
Abstract
Background:
Treatment of advanced and metastatic colorectal cancer with irinotecan is hampered by severe toxicities. The active metabolite of irinotecan, SN-38, is a known substrate of drug-metabolising enzymes, including UGT1A1, as well as OATP and ABC drug transporters.
Methods:
Blood samples (n=127) and tumour tissue (n=30) were obtained from advanced cancer patients treated with irinotecan-based regimens for pharmacogenetic and drug level analysis and transporter expression. Clinical variables, toxicity, and outcomes data were collected.
Results:
SLCO1B1 521C was significantly associated with increased SN-38 exposure (P<0.001), which was additive with UGT1A1*28. ABCC5 (rs562) carriers had significantly reduced SN-38 glucuronide and APC metabolite levels. Reduced risk of neutropenia and diarrhoea was associated with ABCC2–24C/T (odds ratio (OR)=0.22, 0.06–0.85) and CES1 (rs2244613; OR=0.29, 0.09–0.89), respectively. Progression-free survival (PFS) was significantly longer in SLCO1B1 388G/G patients and reduced in ABCC2–24T/T and UGT1A1*28 carriers. Notably, higher OATP1B3 tumour expression was associated with reduced PFS.
Conclusions:
Clarifying the association of host genetic variation in OATP and ABC transporters to SN-38 exposure, toxicity and PFS provides rationale for personalising irinotecan-based chemotherapy. Our findings suggest that OATP polymorphisms and expression in tumour tissue may serve as important new biomarkers.
Similar content being viewed by others


Main
Irinotecan (CPT-11) is currently approved for the treatment of advanced or metastatic colorectal cancer as a single agent or in combination chemotherapy regimens (Paulik et al, 2012; Bekaii-Saab and Wu, 2014). CPT-11 is a semi-synthetic camptothecin analogue capable of blocking DNA replication and transcription through topoisomerase-I inhibition (Marsh and Hoskins, 2010). This prodrug has a complex metabolic pathway where the first step is rapid bioactivation to its active metabolite SN-38, by carboxylesterases (CES1 and CES2) expressed in the liver, as well as to a lesser extent in plasma, gastrointestinal (GI) tract, and tumour tissue (Paulik et al, 2012). SN-38 is subsequently detoxified by uridine disphosphate glucuronosyltransferases (UGTs), mainly UGT1A1, to form the inactive metabolite SN-38 glucuronide (SN-38G). CPT-11 also undergoes CYP3A4/5-dependent oxidation to form the inactive metabolites M4, APC, and NPC with the latter available for further conversion to SN-38 (Kweekel et al, 2008). Increased systemic exposure to SN-38 places patients at an elevated risk of developing severe neutropenia as well as diarrhoea, which is thought to involve reconversion of SN-38G to SN-38 via intestinal β-glucuronidases (Paulik et al, 2012). Currently, although UGT1A1 polymorphisms are considered important, pharmacogenomic variables are not routinely considered when prescribing CPT-11, and dosing tends to be based on body surface area, clinical variables, and history or onset of adverse events (Kweekel et al, 2008).
The majority of CPT-11 pharmacogenetic studies have focused on the role of UGT1A1. Elevation in the level of SN-38, among patients with UGT1A1*28 polymorphism, has been linked to severe CPT-11-induced toxicities including neutropenia and diarrhoea (Innocenti et al, 2009; Sai et al, 2010). Results from a meta-analysis suggest the risk of severe haematological toxicities in homozygous UGT1A1*28 patients is dose dependent with those receiving high doses (>250 mg m−2) at the greatest risk. Although variant patients receiving intermediate doses (150–250 mg m−2) of CPT-11 are at significantly increased risks compared with patients with at least one wild-type allele, the odds of toxicity may be within an acceptable range if no other risk factors for neutropenia are present (Hoskins et al, 2007), questioning the usefulness of genotyping patients prescribed low-to-intermediate doses of CPT-11 before administration. Although it appears UGT1A1 has an important role in the development of CPT-11-related adverse events in some patients, there are many lacking known UGT polymorphisms who suffer from dose-limiting toxicities during CPT-11 therapy. Indeed, the interplay of numerous genes in the CPT-11 metabolism and disposition pathways is likely the basis for often unpredictably severe toxicity during CPT-11 therapy.
There is an increasing appreciation of the role of drug uptake and efflux transporters, both in organs such as the intestine and liver, as well in tumour, to the disposition and response to drugs in clinic use. We know that uptake transporters, such as OATP1B1 (SLCO1B1) are required for hepatic influx of SN-38, whereas ABC transporters including ABCC1, ABCC2, ABCB1, and ABCG2 regulate the hepatic and biliary efflux of CPT-11 metabolites (Fujiwara and Minami, 2010). Polymorphisms within SLCO1B1, ABCB1, and ABCC2 have recently been associated with modulation of CPT-11 and SN-38 exposure (Innocenti et al, 2009; Sai et al, 2010). Additional polymorphisms in ABCG2 and ABCC5 have recently been correlated with non-haematological toxicities (Di Martino et al, 2011; De Mattia et al, 2013). However, pharmacogenetic studies have demonstrated mixed results regarding the role of drug transporters to adverse events and response (Mathijssen and Gurney, 2009; Fujiwara and Minami, 2010). CPT-11 dose and schedule as well as ethnic differences in the populations studied provide plausible explanations for the wide variation in results.
