
Abstract
The multicenter observational BiRD study investigated the real-world effectiveness and safety of ibrutinib in patients with chronic lymphocytic leukemia (CLL), mantle cell lymphoma (MCL) and Waldenström’s macroglobulinemia (WM) in Belgium. This interim analysis reports results for patients with CLL, with a median follow-up of 34 months. Overall, patients had predominantly relapsed/refractory disease (73%) and were elderly (median age 72 years) with high-risk features such as del17p and/or TP53 mutations (59%). Patients were included either prospectively or retrospectively, and the total patient population effectiveness results were adjusted with left truncation. In the effectiveness population (N = 221: prospective, n = 71; retrospective, n = 150), the overall response rate was 90.0%. Median progression-free survival was 38.3 months (prospective, not estimable; retrospective, 51.5 months) and median overall survival was not yet estimable in the total, prospective and retrospective groups. Treatment-emergent adverse events (TEAEs) for the prospective and retrospective groups are reported separately. Any-grade TEAEs of interest in the prospective/retrospective groups included infections (67.1%/60.1%), diarrhea (20.5%/10.5%), hypertension (16.4%/9.8%) and atrial fibrillation (12.3%/7.2%). Major bleeding was reported in 5.5%/3.3% of prospective/retrospective patients, with little difference observed between those receiving versus not receiving antithrombotic treatment. Discontinuations due to toxicity were reported in 10.5% of patients. Results from this interim analysis show treatment with ibrutinib to be effective and tolerable, with no new safety signals observed. Future analyses will report on longer-term follow-up.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Ibrutinib is a first-in-class, once-daily oral inhibitor of Bruton’s tyrosine kinase (BTK) approved in Europe as monotherapy for the treatment of adult patients with previously untreated and relapsed/refractory (R/R) chronic lymphocytic leukemia (CLL) [1].
Ibrutinib has demonstrated efficacy in treating patients with CLL in a number of phase 3 trials. Ibrutinib monotherapy showed improved progression-free survival (PFS) and overall survival (OS) versus chlorambucil in previously untreated patients with CLL aged ≥ 65 years (RESONATE-2™) [2], and also versus ofatumumab in patients with R/R CLL (RESONATE™) [3]. When used as a single agent or in combination with rituximab, ibrutinib therapy showed improved PFS compared with bendamustine plus rituximab in previously untreated patients with CLL aged ≥ 65 years (Alliance) [4]. It also improved PFS and OS as part of combination therapy with rituximab versus fludarabine, cyclophosphamide and rituximab (FCR) in previously untreated patients aged ≤ 70 years (E1912) [5]. Improved PFS was also shown for ibrutinib in combination with obinutuzumab versus chlorambucil plus obinutuzumab in previously untreated CLL (iLLUMINATE) [6] and in combination with bendamustine and rituximab (BR) versus placebo plus BR in patients with R/R CLL (HELIOS) [7, 8].
The Belgian ibrutinib Real-World Data study (BiRD) was designed to investigate the effectiveness and safety of ibrutinib treatment in real-world clinical practice in patients with CLL, MCL or Waldenström’s macroglobulinemia (WM) in Belgium. Results from first and second interim analyses from BiRD have been presented previously in patients with CLL and MCL [9,10,11], and some of the third interim analysis results in patients with CLL and MCL have also been presented [12]. Here we report the results for the cohort of patients with CLL from the third interim analysis of the BiRD study.
2 Patients and Methods
2.1 Study Design
BiRD is a retrospective and prospective, multicenter, non-interventional observational study of adult patients with a confirmed diagnosis of CLL, MCL or WM from 26 sites in Belgium who initiated reimbursed ibrutinib therapy on or after its commercial availability in Belgium (August 1, 2015 for R/R CLL and MCL, September 1, 2016 for WM and May 1, 2017 for first-line CLL), or who participated in the Medical Need Program for CLL, WM or MCL, or received free of charge ibrutinib as a single-patient request for WM and switched to reimbursed ibrutinib. Patients who initiated reimbursed ibrutinib therapy prior to the inclusion visit were eligible for retrospective enrollment in the study, regardless of whether the ibrutinib therapy was ongoing at the time of inclusion (retrospective inclusion). Patients who had begun ibrutinib therapy at the time of inclusion in the study were added prospectively. This is the third of four planned interim analyses. The first occurred after the last patient was enrolled, the second after 18 months, this present and third at 3 years and the fourth is planned for early 2023. However, ad hoc interim analyses may also be conducted by the sponsor, based on specific questions from the medical field or from health authorities. Only patients with CLL are included in this analysis, and the cutoff date was March 31, 2020.
