
Abstract
Geological and ecological features restrict dispersal and gene flow, leading to isolated populations. Dispersal barriers can be obvious physical structures in the landscape; however microgeographic differences can also lead to genetic isolation. Our study examined dispersal barriers at both macro- and micro-geographical scales in the black-capped chickadee, a resident North American songbird. Although birds have high dispersal potential, evidence suggests dispersal is restricted by barriers. The chickadee’s range encompasses a number of physiological features which may impede movement and lead to divergence. Analyses of 913 individuals from 34 sampling sites across the entire range using 11 microsatellite loci revealed as many as 13 genetic clusters. Populations in the east were largely panmictic whereas populations in the western portion of the range showed significant genetic structure, which often coincided with large mountain ranges, such as the Cascade and Rocky Mountains, as well as areas of unsuitable habitat. Unlike populations in the central and southern Rockies, populations on either side of the northern Rockies were not genetically distinct. Furthermore, Northeast Oregon represents a forested island within the Great Basin; genetically isolated from all other populations. Substructuring at the microgeographical scale was also evident within the Fraser Plateau of central British Columbia, and in the southeast Rockies where no obvious physical barriers are present, suggesting additional factors may be impeding dispersal and gene flow. Dispersal barriers are therefore not restricted to large physical structures, although mountain ranges and large water bodies do play a large role in structuring populations in this study.
Similar content being viewed by others


Introduction
Dispersal is the ecological process where individuals move from one population to another to reproduce. This process facilitates gene flow and is essential for the persistence of populations and species. However, ecological and geological features can affect the ability of individuals to move across landscapes and those that restrict dispersal are termed a ‘barrier’. Barriers therefore play a key role in the genetic structuring of populations by influencing important evolutionary processes such as gene flow and adaptation.
Over the last decade, landscape genetics has contributed to our understanding of how contemporary landscapes influence the spatial distribution of genetic variation in a variety of organisms (Manel and Holderegger, 2013). Topographical features (Smissen et al., 2013), unsuitable habitat (Piertney et al., 1998) and anthropogenic disturbance to the landscape (Young et al., 1996) have all been identified as factors strongly influencing population genetic structure in previous studies. Examining the effects of landscape features and environmental variables on current genetic patterns will provide us with a better understanding of how species interact with their environment. Not only does landscape genetics allow us to assess the environmental contributors of population structuring, it also compliments phylogeographic studies allowing researchers to tease apart the effects of historical and contemporary processes on gene flow in complex landscapes.
During the Quaternary period, severe climatic oscillations played a major role in shaping current landscapes and a number of genetic studies have documented the effects of climatic fluctuations on species distributions since the Last Glacial Maximum, approximately 18–21 thousand years ago (Hewitt, 1996, 2004; Carstens and Knowles, 2007). While these historical processes may have contributed to how species are distributed today, many physical structures influenced the dispersal routes of new colonisers, some of which still exist in contemporary landscapes and continue to restrict movement. For example, mountain ranges provide an elevational limit to dispersal and large bodies of water may be perceived as too risky or energetically costly to cross. Barriers can also be climate related (for example, large arid regions) or occur at microgeographic scales (for example, habitat fragmentation). So, although historical processes are important to consider when assessing the genetic integrity of populations, contemporary processes ultimately impact the spatial distribution of genetic variation seen today.
The black-capped chickadee (Poecile atricapillus) is a small, generalist songbird common throughout North America (Figure 1). They are an ideal model for understanding how landscape features influence dispersal and gene flow as their current distribution encompasses a wide and diverse geographic region. Although geographically widespread, they are year-round residents with localised distributions. Only juveniles engage in limited dispersal (approximately 1.1 km; Brennan and Morrison, 1991) creating the potential for restricted gene flow. Owing to their generalist nature, suitable habitat is not limited but they do exhibit preference for different types of woodland varying from deciduous and coniferous woodland to forested wetlands, favourable riparian communities, deciduous shrubs and even urban, suburban and disturbed areas (Smith, 1993). As cavity nesters, they are however, dependent on trees or snags with advanced decay, particularly of those found in mature forest. They also show a varied diet, feeding on mixed berries, seeds and insects in winter months, switching to a completely insectivorous diet in the breeding season (Runde and Capen, 1987; Smith, 1993). Thus, habitat quality is important for reproductive and foraging success of this species (Fort and Otter, 2004). Although black-capped chickadee behaviour is extensively studied in North America, little is known about the roles barriers play in structuring populations. Previous research focused primarily on hybridisation between the black-capped chickadee and other chickadees, (for example, P. carolinensis (Davidson et al., 2013); P. hudsonicus (Lait et al., 2012) and P. gambeli (Grava et al., 2012)), vocalisations (Guillette et al., 2010) and winter survival (Cooper and Swanson, 1994). Geographical variation in song, plumage and morphology (Smith, 1993; Roth and Pravosudov, 2009) in addition to differences in hippocampal gene expression profiles (Pravosudov et al., 2013) are suggestive of divergence among populations. Moreover, previous studies using high-resolution genetic data (Gill et al., 1993; Pravosudov et al., 2012; Hindley, 2013) have all identified genetically distinct populations of the black-capped chickadee over a large geographical range. Hindley’s (2013) study showed the most comprehensive sampling design, but was limited by the use of a single maternally inherited locus (mitochondrial DNA control region). By creating a picture of the overall genetic structure of the black-capped chickadee across a wide range of environments, this current study can help provide additional insights into other ecological patterns found in this species. For example, do patterns in song and morphology reflect differences in genetic patterns and therefore different selective pressures?

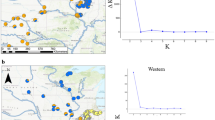
Map illustrating the current geographical distribution of the black-capped chickadee (Poecile atricapillus) across North America with sampling locations (see Table 1 for abbreviations) projected in ArcGIS v.10 (ESRI). A full color version of this figure is available at the Heredity journal online.
The aims of this study are to investigate how contemporary landscapes have shaped the spatial patterns of genetic variation and population structuring of the black-capped chickadee and to identify potential barriers to dispersal providing additional insights into their ecological and evolutionary potential using microsatellite markers. Birds can be used as mobile indicators of habitat quality, so as a common, widely distributed songbird that responds relatively quickly to environmental change (for example, in insect outbreaks (Gray, 1989)) the black-capped chickadee is an ideal model organism for investigating population structure and gene flow in contemporary landscapes at both large and small geographical scales.
In this study, we aim to answer the following questions:
-
1
Do mountain ranges and large bodies of water restrict gene flow across the black-capped chickadee’s range? Mountain ranges have been found to restrict dispersal in a number of organisms (for example, the downy woodpecker Picoides pubescens (Pulgarín-R and Burg, 2012); the hairy woodpecker Picoides villosus (Graham and Burg, 2012) and the tundra vole Micotus oeconomus (Galbreath and Cook, 2004)) producing in some cases a clear east/west divide corresponding to the Rocky and/or Cascade Mountains. We predict significant genetic differences among samples collected on either side of mountain ranges. The most prominent ranges include the Rocky Mountains, the Alaskan Mountain range and the Cascade Mountains. Black-capped chickadees are notably absent from Vancouver Island, Haida Gwaii (also known as the Queen Charlotte Islands) and the Alexander Archipelago, suggesting large expanses of water are also significant dispersal barriers. The island of Newfoundland is separated from continental populations by the Strait of Belle Isle and Cabot Strait and mitochondrial DNA (mtDNA) studies show restricted maternal gene flow between Newfoundland and the mainland in black-capped chickadees (Gill et al., 1993; Hindley, 2013). As such, we predict populations on Newfoundland will be genetically distinct from those on the mainland.
-
2
Are fine-scale genetic differences present within the black-capped chickadee populations? We predict finer scale differences in population structure will be found (in comparison with previous mtDNA and amplified fragment length polymorphism marker studies) using high-resolution microsatellite markers as the result of ecological differences across the species’ range. Restricted gene flow can result from recent modifications to the landscape creating small-scale barriers (for example, change in habitat composition). Habitat loss and associated fragmentation can reduce connectivity and create small, isolated populations leading to increased genetic differentiation (Young et al., 1996).
