
Abstract
Human serum albumin (HSA): (i) controls the plasma oncotic pressure, (ii) modulates fluid distribution between the body compartments, (iii) represents the depot and carrier of endogenous and exogenous compounds, (iv) increases the apparent solubility and lifetime of hydrophobic compounds, (v) affects pharmacokinetics of many drugs, (vi) inactivates toxic compounds, (vii) induces chemical modifications of some ligands, (viii) displays antioxidant properties, and (ix) shows enzymatic properties. Under physiological and pathological conditions, HSA has a pivotal role in heme scavenging transferring the metal-macrocycle from high- and low-density lipoproteins to hemopexin, thus acquiring globin-like reactivity. Here, the heme-based catalytic properties of HSA are reviewed and the structural bases of drug-dependent allosteric regulation are highlighted.
Similar content being viewed by others

Facts
-
Human serum albumin (HSA) is the most abundant protein in plasma.
-
HSA binds endogenous and exogenous ligands at multiple sites.
-
HSA has a pivotal role in heme scavenging.
-
Human serum heme-albumin (HSA-heme) displays metal-based multienzymatic properties.
-
Heme-based catalytic properties of HSA are modulated allosterically by endogenous and exogenous ligands, including drugs, representing a pivotal issue in the pharmacological therapy management.
Open questions
-
In spite of the multifaced roles of HSA, humans almost totally lacking this protein safely survive. Are HSA actions essential?
-
α-Fetoprotein, the fetal homolog of HSA, is recognized by its specific receptor. Does a specific receptor also occur for human serum albumin?
-
HSA-heme may represent a heme-based enzyme and a blood substitute.
HSA, the most represented plasma protein (~7.5×10−4 M), (i) modulates the plasma oncotic pressure, (ii) regulates fluid distribution between the body compartments, (iii) represents a depot and a transporter of endogenous and exogenous compounds (e.g., fatty acids (FA), metal ions, drugs, hormones, toxins and metabolites), (iv) increases the apparent solubility and lifetime of hydrophobic compounds, (v) affects pharmacokinetics of many drugs, (vi) induces chemical modifications of some ligands (e.g., drugs), (vii) inactivates some toxic compounds, (viii) displays antioxidant properties, and (ix) shows enzymatic properties.1,2
Under physiological and pathological conditions, HSA has a pivotal role in heme scavenging.3,4 In fact, although heme regulates gene expression and is the prosthetic group of heme proteins under physiological conditions, high levels of free heme: (i) catalyze the synthesis of toxic free hydroxyl radicals, (ii) affect the integrity of erythrocyte membranes, (iii) induce the enrollment to the vascular endothelium of red blood cells, platelets, and leukocytes, and (iv) cause the oxidation of low-density lipoproteins.5–9 HSA takes out heme from high- and low-density lipoproteins and transfers the metal-macrocycle to hemopexin. After endocytosis of the hemopexin-heme complex into the hepatic parenchymal cells through the CD91 receptor, hemopexin releases heme intracellularly. After heme delivery, hemopexin is released intact into the bloodstream, and the heme is degraded.3,4,10 Therefore, HSA acquires time-dependent globin-like properties, representing a case for ‘chronosteric effects’.2,11 Interestingly, heme-based catalytic properties of HSA are modulated allosterically by drugs, representing a pivotal issue in the pharmacological therapy management.2
Here, the heme-based catalytic properties of HSA are reviewed and the structural bases of drug-dependent allosteric regulation are highlighted.
The HSA structural organization
HSA is an all-α-helical protein consisting of three structurally similar domains usually indicated as I (amino acids 1–195), II (amino acids 196–383), and III (amino acids 384–585). The three domains of HSA assemble asymmetrically and resemble a heart shape. Each domain includes 10 α-helices that are packed in two separate subdomains (named A and B) comprising six (h1–h6) and four (h7–h10) α-helices, respectively; the subdomains are connected by a long extended loop. Terminal regions of the sequential domains contribute to the formation of interdomain regions, 9-turn-long α-helices link domain IB to IIA (residues 173–205) and IIB to IIIA (residues 336–398). The HSA fold is stabilized by 17 intrasubdomain disulfide bridges.2
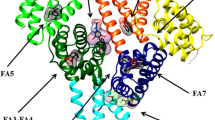
The multidomain organization of HSA is at the root of its extraordinary ligand-binding ability and capacity.1,2,12 The most relevant clefts hosting ligands (e.g., heme, FAs, bilirubin, and drugs) are the so-called FA binding sites (named FA1 to FA9). Bacterial protein recognition cleft(s), thyroxine-binding sites, and metal ion recognition sites also participate to HSA actions.1,2,12–16 (Figure 1).

