
Abstract
Various tools are available to assess atherosclerosis, arterial stiffening, and endothelial function. They offer utility in the assessment of hypertensive phenotypes, in cardiovascular risk prediction, and as surrogate endpoints in clinical trials. We explore the relative influence of participant genetics, with reference to large-scale genomic studies, population-based cohorts, and candidate gene studies. We find heritability estimates highest for carotid intima-media thickness (CIMT 35–65%), followed by pulse wave velocity as a measure of arterial stiffness (26–43%), and flow mediated dilatation as a surrogate for endothelial function (14–39%); data were lacking for peripheral artery tonometry. We furthermore examine genes and polymorphisms relevant to each technique. We conclude that CIMT and pulse wave velocity dominate the existing evidence base, with fewer published genomic linkages for measures of endothelial function. We finally make recommendations regarding planning and reporting of data relating to vascular assessment techniques, particularly when genomic data are also available, to facilitate integration of these tools into cardiovascular disease research.
Similar content being viewed by others

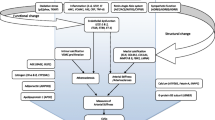

1 Introduction
Hypertension is a major risk factor for Cardiovascular Disease (CVD); in turn CVD is the underlying cause of more than a quarter of deaths in the UK [1]. There are no validated tests that can identify early in the disease process which individuals will develop hypertension-mediated organ damage. Dysfunctional vascular traits represent key pathophysiological processes in the development of hypertension and cardiovascular disease, with both inherited and reversible elements. These traits include stiffness of the large arteries, microvascular abnormalities, endothelial dysfunction, and atherosclerosis, phenotypes often apparent prior to established hypertension or organ damage. Hence the interest in measuring vascular function, and in understanding the relationship between measurement techniques and hypertensive phenotypes, including the relative influence of participant sex and genetics. We explore this topic with reference to large scale genomic studies, population-based cohorts, and candidate gene studies.
1.1 Definitions for the Non-expert
Genome: complete set of genes in an organism including introns (non-coding sequences) and exons (coding sequences).
Genome-wide association study: entire genome surveyed for genetic variants occurring more frequently in cases than in controls.
Candidate gene study: specify fewer variants of interest a priori, and aim to establish if a disease association can be confirmed.
Epigenetics: genetic modification without mutations of the DNA sequence; occur in normal development or induced by environmental factors.
Exome: complete set of exons present in an organism which accounts for all the coding regions of genes present.
SNP: single nucleotide polymorphism—DNA sequence variations with a single nucleotide (adenine, thymine, cytosine, or guanine) in the genome sequence altered.
Common variants: SNPs with minor allele frequency (MAF) of greater than 1%, accounting for over 90% of genetic variation between individuals.
Mendelian Randomization: method of using measured variation in genes of known function to examine the causal effect of a modifiable exposure on disease.
2 Assessment of Vascular Function and Disease
Cardiovascular outcome measures in clinical trials generally relate to coded events such as myocardial infarction or death; alternatively, research trials may employ surrogate markers such as vascular stiffness and endothelial dysfunction—early functional traits known to be predictors of more advanced structural changes and development of cardiovascular disease. Assessment techniques quantifying such traits reflect different aspects of vascular health, assessed in the European Society of Cardiology Working group position paper [2]. First, carotid ultrasound to measure intima-media thickness (CIMT) has clinical utility in diagnosing carotid atherosclerotic vascular disease [3]–[5], but is also linearly associated with blood pressure (BP) [6] and adds prognostic value in the prediction of cardiovascular events and mortality, see Sect. 3.1 [7, 8]. Second, pulse wave analysis (PWA), PWA-derived augmentation index (AIx), and carotid-femoral pulse wave velocity (cfPWV) assess for arterial stiffness, a process characterised by functional changes and structural remodelling within the arterial wall, with associated fibrosis and calcification. These measures of arterial stiffening are independent and reliable predictors of hypertension, myocardial infarction and stroke [9]–[11], with meta-analyses of individual patient data showing the alternative brachial-ankle PWV method also associated with cardiovascular complications [12], and stiffening of the carotid artery with incident stroke [13]. The predictive strength of arterial stiffness is, however, greater in subjects with an established cardiovascular risk [14]. Finally, endothelial function refers to its’ ability to detect physical (shear stress) and biochemical signals, and respond through expression of surface molecules and production of vasoactive and inflammatory mediators. Endothelial dysfunction precedes structural micro-circulatory changes. Hypertension can be both cause and consequence of microcirculatory dysfunction, closely tied to peripheral vascular resistance, with vascular tone in turn regulated by many systems (sympathetic nervous system, endocrine, and local autoregulation), each with polygenic influences [15]. Endothelial function can be assessed using ultrasound of the brachial artery with ‘flow-mediated dilation’ (FMD), dilation predominantly mediated by nitric oxide release from endothelial cells. Alternatively, peripheral arterial tonometry (PAT), commonly quantified by the Endo-PAT2000 device (Itamar Medical) also assesses microcirculatory and endothelial function by measuring arterial tone or ‘hyperaemic response’ in the fingertips in response to proximal occlusion. EndoPAT-2000 device also generates an augmentation index adjusted to a heart rate of 75 bpm (AI@75), similar to PWA but derived from peripheral vessels. Hence, these techniques not only reflect different aspects of the pathophysiology of hypertension and cardiovascular disease but may aid identification of different hypertensive phenotypes [16]–[18]. They are also well accepted as being influenced by age, BP, and sex; factors that should be accounted for when comparing techniques. Less well defined are the effects of underlying genetic differences, i.e. the inherited component, or ‘heritability’ of data pertaining to techniques measuring vascular health. Genotypic effects on these vascular assessment tools are myriad, but key checkpoints where influence may be hypothesised include vascular endothelial cell sensitivity to extracellular stimuli, intra-cellular signalling cascades, and effects on transcription, ultimately influencing production of vasoactive substances, vascular tone, and remodelling.