Materials and methods
Sampling and DNA extraction
Adult birds were captured using mist nets and call playback over six breeding seasons (2007–2012). Blood samples (<100 μl from the brachial vein) and/or feather samples were collected from across the species’ range (Figure 1 and Supplementary Table S1). Suspected family groups and juveniles were removed from the data. Sampling sites were confined to a 40 km radius where possible and a total of 913 individuals from 34 populations were sampled across North America. Each bird was banded with a numbered metal band to prevent re-sampling. All blood samples were stored in 95% ethanol and, on return to the laboratory, stored at −80 °C. Additionally, museum tissue samples (toe pads and skin) were obtained to supplement field sampling (see Acknowledgements). Museum samples were collected within the last 30 years with the oldest sample obtained in 1983. DNA was extracted from blood ethanol mix (10 μl), tissue (∼1 μg) or feather samples using a modified Chelex protocol (Walsh et al., 1991).
DNA amplification and microsatellite genotyping
A subset of individuals was initially screened with 54 passerine microsatellite loci. In total, 29 microsatellite loci yielded PCR products, of which 18 loci were monomorphic (Aar1 (Hannson et al., 2000), Ase48, Ase56 (Richardson et al., 2000), CE150, CE152, CE207, CETC215, CM014, CM026 (Poláková et al., 2007), CtA105 (Tarvin, 2006), Gf06 (Petren, 1998), Hofi20, Hofi24, Hofi5 (Hawley, 2005), Lox1 (Piertney et al., 1998), NPAS2 (Steinmeyer et al., 2009), Pca2 (Dawson et al., 2000) and VeCr02 (Stenzler et al., 2004)) and 11 were polymorphic (Supplementary Table S2).
DNA was amplified in 10 μl reactions containing MgCl2 (Supplementary Table S2), 0.2 mM dNTPs, 1 μM each primer pair (forward and reverse) and 0.5 U Taq DNA polymerase. All forward primers were synthesised with an M13 sequence on the 5′ end to allow for incorporation of a fluorescently labelled M13 primer (0.05 μM; Burg et al., 2005) during DNA amplification. One percent formamide was added to reactions involving PAT MP 2–14. Among 11 markers, 6 could be multiplexed in three sets of two markers each (PAT MP 2-14/Titgata39, Escu6/Titgata02 and Ppi2/Cuμ28). For multiplex reactions involving loci Escu6 and Titgata02, PCR conditions for Titgata02 were used.
We used a two-step annealing protocol: 1 cycle of 94 °C for 2 min, 50 °C for 45 s and 72 °C for 1 min, followed by 7 cycles of 94 °C for 1 min, 50 °C for 30 s and 72 °C for 45 s, followed by 25 cycles of 94 °C for 30 s, 52 °C for 30 s and 72 °C for 45 s, followed by a final extension step of 72 °C for 5 min. For two loci (PAT MP 2–43 and Titgata02), the second step was increased from 25 to 31 cycles. Subsequently, products were denatured and run on a 6% polyacrylamide gel on a LI-COR 4300 DNA Analyser (LI-COR Inc., Lincoln, NE, USA) and manually scored using Saga Lite Electrophoresis Software (LI-COR Inc.). For each gel, three positive controls of known size were included to maintain consistent allele sizing, and all gels were scored by a second person to reduce the possibility of scoring error.
Genetic diversity
Standard statistical analyses were performed on all individuals unless otherwise indicated. MICRO-CHECKER v2.2.3 was used to detect any errors within the data such as input errors, allelic dropout, stutter or null alleles (van Oosterhout et al., 2004). Allelic richness was calculated in FSTAT v2.9.2.3 (Goudet, 2001) after removing under sampled populations (n⩽5). Tests for deviations from Hardy–Weinberg equilibrium (HWE) and linkage disequilibrium (LD) were performed in GENEPOP v4.0.10 (Raymond and Rousset, 1995) using default Markov chain parameters (100 batches, 1000 iterations and 1000 dememorisation steps). Levels of significance were adjusted for multiple statistical tests within populations using a modified False Discovery Rate (FDR) correction method (Benjamini and Yekutieli, 2001). Finally, to determine the levels of population genetic diversity, both observed and expected heterozygosities were calculated in GenAlEx v6.5 (Peakall and Smouse, 2012).
Genetic clustering analyses
Several Bayesian clustering methods are currently available to infer the spatial structure of genetic data (Latch et al., 2006). Genetic structure was therefore assessed using three approaches (one non-spatial and two spatial): STRUCTURE v2.3.4 (Pritchard et al., 2000), BAPS v5.4 (Bayesian Analysis of Population Structure; Corander et al., 2008) and TESS v2.3 (Chen et al., 2007).
As assignments are based on individual multilocus genotypes rather than population allele frequencies, we included samples from all 34 populations as small population sizes will not bias assignment results. All three programs use a Bayesian clustering approach, which assigns individuals to clusters by maximising HWE and minimising LD. They differ in their underlying model and assumptions (reviewed in François and Durand, 2010) and some include the type of algorithm used and how the true number of clusters (K) is determined. For example, STRUCTURE and TESS use a Markov chain Monte Carlo (McMC) simulation and complex hierarchical Bayesian modelling, whereas BAPS models genetic structure using a combination of analytical and stochastic methods, which is computationally more efficient, particularly for large data sets (Corander et al., 2008). Ultimately, STRUCTURE uses a non-spatial prior distribution; relying purely on the genetic data, whereas BAPS and TESS explicitly incorporate spatial information (that is, geographic coordinates) from genotyped individuals to infer genetic clusters. All three programs work well when genetic differentiation among clusters is low (FST ⩽0.05; Latch et al., 2006).
STRUCTURE was run using the admixture model, correlated allele frequencies (Falush et al., 2003) and locations as priors (locpriors). Ten independent runs for each value of K (1–10) were conducted to determine the optimal K. Runs were performed using 50 000 burn in periods followed by 100 000 Markov chain Monte Carlo repetitions. The results from replicate runs were averaged using STRUCTURE HARVESTER v0.6.6 (Earl and vonHoldt, 2012). Both delta K (ΔK; Evanno et al., 2005), LnPr(X|K) and Bayes factor (Pritchard et al., 2000) were used to determine K. Following the initial run, subsets of the data (that is, individuals who formed a single cluster from the initial runs) were re-run to establish if further structure was present using the same parameters and five runs for each value of K. Individuals that showed mixed ancestry to two clusters (Q<60%) were re-run together with a subset of individuals from each of the two groups to confirm assignment.
BAPS was run with the option ‘clustering of individuals’ followed by ‘clustering of groups of individuals’, both for KMAX=34. BAPS searches for all values of K up to the value given for KMAX and gives a final K for the maximum log (marginal likelihood). The ‘spatial clustering of groups’ option was then used on all individuals and their corresponding group geographic coordinates (weighted mid-point values for each population projected in DIVA GIS v7.5 (Hijmans et al., 2012)). This option has been shown to increase the power to detect underlying population structure and allows the user to visually investigate population structure using Voronoi tessellations.
Using the number of clusters inferred from STRUCTURE, TESS was run using 100 000 sweeps and 50 000 burn-in sweeps for KMAX (2–13) to identify which K produced the highest likelihoods. The CAR (conditional autocorrelation) admixture model based on the Delaunay tessellation was used and a deviance information criterion (DIC), a measure of model fit, is computed for each run. We conducted 10 replicates for each value of KMAX with an interaction parameter (Ψ; the degree to which the geographical information influences individual assignment) of 0.6 as described in Chen et al. (2007). To determine the true number of clusters, we retained 20% of the lowest DIC to identify which K produced the highest likelihood (KMAX) and lowest DIC. We also averaged DIC over all 10 runs for each value of KMAX as often the optimum cluster is the value that coincides with the plateau of the DIC curve.