Three-dimensional structure of HSA complexed with endogenous and exogenous ligands bound to the FA sites. The subdomains of HSA are rendered with different colors (domain IA, in blue; domain IB, in cyan; domain IIA, in forest green; domain IIA, in green; domain IIIA, in yellow; domain IIIB, in orange). The FA1-salicylic acid (PDB ID: 2I30),36 F2-capric acid (PDB ID: 1E7E),17 FA3-FA4-ibuprofen (PDB ID: 2BXG),15 FA5-propofol (PDB ID: 1E7A),35 FA6-halothane (PDB ID: 1E7C),35 FA7-warfarin (PDB ID: 2BXD),15 F8-capric acid (PDB ID: 1E7E),17 and FA9-thyroxine (PDB ID: 1HK4)39 complexes are highlighted. Three molecules of halothane are bound in the FA6 site. The picture has been drawn with the UCSF Chimera package.94,95
The FA1 binding site (located in subdomain IB) binds very different ligands (e.g., heme, bilirubin, FAs, and exogenous compounds) and has been reported to represent a major drug-binding pocket. Upon ligand binding to the FA1 site, a conformational change(s) propagates from subdomain IB to subdomains IA, IIA, and IIB.15,17–20 Heme binds to HSA by the IA–IB loop. In particular, Tyr138 and Tyr161 form π–π stacking interactions with the macrocycle ring, the Tyr161 side chain being coordinated to the heme-Fe atom by the phenoxyl O atom, and the Arg114, His146, and Lys190 residues being involved in salt bridges with the heme propionates.21 In contrast, bilirubin binding to FA1 does not need π–π stacking by Tyr138 and Tyr161 for ligand recognition.22 The FA carboxylate head-group is hydrogen bonded to Arg117 and to a water molecule, which is in turn stabilized by hydrogen bonding to the phenoxyl and carbonyl O atoms of Tyr161 and Leu182, respectively.17 Lastly, Tyr138 and Tyr161 have a key role also in drug recognition.18–20
The FA2 site (located at the interface between subdomains IA, IB, and IIA) has been postulated to represent a secondary recognition cleft of ibuprofen and warfarin.2,17,23–26 The FA carboxylate head-group is bound to subdomain IIA through Tyr150, Arg257, and Ser287, whereas the methylene tail is placed in the hydrophobic cleft between subdomains IA and IIA.17 FAs binding to FA2 stabilize the B-conformation of HSA and promote the ligand-induced conformational transition(s).2,17,23–26
The FA3–FA4 cleft (located in subdomain IIIA) is also named Sudlow’s site II and represents a major drug-binding pocket. The FA3–FA4 cleft binds preferentially aromatic carboxylates showing an extended conformation, with ibuprofen representing the prototypical ligand.2,15,27–34 Drugs (e.g., ibuprofen) cluster in the center of the FA3–FA4 cleft, interacting with the hydroxyl group of Tyr411 and forming salt bridges and hydrogen bond interactions with Arg410 and Ser489 residues. The FA carboxylate head-group binds to Ser342 and Arg348 placed in the subdomain IIB and to the Arg485 side chain sited in the subdomain IIIA. In the FA4 site, the FA carboxylate head-group is bound to Arg410, Tyr411, and Ser489 placed externally in the subdomain IIIA, whereas the hydrophobic tail is accommodated into the subdomain IIIA.15,17,34
The FA5 site (positioned in subdomain IIIB) accommodates FAs whose carboxylate head-groups bind to Tyr401 and Lys525.2,15,17,31,35
The FA6 site (placed between subdomains IIA and IIB) represents the secondary binding site of diflunisal, halothane, and ibuprofen.2,15,35 The FA carboxylate head-group binds to Arg209, Lys351, and Ser480, whereas the methylene tail contacts Arg209, Asp324, and Glu354.17,36
The FA7 site (placed in subdomain IIA) is also named Sudlow’s site I and represents a major drug-binding pocket. This pocket binds preferentially heterocyclic compounds, with warfarin representing the prototypical ligand.1,2,15 The FA7 site is also the primary binding site for 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid, azapropazone, di-iodosalicylic acid, indomethacin, iodipamide, oxyphenbutazone, phenylbutazone, and tri-iodobenzoic acid, and represents the secondary binding cleft for diflunisal and indoxyl sulfate.15 The FA7 cleft also houses one aspirin or two salicylic acid molecules.36 Drugs (e.g., warfarin) cluster in the center of the FA7 site, having a planar group pinned snugly between the apolar side chains of Leu238 and Ala291 and making a hydrogen bond interaction with the hydroxyl group of Tyr150. Moreover, warfarin forms hydrogen bonds with His242, and with either Lys199 or Arg222. The FA7 site recognizes FAs by salt bridging the FA carboxylate head-group to Arg257.15,17,31,34,37 Lastly, HSA can covalently bind different FA7 ligands because of the presence of Lys199, which can act as a nucleophile.38
FA8, taking place upon the FA-induced conformational transition(s), is located at the base of the gap between subdomains IA, IB, and IIA on one side and subdomains IIB, IIIA, and IIIB on the other side. Owing to volume restrictions, FA8 only binds short-chain FAs. The Lys195, Lys199, Arg218, Asp451, and Ser454 residues form an open ring that stabilizes the FA carboxylate head-group.17
FA9, occurring upon the FA-induced conformational transition(s), lies in an upper region of the interdomain region paved by subdomains IA, IB, and IIA on one side and subdomains IIB, IIIA and IIIB on the other side. A salt bridge between Glu187 of domain I and Lys432 of domain III contributes to keep the FA in place.17,39
Binding of the thyroid hormone T4 to HSA is modulated allosterically. In FA-free HSA, the thyroid hormone T4 binds to four sites (i.e., Tr-1 to Tr-4). In particular, the Tr-1 site (corresponding to the FA7 site) is positioned in the subdomain IIA, the Tr-2 site (overlapping the FA3–FA4 cleft) is located in the subdomain IIIA, and the Tr-3 and Tr-4 sites (matching the FA5 site) are placed in the subdomain IIIB. Of note, two T4 molecules are accommodated in the FA5 pocket. Although in the presence of myristate binding of the thyroid hormone T4 to the Tr-1 to Tr-4 sites of HSA is impaired, this FA induces conformational change(s) leading to the opening of the fifth T4-binding pocket (i.e., Tr-5), which is located between domains I and III and partially corresponds to the FA9 site. Remarkably, the Arg218 mutation within subdomain IIA greatly enhances the affinity of HSA for the thyroid hormone T4 causing the increase of the serum thyroxine level, which is associated with familial dysalbuminemic hyperthyroxinemia.39
HSA binds several endogenous and exogenous proteins,40 including the protein-G-related albumin-binding module of the anaerobic bacterium Finegoldia magna poly(A)-binding protein in the proximity of the FA6 site.41,42 This protein provides selective advantages to the bacterium, delivering FAs and other nutrients transported by HSA.41,43
