant impact on cellular signaling, whereas a loss-of-function of 1–2% of the total glycolytic activity due to this conversion is negligible on overall cell metabolism [49]. Accordingly, GAPDH-Siah1 protein binding is a key regulatory process that initiates the nuclear-GAPDH (N-GAPDH) cascade, which can be selectively blocked by several compounds without affecting the glycolytic activity of GAPDH. These selective blockers include structural analogs of deprenyl (e.g., CGP 3466B) [51]. We have recently reported that (1 R, 3 R)-1, 3-dimethyl-2-propargyl-1, 2, 3, 4-tetrahydroisoquinoline (designated as RR in this study) is a deprenyl structural analog with the highest potency among the analogs, as far as we are aware, for blocking the GAPDH-Siah1 protein interaction [52]. Our group also reported that the replacement of lysine 225 for alanine (K225A) on GAPDH abolished GAPDH-Siah1 binding [49]. Although this cascade has been mainly studied in the context of cell death [53], activation of the N-GAPDH cascade was also reported in animal models with behavioral deficits that do not accompany cell death [54]. Nevertheless, the mechanism(s) whereby the N-GAPDH cascade mediates stress-induced behavioral alteration remain elusive.
We used a mouse model with repeated administration of low-dose LPS [low-dose cellular stressors (LPS) at multiple times: LSM model]. In rodents, an equivalent of human cognitive inflexibility assessed with the Wisconsin Card Sorting Test is frequently examined with a rule shifting task in which mice are exposed to a shift of dimensions (e.g., a shift of stimulus from odor to texture) [55]. We observed that the LSM model showed deficits in the rule shifting task, together with selective activation of the N-GAPDH cascade in cortical microglia. The cognitive deficits were reverted with pharmacological intervention or microglia-specific genetic intervention targeting the initial steps of the N-GAPDH cascade. Hyperactivation of the prelimbic excitatory neurons, known as a key neural substrate of the deficit in rule shifting tasks, was also normalized with the same set of pharmacological or microglia-specific genetic intervention to the N-GAPDH cascade. At the mechanistic levels, we observed an upregulation in microglial secretion of High-Mobility Group Box (HMGB), which is known to bind with and regulate the NMDA-type glutamate receptors. Consequently, the hyperactivation of the prelimbic layer 5 excitatory neurons, a neural substrate for cognitive inflexibility, was also observed. The upregulation of the HMGB signaling was normalized by the intervention of the N-GAPDH cascade, whereas the neuron hyperactivation was normalized by both N-GAPDH cascade intervention and the application of an antibody against HMGB1. Starting from a specific question of the N-GAPDH stress cascade, the goal of the present study is to demonstrate a molecular/cellular mechanism of stress-induced cognitive inflexibility, a general question in neurobiology and biological psychiatry. We aim to test the hypothesis that an alteration in a specific type(s) of cells inside the brain may sufficiently account for the cognitive phenotype. In summary, we demonstrated that a stress-induced signaling cascade (the N-GAPDH cascade) specifically activated in microglia mediates cognitive inflexibility via a prefrontal microglia-neuron crosstalk (Supplementary Fig. 1b).
Methods
Chemicals
LPS from Escherichia coli O111:B (Sigma-Aldrich, L4391) and RR were reconstituted with saline buffer. The tamoxifen solution (Sigma-Aldrich, T5648, 10 mg/mL) was freshly prepared by sonicating tamoxifen freebase in sunflower seed oil (S5007, Sigma-Aldrich) at room temperature for 10–12 min (with intermittent 20 s vortexing every 3–4 min). This solution was stored at −20 °C for several months.
Antibodies
The following antibodies were used: Anti-sulfonated-GAPDH (Cys152) Antibody (Research Genetics) [45]; Anti-GAPDH Antibody (Santa Cruz Biotech, sc-32233); Anti-HMGB1 Antibody (Abcam, ab18256); Anti-H3 Antibody (Abcam, ab18251); Anti-Siah1 Antibody (Santa Cruz Biotech, sc-5505); Anti-Siah1 Antibody (Santa Cruz Biotech, sc-5506); Anti-NeuN Antibody (Millipore Sigma, MAB377); Anti-Aldh1l1 Antibody (Millipore Sigma, MABN495); Anti-Iba1 Antibody (Wako, 019-19741); Anti-Iba1 Antibody (Wako, 016-20001), Anti-Caspase-3-cleaved Antibody (Cell Signaling, 9661-S); PE Anti-P2ry12 Antibody (BioLegend, 848003); APC Anti-CD45 Antibody (BD Bioscience, 559864); BV421 Anti-CD11b Antibody (BD Bioscience, 562605); True Blot Anti-Rabbit Ig HRP (Rockland, 18-8816-33); True Blot Anti-Goat Ig HRP (Rockland, 605-403-B69); True Blot Anti-Mouse Ig HRP (Rockland, 18-8817-30).
In vitro characterization of RR
MAO activity of RR was measured with a commercially available kit (Peninsula Laboratory). GAPDH enzymatic activity with or without RR was measured in our published protocol [49]. Effects of RR (1 nM) on N-GAPDH cascades were assessed by comparing the levels of GAPDH nuclear translocation in BV2 cells treated with RR only, LPS (1 μg/ml) only, RR and LPS, and no treatment. We used our published protocol for the subcellular fractionation [45]. BV2 cells were purchased from ATCC.
RR pharmacokinetic analysis
Vehicle control or RR treated mice (0.25 mg/kg/day i.p. for 5 days) were euthanized 1 h after the last injection (n = 5 mice). Blood samples were collected in heparinized tubes by cardiac puncture and centrifuged (3000 × g for 10 min) to obtain plasma. Brains were perfused with saline, extracted, and dissected. Both plasma and brains were stored at −80 °C until bioanalysis. RR compound levels in the plasma and brain were measured using high-performance liquid chromatography with tandem mass spectrometry (LC/MS-MS). Briefly, standards were prepared by spiking the compound in naïve mouse plasma or tissue from 0.003-100 nmol/mL or nmol/g. Standards, plasma (20 µL) or weighed brain samples were placed in low retention microcentrifuge tubes and extracted using methanol containing losartan as an internal standard (5 µL methanol/mg tissue). Samples were vortex mixed and centrifuged (16,000 × g for 5 min at 4 °C). The supernatants (80 µL) were transferred to a 96 well plate and 2 µL was injected for analysis. Samples were analyzed on an UltiMate 3000 UHPLC coupled to a Q Exactive Focus orbitrap mass spectrometer (Thermo Fisher Scientific Inc., Waltham MA). Samples were separated on an Agilent EclipsePlus C18 RRHD (1.8 µm) 2.1 × 100 mm column. The mobile phase consisted of water +0.1% formic acid (A), and acetonitrile +0.1% formic acid (B). Separation was achieved at a flow rate of 0.4 mL/min using a gradient run. Quantification was performed in product-reaction monitoring (PRM) mode (RR 200.1434 > 145.1, 82.0650) using Xcalibur software.
