
Abstract
The Eastern Afromontane cloud forests occur as geographically distinct mountain exclaves. The conditions of these forests range from large to small and from fairly intact to strongly degraded. For this study, we sampled individuals of the forest bird species, the Montane White-eye Zosterops poliogaster from 16 sites and four mountain archipelagos. We analysed 12 polymorphic microsatellites and three phenotypic traits, and calculated Species Distribution Models (SDMs) to project past distributions and predict potential future range shifts under a scenario of climate warming. We found well-supported genetic and morphologic clusters corresponding to the mountain ranges where populations were sampled, with 43% of all alleles being restricted to single mountains. Our data suggest that large-scale and long-term geographic isolation on mountain islands caused genetically and morphologically distinct population clusters in Z. poliogaster. However, major genetic and biometric splits were not correlated to the geographic distances among populations. This heterogeneous pattern can be explained by past climatic shifts, as highlighted by our SDM projections. Anthropogenically fragmented populations showed lower genetic diversity and a lower mean body mass, possibly in response to suboptimal habitat conditions. On the basis of these findings and the results from our SDM analysis we predict further loss of genotypic and phenotypic uniqueness in the wake of climate change, due to the contraction of the species’ climatic niche and subsequent decline in population size.
Similar content being viewed by others


Introduction
Fragmented populations have long been assumed to constitute valid ecological models to predict population trajectories in landscapes subject to rapid anthropogenic habitat fragmentation (MacDougall-Shackleton et al., 2011). Indeed, fragmented populations often have smaller effective population sizes, reduced dispersal rates and lower genetic diversity at the population level, compared with panmictic populations (Frankham, 1997; Blanchet et al., 2010). However, in contrast to populations living in historically stable habitat conditions, species exposed to rapid fragmentation of formerly interconnected habitats are rarely able to adapt to these new environmental conditions, in particular as they are often exposed to simultaneous deterioration of the remaining habitat (Stratford and Robinson, 2005; Keyghobadi, 2007; Walker et al., 2008; Habel and Zachos, 2013). Although demographic and genetic effects may be slowed down or counterbalanced by migration and gene flow (Wright, 1951), population connectivity often rapidly decreases when habitat fragmentation increases (Fahrig, 2003). Ultimately, the combination of habitat loss, deterioration and isolation is expected to modify the ecological conditions for populations, which have been shown to be subject to stochastic demographic fluctuations, increased inbreeding, loss of heterozygosity and accumulation of mildly deleterious alleles. Genetic variation might additionally be lost through strong genetic drift in exceedingly small populations (Frankham, 1995; Higgins and Lynch, 2001; Keller and Waller, 2002; Kalinowski and Waples, 2002; Frankham, 2005; Palstra and Ruzzante, 2008). The direction and strength of these effects, however, are expected to vary with both the (evolutionary) history of the landscape and with species-specific life history traits (Callens et al., 2011; MacDougall-Shackleton et al., 2011).
The Eastern Afromontane (EAM) biodiversity hotspot offers a unique setting to study how long-term natural disjunction and short-term anthropogenic fragmentation may affect populations over different temporal and spatial scales. This biodiversity hotspot region consists of widely scattered but biogeographically similar mountains, running from the Arabic Peninsula to Mozambique and Zimbabwe (White, 1978). Strong isolation caused by orographic heterogeneity and relatively stable climatic conditions have led to an accumulation of endemic species and intraspecific lineages (Bowie et al., 2006; Burgess et al., 2007). Many taxa in the region currently occur as geographically restricted units at single mountains, and, as such, comprise naturally fragmented populations with independent evolutionary trajectories (Measey and Tolley, 2011). In addition to these long-term evolutionary dynamics, plant and animal taxa of the EAM biodiversity hotspot are currently affected by severe loss and degradation of their natural habitats over very short timescales. The EAM forests, in particular, have been subjected to variable anthropogenic effects over the last decades that resulted in a wide range of contemporary forest types, that is, (i) both large and small pristine forests; (ii) historically fragmented forests; and (iii) anthropogenically fragmented and highly degraded forests.
In this study, we analysed genetic variation based on 12 polymorphic microsatellite markers and phenotypic variation based on three morphologic traits of the Mountain White-eye, Zosterops poliogaster, sampled at 16 sites across four mountain blocks. Zosterops poliogaster requires cool and moist climatic conditions and is thus largely restricted to the cloud forests on top of the East African mountains. We sampled populations in each of the above mentioned forest types with contrasting habitat history and current characteristics. To study genetic and phenotypic population effects of rapid habitat change, we sampled and measured both museum specimens and current captures, covering past and recent episodes of forest fragmentation (Pellikka et al., 2009). We further performed Species Distribution Models (SDMs) based on bioclimatic variables to project current and future (2080) climatic niches of Z. poliogaster. By integrating results from genetic, biometric and SDM analyses, we addressed the following research questions:
-
i)
How do long-term and short-term processes of habitat isolation and fragmentation affect the genetic and morphological structure of mountain populations of Z. poliogaster?
-
ii)
How do habitat persistence and rapid habitat change affect the population genetic structure in Z. poliogaster?
-
iii)
How may climate change affect the intraspecific structure and uniqueness of Z. poliogaster in the near future?