HSA displays a wide variety of binding sites for several metal ions, including Mg(II), Al(III), Ca(II), Mn(II), Co(II/III), Ni(II), Cu(I/II), Zn(II), Cd(II), Pt(II), Au(I/II), Hg(II), and Tb(III).1,16,44–47 Three major binding sites endowed with appropriate residues matching for the different metal ligand fields have been observed.16 The first site (called the N-terminal binding site) is located at the N-terminus, where Cu(II), Co(II), and Ni(II) are coordinated by nitrogen donor atoms from Asp1, Ala2, and His3.48 The second site is represented by the free Cys34 thiol that binds Au(I), Hg(II), and Pt(II) ions.1,49 The third metal-binding site (called the primary multimetal binding site or cadmium site A) involves His67, Asn99, His247, and Asp249 residues. Owing to its pseudo-octahedral geometry, this site stably allocates different metal ions, representing the primary cleft for Zn(II) and Cd(II) and the secondary site of Cu(II) and Ni(II).50 The secondary site for Cd(II) binding (called as secondary multimetal binding site or cadmium site B) has not been firmly identified.51
Allosteric modulation by drugs of heme-based catalytic properties of HSA
The heme-based catalytic properties of HSA are strictly dependent on the molar fraction of HSA-heme present in the plasma. In fact, because of the low plasma level of HSA-heme in healthy subjects (1.5×10−6 M), the heme-based catalytic properties of HSA appear to be relevant, especially in patients affected with hematologic diseases showing high intravascular hemolysis and high HSA-heme plasmatic levels (reaching 5×10−5 M).25,52 The heme-based catalytic properties of HSA are modulated allosterically by drugs, and, according to linked functions,53 the oxidation state and the axial coordination of the heme-Fe atom affect drug recognition.2
Peroxynitrite scavenging by HSA-heme-Fe(II)-NO
Peroxynitrite reacts with HSA-heme-Fe(II)-NO leading to HSA-heme-Fe(III) and NO, according to Scheme 1.54
In the absence and presence of CO2, values of kon for peroxynitrite scavenging by ferrous nitrosylated HSA-heme, horse heart myoglobin, and human hemoglobin are grossly similar; however, they are lower compared with those reported for ferrous nitrosylated rabbit hemopexin-heme and human neuroglobin by about one order of magnitude (Table 1).54–57 Remarkably, abacavir promotes allosterically peroxynitrite scavenging by HSA-heme-Fe(II)-NO; in fact, the kon value increases from 6.5×103/M/s, in the absence of the drug, to 2.2×104/M/s, in the presence of saturating amounts of abacavir.54 In addition, CO2 accelerates peroxynitrite scavenging by ferrous nitrosylated heme proteins mediated by about one order of magnitude (Table 1).54–57 This reflects the transient formation of the highly reactive CO3●− species following the reaction of peroxynitrite with CO2.58–60 In particular, the value of kon for peroxynitrite detoxification by ferrous nitrosylated HSA-heme increases from 6.5×103/M/s, in the absence of CO2, to 1.3×105/M/s, in the presence of CO2.54 Moreover, abacavir and CO2 cooperate in peroxynitrite scavenging by HSA-heme-Fe(II)-NO, with the value of kon rising to 3.6×105/M/s.54
In the absence and presence of CO2, the rate-limiting step of peroxynitrite detoxification by ferrous nitrosylated heme proteins is represented by NO dissociation from the heme(III)-NO adduct. Moreover, values of hoff are CO2-independent, spanning over two orders of magnitude; of note, the value of hoff for the denitrosylation of the HSA-heme-Fe(III)-NO complex (1.8×10−1/s) is unaffected by abacavir (Table 1).54–57
As a whole, peroxynitrite scavenging with the concomitant release of NO may switch peroxynitrite signaling to NO-based strategies opening new avenues in cell metabolism.
Peroxynitrite detoxification by HSA-heme-Fe(III)
HSA-heme-Fe(III) catalyzes peroxynitrite detoxification according to Scheme 2.24,61–63
Values of lon for peroxynitrite detoxification by ferric mammalian heme proteins range from 1.2×104 to 4.3×105/M/s (Table 2).24,64–67 Peroxynitrite detoxification by HSA-heme-Fe(III) prevents free L-Tyr nitration, possibly displaying a protective role in vivo. Chlorpropamide, digitoxin, furosemide, ibuprofen, imatinib, indomethacin, isoniazid, phenylbutazone, rifampicin, sulfisoxazole, tolbutamide, and warfarin impair allosterically peroxynitrite isomerization in a dose-dependent manner. Indeed, the value of kon for peroxynitrite detoxification by HSA-heme-Fe(III) decreases from 4.3×105/M/s, in the absence of drugs, to 3.5×104/M/s, in the presence of saturating amounts of chlorpropamide, digitoxin, furosemide, ibuprofen, imatinib, indomethacin, isoniazid, phenylbutazone, rifampicin, sulfisoxazole, tolbutamide, and warfarin. The drug-dependent allosteric inhibition of peroxynitrite detoxification by HSA-heme-Fe(III) promotes free L-Tyr nitration, thus modulating protein actions, structure, and metabolism.24,61–63 However, peroxynitrite detoxification by ferric mammalian heme proteins is unaffected by CO2, which in turn facilitates the spontaneous isomerization of peroxynitrite to the harmless NO3−.24,64–67
Interestingly, ibuprofen affects to the same extent peroxynitrite detoxification by both full-length HSA-heme-Fe(III) and truncated HSA-heme-Fe(III) devoided of domain III (Table 2), indicating that domains I and II form the allosteric core of HSA.67
As a whole, the drug-dependent modulation of peroxynitrite detoxification catalyzed by HSA-heme-Fe(III) may affect indirectly signaling pathways involving NO and O2●−,24,61–63 which are the precursors of peroxynitrite.59
The HSA-heme-Fe(II)-catalyzed conversion of NO2− to NO
HSA-heme-Fe(II) catalyzes the conversion of NO2− to NO under anaerobic conditions, according to Scheme 3.68
Values of bon for NO2− conversion to NO2− by mammalian heme proteins range between 1.2×10−2 and 6.0/M/s (Table 3), reflecting the different structural and chemical features of the heme site.68–73 In particular, the low bon values for the conversion of NO2− to NO by ferrous human cytoglobin could reflect the hexa coordination of the heme-Fe(II) atom.73 Moreover, the reactivity of ferrous human neuroglobin reflects the reversible redox-linked hexa- to penta-coordinate transition of the heme Fe(II) atom. In fact, under oxidative conditions, the formation of the Cys46–Cys55 disulfide bridge stabilizes the highly reactive penta-coordinate heme-Fe(II) atom facilitating the reaction, whereas under reductive conditions the cleavage of the Cys46–Cys55 bridge leads to the formation of the low-reactivity hexa-coordinate heme-Fe(II) atom.72 Furthermore, the ferrous HSA-heme-Fe(II)- and human hemoglobin-catalyzed conversion of NO2− to NO is impaired allosterically by warfarin and inositol hexakisphosphate, respectively.68–70 In particular, the bon value for the ferrous HSA-heme-Fe(II)-catalyzed conversion of NO2− to NO decreases from 1.3/M/s, in the absence of warfarin, to 0.1/M/s at saturating drug concentration.68