Each patient was treated according to the standard of care; their decision to take part in the observational study did not influence their medical care. Therapy decisions were made at the discretion of the participating physician, according to routine clinical practice.
Data were collected every 3 months during the first year and every 6 months thereafter for a period of 5 years. Data were collected for ibrutinib treatment until discontinuation, after which the follow-up period for response started with any subsequent therapy. If patients received no further treatment, they were followed up for survival only. Safety data were collected for some patients who began ibrutinib before inclusion and yet continued with ibrutinib therapy; therefore, there was a retrospective/prospective safety data collection period for these patients—defined within the retrospective group as “before inclusion” and “after inclusion.”
The study protocol was approved by the institutional review boards or independent ethics committees of participating centers. All patients received oral and written information on the study and provided informed consent to data collection and source data verification.
2.2 Inclusion and Exclusion Criteria
Patients had to be aged ≥ 18 years, with a confirmed diagnosis of CLL, and eligible for reimbursed ibrutinib treatment, according to the National Institute for Health and Disability Insurance (RIZIV/INAMI). This was both before and after the commercial availability of ibrutinib and, therefore, also included patients participating in a Medical Need Program who were switched to reimbursed ibrutinib treatment when it became available (August 1, 2015). Previously untreated patients without del17p or TP53 mutation who were fit to receive fludarabine-based regimens were not eligible for ibrutinib treatment according to Belgian treatment guidelines at the start of the study. Reimbursement criteria for CLL are based on the International Workshop on Chronic Lymphocytic Leukemia (iwCLL) criteria [13] and include del17p or TP53 mutation or R/R disease. Patients were excluded if they were participating in another investigational or clinical study or in any expanded access program during the ibrutinib treatment period covered by this observational study.
2.3 Outcome Measures
The primary outcome measures were investigator-assessed overall response rate (ORR) and PFS. Secondary outcome measures included OS and safety, which includes treatment exposure and treatment-emergent adverse events (TEAEs).
PFS was defined as the time from the start of ibrutinib treatment to progression or death from any cause. OS was defined as the time in months from initiation of ibrutinib to death. ORR was the sum of complete response (CR), partial response (PR) and partial response with lymphocytosis, as assessed by the investigator. Although confirmation of CR by minimal residual disease analysis of bone marrow was possible, it is not routinely performed in real-world practice, and this definition of CR should be considered unconfirmed in most cases. After discontinuation of ibrutinib therapy, during the treatment-free period, patients were followed for response or disease progression every 6 months ± 2 weeks. If no visit was conducted within this recommended period, data from the visit conducted as close as possible to the protocol-specified time point were recorded.
All TEAEs were collected prospectively, whereas only adverse drug reactions considered related to ibrutinib were collected retrospectively.
2.4 Statistics
This study describes everyday ibrutinib treatment practice for CLL. The effectiveness population included all patients who met the inclusion criteria and who received at least one dose of ibrutinib and the safety population included all patients who received at least one dose of ibrutinib.
All time-to-event variables were analyzed using standard survival analysis methods, including Kaplan–Meier product-limit survival curve. The median time to event with two-sided 95% confidence intervals (CIs) was estimated. All continuous variables were summarized using descriptive statistics, which included the number of patients, mean, standard deviation, median, interquartile range (IQR) minimum and maximum and 95% CI. Data were reported for the prospective, retrospective and/or total patient populations for the effectiveness population. Owing to the differences in adverse event collection between the retrospective and prospective groups, these data are presented separately and are all referred to as “TEAEs” for ease of notation. The number and percentage of patients who experienced at least one occurrence of each TEAE (presented as preferred term and categorized by system organ class) were summarized. TEAEs were not graded but were summarized by severity for serious and severe TEAEs, according to the worst experienced. Patients who died before enrollment in the trial could not be included according to Belgian law, as no informed consent could be obtained. For this reason, the total population effectiveness results were adjusted with left truncation, which reduces part of the follow-up time of the retrospective patients to avoid any mortality bias. However, this shortened the median time to event and, as the retrospective group is larger than the prospective group, this also affected the median time to event of the combined analysis.
