
Abstract
Primary care pediatricians and a variety of specialist physicians strive to define an accurate diagnosis for children presenting with impairment of expressive speech and delay in achieving developmental milestones. Within the past two decades, a group of disorders featuring this presentation have been identified as cerebral creatine deficiency syndromes (CCDS). Patients with these disorders were initially discerned using proton magnetic resonance spectroscopy of the brain within a magnetic resonance imaging (MRI) examination. The objective of this review is to provide the clinician with an overview of the current information available on identifying and treating these conditions. We explain the salient features of creatine metabolism, synthesis, and transport required for normal development. We propose diagnostic approaches for confirming a CCDS diagnosis. Finally, we describe treatment approaches for managing patients with these conditions.
Similar content being viewed by others


Main
The diagnosis and care of children presenting with significant speech deficits and delay in achieving developmental milestones challenges the primary care pediatrician, child neurologist, developmental pediatrician and other health care providers. General prevalence rates worldwide are estimated at 10.4/1,000 for intellectual disabilities and an estimated 6.2/1,000 for pervasive developmental disorders including autism spectrum disorders (1,2). Recently, a group of disorders referred to as cerebral creatine deficiency syndromes (CCDS) have been identified which feature this presentation (3,4,5,6,7). Given the purported prevalence of CCDS in males with intellectual disabilities is about 2%, there is a significant need to increase awareness of how to screen for these conditions (8,9,10,11,12,13,14,15,16,17,18,19). The goal of this review is to provide the clinician with an overview of the current information available regarding CCDS. We begin by presenting the key features of creatine metabolism, synthesis and transport. Next, the clinical features of three creatine deficiency syndromes are summarized. Diagnostic approaches are presented to distinguish the CCDS from other disorders and among themselves. Finally, we describe treatment approaches for managing patients with these conditions.
Creatine Metabolism, Synthesis, and Transport
Synthesis of Creatine
For mammals, dietary creatine provides about half of the daily requirement with endogeneous synthesis providing the remainder. Creatine is synthesized in a two-step reaction as outlined in Figure 1 . The first step (reaction step 1), catalyzed by arginine glycine acyl transferase (AGAT, chromosomal location 15q15.1), occurs with arginine and glycine combining to form guanidinoacetic acid (GAA) and ornithine. The GAA produced at this stage has only one end point: to form creatine, as there is no known physiological use for GAA in humans. The second step (reaction step 2) occurs as guanidinoacetic acid methyl transferase (GAMT, chromosomal location 19p13.3) methylates GAA to yield creatine. This step also uses the traditional S-adenosyl methionine (SAM) reaction. Most of the creatine fashioned in this pathway is produced in the liver, as the liver contains little to no creatine kinase. The liver makes 1–2 g of creatine per day in an adult (20). As creatine circulates in the blood stream, tissues such as heart, muscle and brain take creatine into the cells to be phosphorylated and become part of the creatine kinase system.

Creatine system. A schematic summary of the reactions required for creatine synthesis, uptake, metabolism and excretion with the organ or fluid sites involved noted on the right. Solute carrier family 6, member 8 (SLC6A8), also known as CRTR and CT1, represent the creatine transporter. ADP, adenosine diphosphate; AGAT, arginine glycine acyl transferase; ATP, adenosine triphosphate; CK, creatine kinase; GAMT, guanidinoacetic acid methyl transferase.
Metabolism of Creatine
Creatine is an essential metabolite for normal brain, heart and muscle function. It is converted to creatine phosphate via the reversible enzymatic reaction catalyzed by creatine kinase.
Creatine phosphate can buffer the ATP concentration in cells with high and/or dynamic energy demands. When this system is dysfunctional, the change in energy metabolism, especially in the brain, leads to a host of cognitive changes that are discussed below.
Creatinine is formed in the human body by the spontaneous, nonenzymatic, and irreversible cyclization of phosphocreatine and creatine. Because total body phosphocreatine and creatine is relatively constant, the production of creatinine is also constant. Creatinine is not metabolically active and is excreted in the urine. There are no known reuptake mechanisms for creatinine, whereas creatine is reabsorbed from the distal tubules. Urinary creatinine is therefore often used as an indicator of kidney function with the caveat that total body phosphocreatine and creatine metabolism is stable and normal. If creatine levels were low due to a synthesis defect, creatinine would also be low. A comprehensive review of creatine is found in Wyss and Kaddurah-Daouk (21).
Transport of Creatine
Creatine circulating in the blood stream is generally about 50 µmol/l in concentration and is transported into cells via the creatine transporter protein (a transmembrane protein referred to as: SLC6A8). This electrogenic protein pump actively transports creatine into the cells and concentrates creatine in the cells with a gradient of ~100 to 1. The transporter utilizes the sodium (Na+) gradient when transporting creatine. There are at least two genes for the creatine transporter, but the one most widely expressed in the human body is found on the X chromosome (Xq28); SLC6A8 (22).