The complexity of predicting response to CPT-11 therapy may be further hampered by the lack of ability to assess active drug concentrations within tumour tissue. Influx of SN-38 to the tumour may be dependent on the SN-38 uptake transporter OATP1B3 (SLCO1B3), which our group and others had shown to be highly upregulated in various cancer tissues including colon (Lee et al, 2008; Yamaguchi et al, 2008; Obaidat et al, 2012; Fujita et al, 2014). Recently, a cancer-specific isoform of OATP1B3 (csOATP1B3), lacking the first 28 amino acids in colon cancer tissues, has been identified (Nagai et al, 2012; Thakkar et al, 2013). Expression of csOATP1B3 appears to be induced under hypoxic conditions that are often associated with advanced solid cancers and poorer clinical outcomes (Han et al, 2013). It remains unknown if csOATP1B3 expression is predictive of response to CPT-11 therapy.
Determining the overall contribution of genetic variation in drug transporters as well as metabolising enzymes will be essential to tailoring CPT-11 doses for individual patients. In this study, our primary objective was to better correlate the effects of genetic variation in drug transporters, as well as enzymes such as UGT1A1, with measured CPT-11 and metabolite levels among cancer patients receiving irinotecan-based chemotherapy regimens in a prospective manner. Our secondary objective was to assess the impact of pharmacogenetic factors to the development of adverse events and progression-free survival (PFS). Finally, tumour tissues obtained from a cohort of these patients were analysed for OATP1B3 expression and its effect on PFS.
Materials and Methods
Study population
Metastatic colorectal cancer and advanced or metastatic pancreatic cancer patients (n=127) being treated with CPT-11-based chemotherapy regimens were prospectively recruited between January 2010 and November 2012 from the London Regional Cancer Program, London Health Sciences Centre, London, Ontario, Canada. The majority of patients were prescribed CPT-11 at 180 mg m−2 biweekly in combination with 5-fluorouracil and leucovorin (FOLFIRI regimen) with or without bevacuzimab. Patients were included if they were aged 18 years or above with an ECOG (Eastern Cooperative Oncology Group) performance status ⩽2. Exclusion criteria included: >35 μmol l−1 total bilirubin, >3X upper normal limit AST or ALT without liver metastases or >5X with liver metastases, known hypersensitivity to CPT-11, known history of Gilbert’s syndrome, and concurrent use of ketoconazole. All participants provided written informed consent. The study was approved by the Research Ethics Board at Western University.
Chart review
Paper and electronic record chart review for each consented patient was conducted by a single reviewer. Age recorded was the age at initiation of CPT-11-based chemotherapy. A cycle of CPT-11 was defined as a single administration of CPT-11 alone or in combination, irrespective of the chemotherapy regimen used. Treatment-related toxicities were recorded for the cycle where the blood sample was obtained in addition to any toxicities occurring throughout the duration of treatment with CPT-11. The blood sample cycle was considered to extend to the subsequent measurement of basic laboratory values and/or clinical assessment. Toxicities during this period were not considered to be associated with the particular treatment if they were documented more than 3 weeks after sample blood drawn. NCI-CTCAE version 3.0 (Bethesda, MD, USA) was used to grade toxicities. Toxicity grade was determined using subjective measures (described in clinical notes) when exact grading was not documented.
Responders were defined as having a stable or reduced tumour size on the first CT scan following CPT-11-based chemotherapy initiation. The interval between CT scans was at the discretion of the treating oncologist. Progression was defined as the date the CT or MRI reported an increased tumour size. Progression-free survival was considered to be the time from initiation of CPT-11-based chemotherapy to the date of progression, death, last contact, or censor date (13 August 2013), whichever occurred first.
Genotyping
DNA was extracted from whole blood using the Gentra Purgene Blood Kit (Qiagen, Toronto, Ontario, Canada). The following TaqMan allelic discrimination assays (Applied Biosystems, Carlsbad, CA, USA) were used for genotyping: ABCB1 (c.3435C>T, rs1045642), ABCG2 (c.421C>A, rs2231142; c.34G>A, rs2231137), ABCC2 (c.-24C>T, rs717620; c.1249G>A, rs2273697), ABCC5 (T>C, rs562), SLCO1B1*1b (c.388A>G, rs2306283), SLCO1B1*5 (c.521T>C, rs4149056), SLCO1B3 (g.699G>A, rs7311358), UGT1A1*28 (TA(6/7), rs8175347), CES1 (g.14506G>A, rs71647871; g.27467A>C, rs2244613), CYP3A4*22 (intron 6 C>T, rs35599367), and CYP3A5*3 (g.6986A>G, rs776746). Hardy–Weinburg equilibrium was assessed for all genotypes using the χ2 goodness-of-fit test.
Drug levels
Blood samples were obtained by venipuncture of the opposite arm immediately following the end of the CPT-11 infusion (90 min) during any cycle of CPT-11-based chemotherapy. Plasma was collected and stored at −80 °C until analysis. Plasma concentrations of CPT-11 and metabolites SN-38, SN-38G, and APC (7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino] carbonyloxycamptothecin) were measured by liquid chromatography-tandem mass spectrometry. Standards were obtained from Sigma-Aldrich (Oakville, Ontario, Canada; CPT-11, SN-38) and Toronto Research Chemicals (Toronto, Ontario, Canada; SN-38-glucuronide, APC). Plasma samples (100 μl) were precipitated upon addition of 3 volumes of acetonitrile and 0.1% formic acid (FA) spiked with 15 μl internal standard (camptothecin, 500 ng ml−1, Toronto Research Chemicals). Samples were vortexed, centrifuged, and diluted with H2O (0.1% FA). Analytes were injected (30 μl) into the liquid chromatograph (Agilent 1200) and separated on a reverse-phase column (Hypersil Gold, 50 × 5 mm, 5 μ M particle size) over 6 min using gradient elution with H2O (0.1% FA) and acetonitrile (0.1% FA) (10–90%). Standard curves and quality controls (co-efficient of variation (%), high (13.5), med (2.6), low (3.7)) were prepared in drug-free plasma. The mass spectrometer (Thermo TSQ Vantage, Burlington, ON, Canada) with heated electrospray ionisation source was set in positive mode for detection of CPT-11, SN-38, SN-38G, APC, and camptothecin with transitions 587→124 m/z, 393→349.3 m/z, 569→393.3 m/z, 691→227 m/z, 349→305 m/z, respectively.