3 Results
3.1 Patient Demographics and Baseline Characteristics
A total of 221 evaluable patients with CLL were included in this interim analysis for effectiveness (retrospective, n = 150; prospective, n = 71). More than 80% of patients initiated reimbursed ibrutinib, with the remainder participating in the Medical Need Program. The median age at ibrutinib initiation was 72 (range 38–90) years, 64.3% of patients were male, 86.6% had an Eastern Cooperative Oncology Group performance status of 0–1, and 17.8% had a history of significant cardiovascular (CV) disease. Ongoing atrial fibrillation (AF) and other ongoing CV disease were both reported by 14/38 patients. Table 1 details the patient characteristics at baseline.
The median time from diagnosis to ibrutinib initiation was 5.8 years and 72.6% patients had at least one prior line of therapy. In total, 78 of 132 assessed patients (59.1%) had del17p and/or TP53 mutation, and 72.5% of only 80 patients assessed had unmutated IGHV. In line with reimbursement criteria, 43.6% of patients with del17p and/or TP53 mutations had no prior lines of therapy, whereas 71.7% of patients without del17p and TP53 mutations had at least one prior line of therapy.
The majority of patients (88.1%) initiated ibrutinib therapy at the recommended dose of 420 mg daily.
3.2 Effectiveness
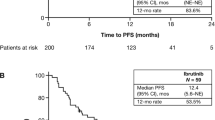
The median (range) follow-up was 34.3 (0.2–69.9) months in the total population. Table 2 details the main effectiveness outcomes. The cumulative ORR (best response at 60 months) was 90.0% (CR 16.7%; PR 51.6%; PR with lymphocytosis, 21.7%). The median (Q1–Q3) PFS for CLL was 38.3 (12.2–66.4) months for the total population, not estimable (NE) (15.7–NE) in the prospective group and 51.5 (29.1–NE) months in the retrospective group (Fig. 1). PFS was longer in patients without del17p and TP53 mutations than in those with del17p and/or TP53 mutations (49.9 versus 41.5 months; Table 2). There was little difference in PFS in patients with mutated or unmutated IGHV (49.9 versus 53.5 months). PFS varied slightly by line of therapy, but the variation did not follow any particular pattern. PFS was longer in patients younger than 65 years compared with those older than 65 years (49.5 versus 36.8 months). Median (Q1–Q3) OS was not estimable (21.3–NE) in the total population and was also not estimable in the prospective and retrospective groups (Fig. 2). The 36-month OS rate was 62.9% overall and 60.6% and 76.3% in the prospective and retrospective groups, respectively. Median time to next treatment was 35.7 months overall and not estimable and 51.5 months in the prospective and retrospective groups, respectively. The numbers obtained for the total population are a result of the statistical analysis used, as detailed in the methods.

a PFS for total population corrected with left truncation. b PFS for prospective and retrospective populations

a OS for total population corrected with left truncation. b OS for prospective and retrospective populations
3.3 Safety
There were 226 patients in the safety analysis population, 73 in the prospective group and 153 in the retrospective group. The median (range) duration of ibrutinib treatment in the total safety population was 25.5 (0.1–70.0) months; it was longer in the retrospective (28.6 [0.4–70.0]), than in the prospective group (19.6 [0.1–45.1]) months. At treatment initiation, most patients (88.1%) were prescribed a daily dose of 420 mg ibrutinib and this was comparable in the prospective and retrospective groups (88.9% and 86.3%, respectively).
A total of 123 (54.4% of 226) patients discontinued treatment during the analysis period, 38 in the prospective and 85 in the retrospective group. The overall reasons for discontinuation, when given, were disease progression (n = 29; 12.8%), toxicity (n = 26; 11.5%), death (n = 21; 9.3%), other (n = 18; 8.0%), comorbidities (n = 6; 2.7%), physician preference (n = 5; 2.2%) and one each (0.4%) of concomitant medication, patient preference and surgery. None of the deaths were reported as sudden cardiac deaths, and the one cardiac-related death (cardiac arrest) reported was considered not related to treatment by the investigator. Only two deaths were considered very likely related to ibrutinib by the investigators, pneumonia and aspergillus infection.