Cerebral Creatine Deficiency Syndromes
There are three recognized syndromes in humans associated with defects in creatine metabolism. Two syndromes are focused on creatine synthesis and one syndrome with transport. The synthesis disorders arise from either a defect in the AGAT or GAMT enzymes as noted in Figure 1 . However, when SLC6A8 is defective, transport of creatine into the brain is impaired resulting in creatine transporter deficiency (CTD). As of 2014, there are ~14 reported patients with AGAT deficiency (23,24) and an estimated 87 with GAMT deficiency (25). The incidence of GAMT is reported as 1/114,072 in the state of Utah within the United States (26). AGAT and GAMT deficiency are inherited in an autosomal recessive manner. CTD is an X-linked disorder estimated to be the second most common cause of X-linked intellectual disabilities with over 150 individuals internationally diagnosed (8,10,11,12,13,14,15).
Case reports and series of patients with CCDS in the literature include a general observation of autistic behaviors and for some, the diagnosis of autism. Unfortunately, the basis for the diagnosis of autism, specifically, the neurobehavioral instrument is usually not described. However, to our knowledge, only one formal study has been conducted to determine the incidence of CCDS in a population diagnosed with autism. Newmeyer et al. found no definitive patients in a cohort of 100 males, ages 3–18 y identified from the Autism Genetic Resource Exchange database (27). There is another study, which evaluated patients with unexplained neurological symptoms from six major French university hospitals over a 28-mo period for CCDS (19). They found the prevalence of CCDS of 0.25% (0% for AGAT, 0.09% for GAMT, and 0.16% for CTD. There are incidence studies for CTD evaluating cohorts with diagnoses of autism and/or intellectual delays with urine screening confirmed by DNA sequencing or MRS (8,10,11,12,13,14,15). Some of these have a “pure” focus of X-linked intellectual delays for CTD. The consensus of these various cohort studies suggests prevalence rates between 0.4 and 1.4%
Individuals with CCDS, who are untreated, are generally unable to care for themselves at any age. CCDS typically present with developmental delay, intellectual deficiency, and severe speech deficits. Developmental delay is often observed prior to 1 y of age with many developmental milestones missed. The inability to speak is fairly profound as are the receptive language deficiencies. Usually, children with CCDS have limited language comprehension and are very poor at communication often only having a hand full of words or communicative vocalizations. van de Kamp et al. reports males with CTD between 0–4 y about 42% have no speech, 47% have only single words, and 11% speak in sentences (28). By 10 y of age and above, 14% have no speech, 55% have only single word, and 31% speak in sentences. Impairment of expressive speech is often confounded with the global development and intellectual delays (29). Notwithstanding, speech difficulty is considered a hallmark of these disorders. Some patients are often diagnosed with an autism spectrum disorder, in part, because of the communication and cognitive deficiencies. Seizure disorder and musculoskeletal delays are common, but dysmorphic features are not typical. Individuals with a CCDS are likely to have a seizure disorder diagnosed (11% for AGAT, 59% for CTD, and 75–90% for GAMT) (23,25,28,30). Often, these are generalized tonic-clonic seizures, but absence seizures have also been observed. In the GAMT deficiency patients, seizure disorder is often controlled with monotherapy, however, about 30% are intractable (25). For patients with GAMT, electroencephalography (EEG) typically shows slow background activity and multifocal spike slow waves (26). Upon treatment with creatine monohydrate, seizure frequency decreases with essentially complete control upon the addition of ornithine (29). There is a report of a seizure disorder in a CTD female, which was successfully treated with creatine supplementation (31). The first CTD patient reported demonstrated multifocal epileptiform discharges on EEG, however, other CTD patients have normal EEG patterns (5). Some patients, especially those with creatine synthesis defects, are suspected to have a metabolic disorder and worked up for mitochondrial myopathies requiring biopsies as part of the diagnostic paradigm. These are generally equivocal. For the physician, the clinical hallmarks to look for are profound speech deficits and intellectual disabilities.
The presentation at time of diagnosis may also depend on the patient gender. For CTD, males are primarily affected; however, some case reports report a milder phenotype than the initially described severe phenotypes (32). Heterozygous carrier females with a family mutation vary in their phenotypes. Female carriers within the first families identified with CTD presented with a mild to moderate phenotype (33,34). However, a severe phenotype was described in a female with CTD deficiency (31). There was no evidence of skewed X-inactivation in peripheral blood cells, which suggests brain specific skewed X-inactivation for this female patient. For patients with AGAT and GAMT defects, the influence of sex on disease presentation is not distinguishable with approximately equivalent numbers of the reported affected male and female patients. For carriers of the autosomal recessive defects of AGAT and GAMT, there is no risk for developing the syndrome.
Brain imaging of patients with developmental delays, intellectual deficiency, and severe speech deficits has limited value in recognizing CCDS. In certain patients with the GAMT defect, pathological signal abnormalities have been described within the globus pallidus of the basal ganglia (29). For some patients with CTD, volume loss has been described (35,36). In the seven CTD patients followed in Cincinnati, five are noted on initial imaging studies to demonstrate mild volume loss.
