
Abstract
Post mortem neuropathology suggests that astrocyte reactivity may play a significant role in neurodegeneration in Alzheimer’s disease. We explored this in vivo using multimodal PET and MRI imaging. Twenty subjects (11 older, cognitively impaired patients and 9 age-matched healthy controls) underwent brain scanning using the novel reactive astrocyte PET tracer 11C-BU99008, 18F-FDG and 18F-florbetaben PET, and T1-weighted MRI. Differences between cognitively impaired patients and healthy controls in regional and voxel-wise levels of astrocyte reactivity, glucose metabolism, grey matter volume and amyloid load were explored, and their relationship to each other was assessed using Biological Parametric Mapping (BPM). Amyloid beta (Aβ)-positive patients showed greater 11C-BU99008 uptake compared to controls, except in the temporal lobe, whilst further increased 11C-BU99008 uptake was observed in Mild Cognitive Impairment subjects compared to those with Alzheimer’s disease in the frontal, temporal and cingulate cortices. BPM correlations revealed that regions which showed reduced 11C-BU99008 uptake in Aβ-positive patients compared to controls, such as the temporal lobe, also showed reduced 18F-FDG uptake and grey matter volume, although the correlations with 18F-FDG uptake were not replicated in the ROI analysis. BPM analysis also revealed a regionally-dynamic relationship between astrocyte reactivity and amyloid uptake: increased amyloid load in cortical association areas of the temporal lobe and cingulate cortices was associated with reduced 11C-BU99008 uptake, whilst increased amyloid uptake in primary motor and sensory areas (in which amyloid deposition occurs later) was associated with increased 11C-BU99008 uptake. These novel observations add to the hypothesis that while astrocyte reactivity may be triggered by early Aβ-deposition, sustained pro-inflammatory astrocyte reactivity with greater amyloid deposition may lead to astrocyte dystrophy and amyloid-associated neuropathology such as grey matter atrophy and glucose hypometabolism, although the evidence for glucose hypometabolism here is less strong.
Similar content being viewed by others


Introduction
Astrocytes are integral to normal brain function, playing important roles in neurogenesis, synaptogenesis, control of blood-brain barrier permeability and maintaining extracellular homeostasis [1]. In Alzheimer’s disease (AD), astrocytes can assume a reactive phenotype in response to disease by undergoing morphological, molecular and functional remodeling [2]. While astrocyte reactivity is associated with amyloid beta (Aβ) plaques [3], the precise role of this astrocyte reactivity in neurodegeneration is still unclear. It is suggested that astrocytes could have a beneficial and detrimental role, and this could also depend on the pathological insult and the susceptibility of the host [4]. It is proposed that with higher levels of Aβ, astrocyte reactivity can produce neurotoxic reactive oxygen species and inflammatory cytokines [5]. Astrocytes in AD also can lose normal neuroprotective capabilities as they become dystrophic with the progression of AD pathology [4, 6].
Glucose hypometabolism, measured using 18F-fluorodeoxyglucose (18F-FDG) PET, and brain atrophy, measured using MRI, are two of the earliest neuroimaging markers of neurodegeneration developed for AD [7]. However, both measures are sensitive to changes in astrocyte reactivity [8, 9], and the contribution of FDG signal from astrocyte reactivity is yet to be fully elucidated. By contributing to synaptic loss and neurodegeneration, pro-inflammatory and dystrophic astrocytes may be associated with accelerated grey matter (GM) atrophy [10]. Astrocytes are also necessary for metabolic support of neuronal activity [11], so AD-related changes in astrocytes might contribute directly to the brain glucose hypometabolism characteristic of AD [7].
The novel PET tracer 11C-BU99008 has high specificity and selectivity for binding sites of type-2 imidazoline receptors (I2-BS), which are expressed primarily within astrocytes and are upregulated with reactivity [12]. This tracer thus allows the study of astrocyte reactivity in vivo [13,14,15,16,17,18]. Pathologically increased 11C-BU99008 PET signal recently has been demonstrated in neurodegenerative disorders including AD [19] and Parkinson’s disease [20]. Currently, the only available PET tracer which can measure astrocyte reactivity in vivo is 11C-deuterium-L-deprenyl (11C-DED) [21, 22]. However, this tracer binds to monoamine oxidase-B (MAO-B), which is reduced in the presence of late stage Aβ-deposition. Therefore, it remains unclear if the lower 11C-DED binding observed in late-stage compared to early-stage AD subjects reflects a reduction in astrocyte reactivity or simply lower levels of MAO-B [23]. The higher specific binding of 11C-BU99008 than 11C-DED to detect astrocyte reactivity has recently been demonstrated in post mortem brain tissue from AD patients [24]—likely due to the fact they will be detecting different astrocyte subtypes—and thus 11C-BU99008 warrants further study in this clinical population. The aim of this study was to evaluate the relationship between astrocyte reactivity, using 11C-BU99008 PET, and glucose metabolism, GM atrophy and Aβ-deposition in cognitively impaired patients with a clinical diagnosis of AD-related dementia or Mild Cognitive Impairment (MCI).
Materials and methods
We recruited 20 subjects for this pilot study. Ethical approval was obtained from the local and regional Research Ethics Committee, whilst approval to administer radiotracers was obtained from the Administration of Radioactive Substances Advisory Committee UK. The human biological samples sourced from participants were obtained ethically and their research use was in accordance with the terms of the informed consent.
Subjects
Subjects were recruited from memory clinics, research registries and advertisements. We included 11 cognitively impaired patients with a clinical diagnosis of AD-related dementia or MCI (6 clinically diagnosed AD, 5 MCI; Mini-Mental Status Examination (MMSE) score [mean ± SD] = 22.6 ± 4.1) and 9 age-matched healthy controls (MMSE score [mean ± SD] = 29.1 ± 1.27) without a history of brain disease (Table 1). The inclusion criteria for cognitively impaired patients included the ability to give informed consent, an MMSE score ≥17 and at least 8 years of education. Exclusion criteria for all participants included contradictions to MRI and any evidence of significant small vessel or vascular disease on MRI. AD patients were defined according to a clinical diagnosis based on previous MRI and/or FDG PET, and Aβ imaging from this study was not used as an inclusion/exclusion criteria, nor were AD patients excluded who were Aβ-negative according to our analysis. All subjects underwent medical and detailed cognitive assessments using the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS), as well as 11C-BU99008, 18F-FDG and 18F-florbetaben PET and T1-weighted structural MRI. Aβ-positivity was defined by using a whole brain uptake cut-off of 1.43 [25].