As TEAEs consisting of all events related and unrelated to treatment that occurred during the study were reported for the prospective group, and only adverse events considered related to ibrutinib were reported for the retrospective group, they are reported separately for the two patient groups. TEAEs of interest are detailed in Table 3. For the prospective and retrospective groups, respectively, 100% and 83.7% of patients had at least one TEAE, while 58.9% and 43.8% of patients had at least one serious TEAE. In the prospective and retrospective groups, respectively, TEAEs led to ibrutinib dose reduction in 21.9% and 10.5% of patients, dose interruption in 39.7% and 23.5%, and withdrawal (which includes any discontinuations related or unrelated to ibrutinib therapy, including death, toxicity, comorbidities, progressive disease and physician preference) in 26.0% and 19.6%.
Some patients were taking concomitant antithrombotic therapy during the study, 28 in the prospective and 42 in the retrospective group. Bleeding was more frequent in patients on such therapy compared with those not, 78.6% versus 31.1% in the prospective and 42.9% versus 25.2% in the retrospective group, and major bleeding was infrequent, 5.5% in the prospective group and 3.3% in the retrospective group. The most common antithrombotic therapy was an antiplatelet agent, 46.4% in the prospective group and 45.2% in the retrospective group, followed by a non-vitamin K oral anticoagulant, 10.7% in the prospective group and 21.4% in the retrospective group, vitamin K antagonist, none in the prospective group and 4.8% in the retrospective group, or other therapy, 14.2% in the prospective group and 9.5% in the retrospective group. More than one antithrombotic treatment was used by 28.6% of patients taking antithrombotic therapy in the prospective group and by 19.0% in the retrospective group; this included patients who switched therapies and, therefore, these patients were not all taking several antithrombotic agents concomitantly.
4 Discussion
BiRD is an ongoing real-world study that aims to evaluate the effectiveness and safety of ibrutinib in patients with CLL in Belgium. Those enrolled in BiRD represent the clinical spectrum of a real-world CLL population, as they were mostly elderly (median age 72 years), and approximately three quarters had been treated previously. Almost half of patients with del17p and/or TP53 mutations had no prior lines of therapy, yet nearly three quarters of patients without del17p and TP53 mutations had at least one prior line of therapy, which is driven by the indication and reimbursement criteria in Belgium. It is encouraging that the vast majority of patients (88%) in this real-world population in normal clinical practice received the recommended dose of ibrutinib at treatment initiation. As many patients were included during the early years of ibrutinib market authorization, it is possible that physicians were initially cautious with dosing, starting at a dose lower than recommended, and titrating up to ensure tolerability.
In this third interim analysis, the median follow-up for effectiveness was 34.3 months although there was a wide range, up to 70 months. The cumulative ORR up to 60 months was 90.0%, a finding similar to the ORR reported in the second interim analysis at 12 months (86.9%) [9], and also similar to some clinical trials with high-risk populations. In the RESONATE™ trial of single-agent ibrutinib in patients with R/R CLL, which had a comparable percentage of high-risk patients (32% with del17p, 51% with TP53 mutation; 86% considered high risk in the ibrutinib arm), the cumulative ORR for ibrutinib at the 6-year follow-up was 91% [14], and in a phase 2 study with high-risk patients (63% with del17p and/or TP53 mutation), the ORR for ibrutinib was 96% at a follow-up of 6 months [15]. High rates of response have also been noted with ibrutinib treatment in clinical trials without such a high-risk population, such as the RESONATE-2™ study in previously untreated patients without del17p over the age of 65, which reported an ORR of 92% with up to 66 months follow-up [16]. Other real-world studies have also produced similar levels of response to ibrutinib, including the FIRE real-world study, which is a similar design to our BiRD study, yet conducted in France. FIRE reported an ORR of 89.6% in the overall CLL patient group at a follow-up of 21.6 months in a population of patients with 58.7% del17p and/or TP53 mutations [17]. In addition, a Danish retrospective cohort study in 205 patients, 72.1% of whom had del17p or TP53 mutations, with a median follow-up of 21.4 months, reported an ORR of 76.4% [18], and an analysis of 95 Swedish patients, 62% with del17p or TP53 mutations, treated with ibrutinib in a compassionate use program reported an ORR of 84% [19].
