
Abstract
Glycopeptide antibiotics have been a key weapon in the fight against bacterial infections for over half a century, with the progenitors, vancomycin (1) and teicoplanin (2), still used extensively. The increased occurrence of resistance and the effectiveness of these ‘last resort’ treatments for Gram-positive infections has led to the discovery and clinical development of second generation, semisynthetic lipoglycopeptide derivatives such as telavancin (3), dalbavancin (4) and oritavancin (5), which all possess broader spectra of activity and improved pharmacokinetic properties. Two of these new antibiotics, telavancin (3) and dalbavancin (4), were approved in the past 5 years and the third, oritavancin (5), is awaiting regulatory approval. In this review, the discovery, development and associated resistance of vancomycin (1) and teicoplanin (2), and semi-synthetic glycopeptides, telavancin (3), dalbavancin (4) and oritavancin (5), are detailed. The clinical implications of glycopeptide resistance, especially vancomycin (1), as well as the future prospects for current glycopeptide drugs and the development of new glycopeptides are discussed.
Similar content being viewed by others


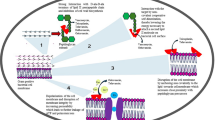
Introduction
The discovery and clinical development of antibiotics was one of the major achievements of the twentieth century, beginning with the sulfonamide and penicillin antibiotics in the 1930s. This heralded the ‘Golden Age of Antibiotics’ that lasted until the 1960s, during which time most of the antibiotic classes used in the clinic today were discovered. Although there has been a small resurgence in new antibiotic classes launched for the treatment of Gram-positive infections in the last 15 years (linezolid (first approved in 2000), daptomycin (2003), retapamulin (2007), fidaxomicin (2011) and bedaquiline (2012)),1, 2 there is still an urgent need for the development of new antibiotics with activity against drug-resistant bacteria, especially Gram-negative bacteria, which are more difficult to kill.2, 3, 4, 5, 6, 7, 8, 9
Glycopeptide antibiotics are actinomycete-derived antibiotics with unique tricyclic or tetracyclic heptapeptide cores that are usually glycosylated and sometimes have additional lipophilic fatty acid side chains (Figure 1). The first member of the glycopeptide class of antibiotics, vancomycin (1), was discovered in the 1950s by Eli Lilly & Co (Indianapolis, IN, USA) and still has an integral role today in the treatment of infections caused by Gram-positive pathogens, especially methicillin-resistant Staphylococcus aureus (MRSA) and the gut anaerobe Clostridium difficile.10, 11 The naturally occurring teicoplanin complex (for example, Factor A2-2 2) is used to treat similar infections, and was discovered in the Lepitit Research Center (Milan, Italy), the same center where Piero Sensi discovered and developed rifamycins.12 In September 2009, the first next generation semisynthetic glycopeptide telavancin (3) was launched, while another, dalbavancin (4), was approved in May 2014 with a third, oritavancin (5) scheduled for an approval decision before the end of 2014 (Figure 2).13 The avoparcin complex was used as a growth promoter in animals in the 1980s, but its use has been halted in most countries around the world because of the fear of drug resistance transfer, especially vancomycin resistant enterococci (VRE).14


Structures of the semisynthetic glycopeptides telavancin (3), dalbavancin (4), oritavancin (5) and [19F]-oritavancin (8).
Glycopeptide antibiotics were divided into four distinct structural subclasses (I–IV) by Lancini15 in 1989 and summarized more recently by Nicolaou et al.16 in 1999, according to the substituents and the type of residues at positions 1 and 3 of the heptapeptide. Glycopeptides with valine-1 and asparagine-3/glutamine-3 residues such as vancomycin (1) are classified as Type I, whereas β-avoparcin (6) is an example of a Type II glycopeptide that have aromatic residues at positions 1 and 3 that are not linked (Figure 1). Type III glycopeptides have the position 1 and 3 aromatic residues linked via an aryl ether, whereas Type IV glycopeptides such as teicoplanin A2-2 (2) are a subclass of Type III with an additional fatty acid component attached to an amino sugar (Figure 1). Nicolaou et al. also designated a Type V class of glycopeptide aglycones such as complestatin (7) that have a tryptophan17, 18, 19 in place of a phenyl group in the heptapeptide core (Figure 1). Analogs with an oxindole-alanine in place of the tryptophan such as neoprotectin A and B have also been reported.20, 21, 22 The biosynthesis of glycopeptides has been recently reviewed by Wright and coworkers23. The heptapeptide backbone of the glycopeptide antibiotics binds to the C-terminal L-Lys-D-Ala-D-Ala subunit of the peptidoglycan precursor Lipid II via five H-bonds (Figure 3), inhibiting the transglycosylation and/or transpeptidation steps involved in cell wall biosynthesis.24, 25

Schematic of the (a) five hydrogen bonds formed between the heptapeptide core and the Lipid II mimic N-acetyl-D-Ala-D-Ala, and (b) the repulsion caused by the resistant Lipid II mimic N-acetyl-D-Ala-D-Lac, which leads to 1000-fold decrease in binding with vancomycin, and the amidine isostere 12 that reintroduces the H-bond and rescues the activity.
The distinguishing feature of the Type IV glycopeptides and semisynthetic derivatives such as telavancin (3) and oritavancin (5) is the appendage of lipophilic aryl or alkyl groups onto the amino sugar subunit through amidation or alkylation. The increased potency of these derivatives is due to membrane anchoring of the lipophilic group, which facilitates binding to Lipid II through an increase in the local concentration at the membrane surface, with possible secondary effects through perturbation of the membrane potential with concomitant membrane leakage.26, 27, 28, 29, 30, 31, 32, 33 Furthermore, the hydrophobic chains may assist in back-to-back homodimerization, in turn enhancing ligand binding through a cooperative effect,34 although this is still controversial. Antibiotics with high dimerization constants generally display high in vitro potencies, and the cooperativity between dimerization and ligand binding has been proposed to correlate with enhanced antibacterial activity.33, 34 In an aqueous solution, vancomycin (1) and oritavancin (5) cooperatively dimerize in the presence of substrates bearing the L-Lys-D-Ala-D-Ala epitope. On the other hand, the dimerization of dalbavancin is anti-cooperative with respect to the same substrate,35 whereas teicoplanin, which already possesses a membrane-anchoring ‘tail’, is exceptional among the vancomycin-type class in that it does not dimerize.33, 35, 36
In this review, the discovery, development and associated resistance of the naturally occurring glycopeptides vancomycin (1) and teicoplanin (2), and semisynthetic glycopeptides, telavancin (3), dalbavancin (4) and oritavancin (5) are described. In addition, clinical implications of glycopeptide resistance, especially vancomycin (1), as well as the future prospects for the use of existing glycopeptide drugs and the development of new glycopeptides is reviewed.
Vancomycin
Discovery of vancomycin
Vancomycin (1) was first identified during an antibiotics discovery program at Eli Lilly37 (Indianapolis, IN, USA) and its discovery and development has been well documented.38, 39, 40, 41 Briefly, the project had its origins in a friendship between a Reverend William Conley and Dr. E. C. Kornfield who first met in 1944 when the US 82nd Airborne Division parachuted into Sicily. Kornfield went on to work as an organic chemist at Eli Lilly, while Conley became a missionary in Borneo. The two exchanged letters, and Kornfield, looking for novel sources of biodiversity in the hunt for new antibiotics for Eli Lilly, sent sterile vials to Conley requesting he return soil samples ‘collected off the beaten track’. There were no Material Transfer Agreements or Non-Disclosure Agreements in those days to stifle the spirit of open collaboration! An actinomycete, Streptomyces orientalis (now called Amycolatopsis orientalis),42 was isolated from the soil sample delivered to Eli Lilly in 1953, and Kornfield demonstrated it could produce a new antibiotic. Over the next few years, two additional strains from India were identified that also produced vancomycin, and optimization of the fermentation and isolation conditions led to production of an enriched fraction (called compound 05865) that was evaluated in patients from 1955.39, 43, 44 The results were promising enough for the United State Food and Drug Administration (FDA) to approve vancomycin as a ‘certifiable antibiotic’ in November 1958 for the treatment of penicillin-resistant staphylococcal infections.
It is amazing to think today that the structure of vancomycin was not fully elucidated until 1982 when Harris and Harris45 corrected the assignment of an isoasparagine residue to the correct asparagine regioisomer. Previously, Marshall had described approximately half of the structural components of 1 using classical degradation methods in 1965.46 The next breakthroughs were Williams and Kalman’s47 partial structure assignment using nuclear magnetic resonance spectroscopy in 1977 and Smith and coworkers’48 X-ray crystal structure of a crystalline degradation product (CDP-I) in 1978. The structure elucidated by Harris and Harris was confirmed by X-ray crystallography in 1996 by Sheldrick and coworkers49 and by total synthesis of vancomycin aglycone in 199850, 51 and vancomycin in 1999.50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60 Further details on the synthesis of glycopeptides can be found later in this review.