3 Genetics of Hypertension
Familial and twin studies estimate that the heritable component of BP lies between 22 and 65% [19]–[22]. BP is a complex trait with no single gene playing a dominant role; instead multiple genes demonstrate minor additive effects. These genes encode for a variety of proteins, ion channels, receptors, and enzymes involved in endocrine, cardiac, renal, vascular and neural systems that influence BP regulation. This complexity is illustrated by the heterogeneity of underlying pathology in the (rare) monogenic cases of secondary hypertension, examples of which are discussed in Sect. 2.1.1. Other genes are identified only by genome wide association studies (GWAS); an illustrative example follows in Sect. 2.1.2.
3.1 Single Gene Disorders
Monogenic causes of hypertension are rare and mechanisms varied. For example, children with homocystinuria and familial hypercholesterolaemia develop premature atherosclerosis and early endothelial dysfunction [23]; in AD glucocorticoid-remediable aldosteronism, chimeric genes encoding steroid 11ß-hydroxylase (CYP11B1) and aldosterone synthase (CYP11B2) lead to aldosterone regulation by ACTH rather than angiotensin II [24, 25], salt and water retention, and elevation in BP [26]. Finally, AD hypertension with brachydactyly syndrome results from a gain of function mutation in PDE3A, encoding phosphodiesterase 3A and resulting in cerebral vascular anomalies and baroreceptors hypersensitivity [27]. For an in-depth review of monogenic hypertensive syndromes, we would highlight Burrello et al. [28].
3.2 GWAS
GWAS have identified multitudes of genetic loci associated with BP, covered in-depth elsewhere [29]. For example ATP2B1 encoding PMCA1, a plasma membrane ATPase expressed in vascular endothelium and involved in calcium pumping from the cytosol to the extracellular compartment. GWAS can, however, be susceptible to false positive associations if statistical analysis lacks rigour, if the panel fails to reflect genomic variation, or the study lacks statistical power; points to remain cognizant of.
3.3 Epigenetics
Processes of epigenetic modification include methylation, post-translational histone modification, and small non-coding RNAs. HSD11B2 gene promoter methylation for example has been associated with hypertension onset [30, 31]; acetylation meanwhile promotes gene transcription of NOS3 (eNOS) and other genes affecting vascular tone and salt and water homeostasis [32, 33]. Finally, small non-coding RNAs (miRNA) may conversely downregulate genes by binding the corresponding mRNA resulting in repression of translation [33]. Population-based studies further support the role of epigenetics in hypertension [34].
3.4 Sex and BP Genetics
The male–female difference in BP, vascular traits, and CVD is complex. Mediating factors include X and Y chromosome differences, sex-hormone influences, renin–angiotensin–aldosterone system divergence [35], societal and behavioral impacts, and even epigenetic differences, with females receiving genetic imprints from each parent’s X chromosome, random X inactivation leading to further genetic heterogeneity. Gene-by-sex interactions, and age (menopause)-dependent effects further complicate interpretation.
3.5 Summary
Bringing together the evidence of different phenotypes of hypertension [16,17,18, 36, 37], determined by pathophysiology but characterised by the aforementioned vascular traits; and considering the exponentially increasing data regarding hypertension risk alleles; it becomes important to explore the genotypic and sex associations with vascular techniques used to measure these hypertensive phenotypes.