Mice
Male C57BL/6 J (The Jackson Laboratory, #000664), Cx3cr1-CreER (The Jackson Laboratory, #021160), VGlut1-Cre (The Jackson Laboratory, #023527), VGAT-Cre (The Jackson Laboratory, #016962), Thy1-GCaMP6s (The Jackson Laboratory, #025776), GCamMP6 floxed (The Jackson Laboratory, #024106) were purchased from the Jackson Laboratory. K225A-GAPDH floxed mice were recently developed [52]. All mice were on a C57BL/6 background. Only male mice were used for all the experiments. Eight to ten weeks old mice were used for all experiments. Mice were housed (5 mice maximum per cage) and maintained under a 12-h light: 12-h dark cycle, at a constant temperature and humidity (20–24 °C, 35–55%), with food and water available ad libitum. The Institutional Animal Care and Use Committee at Johns Hopkins University approved all protocols involving mice that were used in this study.
K225A-GAPDH floxed model
A floxed mouse line in which lysine 225 (K) responsible for the binding between GAPDH and Siah1 was replaced with an alanine (A) residue in exon 5 of the mouse Gapdh gene, were generated using a homologous recombination approach [52]. In this mouse model GAPDH is not able to translocate to the nucleus, therefore the GAPDH nuclear pathway is blocked [56]. These animals were crossed with Cx3cr1-CreER promoter line in order to target microglia (Supplementary Fig. 5). More specifically, mice aged 4–5 weeks were administered with tamoxifen by an oral gavage at 100 mg/kg body weight, once a day for two consecutive days to drive the Cx3cr1-CreER promoter. To eliminate peripheral macrophages and monocytes with the induced K225A-GAPDH mutant, all experiments were performed 5 weeks after the last tamoxifen injection, because the peripheral cells are replaced with non-induced macrophages and monocytes from the bone marrow within 5 weeks [57, 58]. Although the K225A-GAPDH mutation may possibly impact border-associated macrophages (BAMs), the number of BAMs is much smaller than that of microglia (the ratio only at 3–4%, according to the literature [59]). Altogether, we believe that the influence of BAMs may be relatively smaller or almost negligible, compared with that of microglia.
LSM model
We used the “low-dose cellular stressors (LPS) at multiple times” (LSM) model. LSM elicits oxidative stress and activation of microglia, but sickness response is known to be minimal [39]. Mice were injected with saline or RR (0.25 mg/kg/day i.p) 1 day prior to a low-dose LPS treatment (1.5 × 105 EU/kg/day i.p). Then, mice were co-injected with RR (or saline) and LPS once a day for 4 consecutive days (refer to Figs. 1a and 2a). The low dose of LPS corresponds to 0.05 mg/kg/day according to the LPS potency data in the Certificates of Analysis (COA) from the company’s website.

a Schematic diagram of the mouse paradigm and behavioral task. Eight to ten week-old mice were adapted to a reverse light/dark cycle. Drug injections were made intraperitoneally (i.p). Mice were food restricted for 72 h before starting habituation, then habituated for 2 consecutive days to the bowls, digging media, and food reward before testing began. Mice that reached criteria were tested with the behavioral task on the following day. Black arrowhead, injection; red arrowhead, calorie restriction; yellow arrowhead, habituation; and brown arrowhead, behavioral test. b Schematic diagram illustrating the rule-shifting task. Two bowls were presented to mice, each baited by an odor (O1 or O2) and a textured digging medium (TA or TB). Mice had to find a food reward that was associated with a particular stimulus (indicated in red) buried in the digging medium. Mice first learned an initial association; once mice reached the learning criterion (eight correct out of ten consecutive trials), this association underwent an extra-dimensional rule shift. c Performance of Control, RR, LSM, and RR LSM mice in the rule shifting task. Each dot represents data from an individual mouse. Two-way ANOVA (RR x LSM) revealed a significant main effect of RR (F(1,37) = 4.68, p = 0.037), a significant effect of LSM (F(1,37) = 7.33, p = 0.010), and a non-significant interaction between factors (F(1,37) = 2.55, p = 0.119). Error bars represent the mean ± SEM. #p = 0.051 *p < 0.05 by two-way ANOVA with Tukey’s multiple comparisons test. d LSM mice performed significantly more perseverant than random errors during the rule-shifting task. Each dot represents data from an individual mouse. Two-way ANOVA (RR x LSM) revealed a significant main effect of RR (F(1,37) = 6.66, p = 0.014), a significant effect of LSM (F(1,37) = 7, p = 0.012), and a non-significant interaction between factors (F(1,37) = 3.12, p = 0.086). Error bars represent the mean ± SEM. *p < 0.05, **p < 0.01 by two-way ANOVA with Tukey’s multiple comparisons test.

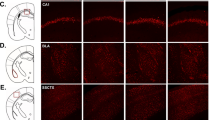
a Schematic diagram showing the mouse intraperitoneal injection paradigm used. b Levels of GAPDH-Siah1 binding (a key indicator of the N-GAPDH cascade activation). Lysates were from cortical microglia and astrocytes isolated from Control and LSM mice using magnetic sorting. “Rest” corresponds to the negative fraction after isolation of microglia and astrocytes. The purity of isolated cells was assessed by using anti-Iba1, NeuN, and Aldh1l1 antibodies that specifically recognize microglia, neurons, and astrocytes, respectively. The Y axis depicts the level of GAPDH-Siah1 binding, which was normalized by the level of Siah1. c Representative confocal images depicting sulfonated GAPDH at cysteine-150 (SulfoGAPDH) co-localized with microglia (Iba1), and not astrocytes (GFAP) or neural cells (NeuN) in the medial prefrontal cortex in LSM mice. Arrows indicate Iba1 and SulfoGAPDH signals, whereas arrowheads indicate colocalization of the Iba1 and SulfoGAPDH signals. Scale bar: 40 μm. d Level of GAPDH-Siah1 binding. Lysates were from cortical and striatal microglia isolated from Control and LSM mice. The Y axis depicts the level of GAPDH-Siah1 binding, which was normalized by the level of Siah1. e Level of GAPDH-Siah1 binding. Lysates were from cortical microglia isolated from Control, LSM, and RR LSM mice. The Y axis depicts the level of GAPDH-Siah1 binding, which was normalized by the level of Siah1. Error bars represent the mean ± SEM. *p < 0.05 by unpaired two-tailed t-test for (b) and (d), and one-way ANOVA with Tukey’s multiple comparisons test for (e). Immunoblots are representative of 5 independent experiments from cells isolated from 10 mice per group [(b), (d), and (e)]. Confocal images are representative of 3 mice per group (c).
Behavioral assays
The Institutional Animal Care and Use Committee at Johns Hopkins University approved all protocols used in this study. The sample size was determined based on our previous publication [3]. We did not use a randomization method to determine how mice were allocated to experimental groups. Experimenters were blinded to the experimental groups.
