
Abstract
Background:
Primary nonsyndromic vesicoureteral reflux (VUR) and VUR with renal hypoplasia/dysplasia (VUR-RHD) are common congenital anomalies of the kidney and urinary tract (CAKUT). Sequence variations of the ROBO2 gene were investigated in children with nonsyndromic VUR or VUR-RHD.
Methods:
Single-strand conformation polymorphism (SSCP) electrophoresis or multiple restriction fragment SSCP (MRF-SSCP), followed occasionally by direct sequencing, was used to screen 103 patients and 200 controls for nucleotide changes. Gene polymorphisms and transposable elements were investigated using bioinformatics.
Results:
Two single-nucleotide polymorphisms were detected: IVS1-53 and IVS5-31. The frequency of A allele of IVS1-53G>A did not differ significantly between patients and controls. IVS1-53 does not affect mRNA splicing according to in silico analysis. IVS5-31A>G substitution was found in one patient, reported here for the first time in VUR. In silico results demonstrated alteration in two serine/arginine-rich (SR) protein-binding sites and two additional acceptor sites. The ROBO2 gene sequence was found to contain 25.9% transposable elements.
Conclusion:
ROBO2 variants were not found to be associated with nonsyndromic VUR or VUR-RHD, providing further evidence for genetic heterogeneity. The role of transposable elements in ROBO2 gene expression in CAKUT needs further investigation since they are generally considered to be mutagens.
Similar content being viewed by others

Main
Congenital anomalies of the kidney and urinary tract (CAKUT) account for up to 30% of all anomalies diagnosed prenatally and constitute the main cause of chronic renal disease in infants and young children. Although the etiology of most of these anomalies has not yet been elucidated, experimental studies have identified several genes that are implicated in nephrogenesis, the derangement of which results in renal mal-development (1).
One of the most common CAKUT is primary nonsyndromic vesicoureteral reflux (VUR). Although the incidence of VUR in healthy children was found to be less than 2% (2), its prevalence in infants with febrile urinary tract infection (UTI) approaches 40% (3). VUR is also diagnosed in ~9–11% of neonates with intrauterine hydronephrosis (4,5). Data from studies in newborn infants with VUR detected during investigation of fetal hydronephrosis documented the presence of VUR in association with congenital renal damage, specifically renal hypoplasia/dysplasia (RHD), in the absence of UTI (4,5).
Recent studies have identified variants of the roundabout, axon guidance receptor, homolog 2 (ROBO2) gene in patients with VUR and VUR accompanied by other CAKUT, but their conclusions are contradictory on whether or not mutations of the ROBO2 gene are a causative factor of VUR or VUR-other CAKUT (6,7,8,9,10,11,12,13,14).
Transposable elements constitute almost half of the mammalian genome (15). There is increasing evidence regarding their involvement in alternative gene regulation, recombination, and creation of new genes and genome restructuring and development (16,17). Transposable elements are capable of mobilization and integration into new genomic sites, and their activity is generally controlled by various cellular defense mechanisms, such as DNA methylation (16). This control mechanism has been shown to be restricted during development and embryogenesis (18). Deregulated activity of transposable elements has been associated with monogenic and multifactorial genetic diseases (16).
The aim of this study was to evaluate the presence of sequence variations in the ROBO2 gene in young children with primary, nonsyndromic VUR and children with VUR-RHD. Comparison was made using as control subjects children with a negative imaging study for CAKUT. In silico estimation of the effect of variants was performed. In addition, for the first time to the knowledge of the authors, the ROBO2 gene sequence was analyzed for the presence of transposable elements that could lead to mutagenesis and a pathological phenotype.
Results
Two SNPs, IVS1-53 (rs9874095 [G>A]) and IVS5-31 (rs76571990 [A>G]), were detected by single-strand conformation polymorphism (SSCP) or multiple restriction fragment SSCP (MRF-SSCP) analysis in the patient group, and these were subsequently characterized by direct sequencing.