Vancomycin resistance and heteroresistance
The semisynthetic penicillin analog methicillin was approved around the same time as vancomycin, whereas the first semisynthetic cephalosporin derivative cephalothin was launched 5 years later in 1964. These and other next-generation β-lactams became the antibiotics of choice for the treatment of Gram-positive infections and relegated vancomycin to a ‘drug of last resort’ status because of its less convenient administration and side effects. However, the constant use of β-lactam antibiotics led to a dramatic rise in drug resistance and the exemplifier of these resistant strains, MRSA was endemic in most hospitals by the early 1990s. Thereafter, community-acquired MRSA infections have frequently emerged in patients without established risk factors.61, 62 This led to the reemergence of vancomycin as a frontline therapy for the treatment of MRSA and also spurred the search for alternative antibiotics, leading to the commercialization of linezolid and daptomycin. During this period, more than 99% of MRSA strains have remained susceptible to vancomycin at the current minimum inhibitory concentration (MIC) susceptibility breakpoints (maximum MIC thresholds for predicting successful treatment) designated by the National Committee for Clinical Laboratory Standards (CLSI) (Table 1).63 Nonetheless, the selection pressure exerted by years of widespread glycopeptide use is becoming apparent, with strains of MRSA now showing reduced susceptibility toward vancomycin. MRSA was recognized as a ‘serious threat’ in a 2013 US CDC report on antibiotic resistance, with over 80 000 severe infections and 11 000 deaths per year in the USA alone.64
A common glycopeptide resistance mutation, especially in enterococci, is a divergent biosynthesis of Lipid II from the D-Ala-D-Ala terminating muropeptide to a D-Ala-D-Lac (vanA, vanB, vanD) or D-Ala-D-Ser (vanC, vanE, vanG) phenotype. The replacement of D-Ala with D-Lac causes a 1000-fold decrease in binding affinity by removal of a key hydrogen bond interaction (Figure 3), whereas D-Ser exerts its effect through a less dramatic conformational change (sevenfold lower binding affinity).24, 25, 65, 66, 67, 68 Consequently, teicoplanin, vancomycin, telavancin and dalbavancin lose their biological effectiveness against strains that express the D-Ala-D-Lac epitope, suggesting that they share a common primary molecular mechanism of action and resistance. Indeed, recent nuclear magnetic resonance spectroscopy and surface plasmon resonance (SPR) studies provided further evidence that Ac-L-Lys-D-Ala-D-Lac has negligible affinity for these antibiotics.33 On the other hand, oritavancin is not subject to the same resistance profile, suggesting that it may be able to overcome resistance through a secondary mode of action.28 Solid state nuclear magnetic resonance spectroscopy studies of [19F]-oritavancin (8) (Figure 1) have demonstrated that, in addition to the sequestration of Lipid II through the D-Ala-D-Ala primary binding site, it also interacts with the pentaglycyl-bridge segment of nascent peptidoglycan in S. aureus and Enterococcus faecium, and was reported not to embed into the bacterial membrane.69, 70
Although the lineage of many VRE strains can be traced back to the global overuse of glycopeptides in the livestock industry,71, 72 the presence of these resistance genes has been identified in permafrost samples from >10 000 years ago, indicating that there is also an innate resistance reservoir in the microbiome.73, 74 A recent large-scale metagenomic study, using 71 environmental shotgun metagenomic DNA data sets from soil, ocean and animal sources, searched for 2999 known antibiotic resistance sequences.75 This study found high levels of vancomycin resistance-related genes (vanA, B, C, D, E and G) in soil, ocean and human feces samples, while vanZ was detected in both soil and human feces, overall forming 17% of the global abundance of resistant sequences measured, second only to multidrug resistance efflux pumps.75 These resistance genes were the most abundant reads detected in the human data sets, despite the relatively low usage of vancomycin compared with other antibiotics.75
Vancomycin-resistant strains of S. aureus are designated heterogeneous vancomycin intermediate (hVISA), vancomycin intermediate (VISA) and vancomycin resistant (VRSA), indicating increasing levels of resistance from heteroresistant to intermediate-resistant to fully resistant. High-level resistance to vancomycin in S. aureus (VRSA) through acquisition of the enterococcal vanA operon is still rare. From 2002 to 2012, only 13 strains of S. aureus containing the vanA operon have been reported in the USA, with several additional reports from Europe76 and South America,77 suggesting that this is not a major mechanism of resistance to vancomycin in staphylococci. The clinical impact of VRSA is not considered significant at this time, with the CDC report classifying VRSA as a ‘concerning threat’.64
VISA evolved from vancomycin-susceptible S. aureus (VSSA) via the intermediacy of hVISA, predominantly through a series of sequential cumulative mutations that did not involve the acquisition of foreign DNA. Isolates of S. aureus (VISA) with vancomycin MICs of 4–8 μg ml−1 are rare (to date patient-to-patient nosocomial transmission has not been documented in MRSA isolates with vancomycin MIC ⩾4 μg ml−1), while S. aureus (hVISA) isolates with MICs of 2 μg ml−1 are relatively common. Despite the scarcity of VISA strains, the concern is that VISA spontaneously emerges at high frequency among cell populations of hVISA.78 The distinguishing feature of hVISA is that it displays vancomycin MICs within the current CLSI mandated susceptible range (⩽2 μg ml−1) but is sub-populated (⩽10−5 to 10−6) with cells that are able to grow at a vancomycin concentration ⩾4 μg ml−1.79 Although clinical isolates of S. aureus exhibiting the VISA and hVISA phenotypes were first reported in Japan in 1997,80, 81 subsequent retrospective studies of stored isolates have since proven their prevalence as early as the mid-1980s in United States and Europe, and from 1990 in Japan, before the clinical introduction of injectable vancomycin in Japan in 1991.82, 83
hVISA (strain Mu3, (ATCC 700698)) was first documented during the treatment of a 64-year-old patient with MRSA pneumonia.80 It displayed a vancomycin MIC of 3 μg ml−1 and produced a subpopulation of cells (10−5 to 10−6) in a drug-free medium with different but stable resistance phenotypes exhibiting vancomycin MICs ranging from 4 to 8 μg ml−1.78, 80, 84 It was reported that Mu3 was widely disseminated among MRSA clinical isolates collected from a series of Japanese hospitals.80 Approximately 6 months later, the first case of VISA (strain Mu50, (ATCC 700699)) was isolated from an infant patient within the same hospital who had suffered a surgical site MRSA infection following heart surgery.81 Mu50 exhibited a vancomycin MIC of 8 μg ml−1, suggesting that Mu50 was also prevalent among the resistant subpopulations of the Mu3 strain (4–8 μg ml−1). The widespread dissemination of a naturally occurring strain of Mu3 in conjunction with its putative propensity to generate Mu50 in vivo should have altered the Japanese clinical landscape in favor of Mu50-like isolates, especially in the face of continuous vancomycin use in the late 1990s.84 However, a national surveillance study84 of 6625 MRSA clinical isolates obtained from 278 Japanese hospitals published after the report by Hiramatsu et al.80 disproved the existence of a vancomycin-heteroresistant Mu3-type strain capable of constitutively producing subpopulations of cells with stable vancomycin resistance, with MICs above those of the parent strain. Furthermore, the clinical isolates did not reveal any evidence of Mu50-type intermediately vancomycin-resistant MRSA, nor the dissemination of any Mu3-type strain and Mu50-type strain.84 Controversially, the original population analysis conditions described by Hiramatsu et al.80 to detect hVISA may have led to the misinterpretation of the data because of poor reproducibility.79, 84 The genetic differences between Mu3 and Mu5 have been studied in detail.85, 86, 87, 88 The Mu3 strain is currently used as a standard reference for the detection of hVISA isolates, despite the fact that the latest CLSI breakpoints actually classify Mu3 as VISA and not hVISA.89
Although VISA and hVISA phenotypes are globally recognized, accurate epidemiologic data are confounded by a lack of standardized criteria for their precise definition and detection. A molecular-based assay for their discrimination and detection does not exist because the genetic determinants of hVISA and VISA are poorly understood. hVISA and VISA may revert to lower levels of resistance during in vitro passage or during longer-term storage, further confounding their accurate detection.90, 91, 92 The CLSI-approved broth MIC method is currently used to define VISA.63 In contrast, there is currently no standardized method for the accurate detection of hVISA, with standard CLSI broth and agar-based testing methods ineffective because of masking by the dominant susceptible population. Detection of the resistant subpopulation of interest necessitates the use of population analysis profile testing in comparison to a known hVISA control strain (Mu3), which is costly and time consuming, rendering it unsuitable for routine automated testing.89 Uncertainties concerning the incidence, definition, phenotype stability and optimal detection of hVISA are reflected in the disparity between current estimates of hVISA in MRSA isolates, most recently reported as 1.3% (82 698 MRSA isolates collectively analyzed from 43 studies conducted worldwide)93 compared with other studies reporting significantly higher rates of 0–50% (15 studies conducted worldwide).94
Vancomycin in the clinic: use and significance of resistance
In the early days of its use, vancomycin produced a number of side effects during intravenous (IV) administration, including nephrotoxicity (especially with patients with sub-optimal renal function), red-man syndrome (an initial infusion-related reaction because of histamine release from non-specific mast cell degranulation) and, very rarely, ototoxicity. However, initial preparations of vancomycin were quite impure leading to a nickname of ‘Mississippi Mud’ because of its color. Optimized dosing and purer preparations mean that vancomycin therapy is now considered safe for patients with normal renal function.
Clinically employed vancomycin is still not a pure compound, with the predominant component making up approximately 85% of the dose. In China, desmethylvancomycin (9) (Figure 1), missing the N-methyl group on the N-terminal Leu residue, been used clinically since 1967 and was first isolated from A. orientalis Van-23, which was obtained from a soil sample collected in Guizhou Province, China in 1959.95, 96 Questions have been raised about purity and equivalence of generic vancomycin, with significant decreases in efficacy demonstrated by three generics compared to the innovator compound in a mouse thigh infection model (5.65 log10 reduction in CFU g−1 for innovator versus 2.0, 2.6 and 3.5 for the generics), despite them being undistinguishable based on in vitro assays such as MIC or protein binding.97 The inferior products were then shown to enhance the production of VISA subpopulations within the thigh infection model.98 A subsequent study of six generic vancomycin products (the innovator being no longer available) for treatment of MRSA endocarditis in rabbits found no significant differences.99
Despite the fact that an overwhelming proportion of MRSA strains are sensitive to vancomycin in vitro, increasing evidence suggests that it may be losing its clinical efficacy against serious MRSA infections with MICs at the higher end of the susceptibility range. As the MIC of an isolate within the susceptible range increases, the proportion of hVISA within that isolate also increases.100, 101 The observed gradual upward trend in MICs in S. aureus clinical isolates within the susceptibility range, a phenomenon known as ‘MIC creep’, may manifest as worsened clinical outcomes,102, 103 although the global incidence of MIC creep is contentious as epidemiological and clinical variations between study sites, and non-conformance between automated and manual MIC determination methods lead to conflicting results.104, 105 Nonetheless, infections caused by VSSA displaying subtle reductions in susceptibility to glycopeptides (MIC ⩾1.5 μg ml−1) are reported to be predictive of treatment failure (delayed early response, increased rate of relapse, prolonged hospitalization), irrespective of MIC testing methodology and infection source, whereas strains with MICs ⩾2 μg ml−1 correlate with increased mortality in staphylococcal bacteremia infections.104 A clinical study106 conducted by the Centers for Disease Control that demonstrated vancomycin was equally ineffective against MRSA strains with MICs of either 4 or 8 μg ml−1 prompted the CLSI to revise the breakpoints for vancomycin against S. aureus (Table 1).100, 107 Interestingly, antibiotic choice may not be the sole discriminator of clinical outcome, with a recent prospective study finding that a high MIC was also associated with worse outcomes in patients with MSSA infections not treated with vancomycin.108
Most hVISA and VISA are reported in hospital MRSA strains, arising primarily by in vivo evolution during failed prolonged glycopeptide therapy for reputably vancomycin-susceptible MRSA infections, particularly in patients with high bacterial load (for example, endocarditis, osteomyelitis/septic arthritis, deep abscesses, infection of prosthetic devices) and/or history of prior vancomycin exposure.83 Glycopeptide exposure may not be a unique selection criterion for hVISA. A recent retrospective study from Japan indicates that hVISA was prevalent in Japanese hospitals in the late 1980s before the clinical introduction of vancomycin, for which the mainstay therapy for MRSA at the time comprised β-lactam antibiotics such as imipenem and flomoxef.82 It was suggested that imipenem played a role in the early emergence of hVISA in Japan in 1990, ahead of its emergence in other countries.82 Indeed, in vitro exposure of S. aureus to imipenem selects for hVISA with a similar, but not identical, phenotype to vancomycin-induced hVISA.109
The impact of reduced vancomycin susceptibility is therefore of great concern, highlighting the need for the continued development of alternative Gram-positive therapies. In stark contrast to vancomycin, which enjoyed decades of resistance-free use, alternative therapies like daptomycin, linezolid, telavancin, third generation cephalosporins (for example, ceftaroline) and the glycylcycline tigecycline antibiotic classes have led to resistance soon after their clinical introduction.110
Resistance via impediment of glycopeptide access
A significant body of work has been devoted to the identification of mutations delineating the genetic differences between VSSA and VISA.79, 83, 111, 112 Although considerable genetic and transcriptional variability between different lineages results in a myriad of phenotypic outcomes, cell wall aberrations are a feature observed consistently in response to the physiological stress imposed by antibiotic treatment.79, 113 This phenomenon, characterized by altered levels of key biochemical intermediates required for peptidoglycan synthesis, is considered to be a major contributor to resistance through the impeded access of cell wall active antibiotics to sites of cell wall biosynthesis in the division septum. Fluctuations in key enzymatic processes that regulate peptidoglycan precursor synthesis and cross-linking ultimately lead to the accumulation of monomeric muropeptides with redundant D-Ala-D-Ala termini that serve as non-productive diversion points for glycopeptides. The accumulation of decoy targets bearing free D-Ala-D-Ala termini may occur by a number of pathways related to altered regulation of transpeptidase activity, cell wall precursor synthesis, cell wall turnover and cell wall cross-linking.114, 115, 116 Functionally, the net result is the same: to sequester the glycopeptide within the bulk peptidoglycan and hinder its access to sites of cell wall biosynthesis at the membrane surface in the division septum (Figure 4).

