
Abstract
A completely new direct voltammetric method has been developed for determination of acetaminophen (APAP), known as popular analgesic drug. The present electroanalytical method is based on anodic oxidation of APAP at the glassy carbon paste electrode modified with reduced graphene oxide (RGO). Key experimental conditions were studied, resulting in a set of optimal conditions: acetate buffer (pH 5.0) as working medium electrolyte, content of RGO, parameters of squarewave voltammetry including the potential step of 5 mV, potential amplitude of 50 mV, and frequency of 40 Hz. If peak area is used for evaluation, a linear range from 1.2 × 10–6 to 2.2 × 10–4 mol L−1 characterized by determination coefficient of 0.9971, limits of quantification and detection, 9.3 × 10–7 mol L−1 and 3.1 × 10–7 mol L−1, respectively, will be obtained. Under validation, the precision was described by relative standard deviation of 2.9% for the model sample analysis. Finally, the developed voltammetric method was compared with a reference high-performance liquid chromatography method in the analysis of commercially available pharmaceutical preparation and human urine collected from five healthy volunteers.
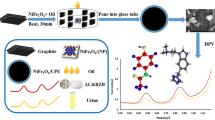
Graphical abstract

Similar content being viewed by others

Introduction
Acetaminophen (APAP), also known as paracetamol, represents the most common analgesic used worldwide and recommended as the first-line therapy in pain conditions by the World Health Organization (WHO) [1]. This medicament can be also used for its antipyretic effect, helping to reduce fever [2], being available in a variety of forms including a syrup form, regular tablet, effervescent tablet, injection, suppository, and other forms [3].
APAP is the major metabolite of phenacetin and acetanilide [2] and mainly metabolized in the liver by 1st order kinetics by three different pathways: conjugation with glucuronide, conjugation with sulfate, and oxidation through the cytochrome P450 enzyme pathway, mainly CYP2E1 to produce a reactive metabolite (N-acetyl-p-benzoquinone imine; NAPQI). High doses of APAP (overdoses) can lead to a hepatic necrosis due to the depletion of glutathione and binding of high levels of the reactive metabolite NAPQI to important parts of liver cells. The afore-mentioned damage of liver can be prevented by the early administration of sulfhydryl compounds, e.g., methionine or N-acetylcysteine [4]. Less than 5% of the total amount is excreted in urine as free (unconjugated) APAP and 90% of the administered dose is excreted within 24 h [2]. In other literature [5], it is possible to learn that about 2% APAP is excreted unchanged; therefore, for 250 mg dose of APAP recommended for children, a minimum detected concentration of ~ 30 μmol per liter of urine might be expected.
Regarding all the facts mentioned, determination of APAP at various concentration levels is of great clinical importance. A wide variety of analytical methods have been developed for identification and quantification of APAP in real samples (biological fluids and pharmaceutical preparations), employing colorimetry [6], various chromatographic [7,8,9,10], or electroanalytical (voltammetric) methods [11,12,13,14,15,16]. In addition, several amperometric biosensors, utilizing aryl-acylamidase, horseradish peroxidase or tyrosinase have been developed [5, 17,18,19].
Graphene, a special 2D-form of carbon (with atoms in the sp2 bonds), has captured great attention in the development of modified electrochemical electrodes because of its large surface area, high electrical conductivity, almost perfect capacitance and ingenuity [20, 21]. It can give rise to graphene oxide (GO) and its reduced form (RGO); the latter being particularly suitable for electrochemical sensors due to a higher conductivity than the original GO and preparable by chemical [22] or even electro-chemical [23] reduction.
This contribution presents a new voltammetric method for the determination of APAP in tablet dosage forms and human urine which is based on anodic oxidation of APAP to NAPQI with the participation of two protons and two electrons [24] at glassy carbon paste electrode (GCPE) modified with 10% (w/w) RGO in acetate buffer of pH 5. Its principles, optimization of key experimental conditions, as well as all important electroanalytical characteristics with a validation by reference method based on reversed-phase high-performance liquid chromatography with spectrophotometric detection (RP–HPLC/UV–Vis) [25, 26], are described in the following sections.