Immunohistochemistry
Archived normal and tumour tissue biopsy slides were obtained from a subset of study participants (n=30) following approval by the Tissue and Archive Committee (Department of Pathology, London Health Sciences Centre). Antigen retrieval was performed with citrate buffer and slides were subsequently incubated with pre-immune serum or anti-OATP1B3 polyclonal antibody (1 : 200) followed by an avidin-biotin immunoperoxidase assay and developed using an AEC (3-Amino-9-ethylcarbazole) staining kit (Sigma-Aldrich) using a modified protocol (Lee et al, 2008). Nuclei were counterstained using Mayer’s haematoxylin (Sigma-Aldrich). Scoring for OATP1B3 expression in normal, normal adjacent, and tumour tissue was performed independently by one pathologist (JP). Staining intensity for OATP1B3 was defined and evaluated using the following semi-quantitative previously published method: (0) no staining, (1) weakly positive, (2) moderately positive, and (3) strongly positive (Lee et al, 2008).
Statistics
The primary objective was to determine covariates associated with interindividual variability of CPT-11 and metabolite plasma concentration. All statistical analysis was performed in GraphPad Prism and the statistical software R. One-way analysis of variance with Bonferroni correction and Student’s t-tests was used to compare drug levels between genotypic groups. Multiple linear regression analysis was performed to determine significant covariates on the interindividual variability of dose-normalised CPT-11 and metabolite plasma concentrations (natural log-transformed). Covariates considered included: age, sex, treatment regimen, ABCB1, ABCG2, ABCC2, ABCC5, SLCO1B1, SLCO1B3, UGT1A1, CES1, CYP3A4, and CYP3A5 genotype. Covariates were assessed individually and were considered for the final model at a significance level of P<0.2. Covariates meeting these criteria were entered into a multiple linear regression model adjusting for age, sex, and treatment regimen and remained in the final model if P<0.1.
Secondary outcomes included assessing covariates associated with toxicity and PFS. Multinomial logistic regression analysis was used to determine association of genotype with toxicity events after adjustment for sex, age, and treatment regimen. Toxicity categories used in the regression analysis were: neutropenia (no event vs low (grade 1 or 2) vs high (grade 3 or 4)) and diarrhoea, nausea/vomiting and oral mucositis (no event vs low (grade 1) vs high (grade 2 or 3)). Univariate analysis was performed for each covariate and only significant genotypes were included in the final model with the exception of adjustment covariates as indicated above. Logistic regression analysis was also performed to determine covariates associated with neutropenia (low (grade 0, 1 or 2) vs high (grade 3 or 4)). Cox regression analysis was used to determine association of covariates with PFS in patients treated with BEV-FOLFIRI or FOLFIRI regimens (excluding patients with pancreatic cancer). Univariate analysis was performed and covariates with a cut value of P<0.2 were included in the final multivariate analysis with the exception of adjusting covariates, age at enrolment, sex and treatment regimen. Kruskal–Wallis with Dunn’s multiple comparative test and Wilcoxon signed rank test was used to compare OATP1B3 pathology scores. Mann–Whitney U-test was used to compare OATP1B3 pathology scores and PFS in patients treated with CPT-11-based chemotherapy regimens scored for OATP1B3 tumour expression (0–1 vs 2–3).
Results
Study population
Patient and tumour characteristics (n=127) are described in Table 1. The treatment profile of patients is presented in Table 2. Approximately half (55%) of the study population had not received previous chemotherapy and the majority of patients (82%) were treated with bevacizumab (BEV)-FOLFIRI or FOLFIRI chemotherapy regimens as first-line therapy (Table 2).
OATP1B1 and ABCC5 are important determinants of SN-38 and SN-38G levels
The primary objective was to determine covariates associated with interindividual variability of plasma concentrations of CPT-11 and metabolites. Plasma concentrations of CPT-11, SN-38, SN-38G, and APC were measured from blood samples collected immediately following the end of CPT-11 infusion. Multiple linear regression was performed on dose-normalised drug levels (natural log transformed) adjusting for age at time of treatment initiation, sex and treatment regimen in the final models. ABCB1 (c.3435 C>T) was significantly associated with CPT-11 exposure as homozygous variant (T/T) carriers had lower levels compared with wild-type patients (P<0.05; Figure 1A and Supplementary Table 1). This model had one significant adjusting covariate (FOLFIRI treatment regimen, P <0.05).

Association between genotype and CPT-11 and metabolite plasma concentrations. (A) Effect of ABCB1 3435C>T genotype on dose-normalised CPT-11 plasma levels. Association between SLCO1B1 521T>C (B) and UGT1A1*28 (C) genotype and dose-normalised SN-38 levels. Effect of increasing number of combined SLCO1B1 521C and UGT1A1*28 variant alleles on dose-normalised SN-38 plasma levels (D) and plasma SN-38-G/SN-38 ratio (E). (F) Association between ABCC5 rs562 T>C genotype and SN-38-G plasma levels. *<0.05, **<0.01, ***<0.001.