Image acquisition
All image acquisition was performed at the Invicro Centre for Imaging Sciences in London, UK. MRI images were acquired using either a 3 Tesla Magnetom Trio or Verio (Siemens Healthcare Sector, Erlangen, Germany) with a 32-receiver channel head matrix coil. All PET imaging was performed on a Siemens Truepoint PET/CT scanner. All three PET scans were completed on separate days, with no longer than 30 days between the first and last scan completed. The order of the PET scans was not fixed and varied between participants depending on the availability of the PET tracers.
MRI
Structural MRI
All subjects underwent a sagittal T1-weighted MPRAGE, acquired with TR = 2400 ms, TE = 3.06 ms, flip angle = 9°, TI = 900 ms, matrix = [256 × 246], a 1 mm isotropic voxel size, anteroposterior phase encoding direction, IPAT factor 2 and a symmetric echo.
PET
11C-BU99008 PET
All subjects underwent 11C-BU99008 PET scanning to assess astrocyte reactivity in the brain. 11C-BU99008 was synthesised on site. An initial CT scan was acquired for attenuation correction of the PET images, before a mean activity of 330 (±30) MBq 11C-BU99008 in 20 ml normal saline was injected into the antecubital vein. Dynamic emission 11C-BU99008 PET images were acquired over 120 min and rebinned into 29 timeframes: 8 × 15 s, 3 × 60 s, 5 × 120 s, 5 × 300 s, and 8 × 600 s. All subjects had arterial blood sampled continuously for the first 15 min, with 12 additional samples taken at 5, 10, 15, 20, 25, 30, 40, 50, 60, 70, 80, and 100 min after injection. A gamma counter was used to measure radioactivity in the whole blood and plasma for each sample. Reverse-phase high-performance liquid chromatography was used to evaluate metabolism of 11C-BU99008 by calculating the relative proportions of parent tracer and metabolites in the blood. Parametric images (Impulse Response Function at 120 min (IRF-120)) of 11C-BU99008 were generated using spectral analysis. This was performed using Modelling, Input Functions and Compartmental Kinetics Parametric Map (MICK-PM) software (available on request from Wolfson Molecular Imaging Centre, University of Manchester, Manchester, UK).
18F-FDG and 18F-florbetaben PET
All subjects also underwent 18F-FDG and 18F-florbetaben PET scanning to assess glucose metabolism and Aβ-deposition in the brain, respectively. Subjects received a target dose of 185 MBq 18F-FDG and 236 MBq 18F-florbetaben as single intravenous boluses in the respective scanning sessions. For 18F-FDG scans, PET acquisition commenced 30 min after tracer injection, and the scans were acquired for 30 min. Using MICKPM, activity over the last 30 min was averaged, resulting in a 3D 30–60 min 18F-FDG add-image. For 18F-florbetaben scans, PET acquisition commenced 90 min after tracer administration and the subjects were scanned for 30 min. Activity over the 30 min acquisition period was averaged, resulting in a 3D 90–120 min 18F-florbetaben add-image.
Image processing
MRI and PET images were pre-processed using SPM12 (Wellcome centre for human neuroimaging, UCL, London, UK) in MATLAB (v2014a). 3D PET data was co-registered to the structural MRI of each subject. The structural MRI was segmented into GM, white matter (WM) and cerebrospinal fluid, and the GM and WM maps were used to generate a study-specific template using Diffeomorphic Anatomical Registration Through Exponential Lie Algebra (DARTEL) [26]. The DARTEL flow fields were then used to normalise each of the co-registered PET images and GM maps to MNI space and an 8 mm FWHM Gaussian kernel was used to smooth the data. Tracer uptake for 11C-BU99008 PET was calculated through spectral analysis (IRF-120 min). Tracer uptake for 18F-FDG and 18F-florbetaben PET was evaluated using the standardised uptake value ratio (SUVr) and the Hammers atlas [27], referenced to the pons GM/WM, and the cerebellar GM, respectively. This was done by dividing the cerebral cortical 18F-FDG and 18F-florbetaben mean images by the uptake value of the relevant reference region, which had been calculated in Analyse 11.0 (developed by the Biomedical Imaging Resource at the Mayo Clinic). This resulted in smoothed, normalised, 18F-FDG, 18F-florbetaben, 11C-BU99008 and GM Voxel-Based Morphometry (VBM) images that were used to assess glucose metabolism, Aβ deposition, astrocyte reactivity and GM atrophy patterns, respectively. This was done through Regions of Interest (ROI) analysis, as well as voxel-wise Statistical Parametric Mapping (SPM) and Biological Parametric Mapping (BPM) analysis. Partial Volume Correction (PVC) was not performed as PVC can increase noise within the data [28], leading to further signal leakage between regions and tissue classes which cannot be corrected for [29]. In addition, when comparing in vivo Aβ PET within subject with post mortem slices in AD subjects, PVC has been shown to have either no effect [30] or reduce [31] the accuracy of quantification of Aβ. Furthermore, as we are looking at increases for both 11C-BU99008 and 18F-Florbetaben, the PVC would have augmented the effect.
Statistical analysis
ROI analysis
Subject-specific object maps were created from the Hammers atlas [27, 32] and were used to sample the ROI radioactivity concentration for the three normalised (not smoothed) PET images, as well as the ROI volume of the VBM images. The ROIs included the frontal lobe, temporal lobe, medial temporal lobe, parietal lobe, occipital lobe, posterior cingulate and the whole brain (made up of the four lobes and the cingulate). Tracer uptake and GM volume for each ROI in cognitively impaired patients was compared against that of the healthy controls using a two-sample Student’s t test (two-tailed), with statistical difference set to p < 0.05. Due to the exploratory nature of this study, multiple comparison corrections were not performed. Correlation between each of the four imaging measures in each of the four lobes and whole brain for Aβ-positive patients was calculated using Pearson’s correlation coefficient in SPSS (v26, released 2019).