Experimental
Reagents and chemicals
Analytical standards of ≥ 99% acetaminophen, ≥ 99.8% acetic acid, ≥ 85% phosphoric acid, ≥ 99.5% boric acid, and a 10 gm commercial package of reduced graphene oxide (RGO) were purchased from Sigma–Aldrich (Cairo, Egypt). Other reagents, namely: ascorbic acid, ≥ 99% uric acid, sodium acetate, and sodium hydroxide (all p.a. grade) were purchased from Lach-Ner (Neratovice, Czech Republic). Ultrapure water was used throughout the work when obtained by passing of the already deionized water through a purification unit ("Milli-Q" system; Merck-Millipore, Darmstadt, Germany).
Preparation and pretreatment of the glassy-carbon paste electrode
The glassy-carbon paste electrode, serving as a substrate for the proper electrode (with the layer of RGO), was prepared by thoroughly hand-mixing of 70%(v/v) glassy carbon micro particles, 20%(v/v) paraffin oil, and 10%(v/v) reduced graphene oxide.
The resultant paste-like mixture was packed into a piston-driven carbon paste holder with Teflon® body (and a cavity of 20 mm in diameter). Whenever needed, the paste in the filled body could be extruded and the active surface renewed by smoothing it with a wet filter paper until a shiny appearance. Then, the working electrode of RGO/GCPE type was ready for electrochemical measurement(s).
Instrumentation and accessories
All electrochemical measurements were carried out in a 50 mL glass cell fixed in mounted head with three-electrode system. It consisted of the working electrode (RGO/GCPE if not stated otherwise), Ag/AgCl/3 M KCl reference and Pt-sheet auxiliary electrodes; all being in connection with an electrochemical analyser (model PGSTAT101, Autolab-Metrohm; Prague, Czech Rep.) that had been controlled by the software ("Nova 1.11" version, Autolab). Whenever needed, the pH values were measured with a pH meter, the glass electrode, and a series of the standards for calibration in an interval of pH 1–11 (all from Metrohm; Prague, Czech Republic).
Square-wave voltammetry
Square-wave voltammetry (SWV) of the analyte of interest provided a single well-developed oxidation peak at about + 0.5 V vs. ref. in an acetate buffer (AcB, pH 5), when applying a potential scan from 0.0 to + 1.0 V at a potential step of 2.5 mV, potential amplitude of 25 mV, and a frequency 40 Hz. The quantification of APAP in tablet dosage forms and in real samples was performed using the calibration-curve method (in an interval of 4–220 μmol L−1 APAP). Each analysis was repeated minimally five times and the respective results are shown as confidence intervals.
Analysis of the tablet dosage form
The calibration curve from 20 to 100 μmol L−1 was used to determine APAP amount in the tablets of a commercial product ("Panadol®500 mg", purchased in an Egyptian pharmacy); each being grinded in a ceramic mortar, mixed, and accurately weighed as the amount equivalent to 1.0 × 10−2 M APAP after dissolution in a 250 mL distilled water. Afterwards, the solution prepared was sonicated for 30 min, filtered, and different dilutions within the corresponding linear range prepared.
An excellent recovery of 99.3% was achieved, indicating the proper accuracy and reliability of the new method and its suitability to quantify APAP in tablet dosage forms and related samples in a simple and rapid way.
Analysis of the urine real sample
As already mentioned, the real sample was taken from five volunteers. More specifically, the sample was collected after 4-h ingestion when the volunteers had taken a pill containing a dosage of 500 mg APAP. The procedure described in detail in the Experimental section was applied in five replicates. Typical voltammograms of the respective analysis are illustrated in Fig. 1S.
Reference chromatographic method
Chromatographic analysis in the reverse-phase HPLC mode and with gradient elution was performed with a system comprising LC-20ADXR binary gradient pump, DGU-20 degassing unit, an SPD-M30A DAD, an SIL-20 AC XR autosampler (all from Shimadzu, Kyoto, Japan), and LCO 102 column thermostat (Ecom, Prague, Czech Rep.). A C18 column ("Ascentis Express" type; 150 mm × 3.0 mm, 2.7 μm) was chosen for achieving the optimum separation at a detection wavelength of 243 nm. The mobile phase composition was 0.3% formic acid in water (A) and methanol (B) with a gradient-elution program from 20 to 40% B for 10 min and at constant temperature of 30 °C. The flow rate was 0.5 mL min−1 and injection volume of 5 μL.
Statistical evaluation
All analyses of selected samples were always made in five replicates (n = 5) and the final results calculated and presented as the confidence intervals x̄ ± st1−α (where x̄ is the arithmetic mean, s the standard deviation, and t1−α the critical value 2.015 of Student's t distribution for five repetitions at a significance level α = 0.05 (with 95% probability).