Material and methods
Study species
The bird genus Zosterops is known as a ‘great speciator’, showing high levels of genetic and phenotypic differentiation across various geographical ranges (Warren et al., 2006; Moyle et al., 2009; Milá et al., 2010). The East African Mountain White-eye, Z. poliogaster is an omnivorous (mostly insectivorous—nectarivorous) flocking bird species that inhabits moist and cool mountain cloud forests across Kenya, Tanzania, Uganda, Somalia, Ethiopia and Eritrea at an elevation of 1500–2500 m (Mulwa et al., 2007; Redman et al., 2009). Populations at geographically isolated mountains show distinct morphological (for example, plumage colouration; Redman et al., 2009), genetic (Habel et al., 2013) and bioacoustic (Habel et al., 2014) variation, which has resulted in the recognition of different taxonomic entities (Borghesio and Laiolo, 2004; Mulwa et al., 2007; Del Hoyo et al., 2008).
Sampling scheme
Between 1990 and 2011, a total of 390 individuals from 16 populations were sampled with mist nets in four mountain ranges across the Kenyan section of the EAM: (i) Mt. Kulal (2 °69′ N; 36 °94′ E; ∼2000 m) and Mt. Kasigau (−3 °82′ N; 38 °65′ E; ∼1500 m) still harbour pristine, interconnected and intact cloud forest, in which 46 and 22 individuals were sampled, respectively; (ii) In the Chyulu Hills (−2 °66′ N; 37 °87′ E; ∼2200 m), cloud forests constitute a forest-grassland mosaic that has at least persisted since the Massai pastoralists caused regular fires many hundreds of years ago. A total of 52 individuals were sampled in two forest fragments located at the northern- and southernmost edges of the mountain range; (iii) In the Taita Hills (−3 °41′ N; 38 °30′E; ∼1800 m), continuous cloud forest has been transformed into isolated and degraded forest remnants mainly due to small-scale subsistence agriculture, with a particularly marked loss, deterioration and isolation of the remaining forest cover since the 1960s (Pellikka et al., 2009). Here, a total of 270 individuals were sampled in 11 forest fragments located along the entire higher elevational range.
Upon capture, each individual was banded with a unique aluminium ring from the East African Ringing Scheme. After measuring its wing length (mm), tarsus length (mm) and body mass (g), a blood sample (stored in pure ethanol and then frozen at −20 °C) or feather sample was collected. To allow analysis of temporal variation, field samples collected during 1990, 1997 and 2000 (Ngangao 1990: N=28; Mbololo 1990: N=27; Mbololo 2000: N=14; Mt. Kulal 1997: N=25) were complemented with 17 museum specimens collected in Chyulu Hills during 1938 (National Museums of Kenya, Nairobi). From these museum specimens, DNA was extracted from blood, feathers or toe pads. The sampling location of each population is shown in Figure 1; further details on locations, sample sizes and genetic parameters are provided in Table 1.

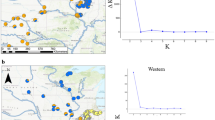
Overview of the 16 sampling locations of Zosterops poliogaster (a). The inset (b) details the Taita Hills sampling sites. 1: Mt. Kasigau; the nine forest fragments from the Dabida mountain block, including 2: TH-Chawia, 3: TH-Fururu, 4: TH-Macha, 5: TH-Mwachora, 6: TH-Ndiwenyi, 7: TH-Ngangao, 8: TH-Vuria, 9: TH-Wundanyi, 10: TH-Yale; and the two forest fragments from the MBololo massif, with 11: TH-Ronge, 12: TH-MBololo; 13: CH-Satellite, 14: CH-Simba valley, 15: Chyulu Hills-1938; 16: Mt. Kulal. Numbers of sampling sites and names of localities coincide with Table 1.CH, Chyulu Hills; TH, Taita Hills.
Genetic analysis
DNA was extracted using the Qiagen DNeasy Tissue Extraction Kit (Hilden, Germany) following the manufacturer’s protocol for blood and toe pads or a user-developed one for feathers (see De Volo et al., 2008). Microsatellite loci were amplified using Thermozyme Mastermix (Molzym, Bremen, Germany). The PCR products were visualised with an automated sequencer (Beckmann Coulter, California, CA, USA). Details of primer-specific PCR conditions and multiplex-assignments are given in Habel et al. (2013). The following 12 microsatellite primers were genotyped: Cu28, LZ44, LZ41, LZ22, LZ45, LZ14, LZ54, LZ35, Mme12, LZ18, LZ50 and LZ2. The forward primer of each pair was 5′-labelled with the fluorescent dyes BMN-6 or CY5. Allele sizes were scored against the internal standard ROX-400SD using GENEMAPPER 3.5 (Applied Biosystems, Grand Island, NY, USA).
Statistics on genetic data
We used the program MICROCHECKER 2.2.3 (Van Oosterhout et al., 2004) to test for patterns indicating stutter bands, large allele dropout or null alleles (Selkoe and Toonen, 2006). The mean number of alleles per population (A), allelic richness (AR) and locus-specific allele frequencies were calculated with FSTAT 2.9.3.2 (Goudet, 1995). Calculations of observed (Ho) and expected (He) heterozygosity, tests for Hardy–Weinberg equilibrium and linkage disequilibrium were calculated with the program ARLEQUIN 3.1 (Excoffier et al., 2005). Recent changes in effective population size were analysed with BOTTLENECK 1.2.02 (Cornuet and Luikart, 1996).