In our study, the median PFS was 38.3 months for the total population, which is unchanged from the second interim analysis at a median follow-up of 20.9 months [9]. Interestingly, the PFS values for the separate prospective and retrospective groups were both longer than this value (NE and 51.5 months, respectively), which is a result of the statistical analysis on the total cohort. As those patients in the retrospective group who died prior to study entry could not be included because they could not provide consent, an adjustment was made with left truncation to avoid bias in the retrospectively included patients on the total population analysis. As the retrospective group is larger than the prospective group of patients, this impacts the results for the total CLL cohort. The PFS values reported in our analysis are all within the range of those reported in clinical trials, such as RESONATE™, with a PFS of 44.1 months at 6 years’ follow-up [14], and RESONATE-2™, with a PFS not estimable after a median follow-up of 60 months [16]. In the FIRE real-world study of similar design, the overall PFS at median follow-up of 21.6 months was 37.7 months, and was not estimable in either the prospective or retrospective patient groups at 15.2 and 29.6 months’ median follow-up, respectively [17]. In the Danish real-world study, the median PFS was 41.2 months [18].
PFS was shorter in patients with del17p and TP53 mutations (42 months compared with 50 months), yet this compares favorably with the PFS of 11 months reported for FCR treatment in previously untreated patients with TP53 alterations after 5 years’ follow-up [20]. Poor responses to chemotherapy in these high-risk patients have been reported previously [21], yet ibrutinib therapy is associated with durable responses [15, 22]. In patients with mutated or unmutated IGHV, the PFS was similar, as reported previously for ibrutinib [16]; however, only 60% of patients in our study underwent cytogenetic testing for del17p or TP53 and even fewer (36%) were tested for IGHV (it must be noted, though, that IGHV testing was not reimbursed for patients over 65 years of age in Belgium at the start of the study, and is currently only reimbursed for patients eligible for chemoimmunotherapy treatment). This highlights a potentially important gap in testing, a finding also noted in another real-world study in the United States [23], which could increase the likelihood of patients not receiving optimal treatment in clinical practice. Cytogenetic testing is now becoming more common, and this will help to improve treatment choices.
As expected, younger patients (aged < 65 years) had longer PFS than those aged ≥ 65 years, and median PFS varied slightly by number of previous lines of therapy. This variation did not follow any particular pattern, partly due to low patient numbers in each group and the long PFS values at this time of follow-up. Interestingly, this non-correlation of PFS with prior therapy line was also noted in a retrospective US real-world cohort analysis [24]. Patients were enrolled into our study at the beginning of ibrutinib use, when practice policies were adapted, and it is therefore likely that early recruits were of higher risk than later ones, and that better results may be expected with longer follow-up, particularly in previously untreated patients.
The median OS in our study was not estimable, as also reported in several other studies at time points shorter than several years [7, 8, 16, 17]. The estimated 36-month OS rate of 62.9% is in the expected range, when considering the OS rate of 63% at 30 months in the Swedish cohort study [19] and in other real-world studies reporting at earlier time points: the Danish cohort study showed an estimated OS of 76.8% at 24 months [18], and the FIRE study estimated a value of 86.9% at the same 24-month time point [17]. It is likely that further follow-up is needed to determine additional information on survival.
In the safety analysis with up to 70 months of follow-up, fewer TEAEs were reported in the retrospective than in the prospective group, as only events considered related to ibrutinib were reported retrospectively. In general, TEAEs were in the range of those previously reported in real-world studies, including FIRE [17], the Denmark cohort [18] and the Swedish cohort [19], and within the range of single-agent ibrutinib clinical trials of CLL [2, 3, 14]. Patients treated in a real-world setting, however, may be expected to have higher rates of TEAEs and treatment discontinuation than those in a clinical trial setting, because a real-world population includes higher risk patients who might be excluded from clinical trials. The rate of discontinuations due to toxicity observed in our study (11.5%) was lower than that reported in other real-world studies (20% in the Swedish retrospective study [19], 22.9% in the Danish retrospective study [18], 20.8% in a US cohort study [24] and 17.5% in a large UK real-world study from 2016 of 315 patients after 16 months median follow-up [25]). These older studies, however, may have included higher-risk populations in the earlier days of ibrutinib use, particularly with compassionate use, potentially leading to higher discontinuations. The frequency of bleeding in our study was similar to that experienced in clinical trials and real-world studies [7, 8, 16, 17]. Bleeding was more frequent in patients receiving antithrombotic therapy, but major bleeding events were too infrequent to determine any correlation. The frequency of TEAEs varied little from the second [10] to the third interim analysis of BiRD, which is to be expected as TEAEs are reported by actual study time and not by treatment course per patient. It is reassuring that no cardiac deaths or sudden deaths were reported during this long follow-up period. Overall, treatment with ibrutinib was tolerable and no new safety signals were observed.