The nitrite reductase activity of mammalian heme proteins may have a relevant role in acidosis and anoxia; in fact, bon increases on pH decrease and on NO2− concentration increase, under anaerobic conditions.68–73
O2-based scavenging of NO-bound HSA-heme-Fe(II)
HSA-heme-Fe(II)-NO is irreversibly oxidized by O2 with the concomitant production of harmless NO3−, according to Scheme 4.74
The NO dissociation rate constant (i.e., doff ) represents the rate-limiting step of the O2-mediated oxidation of HSA-heme-Fe(II)-NO.74 Remarkably, this reaction is modulated allosterically by rifampicin, which accelerates the dissociation of NO. In fact, the value of doff increases from 9.8×10−5/s, in the absence of the drug, to 8.8×10−4/s, in the presence of saturating amounts of the drug.74 This behavior is reminiscent of that reported for the O2-mediated oxidation of ferrous nitrosylated human hemoglobin, wherefore inositol hexakisphosphate affects the reaction by shifting the quaternary equilibrium; consequently, values of doff increase from 3.2×10−4 and 7.0×10−5/s in the R-state to 2.5×10−3 and 9.8×10−5/s in the T-state.75 In contrast, O2 reacts directly with ferrous nitrosylated horse heart myoglobin and rabbit hemopexin-heme, and this leads to the formation of the transient peroxynitrite-bound ferric heme protein species, which precedes the appearance of the final products, that are, ferric heme protein and nitrate.74,76,77 Lastly, a slight rearrangement within the protein structure, which may take place after the formation of ferric human neuroglobin, has been postulated to be the rate-limiting step in O2-mediated oxidation of ferrous nitrosylated neuroglobin.56
Catalase and peroxidase activity of HSA-heme-Fe(III)
HSA-heme-Fe(III) exhibits catalase and peroxidase activity in the oxidation of phenolic compounds related to Tyr (i.e., p-cresol, 3-(p-hydroxyphenyl)propionic acid, tyramine, and tyrosine) and 2,20-azinobis(3-ethylbenzothiazoline-6-sulfonate) according to Scheme 5.78,79 In Scheme 5, HSA-heme-Fe(III)* is the active HSA-heme-Fe(III), S is the substrate, HSA-heme-Fe(III)-S is the HSA-heme-Fe(III) substrate adduct, HSA-heme-Fe(III)*-S is the active HSA-heme-Fe(III) substrate adduct, P is the product, f1 is the rate constant for the formation of active species, f1′ is the rate constant for the transformation of HSA-heme-Fe(III)-S into HSA-heme-Fe(III)*-S, fp is the rate constant for product formation, fc and fc′ are the rate constants for the decomposition of peroxide by HSA-heme-Fe(III)-S and HSA-heme-Fe(III)*-S, respectively, and Fb and Fb′ are the dissociation equilibrium constants for substrate (i.e., S) binding to HSA-heme-Fe(III) and HSA-heme-Fe(III)*, respectively.
In the minimum reaction mechanism depicted by Scheme 5, the binding processes were considered fast with respect to the reaction processes. Values of f1 and of the fp/fc′ ratio, which rule the competition between catalase and peroxidase activity of HSA-heme-Fe(III), range between 4.1×101 and 1.6×102/M/s, and between 5.3×10−3 and 6.3×10−2 M, respectively, depending on the substrate.78,79
The catalytic mechanism of HSA-heme-Fe(III) appears to be different from that of peroxidases. In fact, the rate of formation of the active HSA-heme-Fe(III) species (i.e., HSA-heme-Fe(III)*) is lower compared with its reaction rate with either the substrate (in a peroxidase reaction) or the peroxide (in a catalase reaction) or the heme-Fe(III) itself. The weak catalase and peroxidase activity of HSA-heme-Fe(III) reflects the reduced accessibility of the heme-Fe(III) center, and the lack of an Arg residue in the HSA-heme-Fe(III) pocket that in peroxidases assists the cleavage of bound peroxide and accelerates the formation of the active species.78
Structural bases of HSA allostery
The three-domain structure of HSA is at the root of its allosteric features, which are associated with pH changes, as well as with ligand binding. It is worthy noting that the allosteric modulation of HSA reactivity by drugs is relevant in vivo and represents a pivotal issue in the pharmacological therapy management.2
Domains I and II form the allosteric core of HSA, with the FA1, FA2, FA6, and FA7 sites being functionally linked. In fact, the functional properties of wild-type HSA are superimposable to those of truncated HSA, which contains only domains I and II.67,80 Moreover, contacts between subdomains IA and IIA have been reported to be pivotal in the allosteric modulation of HSA actions.81 Therefore, the C-terminal domain III of HSA, containing the FA3–FA4 and the FA5 sites, has a minor role in the allosteric modulation of ligand-binding and reactivity properties.2,67,80,82–84 However, (i) FAs modulate allosterically and competitively ligand binding to HSA involving domains I, II, and III85, and (ii) the allosteric linkage between the FA3–FA4 cleft (in domain III) and the FA7 site (in domain II) affects benzodiazepines, warfarin, and phenprocoumon enantiomers binding.86,87
Drugs, including ibuprofen and warfarin, modulate allosterically the catalytic properties of HSA-heme affecting the coordination state of the heme-Fe atom. The catalytically active species of HSA-heme display a four- or five-coordinated heme-Fe atom, whereas the HSA-heme-inactive form shows a six-coordinated heme-Fe atom. In particular, Tyr161 coordinates the heme-Fe atom in the five-coordinated species, whereas Tyr161 and His146 are the fifth and sixth coordination ligands of the metal center in the six-coordinated species. Upon drug binding to HSA-heme (most probably to the FA2 site, the sole binding pocket contacting domains I and II), the reorientation of the Glu131-Arg145 α-helix and the axial coordination of the heme-Fe atom by His146 occur; as a consequence, the unreactive six-coordinated HSA-heme species becomes predominant. It is worth noting that the sixth His-Fe coordination bond of six-coordinated HSA-heme is ~2.15 Å in agreement to that observed in heme proteins2,23,24,26,61–63,67,81,88–92 (Figure 2).