Cognitive flexibility tasks
We followed a protocol described by Cho et al. [3] with some adaptations from Heisler et al. [60]. Briefly, mice were single-housed and habituated to a reverse light/dark cycle at least 2 weeks before starting the task. Food intake was restricted 5 days before starting, corresponding to the period in which the mice were adapted to the bowls and reward. At the start of each trial, the mouse was placed in its home cage to explore two bowls, each containing one odor and one digging medium, until it dug in one bowl, signifying a choice. The bait was a piece of a peanut butter chip (approximately 5–10 mg in weight) and the cues, either olfactory (odor) or somatosensory and visual (texture of the digging medium which hides the bait), were altered and counterbalanced. All cues were presented in small animal food bowls identical in color and size. Digging media was mixed with the odor (0.1% by volume) and peanut butter chip powder (0.1% by volume). All odors were ground dried spices (McCormick garlic and coriander). For each trial, while the odor-medium combination present in each of the two bowls may have changed, the stimulus (e.g., a particular odor or medium) that signaled the presence of the food reward remained constant over each portion of the task (initial association, rule shift). If the initial association paired a specific odor with the food reward, then the digging medium would be considered the irrelevant dimension. The mouse was considered to have learned the initial association between stimulus and reward if it made 8 correct choices during 10 consecutive trials. Following the initial association, the rule-shifting portion of the task began, and the stimulus associated with the reward underwent an extra-dimensional shift. The mouse was considered to have learned the extra dimensional rule shift if it made 8 correct choices during 10 consecutive trials. In all portions of the task, after the mouse made an error on a trial or 3 min passed with no choice, the mouse was transferred to the holding (punishment) cage for a minimum of 60 s. If the animal made 6 consecutive no choices (3 min without a choice) this was recorded as a failure to participate.
Forced swim test
Each mouse was put in Plexiglass cylinder tanks measuring 19 cm in height and 13 cm in diameter with a water level of 16 cm. Mice were scored for immobility time over a 6-min uninterrupted test session.
Open field test
An individual mouse was placed near the wall-side of a 50 × 50 cm open-field arena. The movement of the mouse was recorded by laser beam breaks for 30 min using an infrared activity monitor (San Diego Instruments). The data was automatically analyzed with PAS-Open Field software (San Diego Instruments). Time in the center of the field and total activity were measured. The open field arena was cleaned with 70% ethanol and wiped with paper towels between each trial.
Cell biology, biochemistry, and histology
Microglia and astrocytes sorting
CD11b+ or ACSA2+ beads were used to isolate microglia or astrocytes, respectively, by magnetic sorting. The “rest” fraction corresponded with the negative fraction isolated after separating microglia and astrocytes. Microglia were isolated by fluorescence-activated cell-sorting (FACS) using CD45, CD11b, and p2ry12 antibodies (Biolegend).
Immunoblotting
Microglia, astrocytes, or rest cells were lysed in CHAPS buffer (50 mM Tris, 150 mM NaCl, 1% SDS, and 1% Triton X-100) containing a proteinase inhibitor and phosphatase inhibitor cocktails. Six rounds of sonication were performed as follows: 15 s on, 15 s off, and 40% amplitude with a Q125 Sonicator (Q Sonica Sonicators). Lysates were centrifuged for 2 min at 10,000 rpm, the supernatant was collected, quantified using the Pierce BCA Protein assay (Thermo Scientific), aliquoted, and stored at −80 °C until use. Lysates were thawed and 4x NuPAGE LDS sample buffer (Thermo Scientific) and 10% β-mercaptoethanol (Sigma Aldrich) were added. Lysates were heated to 95 °C for 10 min and run on NuPAGE 4–12% bis-tris gels (Invitrogen). Proteins were transferred to PVDF membranes (BioRad) and blocked for 1 h at RT in 5% BSA in Tris buffered saline with 0.05% Tween 20 (TBST). Membranes were incubated in primary antibody overnight at 4 °C, washed and incubated in secondary antibody HRP for 1 h at RT. After washing, membranes were developed using either the Super Signal West Dura or Super Signal West Femto Chemiluminescent Substrate (Thermo Scientific).
Immunoprecipitation (IP)
The GAPDH-Siah1 protein interaction was assessed by our published protocol [49], in which 200 μg of total protein was used for the IP. The total volume was adjusted to 1 ml with supplemented CHAPS buffer, and 1 μg of antibody was added followed by an incubation O/N at 4 °C with continuous agitation. On the next day cell lysates were incubated for 2 h at 4 °C with 30 μL of TrueBlot Anti- Goat or Rabbit IgG beads (Rockland laboratories).
Dot blot
The potency of RR and other deprenyl derivatives to block the binding between GAPDH and Siah1 was evaluated using dot blot. A mixture of GAPDH and Siah1 recombinant proteins was incubated O/N at 4 °C, with each deprenyl derivative (2 nM) or deprenyl (2 nM) or empty solution. The possible protein complexes were immunoprecipitated using a Siah1 antibody as described above, and the immunoprecipitates were loaded onto a nitrocellulose membrane. The membrane was washed and immunoblotted with a GAPDH antibody as described above.
Immunohistochemistry
After perfusion, mice were fixed with 4% paraformaldehyde (PFA) and brains were kept in PFA O/N at 4 °C. The following day brains were transferred to a solution of phosphate buffered saline (PBS) containing 30% sucrose for 1–2 days, then 12–30-μm cryosections were collected on glass slides. Sections were blocked in TBS containing 0.5% Triton (TBS-T) and 10% normal donkey serum (NDS) for 2 h at RT. Sections were then incubated with primary antibodies and kept at 4 °C for 24–48 h. Subsequently, sections were washed three times with TBS-T for 5 min each and incubated with fluorescently conjugated-secondary antibodies for 2 h at RT. After three washes with TBS-T, sections were mounted and coverslipped. Fluorescence images were obtained using a Zeiss LSM 700, Olympus FV1000.
Evans Blue quantification
We followed an established protocol [61]. We used mice treated with a high-dose LPS for three times within one day (1.5 × 107 EU/kg i.p each time) as a positive control of Evans Blue staining [called the blood brain barrier (BBB) impaired model]. Mice were perfused with prewarmed 0.9% NaCl to wash out blood cells, followed by 0.5% Evans Blue in cold 4% PFA. The coronal sections at 40-μm thickness with a vibratome were incubated with DAPI (1:1000), and the fluorescence images were obtained using a Zeiss LSM 800. We measured the Evans Blue signal-positive areas by ImageJ.