Diagnostic Approaches for Identifying CCDS
Magnetic Resonance Spectroscopy
Proton Magnetic Resonance Spectroscopy (MRS) of the human brain was how the first CCDS patients were identified. It also remains the primary diagnostic tool to characterize how GAMT and AGAT deficiency patients respond to treatment. Using magnetic resonance imaging (MRI) scanners, proton MRS performed in the brain is a measurement of neural metabolites. It produces a spectrum with three key signals: one from a neuronal marker (N-acetyl aspartate, NAA), one for a cellular membrane marker (Cholines, Cho) and one representing a combination of creatine and phosphocreatine (Cr). The proton MRS generates signal for the common methyl and methylene groups in both creatine and phosphocreatine. Most clinical MRI scanners have the software capabilities to perform proton MRS; however, not all neuroradiologists are comfortable interpreting the spectra generated from it. Phosphorus MRS is not routinely installed on clinically operated MRI systems. Thus, no phosphorus brain MRS has been reported in creatine deficiency syndromes to date. Research operated MRI scanners are capable; however, they often lack appropriate environments for administration of sedation and anesthesia requisite for this population. It would be useful to determine the phosphocreatine contribution in residual creatine levels and/or treatment response. Proton MRS should be ordered to address a specific question, which in the circumstance of speech deficits and cognitive delay, “is there brain creatine deficiency?” Unlike MRI examinations, where the whole brain is evaluated, MRS is typically performed in the brain to generate spectra for a single region on the order of 8 cc in volume. The brain regions sampled depend on the clinical history and/or regions with signal abnormality, but generally include the basal ganglia or white matter. Each spectral acquisition usually requires 3–5 min, with variations based on the parameters employed by a given imaging center or radiology department. The absence or significant depletion of creatine and phosphocreatine on the proton spectrum is quite striking and is essentially region independent for CCDS. Observing a distinct lack or profound diminution of the combined creatine and phosphocreatine peak on proton MRS is a hallmark of all three CCDS. It is a manifestation of the phenotype of the disorders as well and is only found when one of the three: GAMT deficiency, AGAT deficiency, or CTD is eventually diagnosed. It is highly sensitive and specific for these three syndromes.
In some patients with CTD, residual signal in the location of creatine remains, indicating that small levels of creatine are detected with less than 10% of normative values observed with MRS. ( Figure 2 ) Signal contributions may also arise from the gamma-aminobutyric acid (GABA) coresonating at the same frequency location (three parts per million (ppm)) on MRS. Extracellular creatine in fluid, such as cerebrospinal fluid (CSF), could also be another creatine source. It is known that creatine is present in the CSF of CTD patients (28). Because MRS is not able to distinguish intracellular creatine from extracellular or CSF creatine, it is not clear what contributory source(s) and the metabolic significance the partial creatine peak represents in these patients. Phosphocreatine is generated within functioning cells, so theoretically phosphocreatine may be a better indicator of intracellular creatine levels. The authors maintain that very low (<10% of normative levels) or no visible creatine seen with MRS is a phenotypic confirmation of a creatine deficiency syndrome.

MRI and proton MRS in the first male patient diagnosed with creatine transporter deficiency. Originally discovered at 6 y of age, a long-term follow-up study of the first patient recognized with creatine transporter deficiency (CTD), performed when he was 15 y. (a) Long echo time (288 ms) proton magnetic resonance spectroscopy was acquired within frontal white matter (white box in image). MRS reveals absent/diminished creatine signal noted with the thick arrow. N-acetyl aspartate (NAA) and cholines (Cho) are observed at normal concentrations. (b) Inset shows an axial-orientation, fluid attenuated inversion recovery (FLAIR) image with nonspecific hyperintense signal with the centrum semiovale. Complete MRI revealed mild volume loss. (c) Long echo proton magnetic resonance spectroscopy was acquired within frontal white matter of a healthy child with normal concentrations of NAA, Cho, creatine, and phosphocreatine (Cr).
Intermediate levels of creatine have been observed in the brain using MRS. Female carriers of CTD may show reduced creatine levels in the brain, but they generally do not have the profound cognitive deficits that are apparent in the males and the one reported severely affected female CTD patient (31,33,34,37).
With GAMT and AGAT deficiency patients, brain creatine levels can approach normal levels with creatine supplementation (3,23,25,26,29,38,39,40,41,42,43,44,45,46,47,48,49,50) ( Figure 3 ). In some instances for patients with GAMT deficiency, MRS can also illustrate an elevation of GAA.