Drug-dependent six coordination of the heme-Fe atom of HSA-heme. Superposition of the crystal structure of HSA-heme-Fe(III) (light green and forest green, PDB entry 1O9X)91 and of the binary drug-bound HSA-heme-Fe(III) complex (hot pink and red).26 Heme-Fe(III), His146 and Tyr161 are highlighted. The picture has been drawn using the UCSF Chimera package.94,95
Conclusion
Several pathological conditions such as hematological diseases are characterized by altered heme plasma levels, which in turn switch HSA to HSA-heme and induce heme-based catalysis; this is particularly relevant considering the detoxification role of HSA-heme. Moreover, the heme-based catalytic properties of HSA are modulated allosterically by drugs. In turn, the oxidation state and the axial coordination of the heme-Fe atom affect drug recognition. This represents a pivotal issue in the pharmacological therapy management; in fact, drug association could inhibit allosterically the heme-based catalytic properties of HSA-heme (e.g., the detoxification of reactive nitrogen and oxygen species). Lastly, the heme-based catalytic properties of HSA are time-dependent reflecting the heme shuttling by HSA from low- and high-density lipoproteins to hemopexin, thus representing a case of ‘chronosteric effects’.

Peroxynitrite scavenging by HSA-heme-Fe(II)-NO.

Peroxynitrite detoxification by HSA-heme-Fe(III)

The HSA-heme-Fe(II)-catalyzed conversion of NO2– to NO.

O2-based scavenging of NO-bound HSA-heme-Fe(II).

Catalase and peroxidase activity of HSA-heme-Fe(III).
Abbreviations
- FA:
-
fatty acid
- HSA:
-
human serum albumin
- HSA-heme:
-
human serum heme-albumin.
References
Peters T Jr . All About Albumin: Biochemistry, Genetics and Medical Applications. Academic Press: San Diego, CA, USA and London, UK, 1996.
Fanali G, di Masi A, Trezza V, Marino M, Fasano M, Ascenzi P . Human serum albumin: from bench to bedside. Mol Aspects Med 2012; 33: 209–290.
Miller YI, Shaklai N . Kinetics of hemin distribution in plasma reveals its role in lipoprotein oxidation. Biochim Biophys Acta 1999; 1454: 153–164.
Ascenzi P, Bocedi A, Visca P, Altruda F, Tolosano E, Beringhelli T et al. Hemoglobin and heme scavenging. IUBMB Life 2005; 57: 749–759.
Ryter SW, Tyrrell RM . The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic Biol Med 2000; 28: 289–309.
Jeney V, Balla J, Yachie A, Varga Z, Vercellotti GM, Eaton JW et al. Pro-oxidant and cytotoxic effects of circulating heme. Blood 2002; 100: 879–887.
Kumar S, Bandyopadhyay U . Free heme toxicity and its detoxification systems in human. Toxicol Lett 2005; 157: 175–188.
Balla J, Vercellotti GM, Jeney V, Yachie A, Varga Z, Jacob HS et al. Heme, heme oxygenase, and ferritin: how the vascular endothelium survives (and dies) in an iron-rich environment. Antioxid Redox Signal 2007; 9: 2119–2137.
Tolosano E, Fagoone S, Morello N, Vinchi F, Fiorito V . Heme scavenging and the other facets of hemopexin. Antioxid Redox Signal 2010; 12: 305–320.
di Masi A, Leboffe L, Trezza V, Fanali G, Coletta M, Fasano M et al. Drugs modulate allosterically heme-Fe-recognition by human serum albumin and heme-Fe-mediated reactivity. Curr Pharm Des 2015; 21: 1837–1847.
Ascenzi P, Gianni S . Functional role of transient conformations: rediscovering 'chronosteric effects' thirty years later. IUBMB Life 2013; 65: 836–844.
Fasano M, Curry S, Terreno E, Galliano M, Fanali G, Narciso P et al. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005; 57: 787–796.
Curry S, Mandelkow H, Brick P, Franks N . Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat Struct Biol 1998; 5: 827–835.
Sugio S, Kashima A, Mochizuki S, Noda M, Kobayashi K . Crystal structure of human serum albumin at 2.5 Å resolution. Protein Eng 1999; 12: 439–446.
Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S . Structural basis of the drug-binding specificity of human serum albumin. J Mol Biol 2005; 353: 38–52.
Fanali G, Cao Y, Ascenzi P, Fasano M . Mn(II) binding to human serum albumin: a 1H-NMR relaxometric study. J Inorg Biochem 2012; 117: 198–203.
Bhattacharya AA, Grüne T, Curry S . Crystallographic analysis reveals common modes of binding of medium and long-chain fatty acids to human serum albumin. J Mol Biol 2000; 303: 721–732.