S. aureus cell wall and its interaction with various glycopeptides. (a) Scheme of the cell wall showing the membrane, biosynthetic penicillin binding proteins (PBPs), which act as transglycosylases and/or transpeptidases, Lipid II, nascent and mature peptidoglycan. The mature peptidoglycan is cross-linked via the peptide side chains with a pentaglycyl-bridge, but also contains non-cross-linked residues, terminating in either D-γGln-L-Lys-D-Ala-D-Ala or D-γGlu-L-Lys-D-Ala-D-Ala in vancomycin-sensitive or partially resistant bacteria, or D-γGln/D-γGlu-L-Lys-D-Ala-D-Lac/D-Ser in highly resistant bacteria. These serve as attachment points for teichoic acids in the cell wall, and also act as ‘decoy targets’ for glycopeptides as they diffuse toward the bacterial membrane. (b) Vancomycin is a weak dimer (Kdim=1430 μM), and at clinically relevant concentration, is present in plasma exclusively as monomer. It binds as monomer to free D-Ala-D-Ala residues in bulk peptidoglycan, but this is a fast-on fast-off interaction. Sufficient drug can diffuse to the membrane surface, where it binds cooperatively to two membrane-anchored Lipid II molecules. This much stronger complex does not dissociate because of the chelate, or avidity effect, and vancomycin can inhibit peptidoglycan biosynthesis by sterically occluding the action of the PBPs. In contrast, teicoplanin is monomeric, but it can template on the membrane surface using the C10-acyl chain to insert in the bacterial membrane adjacent to the PBPs. (c) In VISA and hVISA (vancomycin-resistant) bacteria, the peptidoglycan cell wall is considerably thicker, and there are far more ‘decoy target’ L-Lys-D-Ala-D-Ala residues. Here, dalbavancin is effective, as although it also weakly dimerizes like vancomycin,35 it possesses a membrane anchoring C12-acyl chain, which localizes the drug preferentially to the membrane surface to occlude PBP action. (d) In VRE and VRSA, the pendant L-Lys-D-Ala-D-Ala residues are replaced with L-Lys-D-Ala-D-Lac/D-Ser, which lowers the binding affinity with glycopeptides in free solution by 1000-fold. Compounds such as vancomycin, teicoplanin and dalbavancin can no longer bind with sufficient affinity to inhibit PBP action, even when templated on the membrane. However, oritavancin, which possesses a membrane-anchoring 4-chlorobiphenyl moiety and dimerizes much more tightly (Kdim=5 μM),205 gains more benefit from the chelate or avidity effect when binding two resistant phenotype Lipid II molecules at the membrane surface, and can effectively inhibit PBPs.
In an effort to understand how altered peptidoglycan synthesis influences resistance, Hiramatsu and coworkers studied the differences between VISA strain Mu50 and hVISA strain Mu3.113, 114, 115, 117 Both strains exhibit accelerated cell wall synthesis and turnover resulting from enhanced levels of N-acetylglucosamine and the murein monomer precursor UDP-N-acetylmuramyl-pentapeptide, enhanced autolysis and cell wall turnover, and overexpression of penicillin-binding protein 2 (PBP2) and PBP2’, which are involved in both transglycosylation and transpeptidation. The distinguishing feature of Mu50 is that its cell wall is twice the thickness of Mu3, which presumably accounts for its increased level of vancomycin resistance relative to Mu3. The peptidoglycan of Mu50 exhibited an increased capacity for vancomycin binding (1.4-fold) because of an increased amount of muropeptides bearing non-amidated glutamine in the cell wall (that is, stem peptide side chains composed of L-Ala-D-γGlu-L-Lys-D-Ala-D-Ala instead of L-Ala-D-γGln-L-Lys-D-Ala-D-Ala). Given that the peptidoglycan layers of Mu50 are about twice as thick as Mu3, a single Mu50 cell was estimated to bind 2.8 times more vancomycin molecules than a single Mu3 cell.114 It was proposed115 that the non-amidated muropeptides contribute to under-cross-linking of peptidoglycan because of their decreased affinity for PBP, resulting in a surplus of D-Ala-D-Ala residues in the peptidoglycan. Besides causing under-cross-linking of peptidoglycan, non-amidated muropeptides are also implicated in the sequestration of vancomycin because of their greater affinity of binding to vancomycin than that of their amidated counterpart.114, 115 Taken together, the altered peptidoglycan mesh serves to impede glycopeptide access to the desired Lipid II target on the cytoplasmic membrane through a decrease in effective concentration and diffusion.78
In another study, Sieradzki and Tomasz116 described a series of 16 isogenic MRSA isolates obtained from a single bacteremic patient who underwent vancomycin chemotherapy for 2 months. Step-wise comparison of each isolate revealed a continuous spectrum of altered morphologies and biochemical profiles. Resistant isolates grew as multi-cellular aggregates with abnormally thick cell walls, surrounded by large amounts of amorphous extracellular material, and displayed a parallel reduction in both peptidoglycan cross-linking and PBP4. Decreased transpeptidase activity through depletion of PBP4, which catalyzes the cross-linking of the lysine component of one stem peptide to the penultimate D-Ala of a neighboring stem peptide,118 resulted in the accumulation of muropeptides with free D-Ala-D-Ala termini. In this instance, the authors attributed the abnormal cell wall thicknesses to increased amounts of teichoic acid present in the cell wall,118 which is known to interfere with peptidoglycan hydrolysis.116 Teichoic acids are polysaccharides of ribitol or glycerol phosphate that are also present in the staphylococcal cell wall, comprising up to 50% of the dry weight of the cell wall. They are implicated in processes concerning cell wall function, maintenance and turnover. Ribitol teichoic acids (wall teichoic acids) are covalently linked to peptidoglycan and decorated with D-Ala esters and N-acetylglucosamine residues, whereas glycerol teichoic acids (lipoteichoic acids) are linked to glycolipids in the cytoplasmic membrane (Figure 4). In the former case, D-Ala serves to render the teichoic acid zwitterionic by virtue of its free amino group, and the degree of alanylation influences the sensitivity of S. aureus toward glycopeptide antibiotics. Studies of a S. aureus mutant devoid of D-Ala revealed a moderate increased capacity for binding vancomycin compared with the wild type.119 It is therefore postulated that modulation of the net bacterial surface charge by alanylation of teichoic acids serves to promote resistance to cationic antimicrobial molecules through charge repulsion.120 In light of this, it appears unlikely that the D-Ala subunits of teichoic acids are targets for vancomycin binding.119
Clinical limitations of vancomycin
Although it is one of the mainstay parenteral therapies for MRSA infections, vancomycin has relatively poor pharmacokinetic properties, including a short half-life in the range of 4–11 h for healthy adults and poor tissue penetrating properties. During its early years of use, dosing strategies were conservative because of its putative toxicity, resulting in target bacteria exposure to sub-inhibitory drug concentrations. This practice, in combination with its poor tissue penetration properties, is thought to promote selection of resistance.121 In light of the recent advent of S. aureus infections displaying raised vancomycin MICs (⩾2 μg ml−1), achieving therapeutically optimal target trough concentrations is difficult, if not impossible, even with aggressive dosing strategies and constant monitoring.107, 122 The drawbacks associated with vancomycin prompted the development of second generation glycopeptides with improved pharmacokinetic and safety profiles.
Teicoplanin
Discovery and clinical use of teicoplanin
Teicoplanin is a ristocetin-type lipoglycopeptide complex exemplified by its major component teicoplanin A2-2 (2) (Figure 1) from Actinoplanes teichomyceticus, which was derived from an Indian soil sample and first reported by the Lepitit Research Center in 1978.123, 124, 125 The structure of the complex components were elucidated in 1984126, 127 and showed that the teicoplanin A2 complex differed from vancomycin (1) by additional glycosylation, an acyl chain and ether linked 4-hydroxyphenylglycine and 3,5-dihydroxyphenylglycine groups.16 The complex has five related components TA2-1 to TA2-5 that differ only in their fatty acid side chains, as well as the deacylated T-A3-1.126 The ratio of the components can be altered by changing fermentation conditions.128, 129 The difficulties of dealing with an antibiotic consisting of a mixture of complexes has been recently discussed, with calls for tighter regulatory standardization.130 For example, a recent paper reported high variability in teicoplanin protein binding in critically ill patients.131
Teicoplanin (Targocid) was first approved in Europe in 1988 and is available in many other countries around the world with the notable exception of the United States.132 Teicoplanin has a long half-life (30 h), and may be administered IV or intramuscularly as a single daily dose following an initial loading dose, which makes it suitable for outpatient therapy.133, 134 Teicoplanin is better tolerated than vancomycin, in particular causing no histamine release and only rare incidences of red man syndrome, ototoxicity and nephrotoxicity.135
Teicoplanin resistance
Teicoplanin retains activity against strains expressing the vanB operon (a resistant phenotype only induced by vancomycin), but not vanA as it is inducibly resistant to high levels of teicoplanin.136 Most hVISA strains have raised teicoplanin MICs (8–16 μg ml−1) compared with VSSA (⩽4 μg ml−1).78 Clinical isolates of MRSA exhibiting teicoplanin resistance, but sensitivity toward vancomycin, were reported in 1990, 7 years before the first reports of the clinical emergence of VISA and hVISA.137 On the other hand, several clinical MRSA strains exhibiting reduced susceptibility to teicoplanin (8–16 μg ml−1) and cross-resistance to vancomycin have also been reported.138, 139 These strains were characterized by overexpressed levels of PBP2 in addition to the appearance of an unidentified membrane protein, both of which were implicated in the production of the prototypical VISA/hVISA abnormal cell wall phenotype via the excessive production of false targets bearing D-Ala-D-Ala sequences. A later study examining in vitro generated S. aureus mutants supported this view.140 In a recent survey of 9115 S. aureus clinical isolates, the MIC90 of teicoplanin was ⩽2 μg ml−1.141
Other naturally occurring glycopeptides
The ristocetin complex (Spontin) (for example, ristocetin A (10), Figure 1) was first reported in 1956142 by Abbott Laboratories (Chicago, IL, USA) from Amycolatopsis lurida collected in 1951 from Colorado Springs, CO, USA, and was introduced into clinical use in 1957. Ristocetin is a Type II glycopeptide and the structures of its major components were finalized in 1982.143, 144, 145 Unfortunately, ristocetin was later withdrawn from the market after a subset of patients displayed thrombocytopenia, with further studies indicating that ristocetin causes platelet aggregation in patients missing a platelet factor in platelet-type von Willebrand disease.146, 147, 148 The use of ristocetin in the medical community was not lost as it forms the basis of diagnosis of this disease.149
Avoparcin (LL-AV290, Avotan) (for example, β-avoparcin (6), Figure 1) and actaplanin (for example, actaplanin A (11), Figure 1) were first reported in 1968150, 151 and 1976152 and their structures elucidated in 1980153, 154 and 1984,155, 156 respectively. As previously noted, avoparcin was used as a growth promoter in animals, but its use has been halted in most countries.
The increased health issues caused by MRSA in the 1980s led to a resurgence in the interest in the discovery of new antibiotics, including glycopeptides.16, 40 The use of innovative methods to identify new glycopeptide-producing strains40 and the ability to determine their structures led to an explosion in numbers of new glycopeptides identified from 1982 to 1996, which are summarized in reviews by Yao and Crandall40 in 1994 and Nicolaou et al.16 in 1999. To the best of our knowledge, no new naturally occurring glycopeptides have been published since these reports.