MRI and proton MRS of female patient with GAMT deficiency treated with creatine supplementation. (a) T2-weighted imaging of 5-y-old female with guanidinoacetic acid methyltransferase deficiency (GAMT) demonstrates hyperintense signal (noted with arrow) bilaterally within the globus pallidus; (b) Postcreatine supplementation T2-weighted imaging shows normalization of signal (noted with arrow); (c) Long echo time (288 ms) proton magnetic resonance spectroscopy acquisition within basal ganglia of the 5-y-old female patient before creatine supplementation. At 3.0 ppm, the thick arrow indicates where creatine is absent. An elevated guanidinoacetic acid (GAA) peak is also observed at 3.8 ppm; (d) Repeat spectroscopy after supplementation shows the restoration of creatine and normalization of GAA on long echo acquisition repeated after creatine supplementation; (e,f) Axial and coronal images illustrating the site of spectroscopy voxel acquisition.
Blood (Cr, Crn, GAA)
Blood tests reporting low creatine can be used to screen for GAMT and AGAT deficiencies as the lack of functioning synthesis enzymes has the hallmark of low creatine in the blood. If there is not an excess of creatine from an exogenous dietary source, this assumption is valid. There is no definitive evidence to conduct fasting blood creatine level; however, it may help mitigate a false negative result following consumption of creatine. Creatinine may be low in these individuals if they have not been augmenting creatine with their diet or eating significant amounts of meat and fish. The muscles can retain a large amount of creatine and phosphocreatine, which is the source of most creatinine. An absence of GAA in the blood would be indicative of AGAT deficiency, whereas elevated GAA can only be considered as consistent with GAMT deficiency. Very little GAA is taken into the body with the diet, so the main source of GAA in the blood stream would be from synthesis via AGAT.
Creatine, creatinine, and GAA levels in the blood of CTD patients are often normal and reach elevated levels in patients when supplemented with creatine. However, there is currently no evidence supporting transport across the blood–brain barrier without the creatine transporter (51).
In Table 1 , we summarize the relative changes seen with AGAT deficiency, GAMT deficiency and CTD in blood and urine. As different labs determine customized normative values, and employ different units, we have presented this information as descriptive.
Urine (Creatine, Creatinine, GAA)
Levels of creatine, creatinine, and GAA in the urine of GAMT and AGAT deficiency patients are a reflection of what is seen in the blood stream, with all these being low for AGAT deficiency and only GAA being elevated in GAMT deficiency patients (6,23,38,41,42,43,47,48,52).
Creatinine is generally low in GAMT and AGAT deficiencies but normal in CTD. Because of dietary sources of creatine, the urine creatinine level is a very crude screening tool to rule in further testing, but should be considered carefully before ruling out other testing and not used for a definitive diagnosis.
CTD often presents with normal to high metabolite levels in the urine as most patients have creatine and phosphocreatine in nonbrain tissues (5,34,53). There has been some speculation that the ratio of slightly elevated creatine in the urine with low to normal creatinine may be diagnostic for CTD, but that has never been validated and should be avoided as a screening tool (5).
There have been several unpublished reports that urinalysis has led to false positives; this includes CTD and GAMT deficiency. Generally, these occur as part of a traditional urine test, as opposed to a 24-h urine with diet control. Patient advocacy and blog groups have reported having several parents join their groups seeking support after diagnosis and eventually withdraw when the “diagnosis” was refuted or another diagnosis confirmed. Physicians therefore need to be very careful if they choose to use urine tests as a screening tool for creatine deficiency disorders.
Genetics
Genetic testing is a critical component to diagnosing most inborn errors of metabolism including CCDS. Some laboratories chose mRNA for the sequencing, several perform exonic DNA, while others sequence the whole gene for diagnostics. As we are learning more and more about CCDS, we know more about the regions that are mutated. In the CTD gene, for example, there have been over 150 different mutations found with no clear “hot spots” identified (54). With new mutations being identified regularly, it is important to sequence the gene to identify mutations as well as polymorphisms. However, it is also important to demonstrate that a mutation is a disease causing mutation as opposed to a harmless polymorphism, as genetic testing must always be coupled with a phenotypic examination.
There are few established laboratories conducting individual sequencing of one or two creatine deficiency defects. Some laboratories are unable to conduct evaluations for all three creatine deficiency defects and depending upon the methodology chosen, the results might miss some types of mutations. Cincinnati Children’s Hospital Medical Center does a complete sequencing of the DNA for AGAT, GAMT, and CTD. Physicians and patient advocates should be aware of what the genetic test limits are when they choose a laboratory.
In Vitro Diagnostics and Creatine Transport
There have been reports where specialized laboratories have obtained fibroblasts as part of the diagnostic paradigm for assessing CTD specifically to determine if those cells can transport creatine adequately. This is considered a phenotypic confirmation of the disease, but is quite costly, labor intensive, and time consuming (53). Often, these studies have been phenomenological demonstrating a presence or absence of creatine transport that is or is not inhibited by guanidino propionic acid (GPA). GPA is a creatine analog that competes for transport with creatine.
The authors of this work are not aware of any work using in vitro cells to diagnose GAMT deficiency and/or AGAT deficiency. GAMT and AGAT deficiencies tend to be diagnosed by blood, urine, and genetic tests and do not use in vitro diagnostics.