Physiological experiments
Acute brain slice preparation
Mice were anesthetized with ether, perfused using cold N-methyl-D-glucamine (NMDG) based cutting solution containing (in mM): 135 NMDG, 1 KCl, 1.2 KH2PO4, 1.5 MgCl2, 0.5 CaCl2, 10 Dextrose, 20 Choline Bicarbonate, (pH 7.4) as previously described [62]. Brains were dissected and mounted on a vibratome (Leica VT100S). Cortical slices 250 μm thick were cut in ice-cold N-methyl-D-glucamine (NMDG) based cutting solution. Cortical slices were then transferred to artificial cerebral spinal fluid (ACSF) containing (in mM): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgCl2, 1 NaH2PO4, 26.2 NaHCO3, and 11 Dextrose (292–298 mOsm/L) and were maintained at 37 °C for 40 min, and at room temperature thereafter. Both the NMDG solution and ACSF were bubbled continuously with 95% O2/5% CO2. An anti-HMGB1 antibody (Ab) was used in ACSF (1:50 dilution). All slice imaging experiments were carried out at room temperature.
Slice calcium imaging
Fluorescence dynamics arising from GCaMP6s VGlut1, VGAT, or Thy1 positive neurons were recorded using a fixed-stage microscope (BX61-WI, Olympus) with optical lens (40x-0.80, LumPLanFL N), 300 W Xenon lamp using a GFP filter, high speed galvo mirror with filters (DG4-1015, Shutter Instrument Company), and an infrared-sensitive CCD camera (iXon3, ANDOR Technology). Images were processed with digital imaging software (MetaFluor® for Olympus and Metamorph Advanced Molecular Device). All these systems were on an anti-vibration floating table (Technical Management Corp.) and connected to a PC (Windows 7, Microsoft). For imaging analysis, raw videos were pre-processed by applying ×4 spatial down-sampling to reduce file size and processing time, but no temporal down-sampling was applied [63]. To reliably deal with the large fluctuating background, we applied the CNMFe algorithm that is a recent adaptation of the CNMF algorithm [64], enabling us to identify individual neurons, obtain their fluorescent traces, and deconvolve fluorescence signals into neural activity. To define the initial spatial components, candidate seed pixels were manually selected from peak-to-noise (PNR) graphs of the field of view (FOV) [65]. Calcium events with z-scores <8 or those that did not have a >0.5 AUC were excluded from analyses because events of this magnitude did not reliably retain transient, calcium-event characteristics across animals.
Statistical analysis
All data are expressed as the mean ± SEM. Quantification of confocal microscopy, immunoblots, calcium imaging, and slice electrophysiology recordings was performed using Imaris, ImageJ, and MATLAB, and Clampfit respectively. Statistical differences between two groups was determined by a two-tailed unpaired Student’s t-test. For multiple group datasets, two-way or one-way ANOVA analysis was used, followed by Tukey’s multiple comparisons test for more than two groups. For multiple group datasets of slice electrophysiology recordings, repeated measures ANOVA analysis was used, followed by Dunnett’s post-hoc tests. Statistical tests used to measure significance are indicated in each figure legend along with the corresponding significance level (p value).
Results
The N-GAPDH cascade underlies cognitive inflexibility in a stress-induced mouse model
To test whether the N-GAPDH cascade plays a mechanistic role linking stress and behavior at the molecular level, we used the LSM model (Fig. 1a). We observed an excess of oxidative stress and activation of the N-GAPDH cascade in the brains of LSM mice (Supplementary Fig. 2a, b). In contrast, we did not observe any sign of blood brain barrier damage, nor cell death in the brain (Supplementary Fig. 2c–f). Signs of sickness behavior were not present 24 h after treatment [43], when all biochemical or behavioral outcomes were recorded. No changes in locomotor and swimming activity were observed in LSM mice compared to control mice (Supplementary Fig. 2g, h). However, LSM mice presented significant deficits in cognitive flexibility as measured by the rule shifting task, but showed no impairment in learning the initial association portion of the task (Fig. 1b, c and Supplementary Fig. 2i, j). Moreover, we observed that LSM mice performed more perseverant errors than control mice (Fig. 1d). These results allowed us to conclude that LSM mice were less flexible, requiring more attempts in order to disengage from the paradigm that they learned during the initial association.
To test whether the N-GAPDH cascade is involved in the regulation of this cognitive construct, we used the RR pharmacological compound to specifically block activation of the N-GAPDH cascade by interfering with the binding of GAPDH and Siah1, without affecting the glycolytic activity of GAPDH and monoamine oxidase activity (Supplementary Fig. 3a–d). We confirmed in vitro that this compound blocked the GAPDH-Siah1 protein interaction more potently than other drug analogs (Supplementary Fig. 3e). In addition, we demonstrated that RR reached the brain giving nanomolar concentrations (41.5 ± 5.1 pmol/g) when it was systemically injected, and even displayed a unique (3-fold) enrichment in the brain versus serum (Supplementary Fig. 3f). Therefore, we systemically administered the RR compound in LSM mice to evaluate its impact on behavioral flexibility. We confirmed that the initiation of the N-GAPDH cascade in the brain was blocked by the systemic injection of RR (RR LSM mice) (Supplementary Fig. 2b). Interestingly, the RR injection also successfully blocked the behavioral deficits seen in the rule-shifting paradigm. In particular, RR significantly reduced the number of perseverant errors (Fig. 1c, d). These results indicate that the N-GAPDH cascade is activated in response to LSM treatment and underlies cognitive inflexibility.
Stress-induced upregulation of the N-GAPDH cascade occurs selectively in microglia of LSM mouse model
Since activation of the N-GAPDH cascade was responsible for the behavioral inflexibility deficits observed in LSM mice, we decided to determine the cellular specificity of the N-GAPDH activation. We focused on microglia and astrocytes because they are the main cell types involved in the provocation and resolution of inflammatory and oxidative stress related processes in the brain [66,67,68]. Interestingly, we observed a significant upregulation of N-GAPDH only in microglia, as reflected by an increased binding of GAPDH and Siah1 in these cells (Fig. 2a, b). A specific antibody against sulfonation of GAPDH at cysteine-150 (SulfoGAPDH Ab) [49, 56] also confirmed activation of this cascade in microglia in the medial prefrontal cortex (Fig. 2c and Supplementary Fig. 4), but not in astrocytes or neurons. Furthermore, we observed much stronger activation of this cascade in cortical microglia compared to striatal microglia (Fig. 2d). As expected, systemic administration of the RR compound blocked activation of the N-GAPDH cascade in microglia (Fig. 2e).
The N-GAPDH cascade in microglia mediates stress-induced cognitive inflexibility (deficits in a rule-shifting behavior) in LSM mouse model
In addition to the pharmacological intervention by RR, we generated a conditional mouse model designed to disrupt GAPDH-Siah1 binding and thus prevent activation of the N-GAPDH cascade under stressors [52]. We previously published that a point mutation that replaces lysine 225 of GAPDH with alanine (K225A) can abolish the GAPDH-Siah1 binding without affecting other key features of GAPDH, including its glycolytic activity [49, 52]. To specifically block the N-GAPDH cascade in microglia in the central nervous system, we generated GAPDH-K225ACx3cr1-CreER knockin mice (K225A-MG mice) (Supplementary Fig. 5). As expected, we did not observe an upregulation of the N-GAPDH cascade in these knockin mice even when these mice were repeatedly treated with LPS (K225A-MG LSM) (Fig. 3a). Further, this microglia-specific genetic manipulation to block the N-GAPDH cascade successfully prevented the behavioral deficits in the rule-shifting paradigm including a reduced number of perseverant errors in K225A-MG LSM mice (Fig. 3b, c).