Hierarchical and non-hierarchical genetic variance analysis (analyses on molecular variance) were performed with ARLEQUIN 3.1 (Excoffier et al., 2005). Analyses on molecular variance were computed using the microsatellite-specific R-statistics (Slatkin, 1995; Selkoe and Toonen, 2006). The most probable number of genetic clusters without a priori definition of groups was inferred with STRUCTURE 3.1 (Hubisz et al., 2009). The batch run function was applied to carry out a total of 100 runs (10 each for one to ten clusters), that is, K=1 to K=10. Replicate runs allowed us to calculate mean and s.d. for fixed K-values. For each run, burn-in and simulation lengths were 150 000 and 500 000, respectively. As log probability values for K-values were earlier shown to be unreliable in some cases (Evanno et al., 2005), we calculated the more refined ad hoc statistic ΔK, based on the rate of change in the log probability of data between successive K-values. Mantel tests were applied to test for correlations between genetic and geographic distances at a distributional (after merging individuals within each mountain massif and creating four groups, Mt. Kasigau, Taita Hills, Chyulu Hills and Mt. Kulal) and regional (across all 11 Taita populations) scale. Matrices of genetic distances between populations were calculated based on Cavalli-Sforza and Edwards, 1967 distances and as pairwise Rst using ARLEQUIN 3.1. A total of 5000 permutations were performed to infer levels of statistical significance.
To assess the level and direction of gene flow at the same two geographic scales, we estimated the proportion of non-migrants and the source of migrants for each population by using a Markov chain Monte Carlo (MCMC) algorithm in BAYESASS 1.3 (Wilson and Rannala, 2003). We performed 9 × 106 iterations with 3 × 106 iterations discarded as burn-in. Delta values of m=0.30, P=0.15, and F=0.15 yielded an average number of changes within the accepted range (Wilson and Rannala, 2003).
Phenotypic analyses
We measured wing length (mm), tarsus length (mm) and body mass (g) to test for potential phenotypic differentiation among regional population clusters. We tested for autocorrelation among the three characters using a MANCOVA. The obtained residuals were used for subsequent analyses to adjust for potential allometry. As no significant autocorrelation was detected, differences in morphometric characters between populations were analysed by orthogonal squares analysis of variance in combination with post-hoc Tukey tests. We used principal component analysis to reduce the data complexity and to determine traits for which populations were significantly diverged.
Species distribution modelling
Geo-referenced species records were compiled from own field data supplemented with data from specimens housed in the collections of the National Museums of Kenya (NMK, Nairobi, Kenya), the Zoological Research Museum Alexander Koenig (ZFMK, Bonn, Germany), the Zoological Museum Kopenhagen (ZMUC, Denmark), and records from the Global Biodiversity Information Facility (GBIF), as well as from various publications (Zimmerman et al., 1996; Borghesio and Laiolo, 2004; Mulwa et al., 2007; Redman et al., 2009). Confounding effects resulting from spatial autocorrelation were minimised by randomly selecting one species record per 10 arc min grid cell (that is, spatially filtering 195 records). Current local climatic conditions were inferred from the WORLDCLIM 1.4 database with a spatial resolution of 2.5 arc min (Hijmans et al., 2005). Expected climatic conditions for 2080 were derived from four global circulation models (CCCMA-CGCM2, CISRO-MK2, HCCPR HADCM3, NIES99). A2a and B2a scenarios developed by the Inter-governmental Panel on Climate Change (IPCC et al., 2007) were spatially downscaled to 2.5 arc min (Ramirez and Jarvis, 2008). As each climate data set comprised 19 bioclimatic variables (Busby, 1991), we used pairwise squared Pearson’s correlation coefficients to quantify multi-colinearity (Heikkinen et al., 2006). In case of R2>0.75, one variable of each pair that was considered biologically most relevant was retained for species distribution modelling. This procedure resulted in 10 bioclimatic variables: ‘annual mean temperature’ (bio1), ‘isothermality’ (bio3), ‘temperature seasonality’ (bio4), ‘temperature annual range’ (bio7), ‘annual precipitation’ (bio12), ‘precipitation of the wettest month’ (bio13), ‘precipitation of the driest month’ (bio14), ‘precipitation seasonality’ (bio15), ‘precipitation of the warmest quarter’ (bio18) and ‘precipitation of the coldest quarter’ (bio19).
SDMs were computed with MAXENT 3.3.3k, applying default settings and a logistic output format (Phillips et al., 2006; Phillips and Dudík, 2008; Elith et al., 2011) and using a training area enclosed by a 100-km buffer around the species records (see recommendation by Mateo et al. (2010)). To evaluate SDMs through the area under the receiver operating characteristic curve (Swets, 1988), a total of 100 SDMs were computed, each trained with 70% of the species records and tested with the remaining 30%. On the basis of output of these replicate runs, average prediction scores were computed for current and future bioclimatic conditions as suggested by each of the global circulation models. We selected the minimum MAXENT score at a 5% sample omission rate as presence/absence threshold. Non-analogous climatic conditions within projection areas exceeding those that a SDM was trained for, may reduce the reliability of predictions (Fitzpatrick and Hargrove, 2009; Elith et al., 2010; Rocchini et al., 2011). This potential source of uncertainty was quantified in a spatially explicit way in each scenario by using multivariate environmental similarity surfaces (Elith et al., 2010).