Population structure
All populations with a small sample size (n⩽5) were removed from population-level analyses (CoOR n=2; NC n=5 and LAB n=5) unless otherwise indicated. Pairwise FST values were calculated in ARLEQUIN v3.5 (Excoffier and Lischer, 2010) to investigate the degree of genetic differentiation among the predefined populations (significance determined by 1023 permutations). As the theoretical maximum of 1 for FST is only valid when there are two alleles, F’ST standardised by the maximum value it can obtain were also calculated in GenAlEx v6.5 (Peakall and Smouse, 2012).
Since traditional FST is often criticised by its dependency on within-population diversity, sample sizes and its use with highly variable molecular markers such as microsatellites (Meirmans and Hedrick, 2011), we also calculated an alternative diversity measure, Dest (Jost, 2008), using the software SMOGD v1.2.5 (Crawford, 2010). The overall value of Dest is calculated as the harmonic mean across loci for each pairwise population comparison and is suggested to be more accurate for identifying population structure. Measures from both Dest and FST were compared to determine the true level of genetic differentiation. We also assessed the level of concordance between the two estimates by plotting linearised Dest values (Dest/(1−Dest)) against linearised FST values (FST/(1−FST)) using a Mantel test in GenAlEx v6.5. Significance was determined using 9999 permutations. To further assess population structure, a hierarchical analysis of molecular variance was carried out in ARLEQUIN v3.5 on the various groupings produced from both STRUCTURE and BAPS.
Effects of barriers on population structure
Isolation by distance (IBD) was tested using a Mantel test in GenAlEx v6.5 using linearised FST values. Significance was determined using 9999 permutations and geographic distances (km) were calculated using the GEOGRAPHIC DISTANCE MATRIX GENERATOR v1.2.3. (http://biodiversityinformatics.amnh.org/open_source/gdmg/). Straight line distances are not always accurate as barriers can affect dispersal routes and for that reason, we also tested shortest distance through suitable habitat. For example, distance through forest was calculated for populations located on or around the Great Plains (CO, SD, UT, MT, SAB1, SAB2, LETH, CAB, SK, MB, MI, IL and MO).
BARRIER v2.2 uses a geometry approach to compute barriers on a Delaunay triangulation (Manni et al., 2004). Monmonier’s algorithm identifies areas where genetic differences between pairs of populations are the largest. Using a genetic distance matrix (FST), BARRIER identifies the location and direction of barriers to provide a visual representation of how the landscape influences dispersal in comparison with IBD. We computed the first 10 genetic boundaries using an FST distance matrix for all populations (excluding sites with ⩽5 samples: CoOR, NC and LAB).
Finally, we used GIS landscape genetics toolbox (Vandergast et al., 2011) to visualise the distribution of genetic diversity across geographical space. The toolbox is run within the Geographical Information System software package ArcGIS v.9 (ESRI, Redlands, CA, USA) and utilises the population pairwise genetic distances (FST) to produce a genetic divergence raster surface (or heat map). This will help evaluate our hypothesised barriers to movement by plotting values on a map.
Landscape genetics
A landscape genetic approach was used to assess the influence of environmental factors on genetic differentiation in the black-capped chickadee. We used GESTE v2.0 (Foll and Gaggiotti, 2006), a hierarchical Bayesian method, which estimates population-specific FST values and links them to environmental variables using a generalised linear model. It evaluates likelihoods of models that include all the factors, their combinations and a constant (which excludes all variables). Posterior probabilities are used to identify the factor(s) that influence genetic structure. Using a reversible jump Markov chain Monte Carlo method and default parameters, we conducted 10 pilot runs with a burn-in of 50 000 iterations to obtain convergence and a chain length of 2.5 × 105, separated by a thinning interval of 20. A total of six factors were considered, including three environmental variables (annual average temperature, precipitation and elevation) and three related to distance (latitude, longitude and distance to unsuitable habitat). We tested a number of scenarios to determine the models with the highest probabilities. Certain factors were also tested under different environmental scenarios to more closely examine their influence on genetic structuring (as conducted in Wellenreuther et al., 2011). Three environmental scenarios were assessed; spatial, climatic and geographic. In the spatial scenario, we tested latitude and longitude; for the climatic scenario, we tested annual average temperature and precipitation; and with the geographic scenario, we tested elevation and distance to unsuitable habitat. As only two factors are being assessed in these specific scenarios, we added a factor interaction as suggested by Foll and Gaggiotti (2006), and kept all other parameters at their default setting.
Results
Genetic diversity
In total, 913 individuals from 34 populations were successfully genotyped for 11 variable microsatellite loci with the overall number of alleles per locus ranging from 5 to 46 (Supplementary Table S2). Observed heterozygosity ranged from 0.52 (PG) to 0.73 (CoOR) across all loci and expected heterozygosity ranged from 0.39 (NC) to 0.73 (LAB and MI; Table 1). Allelic richness (which accounts for uneven sample size) ranged from 5.26 (AKA) to 8.00 (ON) (Table 1). Nineteen of the 34 populations contained private alleles (Table 1): 16 populations contained 1 or 2 private alleles, whereas NSNB had the highest (10), PG had 5 and FtStJ had 4 private alleles. Evidence of null alleles and homozygote excess was found for locus Pman45. Exclusion of this locus did not change the results and so was included in the final analyses.
Disequilibrium and departures from HWE were detected following corrections for multiple comparisons. Significant LD was detected between Titgata02 and Cuμ28 and between Escu6 and Pman71 within ID (P⩽0.001 and P⩽0.001, respectively); between Escu6 and Titgata02 and Escu6 and Ppi2 within SAB1 (P⩽0.001 and P⩽0.001, respectively); between Titgata39 and Titgata02 within SK (P⩽0.001) and between Titgata02 and Ppi2 within UT (P⩽0.001). LD was not consistent across populations and genotypes showed no association suggesting that LD detected here could be a result of a type 1 error. Significant deviations from HWE were evident for 14 population/loci comparisons: FtStJ at locus PAT MP 2–43; AKA, MI, FtStJ, SOR, NSNB and WV at locus Pman45; SAB2 and MB at locus Ppi2 and PG deviated at PAT MP 2–14, Titgata39, Titgata02, Escu6 and PAT MP 2–43. We checked the data for populations that deviated from HWE at two or more loci for the presence of family groups, which could explain deviations from Hardy–Weinberg expectations. Although a number of individuals were caught at the same location on the same day in PG and NSNB, no evidence of family groups was found.
Bayesian clustering analyses
STRUCTURE estimated 13 clusters (Figures 2 and 3 and Supplementary Figure S1). The initial run of all of the samples resulted in K=3, using mean log likelihood (Pr (X|K)= −34 930) and ΔK, and consisted of: the three Alaskan populations (AKA, AKF and AKW), the Fraser Plateau populations (PG and FtStJ) and all other populations (‘main’; Figure 2a). The two latter clusters showed evidence of further structure. The Fraser Plateau group was subdivided into two groups, PG and FtStJ (Pr (X|K)= −3034; Figure 2b). The ‘main’ cluster produced three clusters: western, central and eastern (Pr (X|K)= −28 689; Figure 2c). Nine of the populations showed evidence of mixed ancestry (NWBC, BCR, LETH, MB, CID, MT, IL, LAB and NC). Each of these populations was run with individuals from the two clusters to which they had high Q values. NWBC, BCR, LETH and MB clustered with the western cluster, MT with the central cluster and the remaining three populations with the eastern cluster (results not shown). These nine populations were then grouped accordingly for additional analyses. Further runs were performed on the western, central and eastern clusters using a hierarchical approach. Subsequent runs of the western group (Figures 2d–g) resulted in a total of five clusters: Canadian Pacific-Prairies (NBC, all AB populations, SK and MB; Pr (X|K)= −14 136), Pacific (WA, SOR, CoOR; Pr (X|K)= −8710), Northwest Rockies (NWBC and BCR; Pr (X|K)= −6614); Idaho (CID and ID) in the Intermountain West and finally NEOR Pr (X|K)= −2383). The central group was subdivided into three clusters: eastern Rockies (MT, SD and UT; Pr (X|K)= −4041), CO and NM (Pr (X|K)= −1096; Figures 2h and i). The eastern cluster was further subdivided into two clusters: NL and eastern mainland (Pr (X|K)= −10 073; Figure 2j). All runs were supported by a Bayes Factor of 1 and ΔK.