Next generation semisynthetic glycopeptides
Telavancin
Telavancin (3) (Vibativ, TD-6424) is derived from vancomycin (1) by addition of a decylaminoethyl lipophilic substituent on the vancosamine amino group, and a hydrophilic (phosphomethyl)aminomethyl moiety at the para position of the aromatic ring on the C-terminal dihydroxyphenylglycine residue. The lipophilic group plays the same role in all lipoglycopeptides, increasing membrane interactions, whereas the hydrophilic group is proposed to promote tissue distribution and clearance and reduce nephrotoxic effects.157, 158 Telavancin was developed at Theravance Inc. (San Francisco, CA, USA), and partnered with Astellas (Tokyo, Japan) in 2005, but Astellas terminated its licensing agreement in 2012. Initial analogs focused on the hydrophobic substituent, but with only this modification, molecules were less soluble than vancomycin, more nephrotoxic, and had reduced urinary clearance. Introduction of the (phosphomethyl)aminomethyl group ameliorated these effects.157, 158 Telavancin has improved potency compared to vancomycin against a range of Gram-positive bacteria, including vancomycin-resistant strains. Two phase-III trials assessed 1794 patients with complicated skin and skin structure infections resulting from suspected or confirmed MRSA, comparing 7–14 days intravenous dosing of 1 g of vancomycin administered every 12 h with 10 mg kg−1 telavancin administered once daily.159, 160 Non-inferiority of telavancin to vancomycin in the clinically evaluable patients was demonstrated, but superiority to vancomycin in the all-treated efficacy population infected with MRSA was not met. FDA approval was granted in 2009 for treatment of complicated skin and skin structure infections caused by susceptible Gram-positive bacteria. Clinical use of telavancin has been subsequently extended to include hospital-acquired and ventilator-associated bacterial pneumonia. Telavancin, with a human half-life of approximately 8 h, has been associated with nephrotoxicity and renal impairment and has a black box warning for fetal risk.160 Cautious use is recommended in patients who are also taking drugs known to prolong the cardiac ventricular polarization (QT) interval.161
Telavancin resistance
Similar to teicoplanin, telavancin retains activity against strains that express vanB, but not vanA as it induces expression of the latter.30 Telavancin has demonstrated a low potential for development of resistance among enterococci and staphylococci (<4.0 × 10−11 to <2.9 × 10−10 at 2 × MIC for VSSA, hVISA and VISA).162 Heteroresistance has not been detected in isolates of S. aureus, nor has resistance been detected in isolates of S. aureus or Streptococcus pneumoniae collected in both clinical trials and through global surveillance.161, 162, 163, 164 MIC90 values of 0.25–0.5 μg ml−1 (S. aureus isolates including MRSA) and 0.03 μg ml−1 (S. pneumoniae) have been reported.161
Dalbavancin
Dalbavancin (4) (Dalvance, BI-397) is a semisynthetic derivative of the teicoplanin-like glycopeptide A40926 Factor B, with a torturous development pathway, eventually approved in May 2014. It was originally developed in the 1990s by the Lepitit Research Centre belonging to Marion Merrell Dow, with the center bought by Hoechst in 1995 and spun out as Biosearch Italia S.p.A. in 1997. In 2003, Versicor acquired Biosearch Italia and renamed the new company as Vicuron Pharmaceuticals, which in turn was acquired by Pfizer (Groton, CT, USA) in 2005. Three phase-III trials were successfully completed between 2003 and 2005, but the FDA required additional non-inferiority data in 2007.30 Durata Therapeutics (Chicago, IL, USA) acquired the program in 2009 and initiated two additional phase-III trials (573 patients at 92 sites and 739 patients at 139 sites) which met their primary endpoint of non-inferiority, comparing two intravenous doses of dalbavancin given 1 week apart with twice-daily vancomycin doses for 14 days.165, 166
Dalbavancin is derived from the A40926 complex from Nonomuraea sp., which is similar in structure and activity to teicoplanin, including a hydrophobic alkyl substituent.167 Like teicoplanin, A40926 is a complex of five closely related homologs, with factor B0 predominating (>80%). Amidation of the C-terminal carboxyl group (based on the precedent that teicoplanin amides were more active than teicoplanin)168 with a dimethylaminopropylamine group produced dalbavancin.167, 169, 170, 171 These modifications led to an extended half-life of over 300 h in humans172 allowing for once weekly dosing.
Dalbavancin resistance
Similar to teicoplanin and telavancin, dalbavancin resistance is observed for strains that express the vanA operon, but not vanB as it only induces expression of the former.173 Prospective worldwide surveillance from 2002 to 2012 of the in vitro potency of dalbavancin from over 150 000 Gram-positive isolates found no evidence of glycopeptide-resistant or glycopeptide-intermediate S. aureus isolates by CLSI criteria (MIC90 was 0.06 μg ml−1 for S. aureus). Isolates from phase-III clinical trials exhibited similar susceptibility profiles to those detailed in the surveillance studies.173, 174, 175 Serial passage of S. aureus ATCC 25923 at sub-MIC concentrations of dalbavancin did not alter susceptibility significantly (twofold increase over parental MIC), in contrast to four and eightfold increases for vancomycin and teicoplanin, respectively, suggesting dalbavancin has a low potential for resistance development.173
Oritavancin
Similar to dalbavancin, oritavancin (5) (LY333328) has had a long development history with a number of companies involved. It was initially discovered and developed by Eli Lilly (Indianapolis, IN, USA) in the 1990s and is a semisynthetic derivative of the Type I glycopeptide chloroeremomycin.176, 177, 178, 179 It is alkylated on the vancosamine amine with a hydrophobic chlorophenyl-benzyl moiety, and possesses an additional aminosugar residue on the phenylserine hydroxyl group. Like dalbavancin, oritavancin has an extended human half-life of over 300 h.177 InterMune, Inc. (Brisbane, CA, USA) acquired the rights in 2001, then Targanta Therapeutics acquired the rights in 2005. Two phase-III trials were successfully completed (3 days of 1.5 or 3 mg kg−1 per day or 200 mg for 3–7 days), with the results disclosed in 2001 and 2003, but the FDA rejected a new drug application (NDA) in December 2008 because of concerns over safety and effectiveness.177 Targanta was acquired by The Medicines Company (Parsippany, NJ, USA) in 2009, with two more phase-III trials for Gram-positive ABSSSI completed (SOLO I, 968 patients at 46 centers, SOLO II, 960 patients at 100 centers), both as a single 1200 mg dose, as well as two phase-I safety trials to assess cardiotoxicity and CYP450 activity. A new drug application was filed with the FDA in Feb 2014, with an action date set for August 2014.13
Oritavancin resistance
Clinical strains of vanA, vanB and vanC enterococci are susceptible to oritavancin, although resistance has been induced in vitro.30 A recent surveillance study of 9115 S. aureus clinical isolates (collected between 2008 and 2012) from the USA and Europe revealed an inhibitory rate of >99.9% at ⩽0.25 μg ml−1 against S. aureus (including those with decreased susceptibility to vancomycin and daptomycin), with an MIC90 of 0.06 μg ml−1.141 A study by Arhin et al.180 examining the in vitro susceptibility against 35 clinical isolates of hVISA, VISA and VRSA revealed MIC90 values of 1, 2 and 0.5 μg ml−1, respectively, with a combined MIC90 value of 1 μg ml−1 across the entire series of isolates.
Glycopeptide synthesis
The total synthesis of the glycopeptide antibiotics is difficult because of a number of synthetic challenges presented by the tricyclic peptide core, including non-natural amino acids, formation of the aryl ether linkages, the presence of atropisomers and further complications presented by the glycosyl substituents. The glycopeptides contain unnatural amino acids including 3′-chlorotyrosine, 4′-hydroxyphenylglycine, 3′,4′,5′-trihydroxyphenylglycine, β-hydroxytyrosine and chlorinated versions of these, further modified by both biaryl ether and biaryl linkages. Total syntheses of vancomycin and the vancomycin aglycon were reported in 1998–1999 by three separate groups50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60 with glycosylations of the aglycon described in 1999,181, 182 total syntheses of the teicoplanin aglycon in 2000183 and 2001,184, 185 of chloropeptin in 2003,186 and ristocetin aglycon in 2004.187
Two different approaches have been taken to forming the macrocyclic peptide/biaryl ether ring systems of vancomycin and related compounds, with the ring system created by either an amidation reaction or by biaryl ether formation. Cycloamidation sequences have been less popular, but a number of researchers have addressed the synthesis of the biaryl ether ring systems via macrocyclization during biaryl ether synthesis, with an extensive review published in 1995.188 Biosynthetic studies of balhimycin, an antibiotic from A. mediterranei with an identical aglycon to vancomycin, indicate that this is the approach nature employs, with linear halogenated peptides synthesized first, followed by oxidative ring closures to form the biaryl ethers and biphenyl linkages, as well as N-methylation and glycosylation.189 Fluorine was introduced into balhimycin via a mutant strain of A mediterranei deficient in β-hydroxy-Tyr biosynthesis, which was supplemented with 2-fluoro-β-hydroxy-Tyr, 3′-fluoro-β-hydroxy-Tyr or 3′,5′-difluoro-β-hydroxy-Tyr. The resulting balhimycin contained fluorine-substituted β-hydroxy-Tyr residues.190 The stereochemical issues and atropisomerism caused by axial and non-planar chirality in the vancomycin aglycon have been discussed in several publications.56, 191, 192 Another study examined thermal atropisomerism of teicoplanin aglycon derivatives, looking at selective equilibration of the DE ring system (Figure 5).193

Summary of glycopeptide modifications.
The difficulties of total synthesis have generally prevented their use in structural modifications. However, over 20 years after the first total synthesis of vancomycin, one of the groups involved has described a total synthesis that replaced the amide carbonyl oxygen of residue 4 in the aglycon with a sulfur and then amine, with the goal of the amidine isostere [ψ[C(dNH)NH]Tpg4] (12) retaining hydrogen bonding to the ester oxygen of the D-lactate residue found in resistant Van A Lipid II (Figure 3). Indeed, the modified glycopeptide retained good activity against Van A bacteria.194, 195, 196
In contrast to the total synthesis route, semisynthetic modifications have been reported for many years, focusing mainly on alkylation/acylation of the vancosamine primary amine group, or amidation of the heptapeptide C-terminal free carboxyl group (Figure 5). A comprehensive review of structural modifications of glycopeptide antibiotics was published in 1997.197 Desmethylvancomycin (9) (Figure 1), which has been employed clinically in China, has been N-alkylated on the vancosamine amino group, as with oritavancin (5).95, 198 The N-terminal amine of teicoplanin has been converted into an azide and used for ‘click’ copper catalyzed 1,3-dipolar cycloadditions with lipophilic alkynes.199
More difficult modifications include preparation of a number of methyl ether derivatives of the vancomycin aglycon.200 The chloro group of vancomycin on residue 2, or on both residues 2 and 6, has been selectively replaced with substituted aryl and vinyl groups by a Suzuki-Miyaura cross-coupling reaction using aryl/alkenyl boronic acids,201 or converted into a reactive boronic acid and functionalized with a range of substituents.202 Site selective bromination of vancomycin on residues 5 and 7 is possible using N-bromophthalimide and various catalysts.203
Conclusion and future outlook
For over half a century, the naturally occurring glycopeptides vancomycin and teicoplanin have been key weapons in the fight against bacterial infections and they still remain one of the first treatment options for MRSA and penicillin-resistant S. pneumoniae infections today. Although widespread clinical resistance to these glycopeptides did not develop for many years, it is now a significant issue that threatens their continued effectiveness. Resistance can be caused by VRE and VRSA strains expressing the vanA and vanB operon that alters the D-Ala-D-Ala binding epitope leading to reduced Lipid II binding, and by VISA and hVISA strains with thickened cell walls that overproduce muropeptides with uncross-linked D-Ala-DAla as false targets. This begs the question as to how these second generation glycopeptides are able to maintain their effectiveness if access to the primary Lipid II binding sites on the cell membrane is restricted; oritavancin has been demonstrated to interact with the bridge peptide component as a secondary mode of action with dimerization and membrane interactions also critical. Whether other glycopeptides bearing lipophilic tails behave similarly to oritavancin remains to be determined. Further exploitation of the bridge peptide binding could lead to the design of new glycopeptide antibiotics with enhanced activity against resistant strains.