a Level of GAPDH-Siah1 binding for cortical microglia. Siah1 immunoprecipitations and immunoblots for GAPDH in Control [Cx3cr1-CreER; WT-GAPDH]; K225A-MG, [Cx3cr1-CreER; K225A-GAPDH]; LSM (LSM on control background); and K225A-MG LSM mice. Two-way ANOVA with Tukey’s multiple comparisons test (genotype x LSM) revealed a significant main effect of genotype (F(1,8) = 14.57, p = 0.005), a significant effect of LSM (F(1,8) = 82.27, p < 0.0001), and a significant interaction between factors (F(1,8) = 54.64, p < 0.0001). Error bars represent the mean ± SEM. ***p < 0.001 by two-way ANOVA with Tukey’s multiple comparisons test. b Performance of Control, LSM, and K225A-MG LSM mice in the rule shifting task. Each dot represents data from an individual mouse. c LSM mice performed significantly more perseverant than random errors during the rule-shifting task. Each dot represents data from an individual mouse. Error bars represent the mean ± SEM. *p < 0.05 by one-way ANOVA with Tukey’s multiple comparisons test for (b) and (c).
The N-GAPDH cascade in microglia mediates hyperactivation of the prelimbic (PrL) layer 5 (L5) excitatory neurons in LSM mouse model
Hyperactivation of the PrL excitatory neurons is known as a key neural substrate of the deficit in rule shifting tasks [3, 69, 70]. We next tested whether the hyperactivation occurred in the LSM mouse model, which may possibly be normalized by the pharmacological and microglia-specific genetic intervention to the N-GAPDH cascade. To address this question, we used mice that selectively expressed GCaMP6s in VGlut1-positive excitatory neurons and specifically focused on the medial prefrontal cortex (the PrL area) [3]. To assess the spontaneous activity of individual excitatory neurons, we used a constrained non-negative matrix factorization (CNMF) algorithm optimized for slice imaging [64] that allowed us to determine the frequency and amplitude of calcium events (Supplementary Video 1). Calcium transients in PrL excitatory neurons were more frequent in LSM compared with control mice (Fig. 4a), whereas amplitude levels were similar (Fig. 4a). These data indicate a plausible increase in neuronal excitability and an overall cytosolic calcium overload in LSM excitatory neurons. As shown before, no neuronal cell death was observed in LSM mice (Supplementary Fig. 2d–f). Treatment of LSM mice with RR ameliorated the increased frequency of calcium transients, suggesting a causal involvement of microglial activation of the N-GAPDH cascade (Fig. 4a). The causal role of microglial N-GAPDH cascade was directly shown by no increase in the frequency of calcium transients in K225A-MG LSM mice that selectively expressed GCaMP6s in excitatory neurons (Fig. 4b). In contrast, the frequency of calcium transients and amplitude levels were not different in the prelimbic L5 VGAT-positive inhibitory neurons between control and LSM mice (Fig. 4c), indicating that the impact of the microglial N-GAPDH cascade is selectively on the excitatory neurons. These data indicate increased excitation in the prelimbic L5 excitatory neurons of LSM mice, which is normalized by genetic intervention with the N-GAPDH cascade in microglia.

a Frequency and peak amplitude for 47 VGlut1-positive cells from 4 Control mice, 57 VGlut1-positive cells from 4 LSM mice, and 89 VGlut1-positive cells from 4 RR LSM mice. b Frequency and peak amplitude for 68 VGlut1-positive cells from 3 Control mice, 238 VGlut1-positive cells from 3 LSM mice, and 171 VGlut1-positive cells from 3 K225A-MG LSM mice. c Frequency and peak amplitude for 169 VGAT-positive cells from 3 Control mice, and 119 VGAT-positive cells from 5 LSM mice. For each panel, error bars represent the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA with Tukey’s multiple comparisons test for (a) and (b). No statistical difference by a two-tailed unpaired Student’s t-test for (c). Representative traces are presented for each group.
High-Mobility Group Box 1 (HMGB1): a potential mediator underlying the microglia-neuron link
Next, we looked for a potential mediator that links microglial activation of N-GAPDH cascade to the neuronal changes. HMGB1 is upregulated in activated microglia and is also known to bind with and regulate the NMDA-glutamate receptors in neurons once released from microglia [71,72,73,74,75]. Thus, we tested whether HMGB1 protein that was augmented in microglia of LSM mice (Fig. 5a) contributed to the microglia-neuron link. The upregulation of HMGB1 was due to the N-GAPDH cascade, because both RR and K225A-MG interventions in LSM led to the amelioration of the level (Fig. 5a). RR did not affect the level of HMGB1 in control mice (Supplementary Fig. 6a) and K225A-MG itself did not alter the level of HMGB1 (Supplementary Fig. 6b), both of which indicated that the effects of RR was via the N-GAPDH cascade. Three-dimensional (3D) reconstruction from confocal images indicated an augmentation of HMGB1 in lysosome in LSM mice, but not in LSM with RR treatment, nor K225A-MG LSM mice (Fig. 5b, Supplementary Fig. 6c). Thus, we further hypothesized that an upregulation of lysosomal HMGB1 as a result of N-GAPDH cascade activation might result in its release from microglia and an activation of adjacent neurons via its binding with the NMDA receptors. To test this hypothesis, we applied an antibody against HMGB1 to the brain slice: we observed an amelioration of the frequency of calcium transients in prelimbic L5 excitatory neurons in LSM mice (Fig. 5c).

a Levels of HMGB1 protein normalized by the levels of H3 protein in lysates from cortical microglia isolated from Control, LSM, RR LSM, and K225A-MG LSM mice. Immunoblots are representative of 6 independent experiments from cortical microglia isolated from 5 mice per group. b Quantification of lysosomal HMGB1 using volumetric analysis of 3D microglia stack reconstructions from Control, LSM, and RR LSM mice. Microglia and lysosomes were stained with an antibody against Iba1 and CD68, respectively. Analysis was performed using Imaris as previously described. Scale bar: 10 μm. Confocal images are representative of 8 z-stack images from 2 mice per group. c Frequency and peak amplitude for 47 VGlut1-positive cells from 4 Control mice, 57 VGlut1-positive cells from 4 LSM mice, and 111 VGlut1-positive cells acutely treated with HMGB1 antibody (Ab) from 4 LSM mice. Representative traces are presented for each group. Control and LSM mice data are same as shown in Fig. 4a. For each panel, error bars represent the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA with Tukey’s multiple comparisons test.