Results and discussion
Electrochemical behaviour of acetaminophen
The aim of work is to present a time saving with simple steps procedure with considerable accuracy and sensitivity that can be applied successfully on real biological samples to have an advantage over already published methods dealing with electrochemical determination of APAP using RGO as they include multi-step and complicated working electrode preparation procedures requiring up to 4 h and limited to spiked samples only [27,28,29,30,31]. Initial SWV measurements were performed in a test solution of 100 μmol L−1 APAP in AcB (pH 5.0) with a series of electrodes, namely, the bare glassy carbon electrode (GCE, a commercial product by Metrohm), bare glassy-carbon paste electrode and its two modified variants: either with multi-walled carbon nanotubes (GCPE and MWCNT/GCPE, resp.), or with the reduced graphene oxide, i.e., RGO/GCPE.
The electrochemical oxidation of APAP gave rise to a single well-defined peak at about + 0.5 V vs. ref. with the bare GCPE and RGO/GCPE and approximately at + 0.6 V when using the GCE or MWCNT/GCE. By comparison of the actual performance at the four electrodes tested it was found that, the most satisfactory signal was obtained for the GCPE/RGO as documented by the corresponding voltammogram in Fig. 1. Therefore, the last-named configuration was the (working) electrode of choice and used in all further measurements.

Square wave voltammograms of 100 μmol L−1 APAP at different bare and surface modified electrodes in 0.1 mol L−1 acetate buffer solution pH 5.0
Effect of pH
Surveying the literature revealed that, APAP is an electrochemical oxidized in a pH-dependent two-electron, two-proton process to N-acetyl-p-quinon-imine (NAPQI).
The electrochemical oxidation of acetaminophen in various pHs using cyclic voltammetry showed that electrochemically generated NAPQI participates in different type reactions based on solution’s pH. It is hydrolyzed in strong acidic media (pH < 5) and hydroxylated in strong alkaline media (pH > 9) [32]. It has been also reported that the stability of NAPQI is influenced by the sample pH with the highest stability in the pH range 4–9 [33, 34]. The pH of a solution affects the electrochemical reaction by shifting the redox potential to more positive or negative directions with changing peak currents response due to the variation of acid dissociation of APAP [35]. The pH-study was performed with a set of Britton–Robinson buffers covering the range of pH 3–11. When considering the overall size and shape of the oxidation peak, as well as the background currents level within the potential range applied (see Fig. 2), the best response was achieved at pH 5.0 used as optimum pH in all next experiments.

Square wave voltammograms of 100 μmol L−1 APAP measured using Britton–Robinson buffer in the pH range (3–11) at GCPE/RGO
Optimization of the parameters of square-wave voltammetric ramp
Selection of optimal SWV conditions was made based on the respective series of comparative measurements, when the effects of potential step, amplitude and frequency were investigated individually at the constant settings of all the remaining parameters.
Performance of the working electrode
During the characterization of the RGO/GCPE working electrode, it has been ascertained that there is a very favourable level of background currents, as well as no distinct memory effects after repeated scans (for both, see Fig. 2S).
Note: The effect of the content of methanol on the electrode response was studied as well, because the use of this organic solvent could improve the solubility of the analyte at high concentrations. However, as known, carbon paste-like electrodes may undergo disintegration in such methanol-containing media [36] although glassy carbon-based mixtures are generally more resistant. Thus, the resistivity of the actual paste was examined in a series of electrolytes having contained 10, 20, 30, 40, and 50%(v/v) methanol in 0.1 mol.L−1 AcB. As expected, it was found that the most satisfactory response could be achieved in the methanol-free buffer (Fig. 3S).
Potential step
The potential step effect was studied while setting potential amplitude and frequency at usually chosen values; i.e., 25 mV and 40 Hz, respectively. Figure 3a reveals that the potential-step value of 5 mV has been the optimum for the determination of APAP; here, the criterion of choice being the highest current obtained.
Potential amplitude
Figure 3b shows the effect of potential amplitude investigated over the range of 5–100 mV. With the increasing values of potential amplitude, the peak heights recorded had exhibited a significant increase up to 50 mV; then, the peaks started to be deformed. Thus, the value of 50 mV was chosen as optimum for all subsequent measurements.