Wang ZM, Ho JX, Ruble JR, Rose J, Rüker F, Ellenburg M et al. Structural studies of several clinically important oncology drugs in complex with human serum albumin. Biochim Biophys Acta 2013; 1830: 5356–5374.
Zsila F, Subdomain IB . is the third major drug binding region of human serum albumin: toward the three-sites model. Mol Pharm 2013; 10: 1668–1682.
Zsila F . Circular dichroism spectroscopic detection of ligand binding induced subdomain IB specific structural adjustment of human serum albumin. J Phys Chem B 2013; 117: 10798–10806.
Wardell M, Wang Z, Ho JX, Robert J, Ruker F, Ruble J et al. The atomic structure of human methemalbumin at 1.9 Å. Biochem Biophys Res Commun 2002; 291: 813–819.
Zunszain PA, Ghuman J, McDonagh AF, Curry S . Crystallographic analysis of human serum albumin complexed with 4Z,15E-bilirubin-IXalpha. J Mol Biol 2008; 381: 394–406.
Nicoletti FP, Howes BD, Fittipaldi M, Fanali G, Fasano M, Ascenzi P et al. Ibuprofen induces an allosteric conformational transition in the heme complex of human serum albumin with significant effects on heme ligation. J Am Chem Soc 2008; 130: 11677–11688.
Ascenzi P, di Masi A, Coletta M, Ciaccio C, Fanali G, Nicoletti FP et al. Ibuprofen impairs allosterically peroxynitrite isomerization by ferric human serum heme-albumin. J Biol Chem 2009; 284: 31006–31017.
Ascenzi P, di Masi A, De Sanctis G, Coletta M, Fasano M . Ibuprofen modulates allosterically NO dissociation from ferrous nitrosylated human serum heme-albumin by binding to three sites. Biochem Biophys Res Commun 2009; 387: 83–86.
Meneghini C, Leboffe L, Bionducci M, Fanali G, Meli M, Colombo G et al. The five-to-six-coordination transition of ferric human serum heme-albumin is allosterically-modulated by ibuprofen and warfarin: a combined XAS and MD study. PLoS One 2014; 9: e104231.
Sudlow G, Birkett DJ, Wade DN . The characterization of two specific drug binding sites on human serum albumin. Mol Pharmacol 1975; 11: 824–832.
Sudlow G, Birkett DJ, Wade DN . Further characterization of specific drug binding sites on human serum albumin. Mol Pharmacol 1976; 12: 1052–1061.
Carter DC, Ho JX . Structure of serum albumin. Adv Protein Chem 1994; 45: 153–203.
Yamasaki K, Maruyama T, Yoshimoto K, Tsutsumi Y, Narazaki R, Fukuhara A et al. Interactive binding to the two principal ligand binding sites of human serum albumin: effect of the neutral-to-base transition. Biochim Biophys Acta 1999; 1432: 313–323.
Petitpas I, Bhattacharya AA, Twine S, East M, Curry S . Crystal structure analysis of warfarin binding to human serum albumin: anatomy of drug site I. J Biol Chem 2001; 276: 22804–22809.
Curry S . Beyond expansion: structural studies on the transport roles of human serum albumin. Vox Sang 2002; 83: 315–319.
Hamilton JA . Fatty acid interactions with proteins: what X-ray crystal and NMR solution structures tell us. Prog Lipid Res 2004; 43: 177–199.
Curry S . Lessons from the crystallographic analysis of small molecule binding to human serum albumin. Drug Metab Pharmacokinet 2009; 24: 342–357.
Bhattacharya AA, Curry S, Franks NP . Binding of the general anesthetics propofol and halothane to human serum albumin. High resolution crystal structures. J Biol Chem 2000; 275: 38731–38738.
Yang F, Bian C, Zhu L, Zhao G, Huang Z, Huang M . Effect of human serum albumin on drug metabolism: structural evidence of esterase activity of human serum albumin. J Struct Biol 2007; 157: 348–355.
Simard JR, Zunszain PA, Hamilton JA, Curry S . Location of high and low affinity fatty acid binding sites on human serum albumin revealed by NMR drug-competition analysis. J Mol Biol 2006; 361: 336–351.
Díaz N, Suárez D, Sordo TL, Merz KM Jr . Molecular dynamics study of the IIA binding site in human serum albumin: influence of the protonation state of Lys195 and Lys199. J Med Chem 2001; 44: 250–260.
Petitpas I, Petersen CE, Ha CE, Bhattacharya AA, Zunszain PA, Ghuman J et al. Structural basis of albumin-thyroxine interactions and familial dysalbuminemic hyperthyroxinemia. Proc Natl Acad Sci USA 2003; 100: 6440–6445.
Gundry RL, Fu Q, Jelinek CA, Van Eyk JE, Cotter RJ . Investigation of an albumin-enriched fraction of human serum and its albuminome. Proteomics Clin Appl 2007; 1: 73–88.
Château M, Björck L . Protein PAB, a mosaic albumin-binding bacterial protein representing the first contemporary example of module shuffling. J Biol Chem 1994; 269: 12147–12451.
Lejon S, Frick IM, Björck L, Wikström M, Svensson S . Crystal structure and biological implications of a bacterial albumin binding module in complex with human serum albumin. J Biol Chem 2004; 279: 42924–42928.
Polticelli F, Caprari S, Gianni S, Ascenzi P . GA/GB fold switching may modulate fatty acid transfer from human serum albumin to bacteria. IUBMB Life 2012; 64: 885–888.
Sokołowska M, Wszelaka-Rylik M, Poznanski J, Bal W . Spectroscopic and thermodynamic determination of three distinct binding sites for Co2+ ions in human serum albumin. J Inorg Biochem 2009; 103: 1005–1013.
Sokołowska M, Pawlas K, Bal W . Effect of common buffers and heterocyclic ligands on the binding of Cu(II) at the multimetal binding site in human serum albumin. Bioinorg Chem Appl 2010; 725153.