Several genotypes were significantly associated with variation in SN-38 levels as part of a model that explained approximately 27% of the variation in exposure (Table 3). SLCO1B1 521C allele carriers had significantly increased systemic exposure to SN-38 (P<0.001, Figure 1B). As expected, heterozygous (*1/*28) and homozygous variant (*28/*28) UGT1A1 carriers had increased SN-38 plasma levels (Figure 1C). Interestingly, a significant increase in SN-38 level was observed with an increasing number of combined SLCO1B1 521C and UGT1A1*28 variant alleles, suggesting an additive effect of polymorphisms in these two genes (Figure 1D). A corresponding decrease in the SN-38G/SN-38 ratio was observed with an increasing number of combined variant alleles (Figure 1E). Together, this suggests that patients heterozygous for both SLCO1B1 521C and UGT1A1*28 may have an equivalent risk of increased SN-38 exposure compared with patients homozygous for either polymorphism, respectively. Carriers of a rare SNP in CES1 (rs71647871, allele frequency, 0.024) had significantly decreased SN-38 levels (P<0.05, Table 3).
Interestingly, SN-38G levels were significantly affected by ABCC5 genotype. Patients harbouring the ABCC5 rs562 C allele had reduced SN-38G plasma exposure compared with wild-type patients (P <0.001, Figure 1F, Supplementary Table 2). In addition, patients expressing CYP3A5 also had significantly reduced SN-38G plasma levels (P <0.05). FOLFIRI and FOLFIRINOX treatment regimens were significantly associated with SN-38G (P<0.001 and P <0.05, respectively).
The model for the inactive metabolite APC explained approximately 24% of the variability in plasma exposure (Supplementary Table 3). SLCO1B3 g.699 and ABCC2 c.1249G>A were positively associated with APC levels (P<0.01). Decreased plasma levels were observed in patients carrying the ABCC5 rs562 C allele compared with wild-type patients (P<0.001). Significant adjusting covariates in this model included sex (P<0.05) and treatment regimen (P<0.01).
Drug transporters predict CPT-11-related toxicities
CPT-11-related adverse reactions and frequencies of grade severity are described in Supplementary Table 4. Multinomial logistic regression analysis was performed to determine association of genotypes to adverse reactions comparing no event vs low-grade vs high-grade toxicity (Table 4). ABCC2 -24C/T carriers had significantly lower risk of grade 3/4 neutropenia compared with wild-type patients (odds ratio (OR)=0.22, 95% CI=0.06–0.85). UGT1A1*28 carriers were at increased risk for severe neutropenia (grade 3/4) compared with wild-type patients following binary logistic regression analysing low- vs high-grade events (OR=3.67; 95% CI=1.19–11.33; Table 4). In addition to their increased SN-38 exposure, patients with two or more combined SLCO1B1 521C and UGT1A1*28 variant alleles were also at significantly increased risk of severe neutropenia (grade 3/4) compared with patients wild-type for both alleles (OR=4.154, 95% CI=1.06–16.36).
Non-haematological toxicity was associated with drug transporters and metabolising enzymes (Table 4). Carriers of a common SNP in CES1 (rs2244613) had significantly lower risk of higher-grade diarrhoea (OR=0.29, 95% CI=0.09–0.89). Patients with ABCB1 3435 C/T and T/T genotypes were much more likely to experience higher-grade nausea/vomiting (OR=9.06, 95% CI=1.03–79.41 and OR=10.52, 95% CI=1.10–100.2, respectively). Higher-grade oral mucositis was observed in patients expressing CYP3A5 (OR=8.10, 95% CI=1.57–41.90), whereas those heterozygous for SLCO1B1 388A/G had a significantly lower risk (OR=0.19, 95% CI=0.05–0.72). No significant adjusting covariates were found in the binary logistic or multinomial logistic regression models.
Biomarkers of PFS
Approximately 73% of patients were considered responders having radiographic evidence of no change or tumour shrinkage during CPT-11-based therapy. At the time of analysis (censor date: 13 August 2013), disease progression was evident in the majority of patients with a median PFS time of 10.5 months (range, 0.2–43 months; Table 2). Cox-regression analysis for PFS was performed on patients (excluding pancreatic cancer patients) treated with BEV-FOLFIRI or FOLFIRI regimens only (n=103, Table 5). Patients homozygous for SLCO1B1 388G/G alleles had a significantly increased PFS compared with wild-type patients (HR=1.60, 95% CI=1.04–2.46). Patients carrying two ABCC2 c.-24T alleles had decreased PFS (HR=0.62, 95% CI=0.40–0.95). Reduced PFS was also observed for UGT1A1*28 (*1/*28, HR=0.69, 95% CI=0.52–0.92; *28/*28, HR=0.60, 95% CI=0.38–0.97). ABCC5 was associated with PFS in the univariate analysis (P=0.05), but did not remain significant following adjustment in the final model. No significant adjusting covariates were found in the multivariate analysis.
Tumour OATP1B3 expression suggests poorer clinical outcomes
The role of OATP1B3 expression, known to transport SN-38, in response to CPT-11-based chemotherapy is unknown. Recently, OATP1B3 expression within colon tumours has been shown to be a cancer-specific isoform that may be expressed primarily as an intracellular protein calling into question the functional relevance of this transporter within the tumour (Thakkar et al, 2013). Paired normal and tumour samples were available for a subset of patients treated with CPT-11-based chemotherapy regimens (n=30: BEV-FOLFIRI, n=22; FOLFIRI, n=5; FOLFIRINOX, n=1; BEV-irinotecan, n=1; cetuximab-irinotecan, n=1). Staining for OATP1B3 was performed and intensity of expression was scored. Tumour tissue had a significantly higher OATP1B3 score compared with paired normal tissue (P<0.05, Figure 2A and C). Progression-free survival was significantly reduced in patients with high (score, 2 or 3) OATP1B3 expression compared with patients with low (score, 0 or 1) expression (Figure 2B), suggesting that OATP1B3 expression may correlate with poorer clinical response to CPT-11 therapy.