There are a number of limitations to consider when evaluating the results from this analysis. Unlike clinical trials, the effectiveness and safety parameters are presented through descriptive data in a real-world setting and assessed by the investigators. The response was assessed by physicians in routine clinical practice, and therefore comparison with clinical trials is challenging. The differences between the retrospective and prospective populations are evident, and adjustments to the overall population aimed to reduce bias, although bias cannot be entirely discounted.
This large, real-world study gives a unique perspective of ibrutinib use in a single country, encompassing most major hematology centers in Belgium. It provides information on the long-term use of ibrutinib in CLL in real-world clinical practice, with up to 7 years (median 34 months) follow-up. It is promising to see that results from randomized trials in more restricted populations have translated into real world with ibrutinib.
5 Conclusions
Results from this large, real-world study show ibrutinib to be effective in this mostly elderly CLL population, with results in alignment with those from randomized controlled trials and with other real-world studies. TEAEs were in line with those from other studies, few patients discontinued due to toxicity and no new safety signals were evident. We await further follow-up from the BiRD study to confirm the longer-term effectiveness and safety of ibrutinib.
Availability of Data and Material
The data-sharing policy of Janssen Pharmaceutical Companies of Johnson & Johnson is available at https://www.janssen.com/clinical-trials/transparency. As noted on this site, requests for access to the study data can be submitted through Yale Open Data Access (YODA) Project site at http://yoda.yale.edu.
Abbreviations
- AF:
-
Atrial fibrillation
- BiRD:
-
Belgian Ibrutinib Real-World Data
- BR:
-
Bendamustine and rituximab
- BTK:
-
Bruton’s tyrosine kinase
- CI:
-
Confidence interval
- CLL:
-
Chronic lymphocytic leukemia
- CR:
-
Complete response
- CV:
-
Cardiovascular
- ECOG PS:
-
Eastern Cooperative Oncology Group performance status
- FCR:
-
Fludarabine, cyclophosphamide and rituximab
- IQR:
-
Interquartile range
- iwCLL:
-
International Workshop on Chronic Lymphocytic Leukemia
- LOT:
-
Line of therapy
- MCL:
-
Mantle cell lymphoma
- NE:
-
Not estimable
- ORR:
-
Overall response rate
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- PR:
-
Partial response
- R/R:
-
Relapsed/refractory
- TEAE:
-
Treatment-emergent adverse event
- WM:
-
Waldenström’s macroglobulinemia
References
European Medicines Agency. Ibrutinib summary of product characteristics. 2021. https://www.ema.europa.eu/en/documents/overview/imbruvica-epar-medicine-overview_en.pdf. Accessed Oct 2022.
Burger JA, Tedeschi A, Barr PM, Robak T, Owen C, Ghia P, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425–37. https://doi.org/10.1056/NEJMoa1509388.
Byrd JC, Brown JR, O’Brien S, Barrientos JC, Kay NE, Reddy NM, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213–23. https://doi.org/10.1056/NEJMoa1400376.
Woyach JA, Ruppert AS, Heerema NA, Zhao W, Booth AM, Ding W, et al. Ibrutinib regimens versus chemoimmunotherapy in older patients with untreated CLL. N Engl J Med. 2018;379(26):2517–28. https://doi.org/10.1056/NEJMoa1812836.
Shanafelt TD, Wang XV, Kay NE, Hanson CA, O’Brien S, Barrientos J, et al. Ibrutinib-rituximab or chemoimmunotherapy for chronic lymphocytic leukemia. N Engl J Med. 2019;381(5):432–43. https://doi.org/10.1056/NEJMoa1817073.
Moreno C, Greil R, Demirkan F, Tedeschi A, Anz B, Larratt L, et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(1):43–56. https://doi.org/10.1016/s1470-2045(18)30788-5.
Chanan-Khan A, Cramer P, Demirkan F, Fraser G, Silva RS, Grosicki S, et al. Ibrutinib combined with bendamustine and rituximab compared with placebo, bendamustine, and rituximab for previously treated chronic lymphocytic leukaemia or small lymphocytic lymphoma (HELIOS): a randomised, double-blind, phase 3 study. Lancet Oncol. 2016;17(2):200–11. https://doi.org/10.1016/S1470-2045(15)00465-9.