Discussion
The present study provides two major findings of neurobiology and cellular signaling, respectively. First, by using LSM mouse model, we show that stress-induced deficits in rule shifting tasks are mediated by a specific molecular cascade (the N-GAPDH cascade) in microglia, which in turn elicits hyperactivation of the prelimbic excitatory neurons. Previously, the involvement of the neuronal changes in cognitive inflexibility was shown in a genetic model that interferes with neuronal development. In contrast, we now demonstrate a pivotal role of microglia and microglia-neuron crosstalk in stress-induced cognitive inflexibility. Second, we also decipher a mechanism whereby the N-GAPDH cascade mediates stress-induced behavioral alteration, which has been an important question in cellular signaling.
We indicate a causal role of the microglial N-GAPDH cascade in neuronal hyperactivation and behavioral changes. Key questions for the next step will include mechanism(s) that links nuclear translocated GAPDH in microglia to downstream neuronal hyperactivity. By choosing a candidate molecular approach, we underscored HMGB1 as a potential mediator for this link. Nuclear translocated GAPDH may systematically influence transcriptional landscape in the microglia, given that GAPDH can reportedly interact with DNAs directly or indirectly [45, 76]. To address the question experimentally, next generation sequencing at the whole genome level will be useful. Whether and how the N-GAPDH cascade is pathologically manifested in a stress-induced manner in patients with brain disorders may be another outstanding question. To address this question properly, we also need to be sensitive to which cells from patients will be selected for the experiments.
Prior to the present study, to our best knowledge, the N-GAPDH cascade in the brain has been studied in three distinct animal models. In one study, a role for the N-GAPDH cascade for cocaine-induced neuronal death in the striatum was studied [77]. Likewise, this cascade also contributes to neuronal death elicited by the middle cerebral artery occlusion [78]. Different from these two acute stress models that result in neuronal cell death, the N-GAPDH cascade is reportedly upregulated in dominant-negative DISC1 transgenic mice in which neurodevelopmental-dependent neuronal dysfunction is reported [54]. In contrast to these three models, the present study reports the activation of N-GAPDH cascade in microglia. Altogether, these indicate the significance of studying the N-GAPDH cascade in a context dependent manner, and the comparison of the N-GAPDH cascade among these models may shed light on mechanistic regulations of this cascade. The present report may also offer a prototype of future studies that explore other mechanisms for cognitive alterations elicited by different stressors. In analogy to the microglia-specific genetic intervention selectively disrupting the initial regulatory step for the N-GAPDH cascade that was used in the present study, mouse models that target a specific molecular cascade in a cell type-specific manner will be crucial for future studies.
Altogether, we provide a novel conceptual framework that an enzyme, which is traditionally considered for cellular homeostasis through glycolysis, regulates microglial function for adaptive behavior in response to stressors. We speculate that GAPDH is not the only enzyme to have such a unique role. The nuclear localization of other glycolytic enzymes has been historically reported [79, 80], however, their role (e.g., transcriptional regulation) remains elusive. We believe that the present study represents the tip of the iceberg for higher-level mechanisms that involve the regulation of homeostasis by bioenergetic enzymes, a mechanism that has probably evolved from cellular to complex systems.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Ionescu T. Exploring the nature of cognitive flexibility. N Ideas Psychol. 2012;30:190–200.
Spellman T, Svei M, Kaminsky J, Manzano-Nieves G, Liston C. Prefrontal deep projection neurons enable cognitive flexibility via persistent feedback monitoring. Cell. 2021;184:2750–66.e17.
Cho KK, Hoch R, Lee AT, Patel T, Rubenstein JL, Sohal VS. Gamma rhythms link prefrontal interneuron dysfunction with cognitive inflexibility in Dlx5/6(+/-) mice. Neuron. 2015;85:1332–43.
Stokes MG, Buschman TJ, Miller EK. The Wiley Handbook of Cognitive Control. Hoboken: Wiley-Blackwell; 2017.
Arnsten AF. Stress signalling pathways that impair prefrontal cortex structure and function. Nat Rev Neurosci. 2009;10:410–22.
Diamond A. Executive functions. Annu Rev Psychol. 2013; 64:135–68.
Plessow F, Fischer R, Kirschbaum C, Goschke T. Inflexibly focused under stress: acute psychosocial stress increases shielding of action goals at the expense of reduced cognitive flexibility with increasing time lag to the stressor. J Cogn Neurosci. 2011;23:3218–27.
Baudic S, Barba GD, Thibaudet MC, Smagghe A, Remy P, Traykov L. Executive function deficits in early Alzheimer’s disease and their relations with episodic memory. Arch Clin Neuropsychol. 2006;21:15–21.
Floresco SB, Zhang Y, Enomoto T. Neural circuits subserving behavioral flexibility and their relevance to schizophrenia. Behav Brain Res. 2009;204:396–409.
Gruber AJ, Calhoon GG, Shusterman I, Schoenbaum G, Roesch MR, O’Donnell P. More is less: a disinhibited prefrontal cortex impairs cognitive flexibility. J Neurosci. 2010;30:17102–10.
Malloy P, McLaughlin NCR. Neuropsychology of Everyday Functioning. New York: Guilford Publications; 2010.
Marshall GA, Rentz DM, Frey MT, Locascio JJ, Johnson KA, Sperling RA, et al. Executive function and instrumental activities of daily living in mild cognitive impairment and Alzheimer’s disease. Alzheimers Dement. 2011;7:300–8.
Coughlin JM, Yang K, Marsman A, Pradhan S, Wang M, Ward RE, et al. A multimodal approach to studying the relationship between peripheral glutathione, brain glutamate, and cognition in health and in schizophrenia. Mol Psychiatry. 2021;26:3502–11.
Kwon HS, Koh SH. Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener. 2020; 9:42.
Wang AM, Pradhan S, Coughlin JM, Trivedi A, DuBois SL, Crawford JL, et al. Assessing brain metabolism with 7-T proton magnetic resonance spectroscopy in patients with first-episode psychosis. JAMA Psychiatry. 2019;76:314–23.
Coughlin JM, Ishizuka K, Kano SI, Edwards JA, Seifuddin FT, Shimano MA, et al. Marked reduction of soluble superoxide dismutase-1 (SOD1) in cerebrospinal fluid of patients with recent-onset schizophrenia. Mol Psychiatry. 2013;18:10–1.
Coughlin JM, Hayes LN, Tanaka T, Xiao M, Yolken RH, Worley P, et al. Reduced superoxide dismutase-1 (SOD1) in cerebrospinal fluid of patients with early psychosis in association with clinical features. Schizophr Res. 2017;183:64–9.
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–34.
Hammond TR, Marsh SE, Stevens B. Immune signaling in neurodegeneration. Immunity. 2019;50:955–74.
Hayes LN, Severance EG, Leek JT, Gressitt KL, Rohleder C, Coughlin JM, et al. Inflammatory molecular signature associated with infectious agents in psychosis. Schizophr Bull. 2014;40:963–72.