SPM analysis
Voxel-level SPM analysis was performed in order to better characterise the spatial distribution of tracer uptake difference between the cognitively impaired patients and the healthy controls. The three smoothed, normalised PET and VBM images of all subjects were entered into four separate two-sample Student’s t tests in SPM12 (two-tailed). Significant clusters were identified using cluster-level family wise error (FWE) corrected p values. Single subject analysis was also performed on each of the four modalities by comparing each patients’ images against a mean healthy control image in further separate two-sample Student’s t tests in SPM12 (two-tailed).
BPM correlation analysis
In order to assess the neuroanatomical relationship between 11C-BU99008 binding and glucose metabolism, Aβ deposition and GM atrophy, Z-score maps for each of the four imaging modalities were created. These represent tracer uptake and GM atrophy patterns relative to the healthy control’s mean and standard deviation for each subject on a voxel-level basis, calculated with the following formulae:
Voxel-level correlations between 11C-BU99008 and the remaining three modalities were estimated for Aβ-positive patients using BPM [33], an SPM toolbox that runs through MATLAB and SPM5. Due to the exploratory nature of this study, multiple comparison corrections were not performed.
Results
ROI and single subject SPM analysis
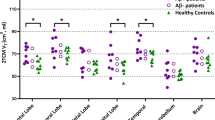
All the healthy controls were Aβ-negative, 7 of the patients were Aβ-positive (4 AD, 3 MCI) and the other 4 patients were Aβ-negative (2 AD, 2 MCI). One of the patients classified as Aβ-positive in the previous publication [19] (whole-brain uptake of 1.43779) was classified as Aβ-negative in this publication following the DARTEL analysis which reduced the whole-brain uptake (1.42195) to beneath the Aβ-positivity threshold used (1.43 3sf). Global tracer uptake (mean uptake ± SD) of 18F-florbetaben was 1.55 ± 0.29 for patients and 1.21 ± 0.06 for controls. Shapiro–Wilk’s tests confirmed data of global tracer uptake were normally distributed for patients (W(11) = 0.948, p = 0.616) and controls (W(9) = 0.956, p = 0.751). Brain 18F-florbetaben uptake was increased in patients compared to healthy controls (p = 0.003; Fig. 1d), with regional increases significant in the frontal (p = 0.004), temporal (p = 0.004), medial temporal (p = 0.045) and parietal (p = 0.003) lobes, as well as the posterior cingulate (p = 0.005) and hippocampus (p = 0.042).

Dot plots demonstrating regional changes in Aβ-positive patients (purple filled circle, N = 7), Aβ-negative patients (purple open circle, N = 4) and healthy controls (green triangle, N = 9): (a) 11C-BU99008 (b) 18F-FDG (c) VBM and (d) 18F-Florbetaben. Difference in tracer uptake and GM volume between Aβ-positive patients and healthy controls was analysed using two-sample Student’s t tests (two-tailed), with statistical difference set to p < 0.05. Multiple comparison corrections were not performed on the ROI results due to the exploratory nature of this pilot study. “Whole Brain” refers to the composite cortex, combining all the four lobes and the cingulate. * denotes p < 0.05.
Global tracer uptake (mean uptake ± SD) of 11C-BU99008 was 82.7 ± 11.5 for patients and 77.7 ± 7.7 for healthy controls. Shapiro–Wilk’s tests confirmed data of global tracer uptake were normally distributed for patients (W(11) = 0.942, p = 0.546) and controls (W(9) = 0.869, p = 0.095). Aβ-positive patients showed increased 11C-BU99008 brain uptake compared to healthy controls (p = 0.021; Fig. 1a). Regional analyses showed increases particularly in the frontal (p = 0.007), parietal (p = 0.018), and occipital (p = 0.039) lobes. An exploratory two-sample Student’s t test was run comparing MCI and AD subjects, demonstrating increased 11C-BU99008 uptake in MCI patients in the anterior (p = 0.023) and posterior (p = 0.007) cingulate.
Global tracer uptake (mean uptake ± SD) of 18F-FDG was 1.38 ± 0.22 for patients and 77.7 ± 7.7 for controls. Shapiro–Wilk’s tests confirmed data of global tracer uptake were normally distributed for patients (W(11) = 0.874, p = 0.114) and controls (W(9) = 0.087, p = 0.137). Aβ-positive patients showed decreased 18F-FDG brain uptake compared to healthy controls (p = 0.021; Fig. 1b). Regional analysis showed decreases in the frontal (p = 0.009), temporal (p < 0.001), medial temporal (p = 0.018), parietal (p < 0.001) and occipital (p < 0.001) lobes, as well as the posterior cingulate (p < 0.001) and hippocampus (p < 0.018).
Whole brain volume (mean volume ± SD) was 568272 ± 37709 for patients and 580365 ± 14566 for controls. Shapiro–Wilk’s tests confirmed data of whole brain volume were normally distributed for patients (W(11) = 0.963, p = 0.856) and controls (W(9) = 0.929, p = 0.513). Aβ-positive patients had lower whole brain GM volume than the healthy controls (p = 0.046; Fig. 1c). Regionally, decreases were significant in the temporal (p = 0.029), medial temporal (p = 0.009) and parietal (p = 0.020) lobes, as well as the posterior cingulate (p = 0.036) and hippocampus (p = 0.032).
SPM single subject analysis showed inter-subject and inter-regional heterogeneity in 11C-BU99008 uptake, and consistently reduced 18F-FDG uptake and GM atrophy in the temporal lobe and hippocampus of Aβ-positive patients (Fig. 2).

Rendered single subject SPM results from two-sample Student’s t tests (two-tailed) for differences in tracer uptake and GM volume between each patient and healthy controls (N = 9): (a) Single subject SPM analysis of Aβ-positive patients for 11C-BU99008, 18F -FDG, VBM and 18F-Florbetaben, rendered at cluster threshold of p < 0.05 with an extent threshold of 50 voxels. b Single subject SPM analysis of Aβ-negative patients for 11C-BU99008, 18F -FDG, VBM and 18F-Florbetaben, rendered at cluster threshold of p < 0.05 with an extent threshold of 50 voxels.