Inferred population structure of the black-capped chickadee (Poecile atricapillus) from 11 microsatellite loci using STRUCTURE v2.3.4 (Pritchard et al., 2000) for (a) K=3; all individuals from 34 populations, (b) K=2; Fraser Plateau (FtStJ and PG), (c) K=3 after removing structured populations from the first run (d) K=2; for all western populations, which resulted in (e) K=2; Canadian Pacific-Prairies (CAB, LETH, SAB1, SAB2, MB, SK, NBC) and Pacific (WA, SOR, CoOR), (f) K=2; NW Rockies (NWBC, BCR) and Intermountain West (CID, ID and NEOR) with further substructuring of NEOR (g). The central and southern Rocky Mountain regions resulted in (h) Eastern Rockies (MT, SD and UT) and (i) substructuring of NM and CO and (j) K=2; Eastern mainland (IL, MI, MO, ON, NSNB, LAB, NC, WV) and Newfoundland (NL). Each vertical line represents one individual and the colour(s) of each line represents the proportion of assignment of that individual to each genetic cluster.

Distribution map illustrating coloured population assignment as inferred from STRUCTURE v2.3.4 for all black-capped chickadee individuals based on 11 microsatellite loci. Also included are the five genetic clusters found using BAPS v5.4 (solid circles; the fifth cluster includes the remaining 25 populations), and the four clusters found using TESS v2.3 (triangles; the fourth cluster includes the remaining 28 populations). Dashed lines and circles represent barriers or genetic boundaries as identified in the program BARRIER v 2.2. On the main figure elevation is indicated with grey shading (darker shades of grey indicate higher elevation) and the inset shows forest cover (dark green=closed forest; mid green=open/fragmented forest; light green=other vegetation types; FAO, 2001).
The two spatial methods were unable to identify finer differences detected in STRUCTURE despite incorporating individual spatial information. BAPS estimated 5 distinct clusters (Figure 3) in comparison with STRUCTURE’s 13. Concordant with groups identified by STRUCTURE, BAPS identified both AK and the Fraser Plateau as being two genetically distinct units in addition to the southern Rockies populations (CO and NM) and Oregon (CoOR and SOR); while the remaining populations formed the fifth cluster. For TESS analyses, the mean DIC plot did not plateau (Supplementary Figure S2). The mean DIC for KMAX of 12 disrupted the curve indicating that the program may have failed to converge. Nevertheless, after comparing runs for various assumed K (2–13), KMAX was estimated from the highest likelihood and lowest DIC run to be 13 (average log likelihood: −33 818; DIC: 68 793.3). The effective number of clusters with this parameter was four (Supplementary Figure S3), detecting the same three groupings as the initial run of STRUCTURE (Figure 2a) and an additional cluster representing Newfoundland, which was not detected by BAPS.
Population structure
Pairwise FST values ranged from −0.014 to 0.148 (Supplementary Table S3) and 318 of the 465 values were significant after corrections for multiple tests. Of the 87 nonsignificant pairwise FST values, 27 were between adjacent sampling sites. Population wide F’ST was 0.231 (Supplementary Table S4). Significant population structure was detected by Dest, which ranged from 0.030 to 0.316 (Supplementary Table S3). Pairwise Dest and FST values shared a significant, positive correlation (r=0.496; P⩽0.001).
Using a hierarchical analysis of molecular variance, the highest among group variance (5.75%) was produced using three groups (AK, Fraser Plateau and all remaining populations). Among group variance decreased once the ‘remaining populations’ were split into western, central and eastern groups, but as these regions were split further into their respective groups identified in the hierarchical STRUCTURE runs, among group variance steadily increased. Once NEOR was split from the Intermountain West group, the amount of variance increased to 4.06% and a final run of all 13 groups from STRUCTURE resulted in 4.12%. Meanwhile, when populations were analysed according to BAPS (K=5) and TESS (K=4) groupings, among group variance was 5.25% and 5.08%, respectively.
Effects of barriers on population structure
The test for IBD among all black-capped chickadee populations using straight line distances was not significant (r2=0.010; P=0.16). However, we did find significant IBD within some clusters identified by STRUCTURE. IBD was significant for the eastern mainland group when NL was included (r2=0.358; P=0.01), but not when NL was removed (r2=0.003; P=0.24). For other populations separated by large geographical barriers (that is, unsuitable habitat), we found a significant effect of IBD using the shortest distance through suitable habitat. For example, when testing populations located around the Great Plains, using the shortest distance through forested habitat resulted in a significant IBD pattern (r2=0.137; P=0.01).
BARRIER identified nine discontinuities. Boundaries detected to the ninth order were considered the most strongly supported for the level of population structure observed in the data, and were overlaid onto a map for visual interpretation (Figure 3). Boundaries detected after the ninth order did not conform to differences observed in previous analyses (for example, pairwise FST and Dest) and so were removed. Overall, populations where barriers exist were significantly different from all other populations (P⩽0.008). Eight of the linear barriers identified were concordant with STRUCTURE results where populations on either side of the barrier belong to different clusters. The ninth barrier that encircles PG and FtStJ was confirmed by STRUCTURE, BAPS and TESS, however, BARRIER failed to identify a genetic discontinuity between these two populations as found in STRUCTURE.
The heat map produced from the GIS toolbox species divergence analysis supports the presence of multiple barriers particularly in the western portion of the range (Figure 4). It shows isolation of Alaska, Pacific, Fraser Plateau and NEOR groups and moderate isolation of Newfoundland. CO and NM are isolated from UT to the west and MO in the east. FST values to MT are modest to low across prairies and ‘around’ the Great Plains.

A heat map of pairwise FST values for 11 microsatellite loci in the black-capped chickadee. Red indicates high FST values and blue indicates low FST values. Each sampling site is represented by a black dot (see Figure 1 for location names).
Landscape genetics
Landscape genetics analyses in GESTE revealed a number of environmental variables influencing genetic structure in the black-capped chickadee. When all factors were run together, GESTE struggled to find the model with the highest probability (results not shown). For all single factor runs, the model including the constant produced the highest posterior probability (Table 2a). However, some single factor runs produced higher probability models than the environmental scenarios with two factors. For example, the highest constant/factor model involved distance to unsuitable habitat (0.481) followed closely by annual mean temperature (0.479). Interestingly, the influence of longitude (east–west) was slightly higher than latitude (north–south) on the genetic differentiation (0.472 and 0.469, respectively).
Of all three environmental scenarios (Table 2b), the model with the highest posterior probability was the spatial scenario, which included latitude, longitude and their interaction term (0.678), suggesting geographic location is an important determinant in the genetic structuring of populations. In the climatic and geographic scenarios, no factors were strongly correlated with pairwise FST values as the models including only the constant outperformed the rest (climate: 0.216; geographic: 0.214). Despite this, the model with the second highest posterior probability in the climate scenario included precipitation (0.204); this factor also displayed the highest sum of probabilities (0.388). In the geographic scenario, the model with the second highest posterior probability included elevation, distance to unsuitable habitat and their interaction term (0.212) and elevation had the highest sum of probabilities (0.384).
Discussion
Microsatellite analyses revealed significant population structuring across the black-capped chickadee’s range. Using clustering programs as many as 13 groups were found supporting the idea of restricted gene flow. The main groups found in this study are: Alaska, Fraser Plateau (which split into FtStJ and PG), eastern Rockies, eastern mainland, Newfoundland, Canadian Pacific-Prairies, Pacific, NW Rockies, southern Rockies (which split into CO and NM), Intermountain West and finally NEOR. The level of genetic structure is much greater in the west, and may reflect the complex landscape of western North America.