Deng B, Wang Y, Zhu P, Xu X, Ning X . Study of the binding equilibrium between Zn(II) and HSA by capillary electrophoresis-inductively coupled plasma optical emission spectrometry. Anal Chim Acta 2010; 683: 58–62.
Duff MR Jr, Kumar CV . The metallomics approach: use of Fe(II) and Cu(II) footprinting to examine metal binding sites on serum albumins. Metallomics 2009; 1: 518–523.
Sadler PJ, Tucker A, Viles JH . Involvement of a lysine residue in the N-terminal Ni2+ and Cu2+ binding site of serum albumins. Comparison with Co2+, Cd2+ and Al3+ . FEBS J 2005; 220: 193–200.
Shaw CF . The protein chemistry of antiarthritic gold(I) thiolates and related complexes. Comments Inorg Chem 1989; 8: 233–267.
Blindauer CA, Harvey I, Bunyan KE, Stewart AJ, Sleep D, Harrison DJ et al. Structure, properties, and engineering of the major zinc binding site on human albumin. J Biol Chem 2009; 284: 23116–23224.
Mothes E, Faller P . Evidence that the principal CoII-binding site in human serum albumin is not at the N-terminus: implication on the albumin cobalt binding test for detecting myocardial ischemia. Biochemistry 2007; 46: 2267–2274.
Muller-Eberhard U, Javid J, Liem HH, Hanstein A, Hanna M . Plasma concentrations of hemopexin, haptoglobin and heme in patients with various hemolytic diseases. Blood 1968; 32: 811–815.
Wyman J Jr . Linked functions and reciprocal effects in hemoglobin: a second look. Adv Protein Chem 1964; 19: 223–286.
Ascenzi P, Fasano M . Abacavir modulates peroxynitrite-mediated oxidation of ferrous nitrosylated human serum heme-albumin. Biochem Biophys Res Commun 2007; 353: 469–474.
Herold S . The outer-sphere oxidation of nitrosyliron(II)hemoglobin by peroxynitrite leads to the release of nitrogen monoxide. Inorg Chem 2004; 43: 3783–3785.
Herold S, Fago A, Weber RE, Dewilde S, Moens L . Reactivity studies of the Fe(III) and Fe(II)NO forms of human neuroglobin reveal a potential role against oxidative stress. J Biol Chem 2004; 279: 22841–22847.
Herold S, Boccini F . NO* release from MbFe(II)NO and HbFe(II)NO after oxidation by peroxynitrite. Inorg Chem 2006; 45: 6933–6943.
Goldstein S, Lind J, Merényi G . Chemistry of peroxynitrites and peroxynitrates. Chem Rev 2005; 105: 2457–2470.
Ascenzi P, Bocedi A, Visca P, Minetti M, Clementi E . Does CO2 modulate peroxynitrite specificity?. IUBMB Life 2006; 58: 611–613.
Papina AA, Koppenol WH . Two pathways of carbon dioxide catalysed oxidative coupling of phenol by peroxynitrite. Chem Res Toxicol 2006; 19: 382–391.
Ascenzi P, Bolli A, Gullotta F, Fanali G, Fasano M . Drug binding to Sudlow’s site I impairs allosterically human serum heme-albumin-catalyzed peroxynitrite detoxification. IUBMB Life 2010; 62: 776–780.
Ascenzi P, Bolli A, di Masi A, Tundo GR, Fanali G, Coletta M et al. Isoniazid and rifampicin inhibit allosterically heme binding to albumin and peroxynitrite isomerization by heme-albumin. J Biol Inorg Chem 2011; 16: 97–108.
Di Muzio E, Polticelli F, Trezza V, Fanali G, Fasano M, Ascenzi P . Imatinib binding to human serum albumin modulates heme association and reactivity. Arch Biochem Biophys 2014; 560: 100–112.
Herold S, Kalinga S . Metmyoglobin and methemoglobin catalyze the isomerization of peroxynitrite to nitrate. Biochemistry 2003; 42: 14036–14046.
Herold S, Kalinga S, Matsui T, Watanabe Y . Mechanistic studies of the isomerization of peroxynitrite to nitrate catalyzed by distal histidine metmyoglobin mutants. J Am Chem Soc 2004; 126: 6945–6955.
Ascenzi P, Ciaccio C, Sinibaldi F, Santucci R, Coletta M . Cardiolipin modulates allosterically peroxynitrite detoxification by horse heart cytochrome c. Biochem Biophys Res Commun 2011; 404: 190–194.
di Masi A, Gullotta F, Bolli A, Fanali G, Fasano M, Ascenzi P . Ibuprofen binding to secondary sites allosterically modulates the spectroscopic and catalytic properties of human serum heme-albumin. FEBS J 2011; 278: 654–662.
Ascenzi P, Tundo GR, Fanali G, Coletta M, Fasano M . Warfarin modulates the nitrite reductase activity of ferrous human serum heme-albumin. J Biol Inorg Chem 2013; 18: 939–946.
Huang KT, Keszler A, Patel N, Patel RP, Gladwin MT, Kim-Shapiro DB et al. The reaction between nitrite and deoxyhemoglobin. Reassessment of reaction kinetics and stoichiometry. J Biol Chem 2005; 280: 31126–31131.
Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE et al. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest 2005; 115: 2099–2107.
Petersen MG, Dewilde S, Fago A . Reactions of ferrous neuroglobin and cytoglobin with nitrite under anaerobic conditions. J Inorg Biochem 2008; 102: 1777–1782.
Tiso M, Tejero J, Basu S, Azarov I, Wang X, Simplaceanu V et al. Human neuroglobin functions as a redox-regulated nitrite reductase. J Biol Chem 2011; 286: 18277–18289.
Li H, Hemann C, Abdelghany TM, El-Mahdy MA, Zweier JL . Characterization of the mechanism and magnitude of cytoglobin-mediated nitrite reduction and nitric oxide generation under anaerobic conditions. J Biol Chem 2012; 287: 36623–36633.