Results
Genetic diversity
The following locality × loci combinations showed significant deviations from Hardy–Weinberg equilibrium due to the presence of null alleles: Chyulu-1938: ZL22; Fururu: ZL44; Ndiwenyi: ZL14, ZL54, Mme12; Ngangao: ZL22, ZL14; Mbololo: ZL14. Samples from Mt. Kulal showed no deviations from Hardy–Weinberg equilibrium, and for Ronge we did not obtain valid results due to small sample sizes. After Bonferroni correction for multiple testing, linkage disequilibrium was not significant in any pair of loci. Locus- and locality-specific allele frequencies are listed in Supplementary Appendix S1. Microsatellite DNA polymorphisms ranged between 2 (ZL41, Mme12, Cu28) and 12 (Zl49) alleles per locus, with a mean of 4.7±2.3 alleles per locus and a total of 61 alleles across all loci. Allele sizes did not significantly differ between recent and historic samples (sampling years are given in the list of allele frequencies in Supplementary Appendix S1). A total of 26 out of 61 alleles (43%) were restricted to single mountains. The proportions of private alleles per population are listed in Table 1.
Mean numbers of alleles (A), allelic richness (AR), proportions of private alleles (AP) and percentages of expected (He) and observed (Ho) heterozygosity did not significantly differ at regional level among the mountain populations (Kruskal–Wallis analysis of variance, all P>0.05). Current and historic estimates (inferred from museum specimens collected in the same forests) did not significantly differ for the following pairwise comparisons in the Taita Hills (Ngangao 1990–2009, Mbololo 1990–2009, Mbololo 2000–2009), Chyulu Hills (1938 to current) and Mt. Kulal (1997–2010) (paired comparisons per fragment including time intervals as co-variable, population-wise U-tests: all P>0.05). The following populations from the Taita Hills showed a significant excess of homozygotes (tested overall loci): Vuria, Ngangao, Ndiwenyi, Chawia, Fururu, Macha (all P<0.05). Population- and mountain-specific values of genetic diversity are listed in Table 1.
Genetic population structuring
The best supported model assigned individuals to two genetic clusters (K=2): a first cluster comprising individuals from the Taita Hills and Mt. Kasigau, and a second one comprising individuals from Chyulu Hills and Mt. Kulal. While this model effectively yielded the strongest statistical support (Figure 2), Hausdorf and Hennig (2010) caution against the validation of clustering genotypes in only few groups. The second best model assigned individuals to seven genetic clusters, thereby (i) splitting individuals from the Taita Hills and Mt. Kasigau, (ii) lumping individuals from the Chyulu Hills and Mt. Kulal and (iii) assigning individuals from the Taita Hills to a very heterogeneous cluster (Figure 2). ΔK-values of all different models are listed in Supplementary Appendix S2.

Bayesian structure analyses of populations from Z. poliogaster performed with STRUCTURE (Hubisz et al., 2009), for K=1–10. Results supported by highest ΔK-values (K=2 and K=7) are presented, distinguishing the mountain population of Mt. Kasigau and Taita Hills (TH), while Chyulu Hills (CH) cluster together with Mt. Kulal. Names of populations coincide with other figures and tables.
At a regional scale, the level of genetic variance among Z. poliogaster populations from Mt. Kasigau, Taita Hills, Chyulu Hills and Mt. Kulal was 11.1878 (RST: 0.5488, P<0.001). Hierarchical variance analysis assigned the strongest genetic split between the Chyulu Hills and the neighbouring Taita Hills (<100 km geographic distance) with a genetic variance of 14.6971 (RCT: 0.6245, P<0.001), while the remote Mt. Kulal and Chyulu populations (>600 km geographic distance) showed comparatively poor genetic differentiation (10.9803, RCT: 0.5079, P<0.001). Within the Taita Hills, the 11 remnant populations showed significant genetic differentiation (0.6885, RST: 0.0757, P<0.001), partitioned over two spatial scales, that is, between the two main mountain isolates Dabida (including forest fragment 2–10) and the Mbololo massif (including the forest fragment 11–12) (0.7642, RCT: 0.0795, P<0.05) and within Dabida (0.4684, RST: 0.0504, P<0.001). In contrast, the southern- and northernmost populations of the Chyulu Hills were not genetically differentiated (−0.0179, RST: −0.0026, P>0.05) despite comparable geographic distances (see Figure 1). The level of genetic differentiation between the two populations from the forest fragments Ngangao and Mbololo increased over a 19-year period from nonsignificant in 1990 to highly significant in 2009 (1990: 0.0044, RCT: 0.0006, P>0.05; 2009: 0.6871, RCT: 0.0930, P<0.001). All values are listed in Table 2.
Mantel tests did not yield significant correlations between genetic and geographic distances at regional (among four mountain groups, Mt. Kasigau, Taita Hills, Chyulu Hills and Mt. Kulal; r=0.32, P=0.13) and local (11 Taita Hills forest fragments; r=−0.20, P=0.90) scales. Results obtained from the program BayesAss underline this lack of spatial differentiation; overall rates of genetic exchange among the four Z. poliogaster mountain groups were low (see Table 3A), and the detected gene flow among the 11 Taita Hills populations was weak. The small Mwachora and Ndiwenyi populations thereby seemed to act as source populations (see Table 3B).
Phenotypic structures
Principal component analysis revealed a morphological split between populations from the Taita Hills and those from the Chyulu Hills and Mt. Kulal (Figure 3a), while individuals from Mt. Kasigau took an intermediate position. Taita individuals were significantly smaller and lighter than individuals from Mt Kulal and the Chyulu Hills (analysis of variance: Tukey post-hoc comparisons P<0.01), while individuals from Mt Kasigau showed intermediate values. Populations from different mountains did not significantly differ in morphology (t-test: P>0.1). A principal component analysis bi-plot showed a strong congruence between morphological and genetic differentiation at a regional level, that is, separating the Chyulu Hills and Mt. Kulal populations from those of the Taita Hills and Mt. Kasigau (Figure 3b). Population mean and s.d. of all phenotypic data are listed in Supplementary Appendix S3.