4 Carotid Intima-Media Thickness
4.1 Heritability
A number of studies have reported heritability estimates for CIMT, though with disparate estimates (21 to 65%) despite similar adjustment for covariates, see Fig. 1 and Table 1 [38, 39]. Sacco et al. for example report 65% heritability in 100 Dominican families (1390 individuals, 61% female, mean age 46 years) after adjustment for age, sex, smoking, and BMI [39]; Cecelja et al. estimate age-adjusted heritability at 49% (95% CI 17–63%) in 762 females of the Twins UK cohort with mean age 58 ± 9 years [40]; whilst only 35% heritability is reported by Sayed-Tabatabaei et al. [41] in their assessment of 930 individuals connected in a single pedigree from an isolated population (participants of the Erasmus Rucphen Family study).

Evidence regarding heritability of techniques assessing vascular function
4.2 Genes
GWAS identifies numerous genetic loci as having possible significance, and studies of candidate genes approximating to these loci have also been widely reported (Table 2); 16 of the 32 identified (50%) also have evidence of association with BP traits. Figure 2 demonstrates that many genes have a role vascular remodelling, such as MMP9 [42] encoding a gelatinase targeting type IV collagen and gelatin; CXCL12 involved in endothelial and epithelial cell proliferation and migration [43]; and VCAN [44] which encodes chondroitin sulfate proteoglycans (extracellular matrix components), thus regulates cell proliferation, differentiation, and survival [45].

Gene polymorphisms relating to techniques measuring vascular health, with genes grouped according to function. Based on data in Table 2
4.3 Interactions
Other genes demonstrate the importance of gene-by-environment interactions in determining CIMT, for example MCPH1 encodes a damage response protein regulating cell cycle [44]. Similarly, gene–gene interactions are apparent, for example genes involved in cholesterol biology and inflammation where high-density lipoprotein composition is altered in an inflammatory state, with apolipoprotein-A-I and –A-II displaced by Serum amyloid A (SAA). SAA SNPs rs2468844 and rs12218 alter binding affinity of SAA proteins [46, 47], with implications for reverse cholesterol transport, CIMT, plaque formation [48], and plaque stability [49].
5 Vascular Stiffness: Pulse Wave Analysis and Pulse Wave Velocity
5.1 Heritability
Genes are estimated to account from 26 to 43% of the variability in vascular stiffness as measured by PWV (see Table 1, Fig. 1), with data derived from both population and twin studies [40, 41, 52, 53]. For example, the Georgia Cardiovascular Twin Study of 388 twins (41% black; 49% male) aged 12–30 years; report 53% (42–62%) heritability for dorsalis pedis (foot) PWV [53], with no sex or race differences; additionally, the aforementioned Twins UK cohort of 762 females, mean age 58 ± 9 report heritability estimate of 38% (95% CI 16–63%) after adjustment, with annual progression interestingly demonstrating higher adjusted heritability estimates of 55% (31–64%) over 5 years follow-up [40].
5.2 Genes
Many studies of the genetics of arterial stiffness focus on parameters other than PWV, such as pulse pressure and forward and reflected wave amplitude, covered in detail elsewhere [142, 143]. Looking specifically at PWV as the most commonly used technique, GWAS of 644 individuals involved in the Framingham Heart Study did not find any variants achieving genome wide significance in the primary analysis [91], despite the Mitchell et al.study of 2127 participants (mean age of 60 years, 57% female) also derived from the Framingham cohort reporting moderate heritability for PWV (h2 = 0.40), with suggestive linkage regions in chromosomes 2, 7, 13, and 15 [52]. Informed by GWAS, and based on UK Biobank data, Zekavat et al. [85] generated a six variant polygenic arterial stiffness score, showing a relationship with SBP and DBP, and Mendelian randomization data supporting causality, with genetic predisposition of arterial stiffness preceding hypertension [85].