Please note, the ROI and single subject SPM results were not corrected for multiple comparisons due to the exploratory nature of the study.
Group-level SPM analysis
Two-sample Student’s t test in SPM contrasting Aβ-positive patients and healthy controls showed distributions of differences in tracer uptake and GM volumes that were consistent with the ROI-analyses (Fig. 3 and Supplementary Table 1). Following cluster-level FWE-correction, Aβ-positive patients had increased 11C-BU99008 uptake particularly in the frontal and occipital lobes (Fig. 3a), reduced 18F-FDG uptake in the temporal, parietal and occipital lobes (Fig. 3b), reduced GM volume in temporal regions, particularly the hippocampi (Fig. 3c), and increased 18F-florbetaben uptake in frontotemporal regions (Fig. 3d). An exploratory two-sample Student’s t test comparing MCI and AD subjects showed increased 11C-BU99008 uptake in MCI patients, particularly in the frontal and temporal regions (Supplementary Fig. 1 and Supplementary Table 1).

Rendered SPM results from two-sample Student’s t tests (two-tailed) for differences in tracer uptake and GM volume between patients (N = 11) and healthy controls (N = 9): (a) Increased 11C-BU99008 uptake in Aβ-positive patients (N = 7), rendered at cluster threshold of p < 0.05 and an extent threshold of 50 voxels. b Decreased 18F-FDG uptake in Aβ-positive patients (N = 7), rendered at cluster threshold of p < 0.001 and an extent threshold of 50 voxels. c Decreased GM volume in Aβ-positive patients (N = 7), rendered at cluster threshold of p < 0.05 and an extent threshold of 50 voxels. d Increased 18F-Florbetaben uptake in all patients (N = 11), rendered at cluster threshold of p < 0.001 and an extent threshold of 50 voxels. Coordinates for significant clusters following FWE-correction can be found in Supplementary Table 1. Colourbar units are contrast estimates representative of Z-scores.
Regional and voxel-wise correlations
Associations of 11C-BU99008 and 18F-FDG
BPM analysis in Aβ-positive patients showed that on a voxel-wise basis reduced 11C-BU99008 uptake was associated with reduced 18F-FDG uptake, particularly in the parietal, frontal and temporal lobes (Fig. 4a). However, these results were not replicated within the ROI correlation analyses (frontal (r = 0.202, p = 0.664), occipital (r = 0.264, p = 0.567), temporal (r = 0.567, p = 0.184) and parietal (r = 0.622, p = 0.135) lobes, and the whole brain (r = 0.478, p = 0.279); Fig. 4b).

a BPM: Reduced 11C-BU99008 uptake correlated with reduced 18F-FDG uptake, rendered at cluster threshold of p < 0.01. b ROI: 11C-BU99008 uptake positively correlated with 18F-FDG uptake in the temporal (r = 0.567) and parietal (r = 0.623) lobes, although these did not reach significance (p = 0.184 and p = 0.135, respectively). c BPM: Reduced 11C-BU99008 uptake correlated with reduced GM volume, shown through sections at cluster threshold of p < 0.01. d ROI: 11C-BU99008 uptake positively correlated with GM volume in the temporal (r = 0.935, p = 0.002) and parietal (r = 0.833, p = 0.02) lobes. e BPM: Increased 18F-florbetaben uptake correlated with reduced 11C-BU99008 uptake, rendered at cluster threshold of p < 0.05. f ROI: 11C-BU99008 uptake positively correlated with 18F-florbetaben uptake in the temporal (r = 0.779) and parietal (r = 0.471) lobes, although only the temporal lobe reached significance (p = 0.039 and p = 0.287, respectively). g BPM: Increased 18F-florbetaben uptake correlated with increased 11C-BU99008 uptake, rendered at cluster threshold of p < 0.05. Colourbar units in (a), (c), (e), and (g) are contrast estimates representative of Z-scores. All BPM correlations are displayed with an extent threshold of 50 voxels. Multiple comparison corrections were not performed on the BPM results due to the exploratory nature of this pilot study.
Associations of 11C-BU99008 and VBM
BPM analysis in Aβ-positive patients showed reduced 11C-BU99008 uptake was correlated with reduced GM volume in the frontal and temporal lobes (Fig. 4c). ROI correlations showed the same correlation (Fig. 4d), showing strong correlations in the frontal (r = 0.808, p = 0.028), temporal (r = 0.935, p = 0.002), parietal (r = 0.833, p = 0.020) and occipital (r = 0.762, p = 0.047) lobes, as well as the whole brain (r = 0.901, p = 0.006).
Associations of 11C-BU99008 and 18F-florbetaben
BPM analysis in Aβ-positive patients described an inverse correlation of increased 18F-florbetaben uptake with reduced 11C-BU99008 uptake in regions such as the temporal lobe and the cingulate (Fig. 4e), whilst increased 18F-florbetaben uptake was positively correlated with increased 11C-BU99008 uptake in primary motor and primary sensory areas (Fig. 4g). ROI analyses showed that reduced 11C-BU99008 uptake was correlated with increased 18F-florbetaben uptake, particularly in the frontal (r = −0.780, p = 0.039), temporal (r = −0.779, p = 0.039) and occipital (r = −0.911, p = 0.004) lobe, as well as the whole brain (r = −0.798, p = 0.032; Fig. 4f).
Please note, the BPM and ROI results were not corrected for multiple comparisons due to the exploratory nature of this pilot study.