Although there is considerable emphasis on the threat posed by Gram-negative bacteria,2, 4 the need for effective antibiotics against Gram-positive strains must not be underestimated or overlooked. The number of infections and deaths per year in the United States from the two main resistant Gram-positive pathogens, MRSA and S. pneumoniae, vastly exceeds those caused by the most serious resistant Gram-negative pathogens Escherichia coli, Pseudomonas aeruginosa, Klebsiella pneumoniae and Acinetobacter baumannii, with 1.28 million versus 49 000 infections and 18 285 versus 3240 deaths per annum, respectively.64 The approval of two second generation antibiotics, telavancin in 2009 and dalbavancin in 2014, and the potential approval of oritavancin later in 2014 will help with the treatment of serious Gram-positive infections and hopefully extend the clinical lifetime of glycopeptide antibiotics for many years to come. Another approach to extend the lifetime of glycopeptide antibiotics is to adopt the same strategy used for β-lactam antibiotics, where co-administration of a β-lactamase inhibitor is used to overcome the primary resistance mechanism. The development of inhibitors of the VanX-induced zinc metallopeptidase dipeptidase involved in the remodeling of bacterial Lipid II D-Ala-D-Ala to the resistant D-Ala-D-Lac, as seen in VRE and VRSA, would allow the glycopeptides to retain efficacy, as has been demonstrated in vitro.204
References
Butler, M. S. & Cooper, M. A. Antibiotics in the clinical pipeline in 2011. J. Antibiot. 64, 413–425 (2011).
Butler, M. S., Blaskovich, M. A. & Cooper, M. A. Antibiotics in the clinical pipeline in 2013. J. Antibiot. 66, 571–591 (2013).
Hede, K. Antibiotic resistance: An infectious arms race. Nature 509, S2–S3 (2014).
Boucher, H. W. et al. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 48, 1–12 (2009).
Lewis, K. Platforms for antibiotic discovery. Nat. Rev. Drug Discov. 12, 371–387 (2013).
Kirst, H. A. Developing new antibacterials through natural product research. Expert Opin. Drug Discov 8, 479–493 (2013).
Butler, M. S. & Cooper, M. A. Screening strategies to identify new antibiotics. Curr. Drug Targets 13, 373–387 (2012).
Shlaes, D. M. & Spellberg, B. Overcoming the challenges to developing new antibiotics. Curr. Opin. Pharmacol. 12, 522–526 (2012).
Jones, D. A guiding hand for antibiotics. Nat. Rev. Drug Discov. 10, 161–162 (2011).
Anstead, G., Cadena, J. & Javeri, H. in Methicillin-Resistant Staphylococcus Aureus (MRSA) Protocols Vol. 1085 Methods in Molecular Biology ed Ji Y., 259–309 Humana Press: New Jersey, (2014).
Venugopal, A. A. & Johnson, S. Current state of Clostridium difficile treatment options. Clin. Infect. Dis. 55, S71–S76 (2012).
Sensi, P. History of the development of rifampin. Rev. Infect. Dis. 5, S402–S406 (1983).
FDA Accepts Filing of The Medicines Company's New Drug Application for Intravenous Antibiotic Oritavancin with Priority Review (Press Release 19 Feb 2014) 〈http://ir.themedicinescompany.com/phoenix.zhtml?c=122204&p=irol-newsArticle&ID=1901078〉 (accessed 15 June 2014.
Phillips, I. Withdrawal of growth-promoting antibiotics in Europe and its effects in relation to human health. Int. J. Antimicrob. Agents 30, 101–107 (2007).
Lancini, G. C. in Progress in Industrial Microbiology Vol. 27 eds Bushell M. E., Graefe U., 283–296 Elsevier: New York, (1989).
Nicolaou, K. C., Boddy, C. N. C., Bräse, S. & Winssinger, N. Chemistry, biology, and medicine of the glycopeptide antibiotics. Angew. Chem., Int. Ed. 38, 2096–2152 (1999).
Kaneko, I., Fearon, D. T. & Austen, K. F. Inhibition of the alternative pathway of human complement in vitro by a natural microbial product, complestatin. J. Immunol. 124, 1194–1198 (1980).
Gouda, H. et al. Stereostructure of (−)-Chloropeptin I, a novel inhibitor of gp120−CD4 binding, via high-temperature molecular dynamics, Monte Carlo conformational searching, and NMR spectroscopy. J. Am. Chem. Soc. 118, 13087–13088 (1996).
Pootoolal, J. et al. Assembling the glycopeptide antibiotic scaffold: The biosynthesis of from Streptomyces toyocaensis NRRL15009. Proc. Natl Acad. Sci. USA 99, 8962–8967 (2002).
Kobayashi, H. et al. Neuroprotectins A and B, bicyclohexapeptides protecting chick telencephalic neuronal cells from excitotoxicity. I. Fermentation, isolation, physico-chemical properties and biological activity. J. Antibiot. 54, 1013–1018 (2001).
Kobayashi, H. et al. Neuroprotectins A and B, bicyclohexapeptides protecting chick telencephalic neurons from excitotoxicity. II. Structure determination. J. Antibiot. 54, 1019–1024 (2001).
Breazzano, S. P. & Boger, D. L. Synthesis and stereochemical determination of complestatin A and B (neuroprotectin A and B). J. Am. Chem. Soc. 133, 18495–18502 (2011).
Yim, G., Thaker, M. N., Koteva, K. & Wright, G. Glycopeptide antibiotic biosynthesis. J. Antibiot. 67, 31–41 (2014).
Kahne, D., Leimkuhler, C., Lu, W. & Walsh, C. Glycopeptide and lipoglycopeptide antibiotics. Chem. Rev. 105, 425–448 (2005).
Arhin, F. F. et alin Antibiotic Discovery and Development eds Dougherty T. J., Pucci M. J., 301–346 Springer US: New York, (2012).
Chmara, H., Ripa, S., Mignini, F. & Borowski, E. Bacteriolytic effect of teicoplanin. J. Gen. Microbiol. 137, 913–919 (1991).
Allen, N. E., LeTourneau, D. L. & Hobbs, Jr J. N. The role of hydrophobic side chains as determinants of antibacterial activity of semisynthetic glycopeptide antibiotics. J. Antibiot. 50, 677–684 (1997).
Allen, N. E. & Nicas, T. I. Mechanism of action of oritavancin and related glycopeptide antibiotics. FEMS Microbiol. Rev. 26, 511–532 (2003).
Higgins, D. L. et al. Telavancin, a multifunctional lipoglycopeptide, disrupts both cell wall synthesis and cell membrane integrity in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 49, 1127–1134 (2005).
Zhanel, G. G. et al. New lipoglycopeptides: a comparative review of dalbavancin, oritavancin and telavancin. Drugs 70, 859–886 (2010).
Belley, A., Harris, R., Beveridge, T., Parr, T. & Moeck, G. Ultrastructural effects of oritavancin on methicillin-resistant Staphylococcus aureus and vancomycin-resistant Enterococcus. Antimicrob. Agents Chemother. 53, 800–804 (2009).
Lunde, C. S. et al. Telavancin disrupts the functional integrity of the bacterial membrane through targeted interaction with the cell wall precursor Lipid II. Antimicrob. Agents Chemother. 53, 3375–3383 (2009).
Trevino, J. et al. New insights into glycopeptide antibiotic binding to cell wall precursors using SPR and NMR spectroscopy. Chem. Eur. J. 20, 7363–7372 (2014).
Beauregard, D. A., Williams, D. H., Gwynn, M. N. & Knowles, D. J. Dimerization and membrane anchors in extracellular targeting of vancomycin group antibiotics. Antimicrob. Agents Chemother. 39, 781–785 (1995).
Cheng, M. et al. Anti-cooperative ligand binding and dimerisation in the glycopeptide antibiotic dalbavancin. Org. Biomol. Chem. 12, 2568–2575 (2014).
Mackay, J. P. et al. Glycopeptide antibiotic activity and the possible role of dimerization: a model for biological signaling. J. Am. Chem. Soc. 116, 4581–4590 (1994).
McCormick, M. H., McGuire, J. M., Pittenger, G. E., Pittenger, R. C. & Stark, W. M. Vancomycin, a new antibiotic. I. Chemical and biologic properties. Antibiot. Annu. 3, 606–611 (1955).
Anonymous. Vancomycin. Lancet 269, 85–86 (1957).
McGraw, D. J. The Antibiotic Discovery Era (1940–1960): Vancomycin as an Example of the Era (PhD thesis, Oregon State University (1976).
Yao, R. C. & Crandall, L. W. in Glycopeptide Antibiotics Vol. Drugs and the Pharmaceutical Sciences. ed. Nagarajan R., 1–27 CRC Press: New York, (1994).
Levine, D. P. Vancomycin: a history. Clin. Infect. Dis 42 (Suppl 1), S5–S12 (2006).
Lechevalier, M. P., Prauser, H., Labeda, D. P. & Ruan, J.-S. Two new genera of nocardioform actinomycetes: Amycolata gen. nov. and Amycolatopsis gen. nov. Int. J. Syst. Bacteriol. 36, 29–37 (1986).
Griffith, R. S. & Peck, Jr F. B. Vancomycin, a new antibiotic. III. Preliminary clinical and laboratory studies. Antibiot. Annu. 3, 619–622 (1955).
Kirby, W. M. M., Perry, D. M. & Bauer, A. W. Treatment of staphylococcal septicemia with vancomycin. New Engl. J. Med. 262, 49–55 (1960).
Harris, C. M. & Harris, T. M. Structure of the glycopeptide antibiotic vancomycin. Evidence for an asparagine residue in the peptide. J. Am. Chem. Soc. 104, 4293–4295 (1982).
Marshall, F. J. Structure studies on vancomycin. J. Med. Chem. 8, 18–22 (1965).
Williams, D. H. & Kalman, J. R. Structural and mode of action studies on the antibiotic vancomycin. Evidence from 270-MHz proton magnetic resonance. J. Am. Chem. Soc. 99, 2768–2774 (1977).
Sheldrick, G. M., Jones, P. G., Kennard, O., Williams, D. H. & Smith, G. A. Structure of vancomycin and its complex with acetyl-D-alanyl-D-alanine. Nature 271, 223–225 (1978).
Schäfer, M., Schneider, T. R. & Sheldrick, G. M. Crystal structure of vancomycin. Structure 4, 1509–1515 (1996).
Evans, D. A. et al. Total syntheses of vancomycin and eremomycin aglycons. Angew. Chem., Int. Ed. 37, 2700–2704 (1998).