Tanaka T, Matsuda T, Hayes LN, Yang S, Rodriguez K, Severance EG, et al. Infection and inflammation in schizophrenia and bipolar disorder. Neurosci Res. 2017;115:59–63.
Ransohoff RM. How neuroinflammation contributes to neurodegeneration. Science. 2016;353:777–83.
Rolstad S, Jakobsson J, Sellgren C, Isgren A, Ekman CJ, Bjerke M, et al. CSF neuroinflammatory biomarkers in bipolar disorder are associated with cognitive impairment. Eur Neuropsychopharmacol. 2015;25:1091–8.
Janelidze S, Mattsson N, Stomrud E, Lindberg O, Palmqvist S, Zetterberg H, et al. CSF biomarkers of neuroinflammation and cerebrovascular dysfunction in early Alzheimer disease. Neurology. 2018;91:e867–e77.
Clark AL, Weigand AJ, Thomas KR, Solders SK, Delano-Wood L, Bondi MW, et al. Elevated inflammatory markers and arterial stiffening exacerbate tau but not amyloid pathology in older adults with mild cognitive impairment. J Alzheimers Dis. 2021;80:1451–63.
Dawson TM, Golde TE, Lagier-Tourenne C. Animal models of neurodegenerative diseases. Nat Neurosci. 2018;21:1370–9.
Gotz J, Bodea LG, Goedert M. Rodent models for Alzheimer disease. Nat Rev Neurosci. 2018;19:583–98.
Landek-Salgado MA, Faust TE, Sawa A. Molecular substrates of schizophrenia: homeostatic signaling to connectivity. Mol Psychiatry. 2016;21:10–28.
Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain Behav Immun. 2008;22:469–86.
Bavley CC, Kabir ZD, Walsh AP, Kosovsky M, Hackett J, Sun H, et al. Dopamine D1R-neuron cacna1c deficiency: a new model of extinction therapy-resistant post-traumatic stress. Mol Psychiatry. 2021;26:2286–98.
Chen S, Zhang H, Pu H, Wang G, Li W, Leak RK, et al. n-3 PUFA supplementation benefits microglial responses to myelin pathology. Sci Rep. 2014;4:7458.
Kroener S, Mulholland PJ, New NN, Gass JT, Becker HC, Chandler LJ. Chronic alcohol exposure alters behavioral and synaptic plasticity of the rodent prefrontal cortex. PLoS One. 2012;7:e37541.
Shin Yim Y, Park A, Berrios J, Lafourcade M, Pascual LM, Soares N, et al. Reversing behavioural abnormalities in mice exposed to maternal inflammation. Nature. 2017;549:482–7.
Tezuka T, Tamura M, Kondo MA, Sakaue M, Okada K, Takemoto K, et al. Cuprizone short-term exposure: astrocytic IL-6 activation and behavioral changes relevant to psychosis. Neurobiol Dis. 2013;59:63–8.
Zhao J, Bi W, Xiao S, Lan X, Cheng X, Zhang J, et al. Neuroinflammation induced by lipopolysaccharide causes cognitive impairment in mice. Sci Rep. 2019;9:5790.
Beier EE, Neal M, Alam G, Edler M, Wu LJ, Richardson JR. Alternative microglial activation is associated with cessation of progressive dopamine neuron loss in mice systemically administered lipopolysaccharide. Neurobiol Dis. 2017;108:115–27.
Chen Z, Jalabi W, Shpargel KB, Farabaugh KT, Dutta R, Yin X, et al. Lipopolysaccharide-induced microglial activation and neuroprotection against experimental brain injury is independent of hematogenous TLR4. J Neurosci. 2012;32:11706–15.
Wendeln AC, Degenhardt K, Kaurani L, Gertig M, Ulas T, Jain G, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556:332–8.
Norden DM, Trojanowski PJ, Villanueva E, Navarro E, Godbout JP. Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia. 2016;64:300–16.
He Y, Wang Y, Yu H, Tian Y, Chen X, Chen C, et al. Protective effect of Nr4a2 (Nurr1) against LPS-induced depressive-like behaviors via regulating activity of microglia and CamkII neurons in anterior cingulate cortex. Pharm Res. 2023;191:106717.
Tian Y, Chen X, Wang Y, He Y, Chen C, Yu H, et al. Neuroinflammatory transcriptional signatures in the entorhinal cortex based on lipopolysaccharide-induced depression model in mice. Biochem Biophys Res Commun. 2022;590:109–16.
Siemsen BM, Landin JD, McFaddin JA, Hooker KN, Chandler LJ, Scofield MD. Chronic intermittent ethanol and lipopolysaccharide exposure differentially alter Iba1-derived microglia morphology in the prelimbic cortex and nucleus accumbens core of male Long-Evans rats. J Neurosci Res. 2021;99:1922–39.
Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56.
Shahani N, Sawa A. Nitric oxide signaling and nitrosative stress in neurons: role for S-nitrosylation. Antioxid Redox Signal. 2011;14:1493–504.
Tristan C, Shahani N, Sedlak TW, Sawa A. The diverse functions of GAPDH: views from different subcellular compartments. Cell Signal. 2011;23:317–23.
Zala D, Hinckelmann MV, Yu H, Lyra da Cunha MM, Liot G, Cordelieres FP, et al. Vesicular glycolysis provides on-board energy for fast axonal transport. Cell. 2013;152:479–91.
Seidler NW. GAPDH and intermediary metabolism. Adv Exp Med Biol. 2013;985:37–59.
Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–51.
Hara MR, Agrawal N, Kim SF, Cascio MB, Fujimuro M, Ozeki Y, et al. S-nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat Cell Biol. 2005;7:665–74.
Sawa A, Khan AA, Hester LD, Snyder SH. Glyceraldehyde-3-phosphate dehydrogenase: nuclear translocation participates in neuronal and nonneuronal cell death. Proc Natl Acad Sci USA. 1997;94:11669–74.
Hara MR, Thomas B, Cascio MB, Bae BI, Hester LD, Dawson VL, et al. Neuroprotection by pharmacologic blockade of the GAPDH death cascade. Proc Natl Acad Sci USA. 2006;103:3887–9.
Zhang M, Kariya T, Genri N, Sasaki H, Sasaki M, Ramos A, et al. Nuclear GAPDH cascade mediates pathological cardiac hypertrophy. bioRxiv. 2024; https://doi.org/10.1101/844902.
Guha P, Harraz MM, Snyder SH. Cocaine elicits autophagic cytotoxicity via a nitric oxide-GAPDH signaling cascade. Proc Natl Acad Sci USA. 2016;113:1417–22.
Johnson AW, Jaaro-Peled H, Shahani N, Sedlak TW, Zoubovsky S, Burruss D, et al. Cognitive and motivational deficits together with prefrontal oxidative stress in a mouse model for neuropsychiatric illness. Proc Natl Acad Sci USA. 2013;110:12462–7.