Discussion
In this study, we used the novel imidazoline receptor PET tracer 11C-BU99008 to test for evidence of a dynamic relationship between astrocyte reactivity and amyloid-associated neurodegeneration based on tissue hypometabolism and atrophy measured using 18F-FDG PET and structural MRI, respectively. We found evidence for increased astrocyte reactivity in Aβ-positive patients, as increased 11C-BU99008 uptake was observed primarily in frontal, parietal and occipital regions. Furthermore, these increases were greater in MCI than AD patients. Voxel-wise correlational analyses showed that lower 11C-BU99008 uptake in Aβ-positive patients was associated with hypometabolism in the parietal, temporal and frontal lobes, even though there was no correlation at ROI level. In addition, lower 11C-BU99008 uptake in Aβ-positive patients was associated with GM atrophy in frontal and temporal lobes both on a regional and voxel-wise basis. Finally, analyses of regional differences in the relationships between PET markers of Aβ-deposition and astrocyte reactivity displayed a striking heterogeneity. Previously, in the same cohort of patients, we evaluated positive correlations between amyloid and 11C-BU99008, and we observed that greater Aβ-deposition was associated with increased 11C-BU99008 uptake in primary motor and primary sensory cortical areas in the parietal cortex [19]. However, as astrocytes can have beneficial and detrimental effects, we performed further evaluation to explore positive and negative associations between amyloid and astrocyte reactivity. We observed greater Aβ-deposition was also associated with decreased 11C-BU99008 uptake in different localised regions in the rest of the cortices; particularly temporal and cingulate regions. These observations are consistent with a hypothetical model (illustrated in Fig. 5) in which astrocyte reactivity is maximal in earlier stages of pathological progression (when it may contribute to the clearance of Aβ plaques [34]), but, with greater Aβ deposition, astrocytes may progressively become dystrophic or transition to assume a neurotoxic phenotype [4], both of which are likely to accelerate neurodegeneration leading to brain atrophy and hypometabolism. Whilst there is regional, temporal and morphological variation with astrocyte reactivity across the AD spectrum [2], our results support previous research and a model which illustrates the rise and fall trajectory of gross levels of astrocyte reactivity through disease progression. This model is also supported by the results from previous studies using other astrocyte markers [21, 22, 35].

Pre-symptomatic: In healthy conditions, astrocytes have several roles in providing neuronal support that contributes to normal neuronal function. Accumulation of Aβ induces astrocyte reactivity, causing the astrocytes to become hypertrophic. Then, the neuroprotective reactive astrocytes release proteases that aid in the cleavage and removal of Aβ plaques. MCI/Early AD: Despite the efforts of the reactive astrocytes, Aβ continues to accumulate and eventually the high levels cause the reactive astrocytes to become neurotoxic, as they release cytokines, chemokines and reactive oxygen species. The switch from neuroprotective to neurotoxic contributes to the early stages of astrocyte and GM atrophy. Advanced AD: Astrocyte reactivity in cortical regions with early amyloid deposition, and therefore early astrocyte reactivity, will experience advanced astrocyte atrophy. This results in the loss of normal function (such as blood flow maintenance) and further new neurotoxic functions (such as excessive glutamate release), both contributing to glucose hypometabolism and advanced GM atrophy which further contribute to cognitive impairment.
11C-BU99008 is a novel PET tracer that binds to I2-BS, expression of which is associated with astrocyte reactivity [36, 37]. Brain I2-BS is upregulated with healthy aging [38], and is further increased in AD [39]. The sensitivity and specificity of 11C-BU99008 to bind to I2-BS expressing reactive astrocytes has been further evidenced in a recent autoradiography study of AD brains where tracer uptake was more significant compared to cognitively normal brains [24]. In line with this, we have previously reported increased 11C-BU99008 uptake in the same cohort of cognitively impaired patients compared to healthy controls [19]. This corroborates with earlier studies using another PET marker of astrocyte reactivity, 11C-DED [35]. Interestingly, another 11C-DED study found increased binding in the frontal lobe in Aβ-positive MCI, but not AD, subjects compared to healthy controls [21], in line with findings in this present study as we observed increased 11C-BU99008 uptake in MCI subjects compared to AD subjects, particularly in the frontal, temporal and cingulate cortices. Both these findings agree with the hypothesis that astrocyte reactivity is predominantly an early event in the progression of AD pathology.
Astrocytes play an essential role in synapse formation along with the maturation of the synapses and synaptic pruning [40]. Astrocytes undergo morphological, molecular and functional remodelling in the presence of injury and disease states to become reactive astrocytes [2]. In the early stages, reactive astrocytes may have a neuroprotective role, aiding in the clearance of Aβ [3]. The spectrum of Aβ species mediating pathogenic changes in astrocytes is broad and complex, but it is hypothesised that Aβ oligomers can be involved in the primary pathogenesis of astrocyte reactivity [41, 42]. In support of this hypothesis, we found increased 11C-BU99008 uptake was associated with high levels of 18F-florbetaben uptake in primary motor areas, regions where amyloid deposition is to happen at a later stage of the disease and therefore would have recently developed. Increased 11C-DED binding has also been found in autosomal dominant AD patients early in their disease progression [43] with recent Aβ deposition [44]. In addition, a recent study demonstrated that plasma levels of glial fibrillary acid protein (GFAP), a marker of astrocyte reactivity, is an early marker of Aβ load, and was associated longitudinally with Aβ accumulation and cognitive decline [45].
Interaction of Aβ with reactive astrocytes has been proposed as a trigger for astrocytes to switch from a neuroprotective to a neurotoxic role. As astrocyte reactivity increases Aβ production [46], a positive feedback loop favouring the formation of neurotoxic, pro-inflammatory astrocytes is initiated. Reactive astrocytes exhibit beneficial and detrimental effects based on their reactivity profile. In mouse models of infection and stroke induced by lipopolysaccharide induction and middle artery occlusion, respectively, resting astrocytes polarise to become reactive astrocytes [47]. Neurotoxic astroglial phenotype is induced by cytokines secreted by activated microglia, which include complement factors (C1q), TNF-α and IL-1α [48]. This leads to astrocytes losing their ability to promote neuronal survival and outgrowth, synaptogenesis, and phagocytosis, and inducing the demise of neurons, oligodendrocytes and even astrocytes themselves. We found evidence of low astrocyte reactivity, perhaps due to astrocyte dystrophy in the presence of high Aβ load in this current study, as decreased 11C-BU99008 uptake was associated with high levels of 18F-florbetaben uptake in the temporal lobe, one of the earliest regions where Aβ deposition occurs [44]. This decreased 11C-BU99008 uptake in the temporal lobe was also associated with greater relative progression of amyloid-associated neuropathology, that is glucose hypometabolism and GM atrophy. However, it is important to note the evidence of an association between reduced 11C-BU99008 and 18F-FDG uptake is less strong as it was observed at a voxel-wise basis, but not on a regional basis. We propose this reduced 11C-BU99008 uptake in the temporal lobe region reflects astrocyte dystrophy [49], that eventually leads to astrocytic and neuronal atrophy [50, 51] and glucose hypometabolism [6] (Fig. 5). Our results are corroborated from previous findings of correlations between regional reductions in 11C-DED and 18F-FDG PET signals, which were associated with regionally more advanced 11C-PIB PET pathology in a longitudinal study of people with autosomal dominant AD or MCI [52].