(a) Principal component analysis based on three biometric characters segregates the individuals from the Taita Hills (open triangles) from Chyulu Hills (Simba and Satellite) (open dots) and the individuals from Mt. Kulal (open squares). Individuals from Mt. Kasigau (black dots) score intermediate between both groups. Axis 1 correlates highly with body mass (r2=0.54, P<0.01). (b) A principal component analysis bi-plot for average morphological (black dots) and genetic RST distances (open circles) segregates populations from Chyulu Hills (Simba, Satellite) and Mt. Kulal from the populations from Taita Hills and Mt. Kasigau.
Species distribution modelling
On the basis of 100 replicates, average training and testing area under the receiver operating characteristic curve scores were 0.932 and 0.871, respectively. Variable bio1 (‘annual mean temperature’) (35.0%) contributed most strongly to the SDMs, followed by bio18 (‘precipitation of the warmest quarter’) (16.9%), bio19 ‘precipitation of the coldest quarter’ (10.1%), bio14 (8.3%), bio3 (8.2%), bio15 (5.6%) and bio7 (5.3%). All other variables contributed <5% on average.
The current potential distribution of Z. poliogaster covers major parts of the East African highlands such as Pare Mts. Mt. Meru, Kilimanjaro, Taita Hills, Chyulu Hills, Central Kenyan Highlands, Cherangani Hills and the northern Kenyan Highlands (Mt. Kulal and Mt. Nyiru) (Figure 4). When projecting the SDMs to future climate change scenarios A2a and B2a as proposed by IPCC et al. (2007) for 2080, a severe retraction in the distribution of the species climatic niche is projected. Particularly strong effects are expected near the southern distribution edge (Eastern Arc Mountains), but also across parts of the Central Kenyan Highlands and northern Kenya. Suitable climatic exclaves might remain in some areas of the Pare and Usambara Mountains, parts of the Central Kenyan Highlands and restricted areas in the Ethiopian highlands (Figure 4).

Projections of the distribution of Zosterops poliogaster from present to future. Warmer colours indicate a higher environmental suitability for the species, unsuitable areas are indicated in light grey. Darker areas indicate areas with non-analogues climatic conditions relative to the training area of the SDMs as identified by multivariate environmental similarity surface analyses. (Upper right) Currently realised and potential distribution of Z. poliogaster in East Africa, as well as average projections of its currently realised niche and future anthropogenic climate change scenarios provided by the fourth IPCC Assessment (A2a and B2a) for 2080. Species records are indicated by black crosses; light grey areas are suggested to be unsuitable; dark grey shading indicates extrapolation of the SDM outside the environmental training range as quantified by multivariate environmental similarity surfaces.
Discussion
Populations of Z. poliogaster are genetically and phenotypically differentiated over both, regional and local scales. Morphological and genetic data indicate that distinct mountain-specific clusters evolved independently from each other. We found no correlation between genetic and geographic distance suggesting that geographic isolation is not a main driver of differentiation in this species. On a regional level (here the Taita Hills forest archipelago), we found significant genetic differentiation even among neighbouring populations (Dabida and Mbololo mountains). Temporal analyses point towards an increase in genetic differentiation over time between the Taita Hills populations, whereas the adjoining Chyulu Hills populations do not show such pattern.
Effects of long-term disjunction
On the basis of our analyses, we identify two major genetic and morphometric clusters within the studied region. A first cluster comprises the two southernmost mountain populations, Mt. Kasigau and the Taita Hills that are separated by 60 km of dry savannah. A second cluster comprises both populations of the Chyulu Hills (60 km distant from the Taita Hills) and the one from Mt. Kulal, located 600 km north (Figure 3b). This intraspecific clustering may reflect the geological history of the mountain massifs: Mt. Kasigau and the Taita Hills represent the northernmost edge of the Eastern Arc Mts., which evolved very early during the Precambrian (White, 1978). In contrast, the other two mountain massifs, Chyulu Hills and Mt. Kulal are geologically much younger (Dimitrov et al., 2012). The influence of these contrasting geological ages on evolutionary processes have been demonstrated for other species, with deep inter- and intraspecific splits detected in taxa inhabiting the Eastern Arc Mts. (Bowie et al., 2006; Fuchs et al., 2011; Tolley et al., 2011; Dimitrov et al., 2012). Yet, to formally test this hypothesis in the genus Zosterops, phylogenetic analyses and robust estimates of divergence times between mountain-specific lineages are required.
A concurrent explanation for the observed within-taxon differentiation is based on climatic fluctuations of the last thousands of years (including the glacial–interglacial cycles). The last glaciation ended about 10 000 years ago and caused major forest contractions. During such glaciation, many forest species most probably expanded their ranges and colonised isolated forest massifs (Hamilton, 1982). The obtained differentiation pattern and the lack of an isolation-by-distance further indicate that the geographic distance is not the most important force driving the genetic and morphological differentiation of these populations.
In addition to this major genetic split, we detected significant genetic differentiation within mountain massifs, for example, between populations from two Taita Hills isolates (Mbololo and Dabida) that are separated by a small valley only (Callens et al., 2011). These findings underline the specific environmental demands (moist and cool climatic conditions, see SDM results) and resulting strong geographical restriction of Z. poliogaster to higher elevations (Mulwa et al., 2007; Redman et al., 2009).