Bayesian analyses comparisons
All Bayesian analyses (STRUCTURE, BAPS and TESS) estimated similar genetic clusters. BAPS failed to separate Newfoundland, or identify substructure in western North America including the differences within the Fraser Plateau and southern Rockies. Although BAPS is computationally more efficient and incorporates the spatial distribution of populations, it struggled to identify key signatures of fine-scale genetic structure. Comparatively, most studies have reported the overestimation of genetic clusters using BAPS (Aspi et al., 2006; Latch et al., 2006) or congruence with STRUCTURE (Canestrelli et al., 2008) rather than the underestimation as found in this study.
Although TESS and STRUCTURE often detect a similar number of genetic clusters (Francois and Durand, 2010), in this study TESS failed to identify the key signatures of genetic differentiation in black-capped chickadees. It did detect the same three genetic clusters (AK, Fraser Plateau and main) as the initial STRUCTURE run when all individuals were included, as well as a fourth cluster involving Newfoundland. This information suggests that when using Bayesian clustering methods to evaluate the spatial genetic structure of organisms, a comparison is essential to detect different levels of population structure and to continue beyond one single run as additional structure can be hidden by noisy data.
Macrogeographic dispersal barriers
A number of prominent landscape features correspond with genetic clusters of black-capped chickadees across North America, including both mountain ranges, particularly in the west, unsuitable habitat in the centre and large water bodies in the east.
In Alaska, a series of three tall mountain ranges (Chugach, Wrangell and Alaska), effectively isolate the three Alaskan black-capped chickadee populations from the rest of their range. Our data support the genetic isolation of the Alaskan populations and confirms previous findings by Pravosudov et al. (2012) and Hindley (2013). Black-capped chickadees in Alaska have larger hippocampus volumes with a subsequent increase in spatial memory and learning capabilities reflecting selective pressures to retrieve cached food items in severe winter climates (Roth and Pravosudov, 2009; Roth et al., 2012). These differences combined with morphological differences support restricted gene flow between Alaska and adjacent populations. Mountains also restrict dispersal in other parts of the chickadee’s range. For example, the Pacific group (WA, CoOR and SOR) and Intermountain West (NEOR, CID and ID), separated by the Cascade Mountains, are genetically distinct (Figures 3 and 4). This pattern is repeated for a number of other populations on either side of the Rocky and Blue Mountains.
Contrary to our earlier prediction, not all mountains are effective dispersal barriers. Populations separated by the northern Rocky Mountains (with the exception of NWBC and BCR) show no evidence of significant population differentiation in either STRUCTURE or FST and Dest comparisons (Figures 2 and 3, Supplementary Table S3). In contrast, populations on either side of the central and southern Rockies are genetically distinct from each other. This was unexpected as the highest tree line elevation; a factor likely to facilitate effective dispersal of forest birds through mountainous valleys and across ranges, actually occurs in the southern Rockies. So although tree line elevation is higher in the American Rockies (3000 m in the eastern Rockies (WY) to 3500 m in the southern Rockies (CO)) than the Canadian Rockies (2400 m) (Körner, 1998), it is possible that lower elevation, treed mountain valleys in the northern Rockies (the lowest elevation being approximately 950 m in comparison with 1500 m in the south) may facilitate dispersal between populations. Overall, mountain topography (particularly elevation) is an effective dispersal barrier to black-capped chickadees and limiting gene flow in the south and has impacted dispersal in a number of organisms such as thin horn sheep (Ovis dalli; Worley et al., 2004). However, mountain ranges are highly heterogeneous environments and low elevation valleys can also increase population connectivity (Pérez-Espona et al., 2008; Hagerty et al., 2010).
Differentiation within the central and southern Rockies cannot solely be explained by contemporary barriers. Historical processes may have also contributed to the genetic structuring in these regions as similar phylogeographic and genetic patterns in north western North America are found in a number of organisms (Avise, 2000). Specifically, the genetic patterns found in our study are concordant with other plant and animal species (Li and Adams, 1989; Nielson et al., 2001; Hindley, 2013). Several hypotheses (such as, biotic distributions, ancient vicariance, dispersal and refugia) have been proposed to explain the genetic concordance observed among diverse taxa (Brunsfeld et al., 2001; Carstens et al., 2005).
Mountain ranges in western North America have undergone a complex history of geological and environmental fluctuations combined with successive glacial–interglacial cycles, which have subsequently influenced ecosystems within and around them. The genetic divergence of coastal (WA, CoOR and SOR) and interior (ID and CID) populations of black-capped chickadees, for both mtDNA and nuclear DNA, may have been influenced by features formed by ‘ancient vicariance’ events such as the uplift of the Cascades combined with the Columbia basin rain shadow; limiting dispersal between these groups (Brunsfeld et al., 2001). The ‘multiple refugia’ hypothesis also helps explain the level of genetic differentiation within the Rocky Mountains (Brunsfeld et al., 2001; Shafer et al., 2010). The Bitterroot crest (located along the north-central Idaho/Montana border) restricts forest connectivity between the eastern and western slopes, and major river canyons have fragmented forest communities throughout the range. In our study, populations in the central/southern Rockies are isolated from each other (for example, CID and ID are differentiated from MT and UT) and from northern populations such as SAB and BCR. This east–west and north–south split is consistent with other studies (Good and Sullivan, 2001) and supports the idea of multiple valley refugia during the Pleistocene.
Black-capped chickadees on Newfoundland are genetically distinct from all continental populations suggesting that large water bodies restrict dispersal. Pairwise FST and Dest values involving NL were all significant (with the exception of MB (n=11)) and relatively high (FST and Dest=0.013 and 0.039 (MB) to 0.108 and 0.221 (PG), respectively, Supplementary Table S3). The Strait of Belle Isle and Cabot Strait have separated Newfoundland from the mainland for approximately 12 000 years (Pielou, 1991). Distances to the mainland are relatively short (18 km to Labrador and 110 km to Nova Scotia); however, oceanic conditions are often harsh. MtDNA data support the presence of genetically distinct groups and show no evidence of maternal gene flow between Newfoundland and continental populations (Gill et al., 1993; Hindley 2013). Large expanses of water are effective barriers to dispersal in a number of other species. Genetically distinct Newfoundland populations have been found in mammals (pine martin Martes americana (McGowan et al., 1999)); plants (red pine Pinus resinosa (Boys et al., 2005)) and other chickadees (boreal chickadee Poecile hudsonicus (Lait and Burg, 2013)) suggesting that long-term isolation of Newfoundland, while not common, is not restricted to black-capped chickadees.
Geographical distance does influence population structuring when distances are measured through suitable habitat. The presence of other dispersal barriers, such as mountains, limits the ability to detect IBD at the rangewide scale using simple straight line distance (McRae, 2006). In the central portion of the black-capped chickadee range lies the Great Plains; a broad expanse of flat land, covered in prairie, steppe and grassland. As a forest-dependent songbird, habitat in this region is unsuitable for dispersal due to lack of trees, necessary for movement. In order for chickadees to move from one side of the Great Plains to the other, they would be required to travel around (through suitable habitat), rather than straight across the unforested landscape. When pairs of populations associated with this region were tested, the effect of geographic distance is clear. Pairwise FST and Dest values are high, and significant, for populations on either side of the Great Plains (Figure 4, Supplementary Table S3). Black-capped chickadee dispersal is therefore limited by geographic distances that are influenced by suitable habitat, which explains why populations to the east of the Great Plains are genetically dissimilar from those to the west.
Population differentiation within continuous habitat
We found additional population structure that cannot be explained by mountain or water barriers. In the southern Rockies, substructuring between CO and NM may reflect large areas of unsuitable habitat in the form of open desert and grassland. A similar pattern was found for the American puma (Puma concolor) across the southwestern US (McRae et al., 2005). Similarly, the unexpected genetic discontinuity of SD and SK, from MB and MO (Figure 3) identified by BARRIER corresponds to the large areas of prairie grasslands (that is, the Great Plains). Although black-capped chickadees are present in the forests surrounding the grasslands, the large geographical distance required to travel in order to circumscribe the unforested area may be impeding movement. Sacks et al. (2004) found that gaps in habitat corresponded to genetically distinct populations in coyotes (Canus latrans). Chestnut-backed chickadees show a similar pattern whereby discontinuities in suitable habitat result in genetically isolated populations (Burg et al., 2005). Animals perceive the landscape at different spatial scales and what appears to be a relatively small break in continuous habitat (for example, 18 km from Newfoundland to Labrador or <10 km between suitable coyote habitat) is perceived by the individual as a large enough risk that dispersal is restricted (Holderegger and Wagner, 2008).