There are obvious limitations to our study. First, only a small number of subjects were able to be imaged, requiring statistical analysis to be applied in a more liberal exploratory nature. However, while this is a pilot study, the explanatory power was enhanced by the design in which uptake of the three PET tracers and brain volume all were assessed in the same people. A second limitation was the cross-sectional design, which we acknowledge; however, post mortem pathology has the same limitation. Our results thus are better interpreted descriptively and as suggestive of a hypothetical model, rather than a strong, independent test. Nonetheless, the consistency of directions of effect observed in this study and the earlier 11C-DED PET studies [21, 52] provides compelling support for the hypothetical model presented (Fig. 5). That is, astrocyte reactivity occurs in response to early Aβ-deposition, aiding in the clearance of Aβ, but following interactions with high levels of Aβ the astrocytes become neurotoxic, contributing to reduced tissue activity and cell death that is associated with cognitive impairment. It also strengthens confidence in the earlier work, which otherwise suffers from uncertainties regarding the specificity of binding of 11C-DED in the brain [21]. Nonetheless, 11C-BU99008 can detect astrocyte reactivity with a greater sensitivity than 11C-DED [24], especially amongst higher levels of amyloid load [19, 53], and thus should be prioritised to take it forward.
In conclusion, this study supports neuropathological observations arguing that astrocyte reactivity with amyloid-related neuropathology is dynamic [54]. We have demonstrated in vivo with the novel PET tracer 11C-BU99008 that astrocyte reactivity is increased in regions presumed to represent earlier stages of pathological progression with low Aβ-deposition loads, and conversely relatively reduced in regions that show signs of more advanced disease progression with greater Aβ-deposition and atrophy. In the absence of molecular imaging markers intrinsically discriminating different reactive astrocyte phenotypes, our multi-modal imaging approach may allow relevant inferences to be made from the relative 11C-BU99008, 18F-FDG and 18F-florbetaben PET signals and brain volume sensitive MRI measures. Future, larger, longitudinal studies are needed to further test this dynamic model and, if supported, interventions developed to arrest progression of the neurotoxic phenotypic transformation of astrocytes in AD.
References
Vasile F, Dossi E, Rouach N. Human astrocytes: structure and functions in the healthy brain. Brain Struct Funct. 2017;222:2017–29.
Escartin C, Galea E, Lakatos A, O’Callaghan JP, Petzold GC, Serrano-Pozo A, et al. Reactive astrocyte nomenclature, definitions, and future directions. Nat Neurosci. 2021;24:312–25.
Ries M, Sastre M. Mechanisms of Aβ clearance and degradation by glial cells. Front Aging Neurosci. 2016;8:160.
Verkhratsky A, Olabarria M, Noristani HN, Yeh CY, Rodriguez JJ. Astrocytes in Alzheimer’s Disease. Neurotherapeutics 2010;7:399–412.
Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007;28:138–45.
Verkhratsky A, Rodrigues JJ, Pivoriunas A, Zorec R, Semyanov A. Astroglial atrophy in Alzheimer’s disease. Neurotherapeutics 2019;471:1247–61.
Márquez F, Yassa MA. Neuroimaging Biomarkers for Alzheimer’s Disease. Mol Neurodegeneration. 2019;14:21.
Zimmer ER, Parent MJ, Souza DG, Leuzy A, Lecrux C, Kim HI, et al. [(18)F]FDG PET signal is driven by astroglial glutamate transport. Nat Neurosci. 2017;20:393–5.
Vilaplana E, Rodriguez-Vieitez E, Ferreira D, Montal V, Almkvist O, Wall A, et al. Cortical microstructural correlates of astrocytosis in autosomal-dominant Alzheimer disease. Neurology 2020;94:e2026–36.
Diniz LP, Tortelli V, Matias I, Morgado J, Araujo APB, Melo HM, et al. Astrocyte transforming growth factor beta 1 protects synapses against Aβ oligomers in Alzheimer’s disease model. J Neurosci. 2017;37:6797–809.
Bélanger M, Allaman I, Magistretti PJ. Brain energy metabolism: focus on Astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–38.
Regunathan S, Feinstein DL, Reis DJ. Expression of non‐adrenergic imidazoline sites in rat cerebral cortical astrocytes. J Neurosci Res. 1993;34:681–8.
Venkataraman AV, Keat N, Myers JF, Turton S, Mick I, Gunn RN, et al. First evaluation of PET-based human biodistribution and radiation dosimetry of 11C-BU99008, a tracer for imaging the imidazoline2 binding site. EJNMMI Res. 2018;8:71.
Tyacke RJ, Myers JFM, Venkataraman A, Mick I, Turton S, Passchier J, et al. Evaluation of 11C-BU99008, a PET Ligand for the Imidazoline2 binding site in human brain. J Nucl Med. 2018;59:1597–602.
Tyacke RJ, Fisher A, Robinson ESJ, Grundt P, Turner EM, Husbands SM, et al. Evaluation and initial in vitro and ex vivo characterization of the potential positron emission tomography ligand, BU99008 (2-(4,5-Dihydro-1H-imidazol-2-yl)-1- methyl-1H-indole), for the imidazoline 2 binding site. Synapse 2012;66:542–51.
Parker CA, Nabulsi N, Holden D, Lin SF, Cass T, Labaree D, et al. Evaluation of 11C-BU99008, a PET Ligand for the Imidazoline 2 Binding Sites in Rhesus Brain. J Nucl Med. 2014;55:838–44.
Kealey S, Turner EM, Husbands SM, Salinas CA, Jakobsen S, Tyacke RJ, et al. Imaging imidazoline-I2 binding sites in porcine brain using 11C-BU99008. J Nucl Med. 2013;54:139–44.