5.3 Interactions
Fifteen of the 24 genes (62.5%) implicated in arterial stiffness have evidence of BP associations, see Table 2. Many candidate gene polymorphisms studied in greater detail relate to the renin angiotensin aldosterone system; in particular angiotensin-converting enzyme (ACE) gene polymorphisms are known to influence vascular tone, fibrosis, and ultimately arterial stiffness, though with discordant results between healthy, diabetic, and hypertensive populations, despite adjustments for demographic and lifestyle factors [104, 106, 142], suggesting either an additional interaction or confounding factor is involved. Similarly, the A1166C polymorphism of angiotensin II type 1 receptor gene (AGTR1) was associated with arterial stiffness in hypertensive participants [103, 108], but not among normotensive participants of the same study, nor the Rotterdam Study population [103, 110]. Study participant age needs to be considered in such publications as combined effects may be apparent, e.g. C allele carriers showing increased PWV, but only beyond 55 years of age [103], though the Rotterdam study population was over 55 years of age but still did not support the association. Additionally, heterogeneous methods of estimating arterial stiffness limit comparisons of studies. Mayer et al. for example find AGTR1 polymorphism significant in femoral-popliteal PWV but not carotid-femoral [108]; Levy et al. conversely report greater heritability estimates for carotid-femoral than for carotid-brachial PWV, consistent with Salvi et al. reporting carotid-femoral techniques are more reliable [91, 144]. This emphasises the need for standardized technique, with the consensus now favouring carotid-femoral PWV [145]. Finally, the importance of ancestry when extrapolating data is highlighted by the concordance of results derived from a common population e.g. Zekavat et al. and Fung et al. reporting UK BioBank data [85, 86], and discordant results in candidate genes and heritability estimates across disparate populations [52, 110].
6 Endothelial Function: Flow Mediated Dilatation and Peripheral Arterial Tone
6.1 Heritability
The influence of genetics on endothelial function as measured by FMD is supported by an Italian cohort of 40 healthy young people (age 6–30, 19 male) with a family history of premature myocardial infarction, demonstrating lower FMD (5.7 ± 5.0% vs. 10.2 ± 6.6% in control subjects; P = 0.001) [146]; and by a cohort of 50 British young people with a family history of coronary artery disease (31 male, mean age 25 years) also suggesting endothelial dysfunction (FMD 4.9 ± 4.6% vs 8.3 ± 3.5% in control group, P < 0.005) [147]. Among 883 participants of the Framingham cohort (53% female; mean age 61), estimated heritability (accounting for covariates) of brachial artery baseline diameter was 0.33 ± 0.07, and FMD% was 0.14 ± 0.06; for FMD%, there was an age-gender interaction (P = 0.01), females showing steeper age-related FMD% decline [56]. Twin studies tend to be preferred above family studies for heritability estimation, as they allow a more precise separation of environmental influences from genetic effects [148], including controlling for such age effects. Twin studies reporting FMD heritability estimates include a Finnish cohort reporting FMD heritability of 24%, derived from 74 male twin pairs (20 monozygous), aged 42–69 years, with monozygous twins demonstrating improved FMD after migrating to Sweden (7.2 ± 4.4 vs 3.7 ± 2.9%, P = 0.003), a country with lower cardiovascular risk [51]. A higher estimate of 39% was reported from 94 male twin pairs from the USA (58 monozygous pairs), mean age 55 ± 2.8 years, 95% Caucasian [57].
6.2 Genes
Candidate genes linked to FMD are included in Table 2, 5 of the 8 (63%) also linked to hypertension, see Fig. 2. Examples include the Asp/Asp genotype of the endothelial nitric oxide synthase (NOS3) Glu298 → Asp polymorphism, which was associated with reduced vascular nitric oxide (NO) generation (a potent vasodilator), decreased brachial artery FMD, and increased CIMT in a group of young healthy individuals free of traditional cardiovascular risk factors [75, 149]. NOS3 regulation involves receptor-mediated mechanisms (e.g. acetylcholine, bradykinin, and substance P) and mechanical stimuli (shear stress). However, NOS3 Asp298 is not unique; more than 100 polymorphisms in NOS3 have been identified [150], with small effect size and significant interaction with other genes and environmental factors [151]. Further elements of the NO system implicated include PDE3A, a phosphodiesterase with a role in the NO/cGMP pathway.
Other genes have more obscure associations, such as PHACTR1 with a role in actin re-organisation but also possibly regulating vasoconstriction via endothelin-1 gene expression [94]; NFKB1 encoding a protein with diverse roles as a transcription regulator [152], and CYBA encoding p22phox, a component of NADPH oxidase involved in vascular ROS generation [134, 135], see Table 2. Yoshino et al. studying the genetics of endothelial dysfunction report coronary vascular responses to Acetylcholine, finding 1563 SNPs connected with cardiovascular physiology and pathology [122]. Variants in adenosine A1 receptor (ADORA 1) were associated with endothelial dysfunction in the entire cohort, while variants in adenosine A3 receptor (ADORA 3) and lipoprotein A (LPA) had the strongest associations with increased risk of endothelial dysfunction in women, again highlighting that sex differences must be considered within this area of research.