Habitat persistence and habitat change
The restriction of gene flow among remnant populations after the break down of landscape connectivity is a commonly observed phenomenon (Knutsen et al., 2000). However, only few studies used historical data to empirically assess the impact of rapid landscape changes on the intraspecific structure of populations. Temporal comparisons of the genetic structure of populations from individuals sampled in the Taita Hills over a 19-year period indicate a significant temporal increase in genetic differentiation. This is likely the result of severe habitat fragmentation, which has taken place in the Taita Hills during the past decades (Pellika et al., 2009).
Several studies have addressed the negative effects of rapid ecosystem change on the viability of populations (Fahrig, 2003), also in the EAM. For instance, Lens et al. (1999) showed that the deviation of bilateral symmetries (i.e., fluctuating asymmetry) in Z. poliogaster (and six sympatric forest bird species) were four to seven times higher for birds in the smallest, most degraded fragments if compared with populations with still intact and large forest. Increased levels of fluctuation asymmetry have been correlated with reductions in growth rates and competitive ability in a range of organisms, as well as with reduced survival probabilities in the critically threatened Taita thrush (Turdus helleri). In the latter species, Galbusera et al. (2000) also reported severe levels of genetic drift in small, degraded populations. A comparable pattern as found for the Mountain White-eye was observed in the Spanish imperial eagle (Aquila adalberti) of which the historic panmictic population network recently collapsed into a scatter of isolated remnant populations (Martinez-Cruz et al., 2007). The fact that Z. poliogaster individuals from the anthropogenically fragmented forest patches of the Taita Hills have a lighter weight might be a plastic response to the lower habitat quality. In contrast, populations from more undisturbed fragments, where habitat conditions remained fairly stable during the recent past, such as in the Chylu Hills, showed higher levels of genetic diversity and weaker genetic differentiation.
Such contrasting intraspecific signatures of habitat subdivision in two adjoining mountain massifs might be explained by two different scenarios. First, while the Taita Hills forest experienced rapid habitat fragmentation and degradation, the Chyulu Hills are covered by forest-grassland mosaics that persisted during the past hundreds of years. While the current degree of forest fragmentation in the Chyulu and Taita Hills is roughly comparable, both areas hence underwent fundamentally different habitat histories. The coinciding patterns of genetic differentiation and habitat history suggest that populations affected from fast habitat change (for example, the Taita Hills) might suffer severely, whereas populations living in a rather stable environment (for example, the Chyulu Hills) are less affected. Thus, fragmented habitats must not always affect species negatively (Vucetich et al., 2001; Habel and Schmitt, 2012; Habel and Zachos, 2013).
Climate warming threats intraspecific diversity
Our biometric and genetic analyses suggest a high level of intraspecific uniqueness for single-mountain areas. In total, 43% of all detected alleles are geographically restricted to single-mountain massifs. This high level of variability and intraspecific endemicity are threatened by a combination of direct (see above) and indirect factors. Strong demographic pressure across major parts of Africa and the increasing need for agricultural products and thus agricultural land lead to increased deforestation (Pellika et al., 2009). In addition, our SDM projections highlight the relevance of cool and moist climatic conditions for this species (the factor ‘annual mean temperature’ contributes 35.0%, ‘precipitation of the warmest quarter’ contributes 16.9% and ‘precipitation of the coldest quarter’ contributes 10.1% to the explanatory power of the suitability of habitats for Z. poliogaster). These models further suggest that climate change will cause warmer and drier conditions leading to a decline of suitable habitats for Z. poliogaster (Figure 4). This might lead to extinction of local populations and result in the loss of intraspecific uniqueness.
Data archiving
Data available from the Dryad Digital Repository: doi:10.5061/dryad.5ms6g.
References
Blanchet S, Rey O, Etienne R, Lek S, Loot G . (2010). Species-specific responses to landscape fragmentation: implications for management strategies. Evol Appl 3: 291–304.
Borghesio L, Laiolo P . (2004). Habitat use and feeding ecology of Kulal white-eye Zosterops (poliogaster) kulalensis. Bird Conserv Int 14: 11–24.
Bowie RCK, Fjeldså J, Hackett SJ, Bates JM, Crowe TM . (2006). Coalescent models reveal the relative roles of ancestral polymorphism, vicariance, and dispersal in shaping phylogeographical structure of an African montane forest robin. Mol Phyl Evol 38: 171–188.
Burgess ND, Butynski TM, Cordeiro NJ, Doggart NH, Fjeldså J, Howell KM et al. (2007). The biological importance of the Eastern Arc Mts. of Tanzania and Kenya. Biol Conserv 34: 209–231.
Busby JR . (1991). BIOCLIM - a bioclimatic analysis and prediction system. In Margules CR, Austin MP (eds) Nature Conservation: Cost Effective Biological Surveys and Data Analysis. CSIRO: Melbourne, Australia. pp 64–68.
Callens T, Galbusera P, Matthysen E, Durand EY, Githiru M, Huyghe JR et al. (2011). Genetic signature of population fragmentation varies with mobility in seven bird species of a fragmented Kenyan cloud forest. Mol Ecol 20: 1829–1844.
Cavalli-Sforza LL, Edwards AWF . (1967). Phylogenetic analysis: models and estimation procedures. Evolution 21: 550–570.
Cornuet J-M, Luikart G . (1996). Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144: 2001–2014.
Del Hoyo J, Elliott A, Christie D . (2008). Handbook of the Birds of the World. Penduline-tits to Shrikes Vol. 13, Lynx Edicions: Barcelona, Spain.