Ascenzi P, Gullotta F, Gioia M, Coletta M, Fasano M . O2-mediated oxidation of ferrous nitrosylated human serum heme-albumin is limited by nitrogen monoxide dissociation. Biochem Biophys Res Commun 2011; 406: 112–116.
Herold S, Röck G . Mechanistic studies of the oxygen-mediated oxidation of nitrosylhemoglobin. Biochemistry 2005; 44: 6223–6231.
Arnold EV, Bohle DS . Isolation and oxygenation reactions of nitrosylmyoglobins. Methods Enzymol 1996; 269: 41–55.
Møller JKS, Skibsted LH . Mechanism of nitrosylmyoglobin autoxidation: temperature and oxygen pressure effects on the two consecutive reactions. Chem Eur J 2004; 10: 2291–2300.
Monzani E, Bonafè B, Fallarini A, Redaelli C, Casella L, Minchiotti L et al. Enzymatic properties of hemalbumin. Biochim Biophys Acta 2001; 1547: 302–312.
Kamal JKA, Behere DV . Spectroscopic studies on human serum albumin and methemalbumin: optical, steady-state, and picosecond time-resolved fluorescence studies, and kinetics of substrate oxidation by methemalbumin. J Biol Inorg Chem 2002; 7: 273–283.
Fanali G, Pariani G, Ascenzi P, Fasano M . Allosteric and binding properties of Asp1-Glu382 truncated recombinant human serum albumin - an optical and NMR spectroscopic investigation. FEBS J 2009; 276: 2241–2250.
Kaneko K, Chuang VT, Minomo A, Yamasaki K, Bhagavan NV, Maruyama T et al. Histidine146 of human serum albumin plays a prominent role at the interface of subdomains IA and IIA in allosteric ligand binding. IUBMB Life 2011; 63: 277–285.
Ascenzi P, Fasano M . Serum heme-albumin: an allosteric protein. IUBMB Life 2009; 61: 1118–1122.
Ascenzi P, Fasano M . Allostery in a monomeric protein: the case of human serum albumin. Biophys Chem. 2010; 148: 16–22.
Fanali G, Fasano M, Ascenzi P, Zingg JM, Azzi A . α-Tocopherol binding to human serum albumin. Biofactors 2013; 39: 294–303.
Chuang VT, Otagiri M . How do fatty acids cause allosteric binding of drugs to human serum albumin?. Pharm Res 2002; 19: 1458–1464.
Domenici E, Bertucci C, Salvadori P, Wainer IW . Use of a human serum albumin-based high-performance liquid chromatography chiral stationary phase for the investigation of protein binding: detection of the allosteric interaction between warfarin and benzodiazepine binding sites. J Pharm Sci 1991; 80: 164–166.
Fitos I, Simonyi M . Stereoselective effect of phenprocoumon enantiomers on the binding of benzodiazepines to human serum albumin. Chirality 1992; 4: 21–23.
Baroni S, Mattu M, Vannini A, Cipollone R, Aime S, Ascenzi P et al. Effect of ibuprofen and warfarin on the allosteric properties of haem-human serum albumin. A spectroscopic study. Eur J Biochem 2001; 268: 6214–6220.
Mattu M, Vannini A, Coletta M, Fasano M, Ascenzi P . Effect of bezafibrate and clofibrate on the heme-iron geometry of ferrous nitrosylated heme-human serum albumin: an EPR study. J Inorg Biochem 2001; 84: 293–296.
Fasano M, Mattu M, Coletta M, Ascenzi P . The heme-iron geometry of ferrous nitrosylated heme-serum lipoproteins, hemopexin, and albumin: a comparative EPR study. J Inorg Biochem 2002; 91: 487–490.
Zunszain PA, Ghuman J, Komatsu T, Tsuchida E, Curry S . Crystal structural analysis of human serum albumin complexed with hemin and fatty acid. BMC Struct Biol 2003; 3: 6.
Fanali G, Fesce R, Agrati C, Ascenzi P, Fasano M . Allosteric modulation of myristate and Mn(III)heme binding to human serum albumin. Optical and NMR spectroscopy characterization. FEBS J 2005; 272: 4672–4683.
Ascenzi P, Bocedi A, Antonini G, Bolognesi M, Fasano M . Reductive nitrosylation and peroxynitrite-mediated oxidation of heme-hemopexin. FEBS J 2007; 274: 551–562.
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC et al. UCSF chimera—a visualization system for exploratory research and analysis. J Comput Chem 2004; 25: 1605–1612.
Meng EC, Pettersen EF, Couch GS, Huang CC, Ferrin TE . Tools for integrated sequence-structure analysis with UCSF Chimera. BMC Bioinformatics 2006; 7: 339.
Acknowledgements
We thank the colleagues as well as past and present members of their laboratories who contributed with data and discussions to the ideas presented here. This work was partially supported by grants from Ministero dell’Istruzione, dell’Università e della Ricerca of Italy (Università Roma Tre, Roma, Italy; CAL 2015 and PRIN 2010-2011 - 20109MXHMR_001 to PA).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ascenzi, P., di Masi, A., Fanali, G. et al. Heme-based catalytic properties of human serum albumin. Cell Death Discovery 1, 15025 (2015). https://doi.org/10.1038/cddiscovery.2015.25
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/cddiscovery.2015.25
- Springer Nature Limited
This article is cited by
-
Lysine reactivity profiling reveals molecular insights into human serum albumin–small-molecule drug interactions
Analytical and Bioanalytical Chemistry (2021)
-
Multi-wavelength analytical ultracentrifugation of human serum albumin complexed with porphyrin
European Biophysics Journal (2018)
-
Exploring the interaction between “site-markers, aspirin and esterase-like activity” ternary systems on the human serum albumin: direct evidence for modulation of catalytic activity of the protein in different inhibition modes
Journal of the Iranian Chemical Society (2018)
-
Heme-albumin: an honorary enzyme
Cell Death & Disease (2015)