We did not find published heritability estimates regarding the EndoPAT assessment tool of peripheral arterial tone, though both race and sex are known to influence results [54, 55]. Numerous candidate genes have been proposed to influence vascular endothelial function, but only six of them reported have specifically been linked to PAT, five of the six (83 percent) had commonality with BP traits. see Fig. 2. The six linked to PAT include NOS3, already discussed in regard to FMD [117]; APO E, ACE [118], and Sphk1 SNPs/alleles [120]. Siedlinski [120] elegantly combine Sphk1 identification through murine transcriptome analysis with in vivo experiments confirming a role in vasoconstriction and endothelial dysfunction, and correlation of human sphingosine-1-phosphate (S1P) serum levels with arterial tonometry.
7 Heritability Study Considerations
BP regulation and vascular function are complex, polygenic traits, additionally influenced by many environmental factors. Molecular genetic analysis is therefore challenging due to the sheer number of relevant genes and their polymorphic effects, as examples in Table 2 illustrate. There are also certain limitations associated with heritability studies, as follows.
7.1 Family Studies
Classical family study design can overlook non-additive genetic effects and shared environmental factors. Additionally, the underlying assumption regarding the genetic relationship is flawed; offspring tend to inherit long segments of DNA resulting in deviations from the expected 50% DNA inheritance from each parent. Furthermore, family studies often recruit based on participant phenotype, with family members then invited to participate. However, techniques to correct for ascertainment bias should be employed, such as Hopper and Mathews method which adjusts the heritability estimate based on the mean and total variance of the genetic and environmental components for each individual family grouping [153].
7.2 Missing Heritability
Another issue is ‘missing heritability’, i.e., the disparity between heritability estimates derived from genotype data (explaining a low proportion of the variance), and from twin studies (estimating significantly higher heritability). Missing heritability is likely a consequence of restriction of many genetic association studies to SNPs—missing rare mutations. Gene-by-gene interactions, epigenetics, and gene-by-environment interactions also contribute to missing heritability, through assumptions that such interactions are minimal, identifiable, and that variance explained by shared environmental factors is identical in pairs. Such assumptions risk inflating heritability estimates by attributing the contribution of environmental factors to genetics.
7.3 Directionality
Directionality is an inherent challenge when assessing genotypic influences effecting vascular traits: differentiating if an identified gene has a direct impact on e.g., PWV, or alternatively elevates BP which in turn leads to arterial remodeling, stiffness, and results in elevated PWV. The high proportion of identified genetic loci and candidate genes common to both vascular phenotypes and hypertension outlined in Table 2 and Fig. 2 highlights this.
7.4 Design
Most data are cross-sectional in nature, from which change over time in vascular function or BP cannot be inferred. One might also hypothesize that SNPs contributing to vascular ageing for example may influence PWV at 60 years of age, but not at 30. Studies that do report heritability of baseline measures and progression, have found discrepancies [40]; therefore, duration of follow up, or population age of cross-sectional data must be reported in detail. Future studies independently confirming heritability of vascular traits and candidate genes, as well as their independence from each other and from BP are required, and will determine the utility of vascular assessment techniques as surrogate endpoints in trials, separate from their use as predictive risk tools.
8 Vascular phenotype
Various genes in Table 2 appear numerous times suggesting effects on multiple vascular function assessment techniques. For example ACE, which cleaves angiotensin I into angiotensin II with vasoconstrictive effects; ACE also stimulates the production of aldosterone, increasing absorption of salt and water in the kidneys; ACE furthermore causes inactivation of the vasoactive mediator bradykinin. It is therefore not surprising that genetic polymorphisms of ACE impact on many of the vascular assessment techniques described. Similarly, NOS3 (endothelial nitric oxide synthase) has been identified as relevant in multiple assessment tools of vascular function, with local vasodilatory regulation of vascular tone and diameter (see Table 2). Other genes or polymorphisms appear specific to the technique or vascular trait, such as SAA1 in CIMT, COL4A in arterial stiffness (PWV), and CYBA encoding p22phox, a component of NADPH oxidase in FMD. Some furthermore show a gene by sex interaction, such as VCAN locus in females, encoding a chondroitin sulfate proteoglycan of the adventitia and intima in CIMT [44], and NOS3 rs1799983 relating to central pulse pressure and forward wave amplitude parameters again only in females [98]. Others appear to only reach significance in those with hypertension, suggesting gene by gene or gene by environment interactions e.g. CYBA T allele associated with higher FMD only in hypertensive individuals [154]. These highlight the importance of comprehensive demographic reporting and consideration of such factors when comparing data from multiple sources. Finally, fewer studies were identified reporting the genetics of measures of endothelial function (FMD and PAT) compared to those relating to vascular stiffness and remodeling/atherosclerosis; we would propose this as an area for future study. Of note, no single gene or SNP discussed here demonstrates a substantial association with the vascular traits and assessment techniques covered. This is to be expected in polygenic traits, but may also reflect features of study design identified above: necessity for standardised technique with these tools, underpowering and lack of external validation cohorts among many studies, gene–gene or gene– environment interactions. Comparisons between different demographic groups are also complicated if age, sex, race, and BP are not fully adjusted for. Researchers should be cognizant of these in future studies.