Another population isolated by unsuitable habitat, and mountains, is NEOR which is a genetically isolated island. Within northeastern Oregon, the Blue Mountains stretch from southeastern Washington towards the Snake River along the Oregon-Idaho border and are associated with the Columbia River Plateau, a flood basalt range located between the Cascade and Rocky mountain ranges. Although mountain ranges may be involved in genetic differentiation, it is possible that the high elevation plateau represents a forested island within the Great Basin; a distinctive natural desert region of western North America bordered by the Sierra Nevada on the west, the Wasatch Mountains (UT) on the east, the Columbia Plateau to the north and the Mojave Desert (CA) to the south. With its rugged north–south mountain ranges and deep intervening valleys, combined with the absence of forested communities in lower elevations, the Great Basin isolates NEOR from nearby populations in Oregon, Idaho and all other populations.
The genetic isolation and differentiation of two central British Columbia populations in the Fraser Plateau was unexpected. The closest sampling site to these two populations is ∼188 km away (NBC) and habitat within the region is continuous. In addition, the further genetic differentiation of PG and FtStJ within the Fraser Plateau, supported by a number of analyses, was surprising given the small geographical distance between these populations (straight line distance ∼120 km). It is possible that a recent change to the habitat composition because of forestry practices both between and encircling these two populations could be impeding movement. Logging in this area and the relative size and abundance of cut blocks may be restricting dispersal and gene flow. Approximately 1–18% of the total cut block area is retained, however, a recent biodiversity assessment in British Columbia stated that it would take over 140 years to recruit appropriate habitat and over 200 years to recruit specific old growth stand structure elements such as large trees and snags (Ministry of Forests, Land and Natural Resource Operations, 2012); the latter being suitable breeding habitat for the black-capped chickadee. Alternatively, the outbreak of the mountain pine beetle (Dendroctonus ponderosae) in British Columbia since the 1950s has led to a huge infestation and devastation of black-capped chickadee habitat (Axelson et al., 2009). At least 17 million hectares of mature and old lodgepole pine (Pinus contorta) stands have been infested (Proulx and Kariz, 2005; Petersen and Stuart, 2014) resulting in huge clearcut operations to recover the infested timber. Although, black-capped chickadees are niche generalists, they are forest dependent and so this infestation combined with the removal of infected trees has an indirect effect on breeding and dispersal.
A large number of private alleles present in both PG and FtStJ suggest that additional factors may also explain structuring in this region (such as hybridisation with other chickadees) but a more advanced landscape genetics approach at a smaller geographical scale is necessary to determine the cause of population structuring in this region.
Landscape genetics analyses
GESTE confirms the influence of latitude and longitude and their interaction on population structuring providing additional support to previous analyses. Although all other factors showed no significant influence on genetic differentiation among black-capped chickadee populations, we cannot rule them out as many exhibited similar posterior probabilities. Populations in this study experience a wide range of different climates (Peel et al., 2007). For example, populations located at high elevation and high latitudes experience harsher polar climates in comparison with coastal populations within temperate climates (with increased precipitation) and those in the south, which experience dry arid climates. Climatic differences result in changes to vegetation, including trees. The complex biogeography may allow black-capped chickadees to adapt to their local environment. In addition, populations located close to unsuitable habitat or barriers have fewer dispersal opportunities (Burg et al., 2005). In this study, many groups (for example, the Alaska and Pacific groups) are highly isolated suggesting that interplay between gene flow and local adaption could explain genetic structure among populations but this is beyond the scope of this study. Further research into adaptive traits and/or loci within this species will allow for a more meaningful interpretation.
Nuclear versus mitochondrial DNA patterns
Using mtDNA restriction fragment length polymorphism data, Gill et al. (1993) first explored population differentiation in the black-capped chickadee. Two groups were found with individuals from Newfoundland being genetically distinct from all continental populations (results not shown). More recently, Hindley (2013) identified five groups with mtDNA sequence data; Newfoundland as well as additional structuring of the continental group (Pacific, Alaska, SE Rockies and main Northeast group; Supplementary Figure S4). A number of these groupings using mtDNA are identical to those in our study, although our microsatellite data identified finer scale differences. Pravosudov et al. (2012) identified four groupings with nuclear amplified fragment length polymorphism data collected from only 10 populations, some of which were used in this study (AK, BC, WA, MT and CO; Supplementary Figure S4). Alaska and Washington were both distinct from other populations; BC and MT formed a cluster and there was also an eastern group. Differences such as BC (PG) clustering with MT, and CO with the eastern populations (MN, KS, IA and ME) in their study are not unexpected. Our groupings match some of those identified using the alternative nuclear marker. Although amplified fragment length polymorphism datas show similar levels of differentiation, microsatellites often show higher levels of within-population diversity because of their codominant, multiallelic nature (Marriette et al., 2001), which may have contributed to the higher levels of genetic structure found in our study. In addition, our study included an additional 24 populations. Overall, two identical groups were identified by all recent data sets: Alaska and Pacific. Our microsatellite data also support the presence of a genetically distinct group on Newfoundland as identified by both Hindley (2013) and Gill et al. (1993) suggesting that Newfoundland may have served as a refugium during the Last Glacial Maximum as previously claimed.
Conclusions
Higher levels of genetic differentiation were found in black-capped chickadee populations across North America using microsatellite markers in comparison with previous studies (for example, mtDNA, amplified fragment length polymorphisms and restriction fragment length polymorphisms), illustrating the sensitivity of microsatellites to detect fine-scale genetic structure. Population differentiation was more prominent in the western portion of the black-capped chickadee range and coincided with a number of landscape features such as mountain ranges and habitat discontinuities. Continued isolation may influence evolutionary processes (gene flow and adaptation) in future generations, particularly in a constantly changing environment. This pattern may also be reflected in other resident organisms. Further study is necessary to detect the locations of genetic breaks among subgroups at the microgeographical scale, particularly within the Fraser Plateau, to help identify the corresponding landscape structures or features restricting dispersal and gene flow among these neighbouring populations.
Data archiving
Genotype data available from the Dryad Digital Repository: doi:10.5061/dryad.k086v. Data file contains the raw data of 11 microsatellite loci for the black-capped chickadee across North America.
References
Aspi J, Roininen E, Ruokonen M, Kojola I, Vilà C . (2006). Genetic diversity, population structure, effective population size and demographic history of the Finnish wolf population. Mol Ecol 15: 1561–1576.
Avise JC . (2000). Phylogeography: the history and formation of species. Harvard University Press: Cambridge, MA, USA.
Axelson JN, Alfaro RI, Hawkes BC . (2009). Influence of fire and mountain pine beetle on the dynamics of lodgepole pine stands in British Columbia, Canada. Forest Ecol Manag 257: 1874–1882.
Benjamini Y, Yekutieli D . (2001). The control of false discovery rate under dependency. Ann Stat 29: 1165–1188.
Boys J, Cherry M, Dayanandan S . (2005). Microsatellite analysis reveals genetically distinct populations of red pine (Pinus resinosa, Pinaceae). Am J Bot 92: 833–841.
Brennan LA, Morrison ML . (1991). Long-term trends of chickadee populations in western North America. Condor 93: 130–137.
Brunsfeld SJ, Sullivan D, Soltis E, Soltis PS . (2001). Comparative phylogeography of northwestern North America: a synthesis. In: Silvertown J, Antonovics J, (eds) Integrating Ecology and Evolution in a Spatial Context. Blackwell Publishing: Williston, VT, USA. pp 319–339.