Effect of OATP1B3 tumour expression on progression-free survival in patients treated with CPT-11-based chemotherapy regimen. (A) Pathology score of OATP1B3 expression in paired samples of normal, normal adjacent, and tumour tissue (n=30). (B) Association between OATP1B3 tumour score and progression-free survival in patients. (C) Representative immunohistochemistry of OATP1B3 expression in normal and tumour colon tissue. *<0.05.
Discussion
Pharmacogenetics of irinotecan therapy has been widely studied and UGT1A1*28 in particular, shown to be of potential clinical relevance especially for patients prescribed high-dose CPT-11 therapy (Hoskins et al, 2007). As severe toxicities are often observed in patients treated with combination regimens at lower CPT-11 doses, we examined pharmacogenetic determinants of CPT-11 and metabolite exposure in this setting.
SN-38 plasma exposure was significantly increased SLCO1B1 521C (*5) allele carriers (P<0.001) (Figure 3) consistent with studies correlating SLCO1B1 521C to higher SN-38 and CPT-11 AUC in both colorectal cancer and non-small-lung cell cancer patients (Han et al, 2008; Innocenti et al, 2009). UGT1A1*28 carriers had significantly increased SN-38 levels compared with non-carriers (P<0.01) consistent with previous reports, which can result in greater toxicity risk (Iyer et al, 2002; Innocenti et al, 2009; Hu et al, 2010; Sai et al, 2010; Cai et al, 2013). Importantly, we demonstrate an additive effect of SLCO1B1 521C and UGT1A1*28 on SN-38 exposure as patients with an increasing number of combined variant alleles had higher plasma levels and a corresponding decrease in the SN-38G/SN-38 ratio. To our knowledge this is the first report of an additive effect in a primarily Caucasian (94%) population, which remained significant upon exclusion of non-Caucasian patients. A similar additive effect has been noted for SN-38 AUC with combined SLCO1B1*15 and UGT1A1*6 or *28 polymorphisms in Japanese patients, likely due to the presence of the SLCO1B1 521C allele (Sai et al, 2010). Together, these results suggest that heterozygous carriers of both SLCO1B1 521C and UGT1A1*28 may reach a toxicity risk comparable to homozygous UGT1A1*28 patients as evidenced by several case reports of SLCO521C and UGT1A1*28 carriers presenting with life-threatening toxicities (Sakaguchi et al, 2009; Takane et al, 2009). Here we observed an OR of 4.15 (95%CI=1.06–16.36) for grade 3/4 neutropenia in patients with two or more combined variant allele. The clinical relevance of the additive effect of these two genes may be underestimated and need to be further examined as a better strategy for personalising CPT-11 therapy.

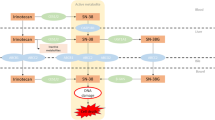
Schematic model depicting known and new insight into the metabolism and transport of CPT-11, SN-38, and SN-38G and the proposed effect on treatment-related toxicities and response. Increased SN-38 exposure due to combined SLCO1B1 and UGT1A1 variants correlates with an increased risk for neutropenia. Increased intestinal SN-38G, due to reduced ABCC5 hepatic efflux, may lead to increased risk for diarrhoea as SN-38G is converted to SN-38 via intestinal β-glucoronidases. Circles with check marks indicate an effect on neutropenia (N), diarrhea (D), and progression-free survival (P).
Although plasma SN-38 levels appear to be the most predictive of toxicity risk, secondary metabolites, SN-38G and APC, may also be important contributors. We observed decreased plasma levels of these metabolites associated with an ABCC5 polymorphism (rs562) that was recently identified as a significant predictor of GI toxicity (Di Martino et al, 2011). These effects may be due to reduced ABCC5 hepatic efflux leading to SN-38G and APC accumulation within the liver (Figure 3). Higher hepatic concentrations may ultimately lead to increased intestinal SN-38G levels, through increased biliary excretion via other ABC transporters, which may then undergo β-glucuronidase-dependent reconversion to SN-38 thereby augmenting GI toxicity risk (Figure 3).
In this study, CPT-11 levels were lower in homozygous ABCB1 3435T patients. ABCB1 is a well-established transporter of CPT-11 and SN-38, but its clinical relevance for CPT-11 therapy remains inconclusive. ABCB1 polymorphisms have been associated with both increased or reduced exposure, decreased clearance and increased toxicity and response, whereas other studies were unable to confirm these results (Mathijssen et al, 2003; Sai et al, 2003; Mathijssen et al, 2004; Cote et al, 2007; Innocenti et al, 2009; Sai et al, 2010; Glimelius et al, 2011). This may be due to the redundancy of other transporters, including ABCC2 and ABCG2, capable of biliary excretion of CPT-11 and metabolites. The lack of consistent evidence suggests that ABCB1 may not be useful in personalising CPT-11 therapy.
As a secondary objective we examined the impact of pharmacogenetic factors on adverse events and PFS. Here, the majority (97%) of patients were prescribed 180 mg m−2 CPT-11 with 17% experiencing severe neutropenia. Neutropenia was associated with UGT1A1*28 as has been reported (Hoskins et al, 2007). More importantly, patients carrying two or more combined SLCO1B1 521C and UGT1A1*28 alleles were at significantly increased risk of myelosuppression, suggesting these SNPs together may provide a more comprehensive strategy for assessing haematological toxicity risk. Heterozygous ABCC2–24C/T but not TT carriers predicted reduced neutropenia risk indicating further validation is needed.