Nicolaou, K. C. et al. Total synthesis of vancomycin aglycon—Part 3: Final stages. Angew. Chem. Int. Ed. 37, 2717–2719 (1998).
Zhang, A. J. & Burgess, K. Total syntheses of vancomycin. Angew. Chem. Int. Ed. 38, 634–636 (1999).
Boger, D. L. et al. Diastereoselective total synthesis of the vancomycin aglycon with ordered atropisomer equilibrations. J. Am. Chem. Soc. 121, 3226–3227 (1999).
Nicolaou, K. C. et al. Total synthesis of vancomycin aglycon—Part 1: Synthesis of amino acids 4–7 and construction of the AB-COD ring skeleton. Angew. Chem. Int. Ed. 37, 2708–2714 (1998).
Nicolaou, K. C. et al. Total synthesis of vancomycin aglycon—Part 2: Synthesis of amino acids 1–3 and construction of the AB-COD-DOE ring skeleton. Angew. Chem. Int. Ed. 37, 2714–2716 (1998).
Boger, D. L. et al. Total synthesis of the vancomycin aglycon. J. Am. Chem. Soc. 121, 10004–10011 (1999).
Nicolaou, K. C. et al. Total synthesis of vancomycin—Part 1: Design and development of methodology. Chem. Eur. J. 5, 2584–2601 (1999).
Nicolaou, K. C. et al. Total synthesis of vancomycin—Part 2: Retrosynthetic analysis, synthesis of amino acid building blocks and strategy evaluations. Chem. Eur. J. 5, 2602–2621 (1999).
Nicolaou, K. C. et al. Total synthesis of vancomycin—Part 3: Synthesis of the aglycon. Chem. Eur. J. 5, 2622–2647 (1999).
Nicolaou, K. C. et al. Total synthesis of vancomycin—Part 4: Attachment of the sugar moieties and completion of the synthesis. Chem. Eur. J. 5, 2648–2667 (1999).
Otto, M. Community-associated MRSA: What makes them special? Int. J. Med. Microbiol. 303, 324–330 (2013).
Sowash, M. G. & Uhlemann, A.-C. in Methicillin-Resistant Staphylococcus Aureus (MRSA) Protocols Vol. 1085 Methods in Molecular Biology ed. Ji Y., 25–69 Humana Press: New Jersey, (2014).
CLSI Methods For Dilution Antimicrobial Susceptibility Tests For Bacteria That Grow Aerobically. Approved standard, 9th ed. CLSI document M7-A9 (CLSI, Wayne, PA, 2011).
U.S. Department of Health and Human Services (Centres for Disease Control and Prevention). http://www.cdc.gov/drugresistance/threat-report-2013/ (accessed on 6 June 2014) (2013).
McKessar, S. J., Berry, A. M., Bell, J. M., Turnidge, J. D. & Paton, J. C. Genetic characterization of vanG, a novel vancomycin resistance locus of Enterococcus faecalis. Antimicrob. Agents Chemother. 44, 3224–3228 (2000).
Depardieu, F., Bonora, M. G., Reynolds, P. E. & Courvalin, P. The vanG glycopeptide resistance operon from Enterococcus faecalis revisited. Mol. Microbiol. 50, 931–948 (2003).
Murray, B. E. Vancomycin-resistant enterococcal infections. New Engl. J. Med. 342, 710–721 (2000).
Rasmussen, R. V., Fowler, V. G. Jr, Skov, R. & Bruun, N. E. Future challenges and treatment of Staphylococcus aureus bacteremia with emphasis on MRSA. Future Microbiol. 6, 43–56 (2010).
Kim, S. J., Tanaka, K. S. E., Dietrich, E., Rafai Far, A. & Schaefer, J. Locations of the hydrophobic side chains of lipoglycopeptides bound to the peptidoglycan of Staphylococcus aureus. Biochemistry 52, 3405–3414 (2013).
Patti, G. J. et al. Vancomycin and oritavancin have different modes of action in Enterococcus faecium. J. Mol. Biol. 392, 1178–1191 (2009).
Manson, J. M., Keis, S., Smith, J. M. B. & Cook, G. M. A clonal lineage of VanA-type Enterococcus faecalis predominates in vancomycin-resistant enterococci isolated in New Zealand. Antimicrob. Agents Chemother. 47, 204–210 (2003).
Phillips, I. et al. Does the use of antibiotics in food animals pose a risk to human health? A critical review of published data. J. Antimicrob. Chemother. 53, 28–52 (2004).
D'Costa, V. M. et al. Antibiotic resistance is ancient. Nature 477, 457–461 (2011).
Wright, G. D. & Poinar, H. Antibiotic resistance is ancient: implications for drug discovery. Trends Microbiol. 20, 157–159 (2012).
Nesme, J. et al. Large-Scale Metagenomic-Based Study of Antibiotic Resistance in the Environment. Curr. Biol. 24, 1096–1100 (2014).
Melo-Cristino, J., Resina, C., Manuel, V., Lito, L. & Ramirez, M. First case of infection with vancomycin-resistant Staphylococcus aureus in Europe. Lancet 382, 205 (2013).
Rossi, F. et al. Transferable vancomycin resistance in a community-associated MRSA lineage. New Engl. J. Med. 370, 1524–1531 (2014).
Hiramatsu, K. in Staphylococci in human disease 2nd Ed., (eds Crossley, K. B. et al.) 193–209 Wiley-Blackwell, (2009).
Howden, B. P., Davies, J. K., Johnson, P. D., Stinear, T. P. & Grayson, M. L. Reduced vancomycin susceptibility in Staphylococcus aureus, including vancomycin-intermediate and heterogeneous vancomycin-intermediate strains: resistance mechanisms, laboratory detection, and clinical implications. Clin. Microbiol. Rev. 23, 99–139 (2010).
Hiramatsu, K. et al. Dissemination in Japanese hospitals of strains of Staphylococcus aureus heterogeneously resistant to vancomycin. Lancet 350, 1670–1673 (1997).
Hiramatsu, K. et al. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J. Antimicrob. Chemother. 40, 135–136 (1997).
Yamakawa, J. et al. Heterogeneously vancomycin-intermediate Staphylococcus aureus (hVISA) emerged before the clinical introduction of vancomycin in Japan: a retrospective study. J. Infect. Chemother. 18, 406–409 (2012).
Howden, B. P., Peleg, A. Y. & Stinear, T. P. The evolution of vancomycin intermediate Staphylococcus aureus (VISA) and heterogenous-VISA. Infect. Genet. Evol. 21, 575–582 (2014).
Ike, Y. et al. Nationwide survey shows that methicillin-resistant Staphylococcus aureus strains heterogeneously and intermediately resistant to vancomycin are not disseminated throughout Japanese hospitals. J. Clin. Microbiol. 39, 4445–4451 (2001).
Matsuo, M. et al. Mutation of RNA polymerase beta subunit (rpoB) promotes hVISA-to-VISA phenotypic conversion of strain Mu3. Antimicrob. Agents Chemother. 55, 4188–4195 (2011).
Cui, L., Neoh, H. M., Shoji, M. & Hiramatsu, K. Contribution of vraSR and graSR point mutations to vancomycin resistance in vancomycin-intermediate Staphylococcus aureus. Antimicrob. Agents Chemother. 53, 1231–1234 (2009).
Cui, L., Lian, J. Q., Neoh, H. M., Reyes, E. & Hiramatsu, K. DNA microarray-based identification of genes associated with glycopeptide resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 49, 3404–3413 (2005).
Neoh, H. M. et al. Mutated response regulator graR is responsible for phenotypic conversion of Staphylococcus aureus from heterogeneous vancomycin-intermediate resistance to vancomycin-intermediate resistance. Antimicrob. Agents Chemother. 52, 45–53 (2008).
Wootton, M. et al. A modified population analysis profile (PAP) method to detect hetero-resistance to vancomycin in Staphylococcus aureus in a UK hospital. J. Antimicrob. Chemother. 47, 399–403 (2001).
Boyle-Vavra, S., Berke, S. K., Lee, J. C. & Daum, R. S. Reversion of the glycopeptide resistance phenotype in Staphylococcus aureus clinical isolates. Antimicrob. Agents Chemother. 44, 272–277 (2000).
Gardete, S. et al. Genetic pathway in acquisition and loss of vancomycin resistance in a methicillin resistant Staphylococcus aureus (MRSA) strain of clonal type USA300. PLoS Pathog. 8, e1002505 (2012).
Plipat, N., Livni, G., Bertram, H. & Thomson, Jr R. B. Unstable vancomycin heteroresistance is common among clinical isolates of methiciliin-resistant Staphylococcus aureus. J. Clin. Microbiol. 43, 2494–2496 (2005).
Belley, A. et al. Oritavancin disrupts membrane integrity of Staphylococcus aureus and vancomycin-resistant enterococci to effect rapid bacterial killing. Antimicrob. Agents Chemother. 54, 5369–5371 (2010).
van Hal, S. J. & Paterson, D. L. Systematic review and meta-analysis of the significance of heterogeneous vancomycin-intermediate Staphylococcus aureus isolates. Antimicrob. Agents Chemother. 55, 405–410 (2011).
Zhang, S.-J., Yang, Q., Xu, L., Chang, J. & Sun, X. Synthesis and antibacterial activity against Clostridium difficile of novel demethylvancomycin derivatives. Bioorg. Med. Chem. Lett. 22, 4942–4945 (2012).
Boeck, L. D., Mertz, F. P., Wolter, R. K. & Higgens, C. E. N-demethylvancomycin, a novel antibiotic produced by a strain of Nocardia orientalis. Taxonomy and fermentation. J. Antibiot. 37, 446–453 (1984).
Vesga, O., Agudelo, M., Salazar, B. E., Rodriguez, C. A. & Zuluaga, A. F. Generic vancomycin products fail in vivo despite being pharmaceutical equivalents of the innovator. Antimicrob. Agents Chemother. 54, 3271–3279 (2010).
Rodriguez, C. A., Agudelo, M., Zuluaga, A. F. & Vesga, O. Generic vancomycin enriches resistant subpopulations of Staphylococcus aureus after exposure in a neutropenic mouse thigh infection model. Antimicrob. Agents Chemother. 56, 243–247 (2012).
Tattevin, P. et al. Comparison of six generic vancomycin products for treatment of methicillin-resistant Staphylococcus aureus experimental endocarditis in rabbits. Antimicrob. Agents Chemother. 57, 1157–1162 (2013).
Tenover, F. C. & Moellering, R. C. The rationale for revising the Clinical and Laboratory Standards Institute vancomycin minimal inhibitory concentration interpretive criteria for Staphylococcus aureus. Clin. Infect. Dis. 44, 1208–1215 (2007).
Musta, A. C. et al. Vancomycin MIC plus heteroresistance and outcome of methicillin-resistant Staphylococcus aureus bacteremia: trends over 11 years. J. Clin. Microbiol. 47, 1640–1644 (2009).
Dhand, A. & Sakoulas, G. Reduced vancomycin susceptibility among clinical Staphylococcus aureus isolates ('the MIC Creep'): implications for therapy. F1000 Med. Rep. 4, 4 (2012).
Gould, I. M. Clinical relevance of increasing glycopeptide MICs against Staphylococcus aureus. Int. J. Antimicrob. Agents 31 (Suppl 2), 1–9 (2008).
van Hal, S. J., Lodise, T. P. & Paterson, D. L. The clinical significance of vancomycin minimum inhibitory concentration in Staphylococcus aureus infections: a systematic review and meta-analysis. Clin. Infect. Dis. 54, 755–771 (2012).
van Hal, S. J. et al. Methicillin-resistant Staphylococcus aureus vancomycin susceptibility testing: methodology correlations, temporal trends and clonal patterns. J. Antimicrob. Chemother. 66, 2284–2287 (2011).