Burg TM, Gaston AJ, Winker K, Friesen VL . (2005). Rapid divergence and postglacial colonization in western North American Steller's jays (Cyanocitta stelleri). Mol Ecol 14: 3745–3755.
Burg TM, Gaston AJ, Winker K, Friesen VL . (2006). Effects of Pleistocene glaciations on population structure of North American chestnut-backed chickadees. Mol Ecol 15: 2409–2419.
Canestrelli D, Cimmaruta R, Nascetti G . (2008). Population genetic structure and diversity of the Apennine endemic stream frog, Rana italica- insights on the Pleistocene evolutionary history of the Italian peninsular biota. Mol Ecol 17: 3856–3872.
Carstens BC, Brunsfeld SJ, Dembroski JR, Good JM, Sullivan J . (2005). Investigating the evolutionary history of the Pacific Northwest mesic forest ecosystems: hypothesis testing within a comparative phylogeographic framework. Evolution 59: 1639–1652.
Carstens BC, Knowles LL . (2007). Shifting distribution and speciation: species divergence during rapid climate change. Mol Ecol 16: 619–627.
Chen C, Durand E, Forbes F, Francois O . (2007). Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes 7: 747–756.
Cooper ST, Swanson DL . (1994). Acclimatization of thermoregulation in the black-capped chickadee. Condor 96: 638–646.
Corander J, Marttinen P, Sirén J, Tang J . (2008). Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinform 9: 539.
Crawford NG . (2010). SMOGD: software for the measurement of genetic diversity. Mol Ecol Res 10: 556–557.
Davidson BS, Sattler GD, Via S, Braun MJ . (2013). Reproductive isolation and cryptic introgression in a sky island enclave of Appalachian birds. Ecol Evol 3: 2485–2496.
Dawson DA, Hanotte O, Greig C, Stewart IAK, Burke T . (2000). Polymorphic microsatellites in the blue tit Parus caeruleus and their cross-species utility in 20 songbird families. Mol Ecol 9: 1941–1944.
Earl DA, vonHoldt BM . (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Res 4: 359–361.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Lischer HEL . (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10: 564–567.
Falush D, Stephens M, Pritchard JK . (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587.
FAO. (2001). Global Forest Resources Assessment 2000. FAO Forestry Paper 140. Rome, Food and Agriculture Organization. http://www.fao.org/forestry/fo/fra/ [Geo-2-402].
Foll M, Gaggiotti O . (2006). Identifying the environmental factors that determine the genetic structure of populations. Genetics 174: 875–891.
Fort KT, Otter K . (2004). Effects of habitat disturbance on reproduction in black-capped chickadees (Poecile atricapillus) in northern British Columbia. The Auk 121: 1070–1080.
François O, Durand E . (2010). Spatially explicit Bayesian clustering models in population genetics. Mol Ecol Res 10: 773–784.
Galbreath KE, Cook JA . (2004). Genetic consequences of Pleistocene glaciations for the tundra vole (Microtus oeconomus) in Beringia. Mol Ecol 13: 135–148.
Gill FB, Mostrom AM, Mack AL . (1993). Speciation in North American chickadees: I. Patterns of mtDNA genetic divergence. Evolution 47: 195–212.
Good JM, Sullivan J . (2001). Phylogeography of the red-tailed chipmunk (Tamis ruficaudus), a northern Rocky Mountain endemic. Mol Ecol 10: 2683–2695.
Goudet J . (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from http://www.unil.ca/izea/softwares/fstat.html. Updated from Goudet (2005)..
Graham BA, Burg TM . (2012). Molecular markers provide insights into contemporary and historic gene flow for a non-migratory species. J Avian Biol 43: 198–214.
Gray LJ . (1989). Response of insectivorous birds to emerging aquatic insects in riparian habitats of a tallgrass prairie stream. Am Midl Nat 129: 288–300.
Grava A, Grava T, Didier R, Lait LA, Dosso J, Koran E et al. (2012). Interspecific dominance relationships and hybridization between black-capped and mountain chickadees. Behav Ecol 23: 566–572.
Guillette LM, Bloomfield LL, Batty ER, Dawson MR, Sturdy CB . (2010). Black-capped (Poecile atricapillus) and mountain chickadee (Poecile gambeli) contact call contains species, sex, and individual identity features. J Acoust Soc Am 127: 1116–1123.
Hagerty BE, Nussear KE, Esque TC, Tracy R . (2010). Making molehills out of mountains: landscape genetics of the Mojave desert tortoise. Landscape Ecol 26: 267–280.
Hansson B, Bensch S, Hasselquist D, Lillandt BG, Wennerberg L, Von Schantz T . (2000). Increase of genetic variation over time in a recently founded population of great reed warblers (Acrocephalus arundinaceus) revealed by microsatellites and DNA fingerprinting. Mol Ecol 9: 1529–1538.
Hawley DM . (2005). Isolation and characterization of eight microsatellite loci from the house finch (Carpodacus mexicanus). Mol Ecol Notes 5: 443–445.
Hewitt GM . (1996). Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linnean Soc 58: 247–276.
Hewitt GM . (2004). Genetic consequences of climatic oscillations in the Quaternary. Philos Trans R Soc Lond B Biol Sci 359: 183–195.
Hindley JA . (2013) Post-Pleistocene dispersal in black-capped (Poecile atricapillus) and mountain (P. gambeli) chickadees, and the effect of social dominance on black-capped chickadee winter resource allocation PhD Thesis University of Lethbridge: AB, Canada.
Hijmans RJ, Guarino L, Mathur P . (2012) DIVA-GIS; Version 7.5 Manual.
Holderegger R, Wagner HH . (2008). Landscape genetics. Bioscience 58: 199–207.
Jost L . (2008). GST and its relatives do not measure differentiation. Mol Ecol 17: 4015–4026.
Körner C . (1998). A re-assessment of high elevation treeline positions and their explanation. Oecologia 115: 445–459.
Lait LA, Burg TM . (2013). When east meets west: population structure of a high-latitude resident species, the boreal chickadee (Poecile hudsonicus). Heredity 111: 321–329.
Lait LA, Lauff RF, Burg TM . (2012). Genetic evidence supports boreal chickadee (Poecile hudsonicus) x black-capped chickadee (Poecile atricapillus) hybridization in Atlantic Canada. Canadian Field-Naturalist 126: 143–147.
Latch EK, Dharmarajan G, Glaubitz JC, Rhodes OE . (2006). Relative performance of Bayesian clustering software for inferring population substructure and individual assignments at low levels of population differentiation. Conserv Genet 7: 295–302.
Li P, Adams WT . (1989). Range-wide patterns of allozyme variation in Douglas-fir (Pseudotsuga menziesii). Can J Forest Research 19: 149–161.
Manel S, Holderegger R . (2013). Ten years of landscape genetics. TREE 28: 614–621.
Manni F, Guérard E, Heyer E . (2004). Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier's algorithm. Hum Biol 76: 173–190.
Mariette S, Changnea D, Zier CLL, Pastuszka P, Raffin A, Plomion C, Kremer A . (2001). Genetic diversity within and among Pinus pinaster populations: comparison between AFLP and microsatellite markers. Heredity 86: 469–479.
McGowan C, Howes LA, Davidson WS . (1999). Genetic analysis of an endangered pine marten (Martes americana) population from Newfoundland using randomly amplified polymorphic DNA markers. Can J Zool 77: 661–666.
McRae BH, Beier P, Dewald E, Huynh Y, Keim P . (2005). Habitat barriers limit gene flow and illuminate historical events in a wide-ranging carnivore, the American puma. Mol Ecol 14: 1965–1977.
McRae B . (2006). Isolation by resistance. Evolution 60: 1551–1561.
Meirmans PG, Hedrick PW . (2011). Assessing population structure: FST and related measures. Mol Ecol Res 11: 5–18.
Ministry of Forests, Land and Natural Resource Operations. (2012) Resource Values Assessment: Biodiversity. Available at: http://www.for.gov.bc.ca/hfp/mountain_pine_beetle/mid-term-timber-supply-project/Biodiversity_summary_june_11.pdf.