The Concept of Phenotype and Genotype
As with many inborn errors of metabolism, AGAT deficiency, GAMT deficiency, and CTD have clinical phenotypes related to the genotype associated with a specific mutation. The phenotype is not just the clinical presentation, but evidence that the protein is not working. The genotype is where the gene is probed or sequenced for mutations, recognizing that a mutation does not always cause a disease. A polymorphism might cause a deletion in the gene or it might result in no change in the protein being synthesized. This occurs because amino acids can be coded for using multiple triplets of DNA sequence.
Diagnosis
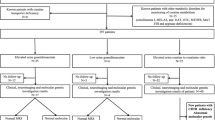
The diagnosis of a CCDS must be done with at least two methods. When a gene mutation confirms a genotype and biochemical evidence shows that the protein is not functioning in a patient with the clinical features, one can make a definitive diagnosis.
For example, if a patient has developmental delay with speech deficit, a physician might at some time order a urinalysis (UA) and or complete blood count (CBC). The UA shows low creatine and elevated GAA. The suspicion would be a GAMT deficiency, so a genetic test is ordered and the genetic test can confirm the GAMT mutation. In another scenario, the patient has severe speech deficits and cognitive impairment. The UA might show nothing or a hint of elevated creatine to creatinine ratio. MRS should be the next test conducted. If MRS shows low or no creatine in the brain and a subsequent genetic analysis confirms a mutation, then the diagnosis of CTD could be given. The MRS is recommended at this point, because it confirms a biochemical deficiency, as the UA did not show biochemical changes in this scenario. Obviously, GAA and creatine are not standard measures in a UA, but they can be done and are essential for assessing creatine deficiency syndromes (28).
Treatments for CCDS
AGAT
Patients with the AGAT deficiency are unable to perform the first step of the creatine synthesis pathway. These patients with AGAT deficiency are often very successfully treated, when found early in life, with oral creatine supplementation. The doses of creatine often used for these individuals are of the order of 100–800 mg/kg/day generally administered via three or four doses daily (23,38,39,41).
GAMT
The GAMT disorder occurs in patients who have a deficiency in the methylation step of creatine synthesis where the GAA is methylated with SAM. In the absence of GAMT activity, GAA accumulates and creatine is not synthesized. GAA levels in GAMT patients can be tenfold higher than normal. When treated early in life, the cognitive deficits of the GAMT disorder can be very well managed with 100–800 mg/kg/day supplementation of creatine. However, the levels of GAA can still remain elevated by two or three times normal. Mitigation of the GAA is managed with additional supplementation with ornithine (100–800 mg/kg/day) and/or sodium benzoate (100 mg/kg/day) (29). There is limited consensus concerning minimizing protein, arginine, and or glycine intake (25,26,29,40,42,43,46,48,55).
Treatment of the GAMT disorders is focused on normalizing cognitive and speech deficits, minimizing seizure activity and keeping GAA as close to normal levels as possible. There can be the incidence of breakthrough seizures in these patients and it is unclear if these are due to the GAA or other sequelae. There is some concern that long-term elevated GAA levels are neurotoxic. More research is needed to better understand the ideal levels of GAA for maintenance in GAMT patients.
CTD
CTD currently has no efficacious treatment available. There are unsuccessful treatment trials with creatine, and precursors to creatine synthesis (5,34,36,49,56,57,58,59,60). Treatment for these patients is currently limited to supportive care, symptom management and controlling seizures. However, creatine supplementation is generally recommended on every new patient in the event there is some residual transporter activity, but no definitive success has been reported. The creatine precursor treatments have not been successful, because there does not appear to be significant creatine synthesis in the brain. There have been no reported attempts at gene therapy for CTD patients.
What is needed for CTD treatment is relatively complicated from a drug development program. A drug that will treat CTD must interact will with both mitochondrial and cytosolic creatine kinases. Furthermore, it must cross the blood–brain barrier to get into brain cells. Thus, blood–brain barrier chaperones of creatine or creatine analogs that work well with both enzymes and could use another transport mechanism are feasible pathways for treating CTD.
It is possible that some metabolic stimulants might improve some of the symptoms of CTD, but these have yet to be identified. Simply stimulating one or more metabolic pathways cannot easily overcome the tremendous energy stored in phosphocreatine and or have the speed with which phosphocreatine can be converted to ATP. So these strategies show less promise for a treatment, but may be beneficial adjuncts with other focused treatments. On the horizon is treatment of CTD with cyclocreatine, a creatine analog demonstrating promise in mice with a mutation of the creatine transporter (51).
Conclusions and Recommendations
CCDS needs to be added to the considerations when clinicians search for a diagnosis in populations with developmental delay, intellectual deficiency, and severe speech deficits. While the etiologies of these conditions are numerous, those with a CCDS can be reliably diagnosed and for some patients, successfully treated.