9 Sex-Differences
Gene-by-sex interaction may not always be captured by GWAS. Efforts to elucidate sex-specific genomic determinants of BP demonstrated in 120 Canadian families found that one quarter of the 539 hemodynamic, anthropometric, metabolic, and humoral traits studied were both age and sex dependent, and one eighth were exclusively age or sex dependent [155].
A vascular phenotypic divide related to participant sex may also exist, demonstrating greater discrimination between normotensive and hypertensive PWV and augmentation index for females than males [16] and supported by our own unit’s experience (unpublished). Conversely, a collaboration establishing reference values for PWV describe apparent sex differences being almost fully accounted for by age and BP differences [156]. Two points therefore to consider if undertaking or analysing vascular function data, is whether the groups were well matched or adjustments for age and BP applied, and we suggest that researchers should also report outcome data stratified by sex to facilitate interpretation.
10 Conclusion
In conclusion, CIMT, PWV/PWA, FMD and PAT offer utility as surrogate markers of atherosclerosis, arterial stiffening, endothelial and microcirculatory function i.e. vascular function, and are predictive of cardiovascular risk. They may also have an increasing role as surrogate endpoints in genomic studies and clinical trials [157], however sex differences remain contentious, and dissecting genetic associations independent from hypertension is challenging. The genetics underlying these vascular assessment techniques have been variably studied, CIMT more so than PAT. The genetics of hypertension has a broad literature base; the next step is to integrate characterization of vascular and hypertensive phenotypes with genotypes as a natural symbiosis in studying the pathophysiology of hypertension and cardiovascular disease, and to better personalize cardiovascular medicine.
Data Availability
All data pertaining to this manuscript are already freely available and full references including DOI have been provided to facilitate access to data.
Abbreviations
- ACE:
-
Angiotensin-converting enzyme
- AD:
-
Autosomal dominant
- AR:
-
Autosomal recessive
- AIx:
-
Augmentation index
- AI@75:
-
Augmentation index adjusted to 75 bpm
- AT II:
-
Angiotensin II
- BMI:
-
Body mass index
- BP:
-
Blood pressure
- CIMT:
-
Carotid intima-media thickness
- cGMP:
-
Guanosine monophosphate
- CVD:
-
Cardiovascular disease
- DBP:
-
Diastolic blood pressure
- DNA:
-
Deoxyribonucleic acid
- DZ:
-
Dizygous
- FMD:
-
Flow-mediated dilatation
- GWAS:
-
Genome wide association studies
- h2 :
-
Heritability
- miRNA:
-
Micro ribonucleic acid
- MZ:
-
Monozygous
- N/A:
-
Not applicable
- NO:
-
Nitric oxide
- PAT:
-
Peripheral artery tonometry
- PWA:
-
Pulse-wave analysis
- PWV:
-
Pulse-wave velocity
- RNA:
-
Ribonucleic acid
- SBP:
-
Systolic blood pressure
- SNP:
-
Single nucleotide polymorphism
- SSA:
-
Serum amyloid A
References
UK, “Office of National Statistics,” www.ons.gov.uk, 2020.
C Vlachopoulos et al. The role of vascular biomarkers for primary and secondary prevention. A position paper from the European society of cardiology working group on peripheral circulation. Endorsed by the association for research into arterial structure and physiology (ARTERY, Atherosclerosis, vol. 241, no. 2. Elsevier, pp. 507–532, 01-Aug-2015.
Bots ML, Hoes AW, Koudstaal PJ, Hofman A, Grobbee DE. Common carotid intima-media thickness and risk of stroke and myocardial infarction: the Rotterdam study. Circulation. 1997;96(5):1432–7.
Stein JH et al. Use of carotid ultrasound to identify subclinical vascular disease and evaluate cardiovascular disease risk: a consensus statement from the american society of echocardiography carotid intima-media thickness task force endorsed by the society for vascular medicine, J Am Soc Echocardiography, 21(2). Mosby Inc., pp. 93–111, 2008.