Kawamura K, Shimoda Y, Yui J, Zhang Y, Yamasaki T, Wakizaka H, et al. A useful PET probe [11C]BU99008 with ultra-high specific radioactivity for small animal PET imaging of I2-imidazoline receptors in the hypothalamus. Nucl Med Biol. 2017;45:1–7.
Calsolaro V, Matthews P, Donat C, Livingston N, Femminella G, Silva Guedes S, et al. Astrocyte reactivity with late onset cognitive impairment assessed in vivo using 11C-BU99008 PET and its relationship with amyloid load. Mol Psychiatry. 2021; (Online ahead of print) https://doi.org/10.1038/s41380-021-01193-z.
Wilson H, Dervenoulas G, Pagano G, Tyacke RJ, Polychronis S, Myers J, et al. Imidazoline 2 binding sites reflecting astroglia pathology in Parkinson’s disease: an in vivo 11C-BU99008 PET study. Brain 2019;142:3116–28.
Carter SF, Schöll M, Almkvist O, Wall A, Engler H, Långström B, et al. Evidence for astrocytosis in prodromal alzheimer disease provided by 11C-deuterium-L-deprenyl: A multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J Nucl Med. 2012;53:37–46.
Carter SF, Herholz K, Rosa-Neto P, Pellerin L, Nordberg A, Zimmer ER. Astrocyte Biomarkers in Alzheimer’s Disease. Trends Mol Med. 2019;25:77–95.
Bellaver B, Ferrari-Souza JP, Uglione da Ros L, Carter SF, Rodriguez-Vieitez E, Nordberg A, et al. Astrocyte Biomarkers in Alzheimer Disease: a systematic review and meta-analysis. Neurology. 2021; (Online ahead of print) https://doi.org/10.1212/WNL.0000000000012109.
Kumar A, Koistinen NA, Malarte ML, Nennesmo I, Ingelsson M, Ghetti B, et al. Astroglial tracer BU99008 detects multiple binding sites in Alzheimer’s disease brain. Mol Psychiatry. 2021; (Online ahead of print) https://doi.org/10.1038/s41380-021-01101-5.
Bullich S, Seibyl J, Catafau AM, Jovalekic A, Koglin N, Barthel H, et al. Optimized classification of 18F-Florbetaben PET scans as positive and negative using an SUVR quantitative approach and comparison to visual assessment. NeuroImage: Clin. 2017;15:325–32.
Ashburner J. A fast diffeomorphic image registration algorithm. Neuroimage 2007;38:95–113.
Hammers A, Allom R, Koepp MJ, Free SL, Myers R, Lemieux L, et al. Three-dimensional maximum probability atlas of the human brain, with particular reference to the temporal lobe. Hum Brain Mapp. 2003;19:224–47.
Erlandsson K, Buvat I, Pretorius PH, Thomas BA, Hutton BF. A review of partial volume correction techniques for emission tomography and their applications in neurology, cardiology and oncology. Phys Med Biol. 2012;57:R119–59.
Aston JA, Cunningham VJ, Asselin MC, Hammers A, Evans AC, Gunn RN. Positron emission tomography partial volume correction: estimation and algorithms. J Cereb Blood Flow Metab. 2002;22:1019–34.
Minhas DS, Price JC, Laymon CM, Becker CR, Klunk WE, Tudorascu DL, et al. Impact of partial volume correction on the regional correspondence between in vivo [C-11]PiB PET and postmortem measures of Abeta load.Neuroimage Clin.2018;19:182–9.
Seo SW, Ayakta N, Grinberg LT, Villeneuve S, Lehmann M, Reed B, et al. Regional correlations between [(11)C]PIB PET and post-mortem burden of amyloid-beta pathology in a diverse neuropathological cohort. Neuroimage Clin. 2017;13:130–7.
Gousias IS, Rueckert D, Heckemann RA, Dyet LE, Boardman JP, Edwards AD, et al. Automatic segmentation of brain MRIs of 2-year-olds into 83 regions of interest. NeuroImage 2008;40:672–84.
Casanova R, Srikanth R, Baer A, Laurienti PJ, Burdette JH, Hayasaka S, et al. Biological parametric mapping: a statistical toolbox for multimodality brain image analysis. NeuroImage 2007;34:137–43.
Nagele RG, D’Andrea MR, Lee H, Venkataraman V, Wang HY. Astrocytes accumulate Aβ42 and give rise to astrocytic amyloid plaques in Alzheimer disease brains. Brain Res. 2003;971:197–209.
Santillo AF, Gambini JP, Lannfelt L, Långström B, Ulla-Marja L, Kilander L, et al. In vivo imaging of astrocytosis in Alzheimer’s disease: an 11C-L-deuteriodeprenyl and PIB PET study. Eur J Nucl Med Mol Imaging. 2011;38:2202–8.
Ruiz J, Martín I, Callado LF, Meana JJ, Barturen F, García-Sevilla JA. Non-adrenoceptor [3H]idazoxan binding sites (I2-imidazoline sites) are increased in postmortem brain from patients with Alzheimer’s disease. Neurosci Lett. 1993;160:109–12.
Li JX. Imidazoline I2 receptors: an update. Pharm Ther. 2017;178:48–56.
Sastre M, Garcia‐Sevilla JA. Opposite Age‐Dependent Changes of α2A‐Adrenoceptors and Nonadrenoceptor [3H]Idazoxan Binding Sites (I2‐Imidazoline Sites) in the Human Brain: Strong Correlation of I2 with Monoamine Oxidase‐B Sites. J Neurochem. 1993;61:881–9.
García-Sevilla JA, Escribá PV, Walzer C, Bouras C, Guimón J. Imidazoline receptor proteins in brains of patients with Alzheimer’s disease. Neurosci Lett. 1998;2:95–98.
Allen NJ, Eroglu C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017;96:697–708.
Wang W, Hou TT, Jia LF, Wu QQ, Quan MN, Jia JP. Toxic amyloid-beta oligomers induced self-replication in astrocytes triggering neuronal injury. EBioMedicine 2019;42:174–87.