CCDS can be diagnosed upon employing two complimentary techniques. The first approach confirms the genetic mutation while the second process characterizes the phenotype and demonstrates the protein is not functioning appropriately. For now, sequencing the whole gene is the safest way to assess a genomic diagnosis. The more research about the genes and mutations, the easier and faster mutation analysis will become in the future. The phenotyping aspect is achieved by determining a lack of creatine. For GAMT and AGAT, a lack of whole body creatine is needed for confirmation such as with urinalysis while controlling for dietary creatine intake. Additional research is needed to understand and validate urinalysis as a “screening tool” versus a “diagnostic tool” for CTD patients. CTD patients, representing the largest creatine deficiency syndrome population, may be missed by this method as the contribution of creatine from the diet may impact the results. For true diagnostic assignment of CTD, a lack of brain creatine can be demonstrated using MRS. Imaging facilities at most major academic health centers and children’s hospitals are capable of performing MRS. This imaging technique should accompany any MRI evaluation of the brain in conditions concerning global developmental delays, expressive speech deficits or even possible metabolic disorders. From our experience in Cincinnati, all of our GAMT and CTD patients were discovered during imaging evaluations for intellectual disability and/or expressive speech delay with MRS added to the protocol.
Further research optimizing treatment for CCDS remains essential. As clinical as well as molecular phenotypical differences are understood, clinicians may be able to tailor treatment regimens, especially for GAMT deficiency. Treatment of CTD remains a significant challenge; however, a variety of promising approaches are being pursued.
Finally, improved strategies for early identification of patients with CCDS are essential towards our goal of improving the lives of these patients, their families, and caregivers.
References
Khan NZ, Gallo LA, Arghir A, et al. Autism and the grand challenges in global mental health. Autism Res 2012;5:156–9.
Elsabbagh M, Divan G, Koh YJ, et al. Global prevalence of autism and other pervasive developmental disorders. Autism Res 2012;5:160–79.
Stöckler S, Holzbach U, Hanefeld F, et al. Creatine deficiency in the brain: a new, treatable inborn error of metabolism. Pediatr Res 1994;36:409–13.
Bianchi MC, Tosetti M, Fornai F, et al. Reversible brain creatine deficiency in two sisters with normal blood creatine level. Ann Neurol 2000;47:511–3.
Cecil KM, Salomons GS, Ball WS Jr, et al. Irreversible brain creatine deficiency with elevated serum and urine creatine: a creatine transporter defect? Ann Neurol 2001;49:401–4.
Item CB, Stöckler-Ipsiroglu S, Stromberger C, et al. Arginine:glycine amidinotransferase deficiency: the third inborn error of creatine metabolism in humans. Am J Hum Genet 2001;69:1127–33.
Salomons GS, van Dooren SJ, Verhoeven NM, et al. X-linked creatine-transporter gene (SLC6A8) defect: a new creatine-deficiency syndrome. Am J Hum Genet 2001;68:1497–500.
Betsalel OT, van de Kamp JM, Martínez-Muñoz C, et al. Detection of low-level somatic and germline mosaicism by denaturing high-performance liquid chromatography in a EURO-MRX family with SLC6A8 deficiency. Neurogenetics 2008;9:183–90.
Caldeira Araújo H, Smit W, Verhoeven NM, et al. Guanidinoacetate methyltransferase deficiency identified in adults and a child with mental retardation. Am J Med Genet A 2005;133A:122–7.
Clark AJ, Rosenberg EH, Almeida LS, et al. X-linked creatine transporter (SLC6A8) mutations in about 1% of males with mental retardation of unknown etiology. Hum Genet 2006;119:604–10.
Lion-François L, Cheillan D, Pitelet G, et al. High frequency of creatine deficiency syndromes in patients with unexplained mental retardation. Neurology 2006;67:1713–4.
Mercimek-Mahmutoglu S, Muehl A, Salomons GS, et al. Screening for X-linked creatine transporter (SLC6A8) deficiency via simultaneous determination of urinary creatine to creatinine ratio by tandem mass-spectrometry. Mol Genet Metab 2009;96:273–5.
Newmeyer A, Cecil KM, Schapiro M, Clark JF, Degrauw TJ . Incidence of brain creatine transporter deficiency in males with developmental delay referred for brain magnetic resonance imaging. J Dev Behav Pediatr 2005;26:276–82.
Puusepp H, Kall K, Salomons GS, et al. The screening of SLC6A8 deficiency among Estonian families with X-linked mental retardation. J Inherit Metab Dis 2010;33:Suppl 3:S5–11.
Rosenberg EH, Almeida LS, Kleefstra T, et al. High prevalence of SLC6A8 deficiency in X-linked mental retardation. Am J Hum Genet 2004;75:97–105.
Kato H, Miyake F, Shimbo H, et al. Urine screening for patients with developmental disabilities detected a patient with creatine transporter deficiency due to a novel missense mutation in SLC6A8. Brain Dev 2014;36:630–3.
Mencarelli MA, Tassini M, Pollazzon M, et al. Creatine transporter defect diagnosed by proton NMR spectroscopy in males with intellectual disability. Am J Med Genet A 2011;155A:2446–52.
Arias A, Corbella M, Fons C, et al. Creatine transporter deficiency: prevalence among patients with mental retardation and pitfalls in metabolite screening. Clin Biochem 2007;40:1328–31.