Non-haematological toxicities including diarrhoea, nausea/vomiting, and oral mucositis were assessed. Most notably, a common SNP in CES1 (rs2244613) was associated with lower risk of diarrhoea, which may be due to reduced conversion of CPT-11 to SN-38. CES1 (rs2244613) was recently associated with reduced trough levels of dabigatran, a new oral anticoagulant drug, and a significantly decreased bleeding risk in patients, suggesting this SNP may be clinically relevant for many CES-dependent drugs (Pare et al, 2013). Interestingly, homozygous ABCB1 3435T carriers had a much higher likelihood of experiencing higher-grade nausea/vomiting (OR=10.52, P<0.05) and patients expressing CYP3A5 had a significantly increased risk of oral mucositis, suggesting higher levels of M4, APC, or NPC metabolites may contribute its development. A potential limitation to this analysis is the concurrent use of 5-fluorouracil with CPT-11 in most patients. Although the side-effect profile of the two drugs is similar, the majority of markers assessed are not specific to 5-fluorouracil, suggesting these correlations are likely due to modulation of irinotecan disposition but require confirmation in a study designed to assess toxicity as a primary objective.
PFS analysis was limited to patients treated with FOLFIRI with or without BEV. SLCO1B1 388(G/G) was associated with longer PFS, suggesting variant carriers may experience better response to FOLFIRI-based regimens. This effect may be due to increased OATP1B1 expression as this variant has recently been correlated to increased expression in Caucasian liver samples (Nies et al, 2013). ABCC2–24TT was associated with reduced PFS, consistent with lower response rates and shorter PFS observed in Japanese patients, but contrary to the lack of association in Caucasian metastatic colorectal cancer patients treated with FOLFIRI regimes (Akiyama et al, 2012). UGT1A1*28 was also associated with reduced PFS, but a recent meta-analysis suggested that UGT1A1*28 status may not be a reliable predictor of PFS (Liu et al, 2013). Our evaluation of PFS may be confounded by patients still undergoing active therapy or surveillance at the date of censoring. Although the genes investigated are not thought to have a role in the other drugs in the FOLFIRI regimen, we cannot rule out the effect of these drugs on PFS.
Importantly, we analysed OATP1B3 tumour expression in a subset of patients. To date, tumour biomarkers of CPT-11 response remain unknown. Our group was the first to note OATP1B3 overexpression in colon tumours and this expression was recently identified exclusively as a cancer-specific (cs)OATP1B3 splice variant (Lee et al, 2008; Han et al, 2013; Imai et al, 2013; Thakkar et al, 2013). The localisation (thought to be intracellular), function, and clinical relevance of csOATP1B3 are under investigation (Imai et al, 2013; Thakkar et al, 2013). Here we show the first evidence, to our knowledge, that higher OATP1B3 expression in colon tumours was significantly associated with reduced PFS (Figure 3). Lack of membrane expression of a functional OATP1B3 transporter may lead to reduced SN-38 tumour influx leading to a poorer clinical response or alternatively, overexpression of csOATP1B3 within the tumour may induce a p53-dependent survival mechanism, which has been demonstrated in WT-OATP1B3 colon cancer cell lines (Lee et al, 2008). Although we are unable to definitively confirm OATP1B3 to be the cancer-specific isoform due to use of an antibody recognising the common c-terminal region, the reported lack of wild-type OATP1B3 expression within colon tumours suggests this is the form expressed in these tumours (Thakkar et al, 2013).
Owing to the complexity of CPT-11 disposition, it is questionable that one gene alone will be useful for personalising therapy and will likely require assessing the right combination of genes. Our data provide new insight regarding transporters, particularly members of the OATP1B subfamily, to the disposition and clinical effects of CPT-11 (summarised in Figure 3). The additive effect of SLCO1B1 521C and UGT1A1*28 on SN-38 exposure and neutropenia risk seen here in patients carrying two or more combined alleles (24.8% of this population) provides rationale for examining the utility of combined genotyping to better predict toxicity risk in CPT-11-based regimens. Future prospective studies should be designed to compare combined genotyping to UGT1A1*28 genotyping alone to advance the goal of personalising irinotecan.
Change history
03 March 2015
This paper was modified 12 months after initial publication to switch to Creative Commons licence terms, as noted at publication
References
R Core Team (2013) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria.
Akiyama Y, Fujita K, Ishida H, Sunakawa Y, Yamashita K, Kawara K, Miwa K, Saji S, Sasaki Y (2012) Association of ABCC2 genotype with efficacy of first-line FOLFIRI in Japanese patients with advanced colorectal cancer. Drug Metab Pharmacokinet 27: 325–335.
Bekaii-Saab T, Wu C (2014) Seeing the forest through the trees: A systematic review of the safety and efficacy of combination chemotherapies used in the treatment of metastatic colorectal cancer. Crit Rev Oncol Hematol 91 (1): 9–34.
Cai X, Cao W, Ding H, Liu T, Zhou X, Wang M, Zhong M, Zhao Z, Xu Q, Wang L (2013) Analysis of UGT1A1*28 genotype and SN-38 pharmacokinetics for irinotecan-based chemotherapy in patients with advanced colorectal cancer: results from a multicenter, retrospective study in Shanghai. J Cancer Res Clin Oncol 139: 1579–1589.
Cote JF, Kirzin S, Kramar A, Mosnier JF, Diebold MD, Soubeyran I, Thirouard AS, Selves J, Laurent-Puig P, Ychou M (2007) UGT1A1 polymorphism can predict hematologic toxicity in patients treated with irinotecan. Clin Cancer Res 13: 3269–3275.