IVS1-53 Polymorphism
Statistical analysis of IVS1-53 polymorphism showed that the A allele was less frequent in the entire group of patients (P = 0.0488) and in the VUR patients (P = 0.0125), compared with the control group. Similarly, the A allele was less frequent in the total of female patients (P = 0.0119) and in the VUR female patients (P = 0.0194) than in the female control group ( Table 1 ). Regarding the genotype distribution, because of the lack of probands with AA genotype in the patient group, the genotypes GA and AA were combined in one group for statistical analysis. The GA/ΑΑ combination was less frequent in the VUR group (males and females) (P = 0.0196), in the total of female patients (with VUR or VUR/RHD) (P = 0.0119) and in female patients with VUR (P = 0.0177) compared with the control group ( Table 1 ). Nevertheless, because of the multiple hypotheses tested on one set of data, the chance of getting a false-positive result is 36.9%. In order to neutralize the risk of type-I error, the Bonferroni correction was used to adjust the P value for each hypothesis to 0.0055. Subsequently, there were no significant differences in the frequency of any of the hypotheses tested.
The genotype frequencies of the study population, patient and control groups, and the subgroups were in Hardy–Weinberg equilibrium.
In silico analysis with SplicePort (19) showed a very low probability for the corresponding splice site to create an alternative splice variant, and ESEFinder (20,21) demonstrated no difference. Overall splicing is probably not affected by IVS1-53.
IVS5-31 Polymorphism
IVS5-31 [A>G] was detected in one patient with VUR in the heterozygous state (AG), while all the other individuals, patients, and controls were homozygous for the ancestral allele (AA). The patient was diagnosed with febrile UTI at the age of 4 mo, and the imaging study revealed VUR grade IV on the left side.
In silico analysis showed alteration of two SR (serine/arginine-rich) protein-binding sites, SC35 and SRp40, due to the polymorphism, using the ESEfinder (20,21) software. Also, in the presence of this polymorphism, two additional acceptor sites were predicted from the SplicePort (19) software, which has been confirmed by ESEfinder (20,21).
Transposable Elements
University of California, Santa Cruz Genome Bioinformatics (22) revealed the presence of transposons in the gene sequence, and RepeatMasker showed the content in transposable elements to be 25.9%, 21.35% of which were retrotrasposons.
Discussion
In this study, ROBO2 screening revealed two SNPs in otherwise healthy children with primary, nonsyndromic VUR or VUR-RHD. The first polymorphism, IVS1-53, has been reported previously in patients with VUR or VUR-other CAKUT (9). In this study, this polymorphism was detected in 27 patients (only in heterozygous state) and 71 control subjects (64 in heterozygous and 7 in homozygous state). The second polymorphism, IVS5-31, reported here is observed for the first time in a CAKUT study group, but it was found in only one patient.
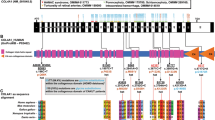
ROBO2 is a member of the immunoglobulin superfamily and is highly conserved from fly to human (23). The gene resides on chromosome 3p12.3 and encodes the transmembrane receptor of SLIT2. The signaling of both genes plays a key role in the formation of the ureteric bud, regulating the expression of glial cell-derived neurothrophic factor (GDNF), the factor that induces the outgrowth of the ureteric bud from the nephric duct. The role of SLIT2-ROBO2 signaling is to restrict GDNF to a single site. In mice, inactivation of either slit2 or robo2 leads to supernumerary ureteric bud development (9,24). The gene codes for two isoforms RΟΒΟ2a and RΟΒΟ2b. The “a” isoform has very restricted expression especially in fetal brain with extremely low level in kidney (13,25).