Narayan P, Holmstrom KM, Kim DH, Whitcomb DJ, Wilson MR, St George-Hyslop P, et al. Rare individual amyloid-beta oligomers act on astrocytes to initiate neuronal damage. Biochemistry 2014;53:2442–53.
Scholl M, Carter SF, Westman E, Rodriguez-Vieitez E, Almkvist O, Thordardottir S, et al. Early astrocytosis in autosomal dominant Alzheimer’s disease measured in vivo by multi-tracer positron emission tomography. Sci Rep. 2015;5:16404.
Zhang X, Fu Z, Meng L, He M, Zhang Z. The Early Events That Initiate beta-Amyloid Aggregation in Alzheimer’s Disease. Front Aging Neurosci. 2018;10:359.
Pereira JB, Janelidze S, Smith R, Mattsson-Carlgren N, Palmqvist S, Teunissen CE, et al. Plasma GFAP is an early marker of amyloid-beta but not tau pathology in Alzheimer’s disease. Brain. 2021; (Online ahead of print) https://doi.org/10.1093/brain/awab223.
Frost GR, Li YM. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017;7:170228.
Zamanian JL, Xu L, Foo LC, Nouri N, Zhou L, Giffard RG, et al. Genomic analysis of reactive astrogliosis. J Neurosci. 2012;32:6391–410.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017;541:481–7.
Olsen M, Aguilar X, Sehlin D, Fang XT, Antoni G, Erlandsson A, et al. Astroglial Responses to Amyloid-Beta Progression in a Mouse Model of Alzheimer’s Disease. Mol Imaging Biol. 2018;20:605–14.
Smale G, Nichols NR, Brady DR, Finch CE, Horton WE. Evidence for Apoptotic Cell Death in Alzheimer’s Disease. Exp Neurol. 1995;133:225–30.
Garaschuk O, Verkhratsky A. GABAergic astrocytes in Alzheimer’s disease. Aging 2019;11:1602–4.
Rodriguez-Vieitez E, Saint-Aubert L, Carter SF, Almkvist O, Farid K, Schöll M, et al. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain 2016;139:922–36.
Rodriguez-Vieitez E, Ni R, Gulyas B, Toth M, Haggkvist J, Halldin C, et al. Astrocytosis precedes amyloid plaque deposition in Alzheimer APPswe transgenic mouse brain: a correlative positron emission tomography and in vitro imaging study. Eur J Nucl Med Mol Imaging. 2015;42:1119–32.
González-Reyes RE, Nava-Mesa MO, Vargas-Sánchez K, Ariza-Salamanca D, Mora-Muñoz L. Involvement of astrocytes in Alzheimer’s disease from a neuroinflammatory and oxidative stress perspective. Front Mol Neurosci. 2017;427:10.
Acknowledgements
The authors thank Invicro Centre for Imaging Sciences for the provision of 11C-BU99008, scanning and blood analysis equipment. The authors also thank Piramal Life Sciences/Life Molecular Imaging for providing the 18F-florbetaben and permission to acquire unlabelled florbetaben. We thank Dementia Platform UK (DPUK) and GSK for the generous funding for this project. This research was co-funded by the NIHR Imperial Biomedical Research Centre and was supported by the NIHR Imperial Clinical Research Facility. The views expressed are those of the authors and not necessarily those of NHS, the NIHR nor the Department of Health. The study was funded by the Dementia Platform UK and the National Institutes of Health Research Imperial College Healthcare Trust Biomedical Research Centre. PE was funded by the Medical Research Council and now by Higher Education Funding Council for England (HEFCE). He has also received grants from Alzheimer’s Research, UK, Alzheimer’s Drug Discovery Foundation, Alzheimer’s Society, UK, Alzheimer’s association, US, Medical Research Council, UK, Novo Nordisk, Piramal Life Sciences and GE Healthcare. PMM gratefully acknowledges generous support from Edmond J Safra Foundation and Lily Safra, the NIHR Investigator programme and the UK Dementia Research Institute.
Author information
Authors and Affiliations
Contributions
PE, DJN and PMM were responsible for the concept of the study. NRL and PE designed the analysis strategy. RJT, DJN, PBW, RP, RNG, EAR, CAP and PSM developed the novel 11C-BU99008 PET tracer. VC collected the data. NRL, SR and JN processed the data. NRL analysed the data. NRL and PE drafted the paper. JM, AVV and SG revised the paper. All authors edited and approved the final version of the paper.
Corresponding author
Ethics declarations
Competing interests
PE is a consultant to Roche, Pfizer and Novo Nordisk. He has received speaker fees from Novo Nordisk, Pfizer, Nordea, Piramal Life Science. He has received educational and research grants from GE Healthcare, Novo Nordisk, Piramal Life Science/Life Molecular Imaging, Avid Radiopharmaceuticals and Eli Lilly. He is an external consultant to Novo Nordisk and a member of their Scientific Advisory Board. PMM acknowledges consultancy fees from Roche, Adelphi Communications, Celgene and Biogen. He has received honoraria or speakers’ honoraria from Novartis, Biogen and Roche and has received research or educational funds from Biogen, Novartis, GlaxoSmithKline and Nodthera.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
Cite this article
Livingston, N.R., Calsolaro, V., Hinz, R. et al. Relationship between astrocyte reactivity, using novel 11C-BU99008 PET, and glucose metabolism, grey matter volume and amyloid load in cognitively impaired individuals. Mol Psychiatry 27, 2019–2029 (2022). https://doi.org/10.1038/s41380-021-01429-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-021-01429-y
- Springer Nature Limited
This article is cited by
-
PET imaging of neuroinflammation: any credible alternatives to TSPO yet?
Molecular Psychiatry (2024)
-
The development status of PET radiotracers for evaluating neuroinflammation
Nuclear Medicine and Molecular Imaging (2024)
-
Tracking reactive astrogliosis in autosomal dominant and sporadic Alzheimer’s disease with multi-modal PET and plasma GFAP
Molecular Neurodegeneration (2023)
-
Astrocytes in human central nervous system diseases: a frontier for new therapies
Signal Transduction and Targeted Therapy (2023)
-
The use of neuroimaging techniques in the early and differential diagnosis of dementia
Molecular Psychiatry (2023)