De Mattia E, Toffoli G, Polesel J, D'Andrea M, Corona G, Zagonel V, Buonadonna A, Dreussi E, Cecchin E (2013) Pharmacogenetics of ABC and SLC transporters in metastatic colorectal cancer patients receiving first-line FOLFIRI treatment. Pharmacogenet Genomics 23: 549–557.
Di Martino MT, Arbitrio M, Leone E, Guzzi PH, Rotundo MS, Ciliberto D, Tomaino V, Fabiani F, Talarico D, Sperlongano P, Doldo P, Cannataro M, Caraglia M, Tassone P, Tagliaferri P (2011) Single nucleotide polymorphisms of ABCC5 and ABCG1 transporter genes correlate to irinotecan-associated gastrointestinal toxicity in colorectal cancer patients: a DMET microarray profiling study. Cancer Biol Ther 12: 780–787.
Fujita K, Sugiura T, Okumura H, Umeda S, Nakamichi N, Watanabe Y, Suzuki H, Sunakawa Y, Shimada K, Kawara K, Sasaki Y, Kato Y (2014) Direct inhibition and down-regulation by uremic plasma components of hepatic uptake transporter for SN-38, an active metabolite of irinotecan, in humans. Pharm Res 31: 204–215.
Fujiwara Y, Minami H (2010) An overview of the recent progress in irinotecan pharmacogenetics. Pharmacogenomics 11: 391–406.
Glimelius B, Garmo H, Berglund A, Fredriksson LA, Berglund M, Kohnke H, Bystrom P, Sorbye H, Wadelius M (2011) Prediction of irinotecan and 5-fluorouracil toxicity and response in patients with advanced colorectal cancer. Pharmacogenomics J 11: 61–71.
Han JY, Lim HS, Shin ES, Yoo YK, Park YH, Lee JE, Kim HT, Lee JS (2008) Influence of the organic anion-transporting polypeptide 1B1 (OATP1B1) polymorphisms on irinotecan-pharmacokinetics and clinical outcome of patients with advanced non-small cell lung cancer. Lung Cancer 59: 69–75.
Han S, Kim K, Thakkar N, Kim D, Lee W (2013) Role of hypoxia inducible factor-1alpha in the regulation of the cancer-specific variant of organic anion transporting polypeptide 1B3 (OATP1B3), in colon and pancreatic cancer. Biochem Pharmacol 86: 816–823.
Hoskins JM, Goldberg RM, Qu P, Ibrahim JG, McLeod HL (2007) UGT1A1*28 genotype and irinotecan-induced neutropenia: dose matters. J Natl Cancer Inst 99: 1290–1295.
Hu ZY, Yu Q, Pei Q, Guo C (2010) Dose-dependent association between UGT1A1*28 genotype and irinotecan-induced neutropenia: low doses also increase risk. Clin Cancer Res 16: 3832–3842.
Imai S, Kikuchi R, Tsuruya Y, Naoi S, Nishida S, Kusuhara H, Sugiyama Y (2013) Epigenetic regulation of organic anion transporting polypeptide 1B3 in cancer cell lines. Pharm Res 30: 2880–2890.
Innocenti F, Kroetz DL, Schuetz E, Dolan ME, Ramirez J, Relling M, Chen P, Das S, Rosner GL, Ratain MJ (2009) Comprehensive pharmacogenetic analysis of irinotecan neutropenia and pharmacokinetics. J Clin Oncol 27: 2604–2614.
Iyer L, Das S, Janisch L, Wen M, Ramirez J, Karrison T, Fleming GF, Vokes EE, Schilsky RL, Ratain MJ (2002) UGT1A1*28 polymorphism as a determinant of irinotecan disposition and toxicity. Pharmacogenomics J 2: 43–47.
Kweekel D, Guchelaar HJ, Gelderblom H (2008) Clinical and pharmacogenetic factors associated with irinotecan toxicity. Cancer Treat Rev 34: 656–669.
Lee W, Belkhiri A, Lockhart AC, Merchant N, Glaeser H, Harris EI, Washington MK, Brunt EM, Zaika A, Kim RB, El-Rifai W (2008) Overexpression of OATP1B3 confers apoptotic resistance in colon cancer. Cancer Res 68: 10315–10323.
Liu X, Cheng D, Kuang Q, Liu G, Xu W (2013) Association between UGT1A1*28 polymorphisms and clinical outcomes of irinotecan-based chemotherapies in colorectal cancer: a meta-analysis in Caucasians. PLoS One 8: e58489.
Marsh S, Hoskins JM (2010) Irinotecan pharmacogenomics. Pharmacogenomics 11: 1003–1010.
Mathijssen RH, de Jong FA, van Schaik RH, Lepper ER, Friberg LE, Rietveld T, de Bruijn P, Graveland WJ, Figg WD, Verweij J, Sparreboom A (2004) Prediction of irinotecan pharmacokinetics by use of cytochrome P450 3A4 phenotyping probes. J Natl Cancer Inst 96: 1585–1592.
Mathijssen RH, Gurney H (2009) Irinogenetics: how many stars are there in the sky? J Clin Oncol 27: 2578–2579.
Mathijssen RH, Marsh S, Karlsson MO, Xie R, Baker SD, Verweij J, Sparreboom A, McLeod HL (2003) Irinotecan pathway genotype analysis to predict pharmacokinetics. Clin Cancer Res 9: 3246–3253.
Nagai M, Furihata T, Matsumoto S, Ishii S, Motohashi S, Yoshino I, Ugajin M, Miyajima A, Chiba K (2012) Identification of a new organic anion transporting polypeptide 1B3 mRNA isoform primarily expressed in human cancerous tissues and cells. Biochem Biophys Res Commun 418: 818–823.