Previous studies have also screened the ROBO2 gene in patients with VUR and VUR-other CAKUT. Lu et al. (7) found a de novo translocation in a man with syndromic VUR, which disrupted the ROBO2 gene. They also studied 124 families with VUR or VUR-other CAKUT and identified two heterozygous missense variants of the gene in two unrelated families (British and Dutch) with VUR-renal hypoplasia, VUR, VUR-duplex kidney, and other CAKUT-small kidney. A study on 95 unrelated Italian patients with VUR or VUR-other CAKUT identified a relatively high frequency of ROBO2 variants (5.1%) in familial cases; 24 variants were discovered, 4 of which led to amino acid substitutions in patients with VUR or VUR-other CAKUT (9). A study on 54 families, mostly of Swedish origin, with different grades of VUR, some accompanied by other CAKUT, detected six ROBO2 variants, including two new variants (10). Dobson et al. (13) performed sequencing in 227 patients with primary VUR and 23 family members and found 55 ROBO2 variants, 20 of which were new. The most recent study analyzed the coding exons of various genes, including ROBO2, in 749 individuals with CAKUT from 650 families, in which the most common phenotypes were VUR, renal hypodysplasia, and unilateral renal agenesis; four mutations of ROBO2 were detected in four unrelated families (14). Three linkage studies, however, have been unable to support these findings. One study on Italian and American kinships with VUR, one on four Dutch families with VUR, and one on UK and Slovenian data sets with VUR found no linkage to the locus of ROBO2 (6,8,11).
In this study, syndromic cases of VUR were excluded, and the cases of primary nonsyndromic VUR accompanied by RHD were studied separately. The frequency of the A allele and GA/AA genotype of IVS1-53 polymorphism did not differ significantly between patients and controls, after the Bonferroni correction. Even though the correction was used to reduce the type I error rate, the difference of not reaching formal significance might also be a false-negative result, as any correction for multiple testing increases the rate of false-negative results. The IVS1-53 polymorphism was found in the homozygous form only in control subjects. The difference in frequencies of the A allele and GA/AA genotype combination in the female subgroup, even though it is not significant, has no clear explanation, besides the large difference in the number of cases and controls (47 female cases vs. 132 female controls), compared to the male subgroup (56 male cases vs. 68 male controls). The second SNP, IVS5-31, was found in only one patient, and no control subject, which is not consistent with the 14% frequency of the G allele given on one registry of the NCBI-SNP database. Since the control subjects had been investigated by imaging studies during routine examination, and all were documented as having no VUR or other CAKUT, there is no obvious explanation for the these findings, apart from the limited number of patients in the study and the method used. The possibility that the absence of nucleotide changes could be due to false-negative results from the SSCP method could not be completely ruled out, since the method’s sensitivity is not 100%. However, using at least two different conditions, as done in this study, it can reportedly reach an efficiency of 97% in fragments up to 300 bp, as is the case here (26,27).
In silico analysis was used to predict possible interference of the SNPs in splicing. IVS1-53 does not appear to affect a splice site, while IVS5-31 displayed alteration in SR protein binding and two additional acceptor sites. IVS5-31 probably affects, therefore, the corresponding splice site, although the SNP itself was only found in one patient.
Based on the above findings, sequence variations of the ROBO2 gene do not appear to play a role in the VUR or VUR-RHD subgroups of the study population. The novelty of this study is that for the first time the control group did not come from a general pediatric population but was recruited from a group of children with negative imaging evaluation for CAKUT. One of the problems of genetic investigation in VUR is that the control subjects coming from the general population may themselves have VUR and possibly VUR-causing mutations, since VUR is common and often asymptomatic in cases of mild to moderate severity. The limited number of patients in the present study, however, even when compared with subjects with documented absence of VUR, does not allow for definitive conclusions.
Retrotransposons can disrupt gene function in many ways, including insertional mutagenesis, alternative splicing, epigenetic effects, and the expression of small noncoding RNAs (28). Two recent studies report that ROBO2 gene is a target for L1 retrotransposons insertional mutagenesis in intronic regions (15% frequency) and was found in a pathogenic cancer phenotype (28,29). The mutational analysis in this study included only the exons and their intronic boundaries, leaving out the noncoding regions of the gene. Even though ROBO2 content in transposable elements is relatively low (15) taking into account the above, along with the fact that retrotransposons are active during embryogenesis (18), it could be speculated that retrotransposon insertions, in addition to a polymorphism such that found here (IVS5-31), which may affect splicing, could lead to alteration of ROBO2 gene expression in patients with CAKUT. The role of retrotransposons in ROBO2 gene expression therefore needs further investigation.