Lorenz MW, Markus HS, Bots ML, Rosvall M, Sitzer M. Prediction of clinical cardiovascular events with carotid intima-media thickness: a systematic review and meta-analysis. Circulation. Jan. 2007;115(4):459–67.
Ferreira JP et al. (2016) Intima-media thickness is linearly and continuously associated with systolic blood pressure in a population-based cohort (STANISLAS cohort study), J. Am. Heart Assoc., 5(6).
Hodis HN et al. (1998) The role of carotid arterial intima - Media thickness in predicting clinical coronary events, Ann Intern Med
Espeland MA, O’Leary DH, Terry JG, Morgan T, Evans G, Mudra H Carotid intimal-media thickness as a surrogate for cardiovascular disease events in trials of HMG-CoA reductase inhibitors, Curr Controlled Trials Cardiovasc Med 6(1). BioMed Central Ltd., 10-Mar-2005.
Laurent S, et al. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. 2001;37(5):1236–41.
Boutouyrie P et al. (2002) Aortic stiffness is an independent predictor of primary coronary events in hypertensive patients: a longitudinal study, Hypertens. (Dallas, Tex. 1979), 39(1): 10–15.
Mitchell GF, et al. Arterial stiffness and cardiovascular events: the framingham heart study. Circulation. 2010;121(4):505–11.
Ohkuma T, et al. Brachial-ankle pulse wave velocity and the risk prediction of cardiovascular disease an individual participant data meta-analysis. Hypertension. 2017;69(6):1045–52.
Van Sloten TT, et al. Carotid stiffness is associated with incident stroke: a systematic review and individual participant data meta-analysis. J Am Coll Cardiol. 2015;66(19):2116–25.
Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness. A systematic review and meta-analysis. J Am Coll Cardiol. 2010;55(13):1318–27.
Kim SK, Massett MP Genetic regulation of endothelial vasomotor function, Front Physiol 7, no. NOV. Frontiers Media S.A., p. 571, 25-Nov-2016.
Nardin C, et al. Cardiovascular phenotype of elevated blood pressure differs markedly between young males and females the ENIGMA study. Hypertension. 2018;72(6):1277–84.
Franklin SS, Wilkinson IB, McEniery CM. Unusual hypertensive phenotypes: What is their significance? Hypertension. 2012;59(2):173–8.
Murray E, Guzik T, Delles C. Early hypertension: exploring the vascular phenotype. J Hypertens. 2021;39(Supplement 1):e44–5.
Tarnoki AD, et al. Heritability of central blood pressure and arterial stiffness: a twin study. J Hypertens. 2012;30(8):1564–71.
van Rijn MJE, et al. Heritability of blood pressure traits and the genetic contribution to blood pressure variance explained by four blood-pressure-related genes. J Hypertens. 2007;25(3):565–70.
Levy D, et al. Evidence for a gene influencing blood pressure on chromosome 17: Genome scan linkage results for longitudinal blood pressure phenotypes in subjects from the Framingham Heart Study. Hypertension. 2000;36(4):477–83.
Alwan H, et al. Heritability of ambulatory and office blood pressure in the Swiss population. J Hypertens. 2015;33(10):2061–7.
Celermajer DS, et al. Non-invasive detection of endothelial dysfunction in children and adults at risk of atherosclerosis. Lancet. 1992;340(8828):1111–5.
Carvajal CA et al. (2011) Familial hyperaldosteronism a new presentation of the chimeric CYP11B1/CYP11B2 gene with low prevalence of primary aldosteronism and atypical gene segregation pattern.
Ise T, et al. A chimeric CYP11B1/CYP11B2 gene in glucocorticoid-insuppressible familial hyperaldosteronism. Clin Endocrinol (Oxf). 2001;55(1):131–4.
Mulatero P, et al. Prevalence and characteristics of familial hyperaldosteronism: The PATOGEN study (Primary aldosteronism in TOrino-GENetic forms). Hypertension. 2011;58(5):797–803.
Jordan J, et al. Severely impaired baroreflex-buffering in patients with monogenic hypertension and neurovascular contact. Circulation. 2000;102(21):2611–8.
Burrello J, et al. Is There a Role for Genomics in the Management of Hypertension? Int J Mol Sci. 2017;18(6):1131.
Padmanabhan S, Joe B. Towards Precision Medicine for Hypertension: A Review of Genomic, Epigenomic, and Microbiomic Effects on Blood Pressure in Experimental Rat Models and Humans. Physiol Rev. 2017;97(4):1469–528.
Friso S, et al. Epigenetic control of 11 beta-hydroxysteroid dehydrogenase 2 gene promoter is related to human hypertension. Atherosclerosis. 2008;199(2):323–7.