Nies AT, Niemi M, Burk O, Winter S, Zanger UM, Stieger B, Schwab M, Schaeffeler E (2013) Genetics is a major determinant of expression of the human hepatic uptake transporter OATP1B1, but not of OATP1B3 and OATP2B1. Genome Med 5: 1.
Obaidat A, Roth M, Hagenbuch B (2012) The expression and function of organic anion transporting polypeptides in normal tissues and in cancer. Annu Rev Pharmacol Toxicol 52: 135–151.
Pare G, Eriksson N, Lehr T, Connolly S, Eikelboom J, Ezekowitz MD, Axelsson T, Haertter S, Oldgren J, Reilly P, Siegbahn A, Syvanen AC, Wadelius C, Wadelius M, Zimdahl-Gelling H, Yusuf S, Wallentin L (2013) Genetic determinants of dabigatran plasma levels and their relation to bleeding. Circulation 127: 1404–1412.
Paulik A, Grim J, Filip S (2012) Predictors of irinotecan toxicity and efficacy in treatment of metastatic colorectal cancer. Acta Medica (Hradec Kralove) 55: 153–159.
Sai K, Kaniwa N, Itoda M, Saito Y, Hasegawa R, Komamura K, Ueno K, Kamakura S, Kitakaze M, Shirao K, Minami H, Ohtsu A, Yoshida T, Saijo N, Kitamura Y, Kamatani N, Ozawa S, Sawada J (2003) Haplotype analysis of ABCB1/MDR1 blocks in a Japanese population reveals genotype-dependent renal clearance of irinotecan. Pharmacogenetics 13: 741–757.
Sai K, Saito Y, Maekawa K, Kim SR, Kaniwa N, Nishimaki-Mogami T, Sawada J, Shirao K, Hamaguchi T, Yamamoto N, Kunitoh H, Ohe Y, Yamada Y, Tamura T, Yoshida T, Matsumura Y, Ohtsu A, Saijo N, Minami H (2010) Additive effects of drug transporter genetic polymorphisms on irinotecan pharmacokinetics/pharmacodynamics in Japanese cancer patients. Cancer Chemother Pharmacol 66: 95–105.
Sakaguchi S, Garcia-Bournissen F, Kim R, Schwarz UI, Nathan PC, Ito S (2009) Prolonged neutropenia after irinotecan-based chemotherapy in a child with polymorphisms of UGT1A1 and SLCO1B1. Arch Dis Child 94: 981–982.
Takane H, Kawamoto K, Sasaki T, Moriki K, Kitano H, Higuchi S, Otsubo K, Ieiri I (2009) Life-threatening toxicities in a patient with UGT1A1*6/*28 and SLCO1B1*15/*15 genotypes after irinotecan-based chemotherapy. Cancer Chemother Pharmacol 63: 1165–1169.
Thakkar N, Kim K, Jang ER, Han S, Kim D, Merchant N, Lockhart AC, Lee W (2013) A cancer-specific variant of the SLCO1B3 gene encodes a novel human organic anion transporting polypeptide 1B3 (OATP1B3) localized mainly in the cytoplasm of colon and pancreatic cancer cells. Mol Pharm 10: 406–416.
Yamaguchi H, Kobayashi M, Okada M, Takeuchi T, Unno M, Abe T, Goto J, Hishinuma T, Mano N (2008) Rapid screening of antineoplastic candidates for the human organic anion transporter OATP1B3 substrates using fluorescent probes. Cancer Lett 260: 163–169.
Acknowledgements
We thank Cameron Ross and Sara Mansell for their technical assistance. We also thank Drs Mary Mackenzie, Ian Kerr, Michael Sanatani, Michael Ong, Mark Vincent, and Shadu Al-Khayyat and all the members of the GI Disease Site Team at the London Regional Cancer Program (LRCP). Dr RB Kim holds the Wolfe Medical Research Chair in Pharmacogenomics at Western. This study was funded by the Cancer Care Ontario (CCO) Research Chair Award (Tier-1) in Experimental Therapeutics (RB Kim). In addition, this study was conducted with the support of the Ontario Institute for Cancer Research through funding provided by the Government of Ontario.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
This work is published under the standard license to publish agreement. After 12 months the work will become freely available and the license terms will switch to a Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License.
Supplementary Information accompanies this paper on British Journal of Cancer website
Supplementary information
Rights and permissions
From twelve months after its original publication, this work is licensed under the Creative Commons Attribution-NonCommercial-Share Alike 4.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Teft, W., Welch, S., Lenehan, J. et al. OATP1B1 and tumour OATP1B3 modulate exposure, toxicity, and survival after irinotecan-based chemotherapy. Br J Cancer 112, 857–865 (2015). https://doi.org/10.1038/bjc.2015.5
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/bjc.2015.5
- Springer Nature Limited
Keywords
This article is cited by
-
Effects of the SLCO1B1 A388G single nucleotide polymorphism on the development, clinical parameters, treatment, and survival of multiple myeloma cases in a Polish population
Molecular Biology Reports (2023)
-
Irinotecan-Induced Toxicity: A Pharmacogenetic Study Beyond UGT1A1
Clinical Pharmacokinetics (2023)
-
Evaluation of pharmacogenomics and hepatic nuclear imaging–related covariates by population pharmacokinetic models of irinotecan and its metabolites
European Journal of Clinical Pharmacology (2022)
-
Hepatobiliary contrast uptake patterns on gadoxetic acid–enhanced MRI in liver metastases from pancreatic ductal adenocarcinoma: can it predict prognosis?
European Radiology (2021)
-
Hepatobiliary phase enhancement of liver metastases on gadoxetic acid MRI: assessment of frequency and patterns
European Radiology (2021)