Overall, the findings of the present study indicate that mutations in ROBO2 gene were not present in the distinct study group of patients with primary nonsyndromic VUR or VUR-RHD, providing further evidence for genetic heterogeneity. Since VUR, VUR-RHD, and other CAKUT are multifactorial disorders, the possibility of a single rare polymorphism, in association with other genetic variations, playing a role in the pathogenic phenotype, could not be ruled out, and further studies, such as investigation of tissue specific transposable elements are needed for safer conclusions to be reached.
Methods
The study was conducted on 103 Caucasian children of Greek origin (56 males, 47 females) diagnosed as having primary nonsyndromic VUR (65 patients, including 10 siblings) or VUR/RHD (38 patients, including 2 siblings) ( Table 1 ). The mean age at the time of diagnosis was 1.48 ± 2.14 y.
The radiological investigation which documented the above diagnoses had been performed because of a history of a first UTI or prenatal hydronephrosis. The imaging studies included urinary tract ultrasound, voiding cystourethrography, and technetium-99m dimercaptosuccinic acid (99mTc-DMSA) scan. In patients with UTI, voiding cystourethrography and DMSA scan were performed at 1–2 mo and 6 mo, respectively, from the onset of the UTI. All the imaging studies were performed in the radiological department of the study hospital. Patients with secondary VUR, syndromal anomalies, or chronic diseases were excluded from the study.
VUR was diagnosed and classified by voiding cystourethrography into grades I–V, according to the International Reflux Classification (2). Renal hypoplasia and dysplasia may coexist and the definitive diagnosis can be confirmed only by renal histology, which is not performed in routine clinical practice. The term renal hypoplasia/dysplasia was used, therefore, for kidneys with reduced renal length and regular outline, with or without cortical hyperechogenicity and loss of corticomedullary differentiation on ultrasound and with persistent (for ≥ 6 mo), general reduction in 99mTc-DMSA uptake. For optimal radiological diagnostic accuracy of RHD, only the severe cases were included (split function ≤ 35%).
The control group consisted of 200 healthy Caucasian children (68 males, 132 females), age matched with the patients. These children had a history of a first UTI and were evaluated during the same period as the patients, by ultrasound, voiding cystourethrography, and DMSA scan in the same pediatric and radiological departments as the patient group. They all had normal renal function, and no urological anomalies or chronic diseases were detected. The mean age of the control subjects at the time of imaging studies was 2.3 ± 3.7 y. They came from the same geographical region as the patients, and there were no ethnic differences between patients and controls.
The study protocol was approved by the Ethics Committee of the University Hospital of Ioannina, Greece, and informed written consent for the genetic study was obtained from the parents of all patients and control subjects.
For the genetic study, peripheral venous blood was collected for genomic DNA extraction, according to the manufacturer’s protocol using QIAamp DNA Mini Kit (Qiagen, Hilden, Germany), at the time of routine blood testing of the patients and control subjects.
PCR Amplification and Nucleotide Change Screening
Genomic DNA (50–100 ng) was amplified in a 25 μl reaction containing 1 U Taq polymerase, 67 mmol/l Tris–HCl (pH 8.8), 16.6 mmol/l (NH4)2SO4, 0.1% Tween 20, 2–6 mmol/l MgCl2 (HyTest, Turku, Finland); 10 pmol from each primer and 0.2 mmol/l dNTPs (Invitrogen, Waltham, MA). Only for exon 26, dimethyl sulfoxide, (Sigma-Aldrich, St Louis, MO) 5% was added to the reaction. All 26 exons of the ROBO2b gene isoform, including their intronic boundaries, were amplified by PCR, using specific primers (7). PCR was performed following the cycling conditions: one initial denaturation step at 95 °C for 3 min, followed by 30 cycles at 95 °C for 30 s, specific annealing temperature for 30 s and 72 °C for 40 s and a final step at 72 °C for 5 min.