Fraser G, Cramer P, Demirkan F, Silva RS, Grosicki S, Pristupa A, et al. Updated results from the phase 3 HELIOS study of ibrutinib, bendamustine, and rituximab in relapsed chronic lymphocytic leukemia/small lymphocytic lymphoma. Leukemia. 2019;33(4):969–80. https://doi.org/10.1038/s41375-018-0276-9.
Janssens A, André M, Berneman Z, Snauwaert S, De Beleyr B, Smet A, et al. Effectiveness and safety of ibrutinib for chronic lymphocytic leukemia (CLL) in routine clinical practice: interim analysis (IA) of the Belgian ibrutinib real-world data (BIRD) study. In: Presented at: 24th Congress of the European Hematology Association (EHA), June 13–16, 2019; Amsterdam, the Netherlands. Poster PF384.
Janssens A, Berneman Z, Bron D, Snauwaert S, De Beleyr B, Smet A et al. Clinical outcomes with single-agent ibrutinib for relapsed/refractory (R/R) mantle cell lymphoma (MCL): interim analysis (IA) of the Belgian ibrutinib Real-World Data (BiRD) study. In: Presented at: 24th Congress of the European Hematology Association (EHA), June 13–16, 2019; Amsterdam, the Netherlands. Poster PF494
Janssens A, Snauwaert S, Wapenaar R, Van Bogaert C, Smet A, Seynhaeve V. Use of ibrutinib for chronic lymphocytic leukemia in routine clinical practice: results from the Belgian ibrutinib Real-World Data (BiRD) study. In: Presented at: 33rd General Annual Meeting of the Belgian Hematology Society, February 2–3, 2018; Brussels, Belgium. Abstract P33
Janssens A, André M, Berneman Z, Snauwaert S, De Beleyr B, Smet A et al. Effectiveness and safety of ibrutinib in chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL) in Belgian routine clinical practice with a 3-year follow-up. In: Presented at: EHA 2021 Virtual, Poster EP654
Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood. 2008;111(12):5446–56. https://doi.org/10.1182/blood-2007-06-093906.
Munir T, Brown JR, O’Brien S, Barrientos JC, Barr PM, Reddy NM, et al. Final analysis from RESONATE: up to six years of follow-up on ibrutinib in patients with previously treated chronic lymphocytic leukemia or small lymphocytic lymphoma. Am J Hematol. 2019;94(12):1353–63. https://doi.org/10.1002/ajh.25638.
Ahn IE, Farooqui MZH, Tian X, Valdez J, Sun C, Soto S, et al. Depth and durability of response to ibrutinib in CLL: 5-year follow-up of a phase 2 study. Blood. 2018;131(21):2357–66. https://doi.org/10.1182/blood-2017-12-820910.
Burger JA, Barr PM, Robak T, Owen C, Ghia P, Tedeschi A, et al. Long-term efficacy and safety of first-line ibrutinib treatment for patients with CLL/SLL: 5 years of follow-up from the phase 3 RESONATE-2 study. Leukemia. 2020;34(3):787–98. https://doi.org/10.1038/s41375-019-0602-x.
Dartigeas C, Feugier P, Ysebaert L, Dilhuydy M-S, Delmer A, Tardy S et al. French Ibrutinib Observational Study (FIRE): real-world study of ibrutinib treatment for chronic lymphocytic leukemia (CLL) in France. In: Presented at: 24th Congress of the European Hematology Association (EHA), June 13–16, 2019; Amsterdam, the Netherlands. Poster PF387
Aarup K, Rotbain EC, Enggaard L, Pedersen RS, Bergmann OJ, Thomsen RH, et al. Real-world outcomes for 205 patients with chronic lymphocytic leukemia treated with ibrutinib. Eur J Haematol. 2020;105(5):646–54. https://doi.org/10.1111/ejh.13499.
Winqvist M, Andersson PO, Asklid A, Karlsson K, Karlsson C, Lauri B, et al. Long-term real-world results of ibrutinib therapy in patients with relapsed or refractory chronic lymphocytic leukemia: 30-month follow up of the Swedish compassionate use cohort. Haematologica. 2019;104(5):e208–10. https://doi.org/10.3324/haematol.2018.198820.