Moore H, Geyer MA, Carter CS, Barch DM. Harnessing cognitive neuroscience to develop new treatments for improving cognition in schizophrenia: CNTRICS selected cognitive paradigms for animal models. Neurosci Biobehav Rev. 2013;37:2087–91.
Sen N, Hara MR, Kornberg MD, Cascio MB, Bae BI, Shahani N, et al. Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat Cell Biol. 2008;10:866–73.
Sahasrabuddhe V, Ghosh HS. Cx3Cr1-Cre induction leads to microglial activation and IFN-1 signaling caused by DNA damage in early postnatal brain. Cell Rep. 2022;38:110252.
McKinsey GL, Lizama CO, Keown-Lang AE, Niu A, Santander N, Larpthaveesarp A, et al. A new genetic strategy for targeting microglia in development and disease. Elife. 2020;9:e54590.
Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci. 2019;22:1021–35.
Heisler JM, Morales J, Donegan JJ, Jett JD, Redus L, O’Connor JC. The attentional set shifting task: a measure of cognitive flexibility in mice. J Vis Exp. 2015;96:51944.
Feng S, Zou L, Wang H, He R, Liu K, Zhu H. RhoA/ROCK-2 pathway inhibition and tight junction protein upregulation by catalpol suppresses lipopolysaccaride-induced disruption of blood-brain barrier permeability. Molecules. 2018;23:2371.
Agarwal A, Wu PH, Hughes EG, Fukaya M, Tischfield MA, Langseth AJ, et al. Transient opening of the mitochondrial permeability transition pore induces microdomain calcium transients in astrocyte processes. Neuron. 2017;93:587–605 e7.
Zhou P, Resendez SL, Rodriguez-Romaguera J, Jimenez JC, Neufeld SQ, Giovannucci A, et al. Efficient and accurate extraction of in vivo calcium signals from microendoscopic video data. Elife. 2018;7:e28728.
Pnevmatikakis EA, Soudry D, Gao Y, Machado TA, Merel J, Pfau D, et al. Simultaneous denoising, deconvolution, and demixing of calcium imaging data. Neuron. 2016;89:285–99.
Murugan M, Jang HJ, Park M, Miller EM, Cox J, Taliaferro JP, et al. Combined social and spatial coding in a descending projection from the prefrontal cortex. Cell. 2017;171:1663–77.e16.
Colombo E, Farina C. Astrocytes: key regulators of neuroinflammation. Trends Immunol. 2016;37:608–20.
Colonna M, Butovsky O. Microglia function in the central nervous system during health and neurodegeneration. Annu Rev Immunol. 2017;35:441–68.
Sochocka M, Diniz BS, Leszek J. Inflammatory response in the CNS: friend or foe? Mol Neurobiol. 2017;54:8071–89.
Enomoto T, Tse MT, Floresco SB. Reducing prefrontal gamma-aminobutyric acid activity induces cognitive, behavioral, and dopaminergic abnormalities that resemble schizophrenia. Biol Psychiatry. 2011;69:432–41.
Paine TA, Slipp LE, Carlezon WA Jr. Schizophrenia-like attentional deficits following blockade of prefrontal cortex GABAA receptors. Neuropsychopharmacology. 2011;36:1703–13.
Lee S, Nam Y, Koo JY, Lim D, Park J, Ock J, et al. A small molecule binding HMGB1 and HMGB2 inhibits microglia-mediated neuroinflammation. Nat Chem Biol. 2014;10:1055–60.
Shi Y, Guo X, Zhang J, Zhou H, Sun B, Feng J. DNA binding protein HMGB1 secreted by activated microglia promotes the apoptosis of hippocampal neurons in diabetes complicated with OSA. Brain Behav Immun. 2018;73:482–92.
Fu L, Liu K, Wake H, Teshigawara K, Yoshino T, Takahashi H, et al. Therapeutic effects of anti-HMGB1 monoclonal antibody on pilocarpine-induced status epilepticus in mice. Sci Rep. 2017;7:1179.
Nestor J, Arinuma Y, Huerta TS, Kowal C, Nasiri E, Kello N, et al. Lupus antibodies induce behavioral changes mediated by microglia and blocked by ACE inhibitors. J Exp Med. 2018;215:2554–66.
Pedrazzi M, Averna M, Sparatore B, Patrone M, Salamino F, Marcoli M, et al. Potentiation of NMDA receptor-dependent cell responses by extracellular high mobility group box 1 protein. PLoS One. 2012;7:e44518.
Colell A, Green DR, Ricci JE. Novel roles for GAPDH in cell death and carcinogenesis. Cell Death Differ. 2009;16:1573–81.
Xu R, Serritella AV, Sen T, Farook JM, Sedlak TW, Baraban J, et al. Behavioral effects of cocaine mediated by nitric oxide-GAPDH transcriptional signaling. Neuron. 2013;78:623–30.
Nakajima H, Kubo T, Ihara H, Hikida T, Danjo T, Nakatsuji M, et al. Nuclear-translocated Glyceraldehyde-3-phosphate Dehydrogenase Promotes Poly(ADP-ribose) polymerase-1 activation during oxidative/nitrosative stress in stroke. J Biol Chem. 2015;290:14493–503.
Boukouris AE, Zervopoulos SD, Michelakis ED. Metabolic enzymes moonlighting in the nucleus: metabolic regulation of gene transcription. Trends Biochem Sci. 2016;41:712–30.
Yu X, Li S. Non-metabolic functions of glycolytic enzymes in tumorigenesis. Oncogene. 2017;36:2629–36.
Acknowledgements
We would like to thank Hao Zhang and the Flow Cytometry Core at JHSPH for providing sorting and analysis services. We thank Dr. Masayuki Sasaki for technical support. We thank Yukiko Lema for figure and manuscript organization. We also appreciate Melissa Landek-Salgado and Richard Farrell for thoughtful comments and editions of this manuscript. This work was supported by the grants from the National Institute of Health (MH-094268 Silvio O. Conte center, MH-105660, MH-107730), as well as the grants from NARSAD, Stanley, S-R/RUSK (to AS). This work was also supported by the Subsidies for Current Expenditures to Private Institutions of Higher Education from the Promotion and Mutual Aid Corporation for Private Schools of Japan (to KI).
Author information
Authors and Affiliations
Contributions
AS (Sawa) conceived the general ideas for this study. AR, KI, and AS (Sawa) designed experiments. AR, KI, AH, HN, LNH, RS, MZ, TK, NE, TP, EC, TT, CC, RR, MN, and AS performed the experiments and data analysis with guidance from SI, BSS, TS, ET, and AS (Sawa). AR, KI, and AS (Sawa) drafted the manuscript. All authors contributed to the discussion of the results and have approved the final manuscript to be published.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ramos, A., Ishizuka, K., Hayashida, A. et al. Nuclear GAPDH in cortical microglia mediates cellular stress-induced cognitive inflexibility. Mol Psychiatry (2024). https://doi.org/10.1038/s41380-024-02553-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41380-024-02553-1
- Springer Nature Limited