Fridkin, S. K. et al. Epidemiological and microbiological characterization of infections caused by Staphylococcus aureus with reduced susceptibility to vancomycin, United States, 1997-2001. Clin. Infect. Dis. 36, 429–439 (2003).
Liu, C. et al. Clinical practice guidelines by the Infectious Diseases Society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin. Infect. Dis. 52, e18–e55 (2011).
Holmes, N. E. et al. Antibiotic choice may not explain poorer outcomes in patients with Staphylococcus aureus bacteremia and high vancomycin minimum inhibitory concentrations. J. Infect. Dis. 204, 340–347 (2011).
Katayama, Y., Murakami-Kuroda, H., Cui, L. & Hiramatsu, K. Selection of heterogeneous vancomycin-intermediate Staphylococcus aureus by imipenem. Antimicrob. Agents Chemother. 53, 3190–3196 (2009).
Sakoulas, G., Guram, K., Reyes, K., Nizet, V. & Zervos, M. Human cathelicidin LL-37 resistance and increased daptomycin MIC in USA 600 (ST45) methicillin-resistant Staphylococcus aureus is associated with increased mortality in a hospital setting. J. Clin. Microbiol. 52, 2172–2174 (2014).
Matsuo, M., Cui, L., Kim, J. & Hiramatsu, K. Comprehensive identification of mutations responsible for heterogeneous vancomycin-intermediate Staphylococcus aureus (hVISA)-to-VISA conversion in laboratory-generated VISA strains derived from hVISA clinical strain Mu3. Antimicrob. Agents Chemother. 57, 5843–5853 (2013).
Mwangi, M. M. et al. Tracking the in vivo evolution of multidrug resistance in Staphylococcus aureus by whole-genome sequencing. Proc. Natl Acad. Sci. USA 104, 9451–9456 (2007).
Cui, L. et al. Cell wall thickening is a common feature of vancomycin resistance in Staphylococcus aureus. J. Clin. Microbiol. 41, 5–14 (2003).
Hanaki, H. et al. Increase in glutamine-non-amidated muropeptides in the peptidoglycan of vancomycin-resistant Staphylococcus aureus strain Mu50. J. Antimicrob. Chemother. 42, 315–320 (1998).
Cui, L., Murakami, H., Kuwahara-Arai, K., Hanaki, H. & Hiramatsu, K. Contribution of a thickened cell wall and its glutamine nonamidated component to the vancomycin resistance expressed by Staphylococcus aureus Mu50. Antimicrob. Agents Chemother. 44, 2276–2285 (2000).
Sieradzki, K. & Tomasz, A. Alterations of cell wall structure and metabolism accompany reduced susceptibility to vancomycin in an isogenic series of clinical isolates of Staphylococcus aureus. J. Bacteriol. 185, 7103–7110 (2003).
Hanaki, H. et al. Activated cell-wall synthesis is associated with vancomycin resistance in methicillin-resistant Staphylococcus aureus clinical strains Mu3 and Mu50. J. Antimicrob. Chemother. 42, 199–209 (1998).
Sauvage, E., Kerff, F., Terrak, M., Ayala, J. A. & Charlier, P. The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis. FEMS Microbiol. Rev. 32, 234–258 (2008).
Peschel, A., Vuong, C., Otto, M. & Gotz, F. The D-alanine residues of Staphylococcus aureus teichoic acids alter the susceptibility to vancomycin and the activity of autolytic enzymes. Antimicrob. Agents Chemother. 44, 2845–2847 (2000).
Weidenmaier, C. & Peschel, A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat. Rev. Microbiol. 6, 276–287 (2008).
Falagas, M. E., Makris, G. C., Dimopoulos, G. & Matthaiou, D. K. Heteroresistance: a concern of increasing clinical significance? Clin. Microbiol. Infec. 14, 101–104 (2008).
van Hal, S. J. & Fowler, V. G. Jr Is it time to replace vancomycin in the treatment of methicillin-resistant Staphylococcus aureus infections? Clin. Infect. Dis. 56, 1779–1788 (2013).
Parenti, F., Beretta, G., Berti, M. & Arioli, V. Teichomycins, new antibiotics from Actinoplanes teichomyceticus nov. sp. I. Description of the producer strain, fermentation studies and biological properties. J. Antibiot. 31, 276–283 (1978).
Parenti, F. Structure and mechanism of action of teicoplanin. J. Hosp. Infect. 7 (Suppl. A), 79–83 (1986).
Goldstein, B. P., Rosina, R. & Parenti, F. in Glycopeptide Antibiotics Vol. Drugs and the Pharmaceutical Sciences ed. Nagarajan R., 273–307 CRC Press: New York, (1994).
Barna, J. C. J., Williams, D. H., Stone, D. J. M., Leung, T. W. C. & Doddrell, D. M. Structure elucidation of the teicoplanin antibiotics. J. Am. Chem. Soc. 106, 4895–4902 (1984).
Coronelli, C. et al. Teicoplanin, antibiotics from Actinoplanes teichomyceticus nov. sp. V. Aromatic constituents. J. Antibiot. 37, 621–626 (1984).
Borghi, A. et al. Isolation and structure determination of two new analogs of teicoplanin, a glycopeptide antibiotic. J. Antibiot. 42, 361–366 (1989).
Borghi, A., Edwards, D., Zerilli, L. F. & Lancini, G. C. Factors affecting the normal and branched-chain acyl moieties of teicoplanin components produced by Actinoplanes teichomyceticus. J. Gen. Microbiol. 137, 587–592 (1991).
Brink, A. J. et al. Multicomponent antibiotic substances produced by fermentation: Implications for regulatory authorities, critically ill patients and generics. Int. J. Antimicrob. Agents 43, 1–6 (2014).
Roberts, J. A. et al. Variability in protein binding of teicoplanin and achievement of therapeutic drug monitoring targets in critically ill patients: Lessons from the DALI Study. Int. J. Antimicrob. Agents 43, 423–430 (2014).
Drugs.com website: Targocid, 〈http://www.drugs.com/international/targocid.html〉 (accessed 27 May 2014).
Brogden, R. N. & Peters, D. H. Teicoplanin. A reappraisal of its antimicrobial activity, pharmacokinetic properties and therapeutic efficacy. Drugs 47, 823–854 (1994).
Pea, F., Brollo, L., Viale, P., Pavan, F. & Furlanut, M. Teicoplanin therapeutic drug monitoring in critically ill patients: a retrospective study emphasizing the importance of a loading dose. J. Antimicrob. Chemother. 51, 971–975 (2003).
Cavalcanti, A. B., Goncalves, A. R., Almeida, C. S., Bugano, D. D. & Silva, E. Teicoplanin versus vancomycin for proven or suspected infection. Cochrane Database Syst. Rev. CD007022, 1–53 (2010).
Périchon, B. & Courvalin, P. in Antibiotic Discovery and Development eds Dougherty T. J., Pucci M. J., 515–542 Springer US: New York, (2012).
Kaatz, G. W., Seo, S. M., Dorman, N. J. & Lerner, S. A. Emergence of teicoplanin resistance during therapy of Staphylococcus aureus endocarditis. J. Infect. Dis. 162, 103–108 (1990).
Shlaes, D. M. & Shlaes, J. H. Teicoplanin selects for Staphylococcus aureus that is resistant to vancomycin. Clin. Infect. Dis. 20, 1071–1073 (1995).
Mainardi, J. L. et al. Decreased teicoplanin susceptibility of methicillin-resistant strains of Staphylococcus aureus. J. Infect. Dis. 171, 1646–1650 (1995).
Sieradzki, K. & Tomasz, A. Suppression of glycopeptide resistance in a highly teicoplanin-resistant mutant of Staphylococcus aureus by transposon inactivation of genes involved in cell wall synthesis. Microb. Drug Resist. 4, 159–168 (1998).
Mendes, R. E., Sader, H. S., Flamm, R. K., Farrell, D. J. & Jones, R. N. Oritavancin activity against Staphylococcus aureus causing invasive infections in U.S. and European hospitals: a 5-year international surveillance program. Antimicrob. Agents Chemother. 58, 2921–2924 (2014).
Philip, J. E., Schenck, J. R. & Hargie, M. P. Ristocetins A and B, two new antibiotics; isolation and properties. Antibiot. Annu 1955–1956, 699–705 (1956).
Harris, C. M. & Harris, T. M. Structure of ristocetin A: configurational studies of the peptide. J. Am. Chem. Soc. 104, 363–365 (1982).
Nahoum, V., Spector, S. & Loll, P. J. Structure of ristocetin A in complex with a bacterial cell-wall mimetic. Acta Crystallogr. Sect. D: Biol. Crystallogr. 65, 832–838 (2009).
Nahoum, V., Spector, S. & Loll, P. Structure of ristocetin A in complex with a bacterial cell-wall mimetic. Corrigendum. Acta. Crystallogr. D. Biol. Crystallogr. 67, 592 (2011).
Jawetz, E. Polymyxins, colistin, bacitracin, ristocetin and vancomycin. Pediatr. Clin. North Am. 15, 85–94 (1968).
Howard, M. A. & Firkin, B. G. Ristocetin–a new tool in the investigation of platelet aggregation. Thromb. Diath. Haemorrh 26, 362–369 (1971).
Jenkins, C. S. P., Meyer, D., Dreyfus, M. D. & Larreu, M. J. Willebrand factor and ristocetin I. Mechanism of ristocetin-induced platelet aggregation. Brit. J. Haematol. 28, 561–578 (1974).
Othman, M., Kaur, H. & Emsley, J. Platelet-type von Willebrand disease: new insights into the molecular pathophysiology of a unique platelet defect. Semin. Thromb. Hemost. 39, 663–673 (2013).
Kunstmann, M. P., Mitscher, L. A., Porter, J. N., Shay, A. J. & Darken, M. A. LL-AV290, a new antibiotic. I. Fermentation, isolation, and characterization. Antimicrob. Agents Chemother. 8, 242–245 (1968).
Redin, G. S. & Dornbush, A. C. LL-AV290, a new antibiotic. II. Antibacterial efficacy in mice and in vitro. Antimicrob. Agents Chemother. 8, 246–248 (1968).
Hamill, R. L., Stark, W. M. & DeLong, D. C., (Eli Lilly & Co)Novel antibiotic and a process for the production thereof. US 3,952,095 Apr 20 (1976).
McGahren, W. J. et al. Structure of avoparcin components. J. Am. Chem. Soc. 102, 1671–1684 (1980).
McGahren, W. J. et al. Components and degradation compounds of the avoparcin complex. J. Antibiot. 36, 1671–1682 (1983).
Debono, M. et al. Actaplanin, new glycopeptide antibiotics produced by Actinoplanes missouriensis. The isolation and preliminary chemical characterization of actaplanin. J. Antibiot. 37, 85–95 (1984).
Hunt, A. H., Elzey, T. K., Merkel, K. E. & Debono, M. Structures of the actaplanins. J. Org. Chem. 49, 641–645 (1984).
Judice, J. K. & Pace, J. L. Semi-synthetic glycopeptide antibacterials. Bioorg. Med. Chem. Lett. 13, 4165–4168 (2003).
Leadbetter, M. R. et al. Hydrophobic vancomycin derivatives with improved ADME properties: discovery of telavancin (TD-6424). J. Antibiot. 57, 326–336 (2004).