For the detection of mutations and polymorphisms, the patients were screened by SSCP electrophoresis or MRF-SSCP, depending on the PCR product size. Two different electrophoresis conditions at room temperature were used, 8% polyacrylamide gel and 8% polyacrylamide gel with 5% glycerol. DNA bands were visualized by silver staining. In samples with abnormal migration patterns, the relevant DNA sequences were re-amplified and submitted to a second electrophoresis. Samples with an unusual pattern were then sequenced by an ABI 3730XL DNA sequencer (Macrogen Europe, Amsterdam, The Netherlands). The normal migration pattern was simultaneously confirmed by direct sequencing.
The control group samples were screened for the nucleotide variants identified in the patients, using restriction fragment length polymorphism analysis or MRF-SSCP methods.
Statistical and In Silico Analysis
Genotype and allele frequencies in patients and controls were compared using the χ2 test. Since nine different hypotheses are tested on the same set of data, the Bonferroni correction was used to adjust the P value for each hypothesis to 0.0055, taking into account the increased possibility of type-I error. Deviation from the Hardy–Weinberg equilibrium for alleles in each SNP was assessed using the χ2 test with one degree of freedom. Additionally, the gender association was evaluated.
In silico estimation of the effect of variants was performed using the bioinformatic tools available online, SplicePort (19) and ESEfinder (20,21), to evaluate the SNP interference in mRNA splicing.
The gene content in transposable elements due to evidence of L1 retrotransposon insertion into intronic regions of the gene was estimated using University of California, Santa Cruz Genome Bioinformatics (Santa Cruz, CA) (22) and RepeatMasker.
Statement of Financial Support
This study was supported by an Award from Procter & Gamble Company Hellas LTD, Athens, Greece (2012–2013 “Child @ Health”).
Disclosure
There are no financial ties to products in the study and no potential/perceived conflicts of interest.
References
Woolf AS. A molecular and genetic view of human renal and urinary tract malformations. Kidney Int 2000;58:500–12.
Report of the International Reflux Study Committee. Medical versus surgical treatment of primary vesicoureteral reflux. Pediatrics 1981;67:392–400.
Hoberman A, Charron M, Hickey RW, Baskin M, Kearney DH, Wald ER. Imaging studies after a first febrile urinary tract infection in young children. N Engl J Med 2003;348:195–202.
Farhat W, McLorie G, Geary D, et al. The natural history of neonatal vesicoureteral reflux associated with antenatal hydronephrosis. J Urol 2000;164:1057–60.
Ismaili K, Hall M, Piepsz A, et al. Primary vesicoureteral reflux detected in neonates with a history of fetal renal pelvis dilatation: a prospective clinical and imaging study. J Pediatr 2006;148:222–7.
Sanna-Cherchi S, Reese A, Hensle T, et al. Familial vesicoureteral reflux: testing replication of linkage in seven new multigenerational kindreds. J Am Soc Nephrol 2005;16:1781–7.
Lu W, van Eerde AM, Fan X, et al. Disruption of ROBO2 is associated with urinary tract anomalies and confers risk of vesicoureteral reflux. Am J Hum Genet 2007;80:616–32.
van Eerde AM, Koeleman BP, van de Kamp JM, de Jong TP, Wijmenga C, Giltay JC. Linkage study of 14 candidate genes and loci in four large Dutch families with vesico-ureteral reflux. Pediatr Nephrol 2007;22:1129–33.
Bertoli-Avella AM, Conte ML, Punzo F, et al. ROBO2 gene variants are associated with familial vesicoureteral reflux. J Am Soc Nephrol 2008;19:825–31.