Frequency
Changes of the value of frequency in a range of 20–100 Hz were investigated as the last key parameter of the SWV ramp. Figure 3c illustrates that height of APAP oxidation peak increases remarkably with higher frequencies, reaching its maximum at 40 Hz, setting frequency at higher values did not lead to anymore increase in peak current response. Hence, it was chosen as optimum value for entire analysis.

Dependence of peak current response of 100 μmol L−1 APAP on a step potential, b potential amplitude and c frequency measured in 0.1 mol L−1 acetate buffer solution pH 5.0 at GCPE/RGO
Analytical performance of the method
The above-optimised experimental conditions and instrumental parameters had resulted in a set of conditions defining the new method whose analytical performance had to be examined experimentally. The calibration measurements were performed in a range of 1 × 10–6 to 5 × 10–4 mol L−1, resulting in a linearity of 4.0 × 10–6 to 2.2 × 10–4 mol L−1, defined by the regression equation Ap(μA V) = 0.0146c(μmol L−1) − 0.0032, R2 = 0.9971 (see Fig. 4), limits of detection (LOD) and quantification (LOQ) to be 0.3 × 10–6 mol L−1 and 0.9 × 10–6 mol L−1, respectively, estimated according to the criteria LOD = 3 s/k and for LOQ = 10 s/k (where k refers to the slope of linear part of calibration curve specified above, s is the standard deviation of minimally five repetitions of a chosen concentration of 4 μmol L−1 APAP and with the average response of Ip = 0.0585 ± 0.0014 μA presented as arithmetic mean and the standard deviation). When testing the new method precision with respect to the repeatability of the signal of interest, a value ± 2.7% as the RSD was obtained. Before analyses of selected tablet-dosage forms, a series of recovery studies with a model sample were performed (with n = 5), yielding the satisfactory result of 103%.

Voltammograms for 4, 8, 12, 16, 20, 60, 100, 140, 180 and 220 μmol L−1 APAP with corresponding calibration curve obtained at GCPE/RGO measured in 0.1 mol L−1 acetate buffer solution pH 5.0 using SWV at potential step 5 mV, potential amplitude 50 mV and frequency 40 Hz
Interference study
Possible interferences were investigated for substances considered to represent typical minor constituents in the samples of interest—i.e., tablet formulation excipients (lactose monohydrate, magnesium stearate, croscarmellose sodium, corn starch) and main matrix of urine (uric acid, ascorbic acid, and urea). As found out, all these compounds did not exhibit any evident electrochemical signals within the working potential range applied, documenting a very good selectivity of the method developed. Finally, to complete the basic characterization of the new method, its comparison with other electrochemical approaches for the determination of APAP is surveyed in Table 1.
Testing the reference RP–HPLC method before its use for validation
The reference chromatographic procedure had to be yet validated before analyzing the samples intended to be suitable for the new voltammetric method—i.e., tablet dosage forms and urine samples. When using chromatographic conditions under Experimental in the respective section, it was found that the peak of APAP could be obtained at retention time of 5 min. 15 s. A typical chromatogram in Fig. 5 shows the respective calibration curve defined by the equation A(mAU) = 2,092.78c(μmol L−1) − 12,036.26 (R2 = 0.9972). Precision of the reference method was determined using five repeated measurements (n = 5), resulting in RSD of 0.79% for a urine sample collected from 5 healthy volunteers. Table 2 shows that the proposed method provides statistically satisfied results comparable to the chosen reference HPLC method as well as with the declared amount by a manufacturer.

HPLC analyses of sample APAP in human urine. Ascentis Express C18 column (150 × 3 mm, 2.7 μm); mobile phase, 0.3% formic acid in water (a) and methanol (b); gradient program from 10 to 35% B for 10 min; flow rate, 0.5 mL min−1; sample volume, 5 μL; temperature, 30 °C; detection at 243 nm
Conclusions
Based on the above presentation of all the important features and characteristics of the newly developed method, one can state that it can offer a simple and quick approach of how to accurately and precisely determine acetaminophen (APAP) in pharmaceutical preparations and in urine samples. The developed procedure is based on the direct voltammetric oxidation of this substance at the RGO/GCPE in 0.1 mol L−1 acetate buffer (pH 5.0) as the supporting electrolyte of choice.
It seems that the method, for the first time described herein, could be offered as an interesting contribution for laboratories equipped with electrochemical instrumentation, or even for those equipped with chromatographic techniques that still represent the prevailing tool for determination of APAP in pharmaceutical and biological samples. In such cases, this new voltammetric procedure could serve as an independent and fully reliable reference method to occasionally check the results being obtained by routine chromatographic analyses.