Corey, G. R., Stryjewski, M. E., Weyenberg, W., Yasothan, U. & Kirkpatrick, P. Telavancin. Nat. Rev. Drug Discov. 8, 929–930 (2009).
Lyseng-Williamson, K. & Blick, S. A. Telavancin. Drugs 69, 2607–2620 (2009).
Theravance Inc. VIBATIV (TELAVANCIN for Injection) NDA briefing document, 29 Nov 2012 〈http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/UCM329480.pdf〉 (accessed 6 June 2014).
Kosowska-Shick, K. et al. Activity of telavancin against staphylococci and enterococci determined by MIC and resistance selection studies. Antimicrob. Agents Chemother. 53, 4217–4224 (2009).
Krause, K. M. et al. In vitro activity of telavancin and occurrence of vancomycin heteroresistance in isolates from patients enrolled in phase 3 clinical trials of hospital-acquired pneumonia. Diagn. Microbiol. Infect. Dis. 74, 429–431 (2012).
Pfaller, M. A., Mendes, R. E., Sader, H. S. & Jones, R. N. Telavancin activity against Gram-positive bacteria isolated from respiratory tract specimens of patients with nosocomial pneumonia. J. Antimicrob. Chemother. 65, 2396–2404 (2010).
Durata Therapeutics Announces Phase 3 Clinical Trial Results for Dalbavancin in the Treatment of ABSSSI (Press release 11 December 2012) 〈http://www.duratatherapeutics.com/news-media/press-releases/detail/333/durata-therapeutics-announces-phase-3-clinical-trial〉 (accessed 2 June 2014)..
Durata Therapeutics Announces Preliminary, Topline Phase 3 Clinical Trial Results for Dalbavancin in the Treatment of ABSSSI (press release 25 February 2013) 〈http://www.duratatherapeutics.com/news-media/press-releases/detail/364/durata-therapeutics-announces-preliminary-topline-phase-3〉 (accessed 2 June 2013)..
Candiani, G., Abbondi, M., Borgonovi, M., Romanò, G. & Parenti, F. In-vitro and in-vivo antibacterial activity of BI 397, a new semi-synthetic glycopeptide antibiotic. J. Antimicrob. Chemother. 44, 179–192 (1999).
Malabarba, A. & Ciabatti, R. Glycopeptide derivatives. Curr. Med. Chem. 8, 1759–1773 (2001).
Malabarba, A. et al. Amides of de-acetylglucosaminyl-deoxy teicoplanin active against highly glycopeptide-resistant enterococci. Synthesis and antibacterial activity. J. Antibiot. 47, 1493–1506 (1994).
Borghi, A. et al. Deacylation of the glycopeptide antibiotic A40926 by Actinoplanes teichomyceticus ATCC 31121. J. Antibiot. 49, 607–609 (1996).
Malabarba, A. & Goldstein, B. P. Origin, structure, and activity in vitro and in vivo of dalbavancin. J. Antimicrob. Chemother. 55, ii15–ii20 (2005).
Anderson, V. R. & Keating, G. M. Dalbavancin. Drugs 68, 639–648 (2008).
Durata Therapeutics International B.V. Dalbavancin for Injection for Treatment of Acute Bacterial Skin and Skin Structure Infections, NDA 021-883 Briefing Document, 31 Mar 2014 〈http://www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/Anti-InfectiveDrugsAdvisoryCommittee/UCM390793.pdf〉 (accessed 6 June 2014)..
Jones, R. N., Sader, H. S. & Flamm, R. K. Update of dalbavancin spectrum and potency in the USA: report from the SENTRY Antimicrobial Surveillance Program (2011). Diagn. Microbiol. Infect. Dis. 75, 304–307 (2013).
Jones, R. N., Sader, H. S., Mendes, R. E. & Flamm, R. K. Update on antimicrobial susceptibility trends among Streptococcus pneumoniae in the United States: report of ceftaroline activity from the SENTRY Antimicrobial Surveillance Program (1998–2011). Diagn. Microbiol. Infect. Dis. 75, 107–109 (2013).
Cooper, R. D. et al. Reductive alkylation of glycopeptide antibiotics: synthesis and antibacterial activity. J. Antibiot. 49, 575–581 (1996).
Bouza, E. & Burillo, A. Oritavancin: a novel lipoglycopeptide active against Gram-positive pathogens including multiresistant strains. Int. J. Antimicrob. Agents 36, 401–407 (2010).
Guskey, M. T. & Tsuji, B. T. A comparative review of the lipoglycopeptides: oritavancin, dalbavancin, and telavancin. Pharmacotherapy 30, 80–94 (2010).
Allen, N. E. From vancomycin to oritavancin: the discovery and development of a novel lipoglycopeptide antibiotic. Anti-Infect. Agents. Med. Chem. 9, 23–47 (2010).
Arhin, F. F., Sarmiento, I., Parr, Jr T. R. & Moeck, G. Comparative in vitro activity of oritavancin against Staphylococcus aureus strains that are resistant, intermediate or heteroresistant to vancomycin. J. Antimicrob. Chemother. 64, 868–870 (2009).
Thompson, C., Ge, M. & Kahne, D. Synthesis of vancomycin from the aglycon. J. Am. Chem. Soc. 121, 1237–1244 (1999).
Nicolaou, K. C. et al. Total synthesis of vancomycin. Angew. Chem. Int. Ed. 38, 240–244 (1999).
Boger, D. L. et al. Total synthesis of the teicoplanin aglycon. J. Am. Chem. Soc. 122, 7416–7417 (2000).
Evans, D. A., Katz, J. L., Peterson, G. S. & Hintermann, T. Total synthesis of teicoplanin aglycon. J. Am. Chem. Soc. 123, 12411–12413 (2001).
Boger, D. L. et al. First and second generation total synthesis of the teicoplanin aglycon. J. Am. Chem. Soc. 123, 1862–1871 (2001).
Deng, H. et al. Total synthesis of anti-HIV agent chloropeptin I. J. Am. Chem. Soc 125, 9032–9034 (2003).
Crowley, B. M., Mori, Y., McComas, C. C., Tang, D. & Boger, D. L. Total synthesis of the ristocetin aglycon. J. Am. Chem. Soc. 126, 4310–4317 (2004).
Rao, A. V. R., Gurjar, M. K., Reddy, K. L. & Rao, A. S. Studies directed toward the synthesis of vancomycin and related cyclic peptides. Chem. Rev. 95, 2135–2167 (1995).
Süssmuth, R. D. et al. New Advances in the Biosynthesis of Glycopeptide Antibiotics of the Vancomycin Type from Amycolatopsis mediterranei. Angew. Chem. Int. Ed. 38, 1976–1979 (1999).
Weist, S. et al. Fluorobalhimycin—A New Chapter in Glycopeptide Antibiotic Research. Angew. Chem. Int. Ed. 41, 3383–3385 (2002).
Evans, D. A. et al. Nonconventional stereochemical issues in the design of the synthesis of the vancomycin antibiotics: challenges imposed by axial and nonplanar chiral elements in the heptapeptide aglycons. Angew. Chem. Int. Ed. 37, 2704–2708 (1998).
Boger, D. L. et al. Vancomycin CD and DE macrocyclization and atropisomerism studies. J. Org. Chem. 64, 70–80 (1998).
Boger, D. L. et al. Thermal atropisomerism of teicoplanin aglycon derivatives: preparation of the P,P,P and M,P,P atropisomers of the teicoplanin aglycon via selective equilibration of the DE ring system. J. Am. Chem. Soc. 122, 10047–10055 (2000).
Xie, J. et al. Total synthesis of [Ψ[C(=S)NH]Tpg4]vancomycin aglycon, [ψ[C(=NH)NH]Tpg4]vancomycin aglycon, and related key compounds: reengineering vancomycin for dual D-Ala-D-Ala and D-Ala-D-Lac binding. J. Am. Chem. Soc. 134, 1284–1297 (2011).
Xie, J., Pierce, J. G., James, R. C., Okano, A. & Boger, D. L. A redesigned vancomycin engineered for dual D-Ala-D-Ala and D-Ala-D-Lac binding exhibits potent antimicrobial activity against vancomycin-resistant bacteria. J. Am. Chem. Soc. 133, 13946–13949 (2011).
Okano, A., James, R. C., Pierce, J. G., Xie, J. & Boger, D. L. Silver(I)-promoted conversion of thioamides to amidines: divergent synthesis of a key series of vancomycin aglycon residue 4 amidines that clarify binding behavior to model ligands. J. Am. Chem. Soc. 134, 8790–8793 (2012).
Malabarba, A., Nicas, T. I. & Thompson, R. C. Structural modifications of glycopeptide antibiotics. Med. Res. Rev. 17, 69–137 (1997).
Chang, J. et al. Design, synthesis, and antibacterial activity of demethylvancomycin analogues against drug-resistant bacteria. ChemMedChem 8, 976–984 (2013).
Pintér, G. et al. Diazo transfer−click reaction route to new, lipophilic teicoplanin and ristocetin aglycon derivatives with high antibacterial and anti-influenza virus activity: an aggregation and receptor binding study. J. Med. Chem. 52, 6053–6061 (2009).
Crane, C. M. et al. Synthesis and evaluation of selected key methyl ether derivatives of vancomycin aglycon. J. Med. Chem. 53, 7229–7235 (2010).
Nakama, Y. et al. Discovery of a novel series of semisynthetic vancomycin derivatives effective against vancomycin-resistant bacteria. J. Med. Chem. 53, 2528–2533 (2010).
Pinchman, J. R. & Boger, D. L. Probing the role of the vancomycin E-ring aryl chloride: selective divergent synthesis and evaluation of alternatively substituted E-ring analogues. J. Med. Chem. 56, 4116–4124 (2013).
Pathak, T. P. & Miller, S. J. Site-selective bromination of vancomycin. J. Am. Chem. Soc. 134, 6120–6123 (2012).
Muthyala, R., Rastogi, N., Shin, W. S., Peterson, M. L. & Sham, Y. Y. Cell permeable vanX inhibitors as vancomycin re-sensitizing agents. Bioorg. Med. Chem. Lett. 24, 2535–2538 (2014).
Cooper, M. A. & Williams, D. H. Binding of glycopeptide antibiotics to a model of a vancomycin-resistant bacterium. Chem. Biol. 6, 891–899 (1999).
Acknowledgements
This paper was prepared with the support of NHMRC grant APP1026922. MSB, KAH and MAB are supported by a Wellcome Trust Seeding Drug Discovery Award (094977/Z/10/Z) and MAC by an NHMRC Principal Research Fellowship (APP1059354).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Butler, M., Hansford, K., Blaskovich, M. et al. Glycopeptide antibiotics: Back to the future. J Antibiot 67, 631–644 (2014). https://doi.org/10.1038/ja.2014.111
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2014.111
- Springer Japan KK
This article is cited by
-
Self-association of the glycopeptide antibiotic teicoplanin A2 in aqueous solution studied by molecular hydrodynamics
Scientific Reports (2023)
-
Synthesis of vancomycin fluorescent probes that retain antimicrobial activity, identify Gram-positive bacteria, and detect Gram-negative outer membrane damage
Communications Biology (2023)
-
Newest perspectives of glycopeptide antibiotics: biosynthetic cascades, novel derivatives, and new appealing antimicrobial applications
World Journal of Microbiology and Biotechnology (2023)
-
Strategic Approaches to Improvise Peptide Drugs as Next Generation Therapeutics
International Journal of Peptide Research and Therapeutics (2023)
-
Glycopeptide antibiotic drug stability in aqueous solution
AAPS Open (2022)