Nielson M, Lohman K, Sullivan J . (2001). Phylogeography of the tailed frog (Ascaphus truei): implications for the biogeography of the Pacific Northwest. Evolution 55: 147–160.
Peakall R, Smouse PE . (2012). GenAlEx 6.5: genetic analysis in excel. Population genetic software for teaching and research—an update. Bioinformatics 28: 2537–2539.
Peel MC, Finlayson BL, McMahon TA . (2007). Updated world map of the Köppen-Geiger climate classification. Hydrol Earth Syst Sci 11: 1633–1644.
Pérez-Espona S, Pérez-Barberia FJ, McLeod JE, Jiggins CD, Gordon IJ, Pemberton JM . (2008). Landscape features affect gene flow of Scottish Highland red deer (Cervus elaphus). Mol Ecol 17: 981–996.
Petersen B, Stuart D . (2014). Explanations of a changing landscape: a critical examination of the British Columbia bark beetle epidemic. Environment and Planning A 46: 598–613.
Petren K . (1998). Microsatellite primers from Geospiza fortis and cross-species amplification in Darwin’s finches. Mol Ecol 7: 1782–1784.
Pielou EC . (1991) After the Ice Age: The Return of Life to Glaciated North America. University of Chicago Press: Chicago, IL, USA.
Piertney SB, MacColl AD, Bacon PJ, Dallas JF . (1998). Local genetic structure in red grouse (Lagopus lagopus scoticus): evidence from microsatellite DNA markers. Mol Ecol 7: 1645–1654.
Poláková R, Vyskočilová M, Martin JF, Mays HL, Hill GE, Bryja J et al. (2007). A multiplex set of microsatellite markers for the Scarlet Rosefinch (Carpodacus erythrinus). Mol Ecol Notes 7: 1375–1378.
Pravosudov VV, Roth TC, Forister NL, Ladage LD, Burg TM, Braun MJ et al. (2012). Population genetic structure and its implications for adaptive variation in memory and the hippocampus on a continental scale in food-caching black-capped chickadees. Mol Ecol 21: 4486–4497.
Pravosudov VV, Roth TC, Forister ML, Ladage LD, Kramer R, Schilkey F et al. (2013). Differential hippocampal gene expression is associated with climate-related natural variation in memory and the hippocampus in food-caching chickadees. Mol Ecol 22: 397–408.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Proulx G, Kariz RM . (2005). Winter habitat use by moose, Alces alces, in central interior British Columbia. Can Field-Nat 119: 186–191.
Pulgarín-R PC, Burg TM . (2012). Genetic signals of demographic expansion in downy woodpecker (Picoides pubescens) after the last North American glacial maximum. PLoS ONE 7: e40412.
Raymond M, Rousset F . (1995). GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86: 248–249.
Richardson DS, Jury FL, Dawson DA, Salgueiro P, Komdeur J, Burke T . (2000). Fifty Seychelles warbler (Acrocephalus sechellensis) microsatellite loci polymorphic in Sylviidae species and their cross-species amplification in other passerine birds. Mol Ecol 9: 2226–2231.
Roth TC II, Pravosudov VV . (2009). Hippocampal volumes and neuron numbers increase along a gradient of environmental harshness: a large-scale comparison. Proc R Soc Lond B Biol Sci 276: 401–405.
Roth TC, LaDage LD, Freas CA, Pravosudov VV . (2012). Variation in memory and the hippocampus across populations from different climates: a common garden approach. Proc R Soc Lond B Biol Sci 279: 402–410.
Runde DE, Capen DE . (1987). Characteristics of northern hardwood trees used by cavity-nesting birds. J Wildlife Manage 51: 217–223.
Sacks BN, Browns SK, Ernest HB . (2004). Population structure of California coyotes corresponds to habitat-specific breaks and illuminates species history. Mol Ecol 13: 1265–1275.
Shafer ABA, Cullingham CI, Cote SD, Coltman DW . (2010). Of glaciers and refugia: a decade of study sheds new light on phylogeography of northwestern North America. Mol Ecol 19: 4589–4621.
Smissen PJ, Melville J, Sumner J, Jessop TS . (2013). Mountain barriers and river conduits: phylogeographical structure in a large, mobile lizard (Varanidae: Varanus varius) from eastern Australia. J Biogeogr 40: 1729–1740.
Smith SM . (1993). Black-capped chickadee (Parus atricapillus). In: Poole A, Gill F, (eds) The Birds of North America. The Birds of North America, Inc.: Philadelphia, PA, USA 39.
Steinmeyer C, Mueller JC, Kempanaers B . (2009). Search for information polymorphisms in candidate genes: clock genes and circadian behaviour in blue tits. Genetica 136: 109–117.
Stenzler LM, Fraser R, Lovette IJ . (2011). Isolation and characterization of 12 microsatellite loci from Golden-winged Warblers (Vermivora chrysoptera) with broad cross-taxon utility in emberizine songbirds. Mol Ecol Notes 4: 602–604.
Tarvin KA . (2006). Polymorphic microsatellite loci from the American goldfinch (Carduelis tristis) and their cross-amplification in a variety of passerine species. Mol Evol Notes 6: 470–472.
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P . (2004). MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol 4: 535–538.
Vandergast AG, Perry WM, Lugo RV, Hathaway SA . (2011). Genetic landscapes GIS toolbox: tools to map patterns of genetic divergence and diversity. Mol Ecol Res 11: 158–161.
Walsh PS, Metzger DA, Higuchi R . (1991). Chelex 100 as a medium for simple extraction of DNA for PCR based typing from forensic material. Biotechniques 10: 506–513.
Wellenreuther M, Sánchez-Guillén RA, Cordero-Rivera A, Svensson EI, Hansson B . (2011). Environmental and climatic determinants of molecular diversity and genetic population structure in a coenagionid damselfly. PLoS ONE 6: e20440.
Worley K, Strobeck C, Arthur S, Carey J, Schwantje H, Veitch A et al. (2004). Population genetic structure of North American thinhorn sheep (Ovis dalli). Mol Ecol 13: 2545–2556.
Young A, Boyle T, Brown T . (1996). The population genetic consequences of habitat fragmentation for plants. TREE 11: 413–418.
Acknowledgements
We gratefully thank funding for this project provided by a Natural Science and Engineering Research Council (NSERC) Discovery Grant and Alberta Innovates (AI) New Faculty Award. We also thank J Hindley, L Lait, K Dohms, A Martin, C MacFarlane, K Gohdes, K Nielson and others for helping with sample collection for this project. We are also grateful to the following for providing additional samples to supplement field sampling; Brian Davidson (Smithsonian Museum), Daniel Mennill, Jenn Foote and Laurene Ratcliffe (Queen’s University Biological Station), Steve Van Wilgenburg (CWS Saskatoon), Ken Otter and Angelique Grava (University of Northern British Columbia), North Carolina Museum of Natural Sciences, University of Michigan, Field Museum of Chicago and the Museum of Southwestern Biology at the University of New Mexico. Finally, we would like to thank the anonymous reviewers and editor for their constructive comments.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Adams, R., Burg, T. Influence of ecological and geological features on rangewide patterns of genetic structure in a widespread passerine. Heredity 114, 143–154 (2015). https://doi.org/10.1038/hdy.2014.64
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.64
- Springer Nature Switzerland AG
This article is cited by
-
Using conservation genetics to prioritise management options for an endangered songbird
Heredity (2023)
-
Evidence for adaptive introgression of exons across a hybrid swarm in deer
BMC Evolutionary Biology (2019)
-
Low population genetic differentiation in two Tamarix species (Tamarix austromongolica and Tamarix chinensis) along the Yellow River
Genetica (2019)
-
The influence of latitude, geographic distance, and habitat discontinuities on genetic variation in a high latitude montane species
Scientific Reports (2018)
-
Influence of landscape features on the microgeographic genetic structure of a resident songbird
Heredity (2016)