Cheillan D, Joncquel-Chevalier Curt M, Briand G, et al. Screening for primary creatine deficiencies in French patients with unexplained neurological symptoms. Orphanet J Rare Dis 2012;7:96.
Andres RH, Ducray AD, Schlattner U, Wallimann T, Widmer HR . Functions and effects of creatine in the central nervous system. Brain Res Bull 2008;76:329–43.
Wyss M, Kaddurah-Daouk R . Creatine and creatinine metabolism. Physiol Rev 2000;80:1107–213.
Nash SR, Giros B, Kingsmore SF, et al. Cloning, pharmacological characterization, and genomic localization of the human creatine transporter. Recept Channels 1994;2:165–74.
Ndika JD, Johnston K, Barkovich JA, et al. Developmental progress and creatine restoration upon long-term creatine supplementation of a patient with arginine:glycine amidinotransferase deficiency. Mol Genet Metab 2012;106:48–54.
Comeaux MS, Wang J, Wang G, et al. Biochemical, molecular, and clinical diagnoses of patients with cerebral creatine deficiency syndromes. Mol Genet Metab 2013;109:260–8.
Mercimek-Mahmutoglu S, Ndika J, Kanhai W, et al. Thirteen new patients with guanidinoacetate methyltransferase deficiency and functional characterization of nineteen novel missense variants in the GAMT gene. Hum Mutat 2014;35:462–9.
Viau KS, Ernst SL, Pasquali M, Botto LD, Hedlund G, Longo N . Evidence-based treatment of guanidinoacetate methyltransferase (GAMT) deficiency. Mol Genet Metab 2013;110:255–62.
Newmeyer A, deGrauw T, Clark J, Chuck G, Salomons G . Screening of male patients with autism spectrum disorder for creatine transporter deficiency. Neuropediatrics 2007;38:310–2.
van de Kamp JM, Betsalel OT, Mercimek-Mahmutoglu S, et al. Phenotype and genotype in 101 males with X-linked creatine transporter deficiency. J Med Genet 2013;50:463–72.
Stockler-Ipsiroglu S, van Karnebeek C, Longo N, et al. Guanidinoacetate methyltransferase (GAMT) deficiency: outcomes in 48 individuals and recommendations for diagnosis, treatment and monitoring. Mol Genet Metab 2014;111:16–25.
Mercimek-Mahmutoglu S, Stoeckler-Ipsiroglu S, Adami A, et al. GAMT deficiency: features, treatment, and outcome in an inborn error of creatine synthesis. Neurology 2006;67:480–4.
Mercimek-Mahmutoglu S, Connolly MB, Poskitt KJ, et al. Treatment of intractable epilepsy in a female with SLC6A8 deficiency. Mol Genet Metab 2010;101:409–12.
Battini R, Chilosi AM, Casarano M, et al. Language disorder with mild intellectual disability in a child affected by a novel mutation of SLC6A8 gene. Mol Genet Metab 2011;102:153–6.
deGrauw TJ, Cecil KM, Byars AW, Salomons GS, Ball WS, Jakobs C . The clinical syndrome of creatine transporter deficiency. Mol Cell Biochem 2003;244:45–8.
deGrauw TJ, Salomons GS, Cecil KM, et al. Congenital creatine transporter deficiency. Neuropediatrics 2002;33:232–8.
Mancini GM, Catsman-Berrevoets CE, de Coo IF, et al. Two novel mutations in SLC6A8 cause creatine transporter defect and distinctive X-linked mental retardation in two unrelated Dutch families. Am J Med Genet A 2005;132A:288–95.
Póo-Argüelles P, Arias A, Vilaseca MA, et al. X-Linked creatine transporter deficiency in two patients with severe mental retardation and autism. J Inherit Metab Dis 2006;29:220–3.
Cecil KM, DeGrauw TJ, Salomons GS, Jakobs C, Egelhoff JC, Clark JF . Magnetic resonance spectroscopy in a 9-day-old heterozygous female child with creatine transporter deficiency. J Comput Assist Tomogr 2003;27:44–7.
Battini R, Alessandrì MG, Leuzzi V, et al. Arginine:glycine amidinotransferase (AGAT) deficiency in a newborn: early treatment can prevent phenotypic expression of the disease. J Pediatr 2006;148:828–30.
Battini R, Leuzzi V, Carducci C, et al. Creatine depletion in a new case with AGAT deficiency: clinical and genetic study in a large pedigree. Mol Genet Metab 2002;77:326–31.
Dhar SU, Scaglia F, Li FY, et al. Expanded clinical and molecular spectrum of guanidinoacetate methyltransferase (GAMT) deficiency. Mol Genet Metab 2009;96:38–43.
Edvardson S, Korman SH, Livne A, et al. l-arginine:glycine amidinotransferase (AGAT) deficiency: clinical presentation and response to treatment in two patients with a novel mutation. Mol Genet Metab 2010;101:228–32.