Fischer K, Bahlo J, Fink AM, Goede V, Herling CD, Cramer P, et al. Long-term remissions after FCR chemoimmunotherapy in previously untreated patients with CLL: updated results of the CLL8 trial. Blood. 2016;127(2):208–15. https://doi.org/10.1182/blood-2015-06-651125.
Gentile M, Zirlik K, Ciolli S, Mauro FR, Di Renzo N, Mastrullo L, et al. Combination of bendamustine and rituximab as front-line therapy for patients with chronic lymphocytic leukaemia: multicenter, retrospective clinical practice experience with 279 cases outside of controlled clinical trials. Eur J Cancer. 2016;60:154–65. https://doi.org/10.1016/j.ejca.2016.03.069.
Allan JN, Shanafelt TD, Wiestner A, Moreno C, O'Brien S, Braggio E, et al. Long-term efficacy of first-line ibrutinib treatment for chronic lymphocytic leukemia with 4 years of follow-up in patients with TP53 aberrations: a pooled analysis from 4 clinical trials. In: Presented at: 2nd ASH Annual Meeting and Exposition, December 5–8, 2020; Virtual. Poster 2219
Mato A, Barrientos J, Sharman J, Brander D, Gutierrez M, Karen K, et al. Real-world prognostic biomarker testing, treatment patterns and dosing among 1461 patients (pts) with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) from the informCLL prospective observational registry. Blood. 2020;136:42–3. https://doi.org/10.1182/blood-2020-133798.
Mato AR, Nabhan C, Thompson MC, Lamanna N, Brander DM, Hill B, et al. Toxicities and outcomes of 616 ibrutinib-treated patients in the United States: a real-world analysis. Haematologica. 2018;103(5):874–9. https://doi.org/10.3324/haematol.2017.182907.
UK CLL Forum. Ibrutinib for relapsed/refractory chronic lymphocytic leukemia: a UK and Ireland analysis of outcomes in 315 patients. Haematologica. 2016;101(12):1563–72. https://doi.org/10.3324/haematol.2016.147900.
Acknowledgements
The authors would like to thank all the patients who were included in this analysis. Writing assistance was provided by Emma Fulkes, Ph.D., of Parexel, and was funded by Janssen Research & Development. Statistical support was provided by Simon Paternotte of ICTA and funded by Janssen Research and Development.
Funding
This study was sponsored by Janssen Pharmaceutica NV, and Pharmacyclics LLC, an AbbVie Company. Writing assistance was provided by Emma Fulkes of Parexel and funded by Janssen Global Services, LLC.
Author information
Authors and Affiliations
Contributions
All authors reviewed and approved the manuscript. AJ, MA, BDB and CVB also provided study design and RW also provided data analysis.
Corresponding author
Ethics declarations
Conflict of Interest
AJ reports consultancy for Janssen, Roche, Gilead, AbbVie, Novartis, Amgen, Sanofi-Genzyme and Celgene; research grant from Janssen; travel grant from Janssen, Celgene, AbbVie and Roche. ZNB reports research grants from Janssen, Roche, Takeda, CAF-DCF. SM reports consultancy for Janssen. DB reports consultancy and research grant from Janssen. FO reports research grants from Janssen. MA reports consultancy for Takeda, Bristol Myers Squibb, Karyopharm, Gilead and Incyte; research grants from Roche, Johnson & Johnson and Takeda; travel grants from Roche, Bristol Myers Squib, Celgene, Gilead and AbbVie. CVB, BDB, AS, LN and RW are employees of Janssen-Cilag. BDB reports stock ownership from Johnson & Johnson and Pfizer. RW reports stock ownership from Johnson & Johnson. IS, GV, PM, SS and IVB ZB have no conflicts to report.
Ethical Approval
The study protocol was approved by the institutional review boards or independent ethics committees of participating centers. All patients received oral and written information on the study and provided informed consent to data collection and source data verification.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Janssens, A., Berneman, Z.N., Offner, F. et al. Effectiveness and Safety of Ibrutinib for Chronic Lymphocytic Leukemia in Routine Clinical Practice: 3-Year Follow-up of the Belgian Ibrutinib Real-World Data (BiRD) Study. Clin Hematol Int 4, 133–143 (2022). https://doi.org/10.1007/s44228-022-00020-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s44228-022-00020-8