References
Z.N. Ennis, D. Dideriksen, H.B. Vaegter, G. Handberg, A. Pottegård, Acetaminophen for chronic pain: a systematic review on efficacy. Basic Clin. Pharmacol. Toxicol. 118, 184 (2016)
B. Bannwarth, F. Péhourcq, Pharmacologic basis for using paracetamol: pharmacokinetic and pharmacodynamic issues. Drugs 63, 5 (2003)
L.L. Mazaleuskaya, K. Sangkuhl, C.F. Thorn, G.A. FitzGerald, R.B. Altman, T.E. Klein, PharmGKB summary: Pathways of acetaminophen metabolism at the therapeutic versus toxic doses. Pharmacogenet. Genom. 25, 416 (2015)
J.A. Forrest, J.A. Clements, L.F. Prescott, Clinical pharmacokinetics of paracetamol. Clin. Pharmacokinet. 7, 93 (1982)
A. Frangu, K. Pravcová, P. Šilarová, T. Arbneshi, M. Sýs, Flow injection tyrosinase biosensor for direct determination of acetaminophen in human urine. Anal. Bioanal. Chem. 411, 2415 (2019)
M.J. Stewart, P.I. Addriaenssens, D.J. Jarvie, L.F. Prescott, Inappropriate Methods for the Emergency Determination of plasma paracetamol. Ann. Clin. Biochem. 16, 89 (1979)
Y. Pegon, J.J. Vallon, A rapid extraction technique for the determination of paracetamol in plasma by gas chromatography. Anal. Chim. Acta 130, 405 (1981)
L.T. Wong, G. Solomonraj, B.H. Thomas, High-pressure liquid chromatographic determination of acetaminophen in biological fluids. J. Pharm. Sci. 65, 1064 (1976)
R.M. Riggin, A.L. Schmidt, P.T. Kissinger, Determination of acetaminophen in pharmaceutical preparations and body fluids by high-performance liquid chromatography with electrochemical detection. J. Pharm. Sci. 64, 680 (1975)
S.S. Al-Obaidy, A. Li-Wan-Po, P.J. McKiernan, J.F. Glasgow, J. Millership, Assay of paracetamol and its metabolites in urine, plasma and saliva of children with chronic liver disease. J. Pharm. Biomed. Anal. 13, 1033 (1995)
I. Navarro, D. Gonzalez-Arjona, E. Roldan, M. Rueda, Determination of paracetamol in tablets and blood plasma by differential pulse voltammetry. J. Pharm. Biomed. Anal. 6, 969 (1988)
M. Li, L. Jing, Electrochemical behavior of acetaminophen and its detection on the PANI–MWCNTs composite modified electrode. Electrochim. Acta 52, 3250 (2007)
M.H. Pournaghi-Azar, A. Saadatirada, Simultaneous determination of paracetamol, ascorbic acid and codeine by differential pulse voltammetry on aluminum electrode modified by thin layer of palladium. Electroanalysis 22, 1952 (2010)
M. Solomon, A. Shimelis, T. Merid, S. Theodros, Voltammetric determination of paracetamol with poly(3,4-ethylenedioxythiophene) modified glassy carbon electrode. Anal. Bioanal. Electrochem. 3, 38 (2011)
W.Y. Su, S.H. Cheng, Electrochemical oxidation and sensitive determination of acetaminophen in pharmaceuticals at poly(3,4-ethylenedioxythiophene)-modified screen-printed electrodes. Electroanalysis 22, 707 (2010)
M.A.T. Gilmartin, J.P. Hart, Rapid detection of paracetamol using a disposable, surface-modified screen-printed carbon electrode. Analyst 119, 2431 (1994)
M. Amare, W. Teklay, Voltammetric determination of paracetamol in pharmaceutical tablet samples using anthraquinone modified carbon paste electrode. Cogent Chem. 5, 1576349 (2019)
M. Tertis, A. Florea, R. Sandulescu, C. Cristea, Carbon based electrodes modified with horseradish peroxidase immobilized in conducting polymers for acetaminophen analysis. Sensors. 13, 4841 (2013)
C. Calas-Blanchard, G. Istamboulié, M. Bontoux, G. Plantard, V. Goetz, T. Noguer, Biosensor-based real-time monitoring of paracetamol photocatalytic degradation. Chemosphere 131, 124 (2015)
Y. Liu, X. Dong, P. Chen, Biological and chemical sensors based on graphene materials. Chem. Soc. Rev. 41, 2283 (2012)