De Volo SB, Reynolds RT, Doublas MR, Antolin MF . (2008). An improved extraction method to increase DNA yield from molted feathers. Condor 110: 762–766.
Dimitrov D, Nogués-Bravo D, Scharff N . (2012). Why do tropical mountains support exceptionally high biodiversity? The Eastern Arc Mountains and the drivers of Saintpaulia diversity. PLoS One 7: e48908.
Elith J, Kearney M, Phillips S . (2010). The art of modelling range-shifting species. Methods in Ecol Evol 1: 330–342.
Elith J, Phillips SJ, Hastie T, Dudík M, Chee YE, Yates CJ . (2011). A statistical explanation of MaxEnt for ecologists. Div Distr 17: 43–57.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Laval G, Schneider S . (2005). Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinf Online 1: 47–50.
Fahrig L . (2003). Effects of habitat fragmentation on biodiversity. Ann Rev Ecol Evol Sys 34: 487–515.
Fitzpatrick MC, Hargrove WW . (2009). The projection of species distribution models and the problem of non-analog climate. Biodiv Conserv 18: 2255–2261.
Frankham R . (1995). Effective population size/adult population size ratios in wildlife: a review. Genet Res 2: 91–107.
Frankham R . (1997). Do island populations have less genetic variation than mainland populations? Heredity 78: 311–327.
Frankham R . (2005). Genetics and extinction. Biol Conserv 126: 131–140.
Fuchs J, Fjeldså j, Bowie RCK . (2011). Diversification across an altitudinal gradient in the Tiny Greenbul (Phyllastrephus debilis) from the Eastern Arc Mountains of Africa. BMC Evol Biol 11: 117.
Galbusera P, Lens L, Schenck T, Waiyaki E, Matthysen E . (2000). Genetic variability and gene flow in the globally critically endangered Taita thrush. Conserv Genet 1: 45–55.
Goudet J . (1995). FSTAT (Version 1.2): a computer program to calculate F-statistics. Heredity 86: 485–486.
Habel JC, Cox S, Gassert F, Mulwa RK, Meyer J, Lens L . (2013). Population genetics of four East African Mountain White-eye congeners on land- and seascape. Conserv Genet 14: 1019–1028.
Habel JC, Schmitt T . (2012). The burden of genetic diversity. Biol Conserv 147: 270–274.
Habel JC, Ulrich W, Peters G, Husemann M, Lens L . (2014). Lowland panmixia versus highland disjunction: genetic and bioacoustic differentiation in two species of East African White-eye birds. Conserv Genet (e-pub ahead of print 2 February 2014; doi:10.1007/s10592-014-0567-2).
Habel JC, Zachos FE . (2013). Past population history versus recent population decline – founder effects in island species and their genetic signatures. J Biogeogr 40: 206–207.
Hamilton AC . (1982) Environmental History of East Africa: a Study of the Quaternary. Academic Press: London, UK.
Hausdorf B, Hennig C . (2010). Species delimitation using dominant and codominant multilocus markers. Sys Biol 59: 491–503.
Heikkinen RK, Luoto M, Araújo MB, Virkkala R, Thuiller W, Sykes MT . (2006). Methods and uncertainties in bioclimatic envelope modeling under climate change. Progress Phys Geogr 30: 751–777.
Higgins K, Lynch M . (2001). Metapopulation extinction caused by mutation accumulation. Proc Nat Acad Sci 98: 2928–2933.
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A . (2005). Very high resolution interpolated climate surfaces for global land areas. Int J Climat 25: 1965–1978.
Hubisz MJ, Falush D, Stephens M, Pritchard JK . (2009). Inferring weak population structure with the assitance of sample group information. Mol Ecol Res 9: 1322–1332.
IPCC. (2007) Summary for Policymakers. In: Solomon SD, Qin D, Manning M, Chen Z, Marquis M, Averyt KB et al. (eds). Climate Change. 2007: The Physical Science Basis. Contribution of Working Group I to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change. Cambridge University Press, Cambridge, United Kingdom and New York, NY, USA. pp 18.
Kalinowski TS, Waples RS . (2002). Relationship of effective to census size in fluctuating populations. Conserv Biol 16: 129–136.
Keller LF, Waller DM . (2002). Inbreeding effects in wild populations. Trends Ecol Evol 17: 230–241.
Keyghobadi N . (2007). The genetic implications of habitat fragmentation for animals. Canad J Zool 85: 1048–1064.
Knutsen JH, Rukke BA, Jorde PE, Ims RA . (2000). Genetic differentiation among populations of the beetle Bolitophagus reticulates (Coleoptera: Tenebrionidae) in a fragmented and a continuous landscape. Heredity 84: 667–676.
Lens L, van Dongen S, Wilder C, Brooks T, Matthysen E . (1999). Fluctuating asymmetry increases with habitat disturbance in seven species of a fragmented afrotropical forest. Proc Roy Soc Lond B 266: 1241–1246.
MacDougall-Shackleton E, Clinchy M, Zanett L, Neff BD . (2011). Songbird genetic diversity is lower in anthropogenically versus naturally fragmented landscapes. Conserv Genet 12: 1195–1203.
Martinez-Cruz B, Godoy JA, Negro JJ . (2007). Population fragmentation leads to spatial and temporal genetic structure in the endangered Spanish imperial eagle. Mol Ecol 16: 477–486.
Mateo RG, Croat TB, Felicísimo ÁM, Munoz J . (2010). Profile or group discriminative techniques? Generating reliable species distribution models using pseudo-absences and target-group absences from natural history collections. Div Distr 16: 84–94.