Mercimek-Mahmutoglu S, Dunbar M, Friesen A, et al. Evaluation of two year treatment outcome and limited impact of arginine restriction in a patient with GAMT deficiency. Mol Genet Metab 2012;105:155–8.
Schulze A, Hess T, Wevers R, et al. Creatine deficiency syndrome caused by guanidinoacetate methyltransferase deficiency: diagnostic tools for a new inborn error of metabolism. J Pediatr 1997;131:626–31.
Schulze A, Hoffmann GF, Bachert P, et al. Presymptomatic treatment of neonatal guanidinoacetate methyltransferase deficiency. Neurology 2006;67:719–21.
Schulze A, Mayatepek E, Bachert P, Marescau B, De Deyn PP, Rating D . Therapeutic trial of arginine restriction in creatine deficiency syndrome. Eur J Pediatr 1998;157:606–7.
Stöckler S, Hanefeld F, Frahm J . Creatine replacement therapy in guanidinoacetate methyltransferase deficiency, a novel inborn error of metabolism. Lancet 1996;348:789–90.
Stöckler S, Marescau B, De Deyn PP, Trijbels JM, Hanefeld F . Guanidino compounds in guanidinoacetate methyltransferase deficiency, a new inborn error of creatine synthesis. Metab Clin Exp 1997;46:1189–93.
Verbruggen KT, Sijens PE, Schulze A, et al. Successful treatment of a guanidinoacetate methyltransferase deficient patient: findings with relevance to treatment strategy and pathophysiology. Mol Genet Metab 2007;91:294–6.
Dunbar M, Jaggumantri S, Sargent M, Stockler-Ipsiroglu S, van Karnebeek CD . Treatment of X-linked creatine transporter (SLC6A8) deficiency: systematic review of the literature and three new cases. Mol Genet Metab 2014;112:259–74.
van de Kamp JM, Mancini GM, Salomons GS . X-linked creatine transporter deficiency: clinical aspects and pathophysiology. J Inherit Metab Dis 2014; 37:715–33.
Kurosawa Y, Degrauw TJ, Lindquist DM, et al. Cyclocreatine treatment improves cognition in mice with creatine transporter deficiency. J Clin Invest 2012;122:2837–46.
Engelke UF, Tassini M, Hayek J, et al. Guanidinoacetate methyltransferase (GAMT) deficiency diagnosed by proton NMR spectroscopy of body fluids. NMR Biomed 2009;22:538–44.
Salomons GS, van Dooren SJ, Verhoeven NM, et al. X-linked creatine transporter defect: an overview. J Inherit Metab Dis 2003;26:309–18.
Betsalel OT, Rosenberg EH, Almeida LS, et al. Characterization of novel SLC6A8 variants with the use of splice-site analysis tools and implementation of a newly developed LOVD database. Eur J Hum Genet 2011;19:56–63.
Stockler S, Schutz PW, Salomons GS . Cerebral creatine deficiency syndromes: clinical aspects, treatment and pathophysiology. Subcell Biochem 2007;46:149–66.
Almeida LS, Verhoeven NM, Roos B, et al. Creatine and guanidinoacetate: diagnostic markers for inborn errors in creatine biosynthesis and transport. Mol Genet Metab 2004;82:214–9.
Anselm IA, Anselm IM, Alkuraya FS, et al. X-linked creatine transporter defect: a report on two unrelated boys with a severe clinical phenotype. J Inherit Metab Dis 2006;29:214–9.
Chilosi A, Casarano M, Comparini A, et al. Neuropsychological profile and clinical effects of arginine treatment in children with creatine transport deficiency. Orphanet J Rare Dis 2012;7:43.
Chilosi A, Leuzzi V, Battini R, et al. Treatment with L-arginine improves neuropsychological disorders in a child with creatine transporter defect. Neurocase 2008;14:151–61.
Fons C, Sempere A, Arias A, et al. Arginine supplementation in four patients with X-linked creatine transporter defect. J Inherit Metab Dis 2008;31:724–8.
Acknowledgements
Disclosure: There is no conflict of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Clark, J., Cecil, K. Diagnostic methods and recommendations for the cerebral creatine deficiency syndromes. Pediatr Res 77, 398–405 (2015). https://doi.org/10.1038/pr.2014.203
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/pr.2014.203
- Springer Nature America, Inc.
This article is cited by
-
The effects of creatine supplementation on cognitive performance—a randomised controlled study
BMC Medicine (2023)
-
Expanding the neuroimaging findings of guanidinoacetate methyltransferase deficiency in an Iranian girl with a homozygous frameshift variant in the GAMT
neurogenetics (2023)
-
SLC gene mutations and pediatric neurological disorders: diverse clinical phenotypes in a Saudi Arabian population
Human Genetics (2022)
-
Metabolic Epilepsy
Indian Journal of Pediatrics (2021)
-
Genomics and Radiogenomics in Inherited Neurometabolic Disorders – A Practical Primer for Pediatricians
The Indian Journal of Pediatrics (2019)