Zu S, Bartik Z, Zhao S, Sillen U, Nordenskjöld A. Mutations in the ROBO2 and SLIT2 genes are rare causes of familial vesico-ureteral reflux. Pediatr Nephrol 2009;24:1501–8.
Cordell HJ, Darlay R, Charoen P, et al.; UK VUR Study Group. Whole-genome linkage and association scan in primary, nonsyndromic vesicoureteric reflux. J Am Soc Nephrol 2010;21:113–23.
van Eerde AM, Duran K, van Riel E, et al. Genes in the ureteric budding pathway: association study on vesico-ureteral reflux patients. PLoS One 2012;7:e31327.
Dobson MG, Darlow JM, Hunziker M, Green AJ, Barton DE, Puri P. Heterozygous non-synonymous ROBO2 variants are unlikely to be sufficient to cause familial vesicoureteric reflux. Kidney Int 2013;84:327–37.
Hwang DY, Dworschak GC, Kohl S, et al. Mutations in 12 known dominant disease-causing genes clarify many congenital anomalies of the kidney and urinary tract. Kidney Int 2014;85:1429–33.
Adekoya E, Ait-Zahra M, et al.; International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature 2001;409:860–921.
Goodier JL, Kazazian HH Jr . Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell 2008;135:23–35.
Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet 2009;10:691–703.
Kano H, Godoy I, Courtney C, et al. L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes Dev 2009;23:1303–12.
Dogan RI, Getoor L, Wilbur WJ, Mount SM. SplicePort–an interactive splice-site analysis tool. Nucleic Acids Res 2007;35:W285–91.
Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ, Krainer AR. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet 2006;15:2490–508.
Cartegni L, Wang J, Zhu Z, Zhang MQ, Krainer AR. ESEfinder: a web resource to identify exonic splicing enhancers. Nucleic Acids Res 2003;31:3568–71.
Kent WJ, Sugnet CW, Furey TS, et al. The human genome browser at UCSC. Genome Res 2002;12:996–1006.
Kidd T, Brose K, Mitchell KJ, et al. Roundabout controls axon crossing of the CNS midline and defines a novel subfamily of evolutionarily conserved guidance receptors. Cell 1998;92:205–15.
Wang H, Li Q, Liu J, Mendelsohn C, Salant DJ, Lu W. Noninvasive assessment of antenatal hydronephrosis in mice reveals a critical role for Robo2 in maintaining anti-reflux mechanism. PLoS One 2011;6:e24763.
Yue Y, Grossmann B, Galetzka D, Zechner U, Haaf T. Isolation and differential expression of two isoforms of the ROBO2/Robo2 axon guidance receptor gene in humans and mice. Genomics 2006;88:772–8.
Hayashi K. PCR-SSCP: a simple and sensitive method for detection of mutations in the genomic DNA. PCR Methods Appl 1991;1:34–8.
Forrest S, Cotton R, Landegren U, Southern E. How to find all those mutations. Nat Genet 1995;10:375–6.
Solyom S, Ewing AD, Rahrmann EP, et al. Extensive somatic L1 retrotransposition in colorectal tumors. Genome Res 2012;22:2328–38.
Lee E, Iskow R, Yang L, et al.; Cancer Genome Atlas Research Network. Landscape of somatic retrotransposition in human cancers. Science 2012;337:967–71.
Acknowledgements
The authors thank Dimitrios Noutsopoulos for the critical reviewing of the manuscript and Nikolaos Monokrousos for his assistance in statistical analysis issues.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Mitsioni, A., Siomou, E., Bouba, I. et al. ROBO2 gene variants in children with primary nonsyndromic vesicoureteral reflux with or without renal hypoplasia/dysplasia. Pediatr Res 80, 72–76 (2016). https://doi.org/10.1038/pr.2016.51
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2016.51
- Springer Nature America, Inc.
This article is cited by
-
GEN1 as a risk factor for human congenital anomalies of the kidney and urinary tract
Human Genomics (2024)