Measey GJ, Tolley KA . (2011). Sequential fragmentation of Pleistocene forests in an East Africa biodiversity hotspot: chameleons as a model to track forest history. PLoS One 6: e26606.
Milá B, Warren BH, Heeb P, Thébaud C . (2010). The geographic scale of diversification on islands: genetic and morphological divergence at a very small spatial scale in the Mascarene grey white-eye (Aves: Zosterops borbonicus). BMC Evol Biol 10: 158.
Moyle RG, Filardi CE, Smith CE, Diamond J . (2009). Explosive Pleistocene diversification and hemispheric expansion of a “great speciator”. Proc Nat Acad Sci USA 106: 1863–1868.
Mulwa RK, Bennun LA, Ogol CPK, Lens L . (2007). Population status and distribution of Taita White-eye Zosterops silvanus in the fragmented forests of Taita Hills and Mount Kasigau, Kenya. Bird Conserv Int 17: 141–150.
Palstra FP, Ruzzante DE . (2008). Genetic estimates of contemporary effective population size: what can they tell us about the importance of genetic stochasticity for wild population persistence? Mol Ecol 17: 3428–3447.
Pellikka PKE, Lötjönen M, Siljander M, Lens L . (2009). Airborne remote sensing of spatiotemporal change (1955–2004) in indigenous and exotic forest cover in the Taita Hills, Kenya. Int J Appl Earth Observ Geoinf 11: 221–232.
Phillips SJ, Anderson RP, Schapire RE . (2006). Maximum entropy modeling of species geographic distributions. Ecol Mod 190: 231–259.
Phillips SJ, Dudík M . (2008). Modeling of species distributions with Maxent: new extensions and a comprehensive evaluation. Ecography 31: 161–175.
Ramirez J, Jarvis A . (2008). High resolution statistically downscaled future climate surfaces. International Centre for Tropical Agriculture (CIAT), Cali, Colombia. Available from: http://gisweb.ciat.cgiar.org/GCMPage (Accessed on 16.7.2012)..
Redman N, Stevenson T, Fanshawe J . (2009) Birds of the Horn of Africa: Ethiopia, Eritrea, Djibouti, Somalia, Socotra. Helm Field Guides, A and C Black Publisher Ltd: London, UK.
Rocchini D, Hortal J, Lengyel S, Lobo JM, Jimenez-Valverde A, Ricotta C et al. (2011). Accounting for uncertainty when mapping species distributions: the need for maps of ignorance. Progr Phys Geogr 35: 211–226.
Selkoe T, Toonen RJ . (2006). Microsatellites for ecologists: a pratical guide to using and evaluating microsatellite markers. Ecol Lett 9: 615–629.
Slatkin MA . (1995). Measure of population subdivision based on microsatellite allele frequencies. Genetics 139: 457–462.
Stratford JA, Robinson WD . (2005). Gulliver travels to the fragmented tropics: geographic variation in mechanisms of avian extinction. Front Ecol Env 3: 85–92.
Swets K . (1988). Measuring the accuracy of diagnostic systems. Science 240: 1285–1293.
Tolley KA, Tilbury CR, Measey GJ, Menegon M, Branch WR, Matthee CA . (2011). Ancient forest fragmentation or recent radiation? Testing refugial speciation models in chameleons within an African biodiversity hotspot. J Biogeogr 38: 1748–1760.
Van Oosterhout C, Hutchinson W, Wills DPM, Shipley P . (2004). MICRO-CHECKER (Version 2.2.3): software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol 4: 535–538.
Vucetich LM, Vucetich JA, Joshi CP, Waite TA, Peterson RO . (2001). Genetic (RAPD) diversity in Peromyscus maniculatus populations in a naturally fragmented landscape. Mol Ecol 10: 35–40.
Walker FM, Sunnucks P, Taylor AC . (2008). Evidence for habitat fragmentation altering within-population processes in wombats. Mol Ecol 17: 1674–1684.
Warren BH, Bermingham E, Prys-Jones R, Thebaud C . (2006). Immigration, species radiation and extinction in a highly diverse songbird lineage: White-eyes on Indian Ocean islands. Mol Ecol 15: 3769–3786.
White F . (1978). The afromontane region. in: Werger MJA (ed) Biogeography and Ecology of Southern Africa. Junk Publishers: The Hague, pp, 463–513.
Wilson GA, Rannala B . (2003). Bayesian inference of recent migration rates using multilocus genotypes. Genetics 163: 1177–1191.
Wright S . (1951). The genetical structure of populations. Ann Eugen 15: 323–354.
Zimmermann DA, Turner DA, Person DJ . (1996) Birds of Kenya and Northern Tanzania. Christopher Helm: London, UK.
Acknowledgements
The research was financed by the German Academic Exchange Service (DAAD) and the Natural History Museum Luxembourg (MNHN Luxembourg). We thank Titus Imboma, Onesmus Kioko (Nairobi, Kenya) and Dirk Louy, Thomas Schmitt and Sönke Twietmeyer (Trier, Germany) for field assistance. We thank Hans Matheve (Ghent, Belgium) and two anonymous referees for improving a draft version of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article
Cite this article
Habel, J., Mulwa, R., Gassert, F. et al. Population signatures of large-scale, long-term disjunction and small-scale, short-term habitat fragmentation in an Afromontane forest bird. Heredity 113, 205–214 (2014). https://doi.org/10.1038/hdy.2014.15
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2014.15
- Springer Nature Switzerland AG