Y. Xu, H. Gao, M. Li, Z. Guo, H. Chen, Z. Jin, B. Yu, Electronic transport in monolayer graphene with extreme physical deformation: ab initio density functional calculation. Nanotechnology 22, 365202 (2011)
Y. Zeng, Y. Zhou, T. Zhou, G. Shi, A novel composite of reduced graphene oxide and molecularly imprinted polymer for electrochemical sensing 4-nitrophenol. Electrochim. Acta 130, 504 (2014)
L. Chen, Y. Tang, K. Wang, C. Liu, S. Luo, Direct electrodeposition of reduced graphene oxide on glassy carbon electrode and its electrochemical application. Electrochem. Commun. 13, 133 (2011)
Y. Li, S.M. Chen, The electrochemical properties of acetaminophen on bare glassy carbon electrode. Int. J. Electrochem. Sci. 7, 2175 (2012)
I. Baranowska, A. Wilczek, Simultaneous RP-HPLC determination of sotalol, metoprolol, alpha-hydroxymetoprolol, paracetamol and its glucuronide and sulfate metabolites in human urine. Anal Sci. 25, 769 (2009)
A.W. Abu-Qare, M.B. Abou-Donia, A validated HPLC method for the determination of pyridostigmine bromide, acetaminophen, acetylsalicylic acid and caffeine in rat plasma and urine. J. Pharm. Biomed. Anal. 26, 939 (2001)
H. Filik, G. Çetintaş, A.A. Avan, S.N. Koç, İ Boz, Electrochemical sensing of acetaminophen on electrochemically reduced graphene oxide-nafion composite film modified electrode. Int. J. Electrochem. Sci. 8, 5724 (2013)
S. Yu, H. Li, G. Li, L. Niu, W. Liu, X. Di, Reduced graphene oxide-supported gold dendrite for electrochemical sensing of acetaminophen. Talanta 184, 244 (2018)
T.S.H. Pham, P.J. Mahon, G. Lai, L. Fu, C. Lin, A. Yu, Cauliflower-like platinum particles decorated reduced graphene oxide for sensitive determination of acetaminophen. Electroanalysis 31, 1758 (2019)
N. Dou, S. Zhang, J. Qu, Simultaneous detection of acetaminophen and 4-aminophenol with an electrochemical sensor based on silver–palladium bimetal nanoparticles and reduced graphene oxide. RCS Adv. 9, 31440 (2019)
V.N. Palakollu, T.E. Chiwunze, C. Liu, R. Karpoormath, Electrochemical sensitive determination of acetaminophen in pharmaceutical formulations at iron oxide/graphene composite modified electrode. Arab. J. Chem. 13, 4350 (2020)
D.J. Miner, J.R. Rice, R.M. Riggin, P.T. Kissinger, Voltammetry of acetaminophen and its metabolites. Anal. Chem. 53, 2258 (1981)
K.G. Madsen, J. Olsen, C. Skonberg, S.H. Hansen, U. Jurva, Development and evaluation of an electrochemical method for studying reactive phase-I metabolites: correlation to in vitro drug metabolism. Chem. Res. Toxicol. 20, 821 (2007)
A. Sarakbi, Z. Aydogmus, T. Sidali, G. Gokce, J. Kauffmann, Simultaneous determination of acetaminophen (paracetamol) and ascorbic acid in pharmaceutical formulations by LC coupled to a screen printed carbon based amperometric detector. Electroanalysis 23, 29 (2010)
D.A.C. Brownson, C. E. Banks, The handbook of graphene electrochemistry, 1st edn. (Springer, London, 2014), pp. 79–126.
I. Švancara, K. Vytřas, F. Renger, M.R. Smyth, Application of carbon paste electrodes in highly methanolic solutions. Electrochim. Acta 37, 1355 (1992)
Acknowledgements
Financial support from faculty of pharmacy, Helwan University is gratefully acknowledged.
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declare no conflict of interest.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Farag, A.S. Voltammetric determination of acetaminophen in pharmaceutical preparations and human urine using glassy carbon paste electrode modified with reduced graphene oxide. ANAL. SCI. 38, 1213–1220 (2022). https://doi.org/10.1007/s44211-022-00150-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s44211-022-00150-2