Hu W, Wang H, Huang H. Analysis of gene expression and preliminary study of methylation about 11β-HSD2 gene in placentas of Chinese pre-eclampsia patients of Han ethnicity. J Obstet Gynaecol Res. 2015;41(3):343–9.
Lee HA, Cho HM, Lee DY, Kim KC, Han HS, Kim IK. Tissue-specific upregulation of angiotensin-converting enzyme 1 in spontaneously hypertensive rats through histone code modifications. Hypertension. 2012;59(3):621–6.
Wise IA, Charchar FJ Epigenetic modifications in essential hypertension, Int J Mol Sci 17(4). MDPI AG, p. 451, 25-Mar-2016.
Niiranen TJ, et al. Risk for hypertension crosses generations in the community: a multi-generational cohort study. Eur Heart J. 2017;38(29):2300–8.
Ramirez LA, Sullivan JC Sex differences in hypertension: Where we have been and where we are going, Am J Hypertension, 31(12). Oxford University Press, pp. 1247–1254, 13-Nov-2018.
Paiva AMG, et al. Impact of hypertension phenotypes on the office and 24-h pulse wave velocity and augmentation index in individuals with or without antihypertensive medication use. Hypertens Res. 2019;42(12):1989–95.
Yan RT, Anderson TJ, Charbonneau F, Title L, Verma S, Lonn E (2005) Relationship between carotid artery intima-media thickness and brachial artery flow-mediated dilation in middle-aged healthy men, J Am Coll Cardiol
North KE, et al. Heritability of carotid artery structure and function: the strong heart family study. Arterioscler Thromb Vasc Biol. 2002;22(10):1698–703.
Sacco RL, et al. Heritability and linkage analysis for carotid intima-media thickness: the family study of stroke risk and carotid atherosclerosis. Stroke. 2009;40(7):2307–12.
Cecelja M, et al. Arterial stiffening is a heritable trait associated with arterial dilation but not wall thickening: a longitudinal study in the twins UK cohort. Eur Heart J. 2018;39(24):2282–8.
Sayed-Tabatabaei FA, et al. Heritability of the function and structure of the arterial wall: findings of the Erasmus Rucphen Family (ERF) study. Stroke. 2005;36(11):2351–6.
Armstrong C, Abilleira S, Sitzer M, Markus HS, Bevan S. Polymorphisms in MMP family and TIMP genes and carotid artery intima-media thickness. Stroke. 2007;38(11):2895–9.
Zabalza M et al. Association Between Coronary Artery Disease Genetic Variants and Subclinical Atherosclerosis: An Association Study and Meta-analysis, Rev. Española Cardiol. (English Ed., 68(10):869–877, Oct. 2015.
Strawbridge RJ et al. Carotid intima-media thickness novel loci, sex-specific effects, and genetic correlations with obesity and glucometabolic traits in UK Biobank, Arterioscler Thromb Vasc Biol 2020.
Wight TN (2002) Versican: A versatile extracellular matrix proteoglycan in cell biology, Curr Opin Cell Biol.
Carty CL, et al. Association of genetic variation in serum amyloid-A with cardiovascular disease and interactions with IL6, IL1RN, IL1β and TNF genes in the cardiovascular health study. J Atheroscler Thromb. 2009;16(4):419–30.
Wang L, Colón W. The interaction between apolipoprotein serum amyloid A and high-density lipoprotein. Biochem Biophys Res Commun. 2004;317(1):157–61.
Xie X, et al. Polymorphisms in the SAA1/2 gene are associated with carotid intima media thickness in healthy han chinese subjects: the cardiovascular risk survey. PLoS ONE. 2010;5(11): e13997.
Johnson BD, et al. Serum amyloid a as a predictor of coronary artery disease and cardiovascular outcome in women: the national heart, lung, and blood institute-sponsored women’s ischemia syndrome evaluation (WISE). Circulation. 2004;109(6):726–32.
Fox CS, et al. Genetic and environmental contributions to atherosclerosis phenotypes in men and women: heritability of carotid intima-media thickness in the Framingham heart study. Stroke. 2003;34(2):397–401.
Jartti L, et al. Population-based twin study of the effects of migration from Finland to Sweden on endothelial function and intima-media thickness. Arterioscler Thromb Vasc Biol. 2002;22(5):832–7.
Mitchell GF, et al. Heritability and a genome-wide linkage scan for arterial stiffness, wave reflection, and mean arterial pressure: The Framingham heart study. Circulation. 2005;112(2):194–9.
Ge D, Young TW, Wang X, Kapuku GK, Treiber FA, Snieder H (2007) Heritability of Arterial Stiffness in Black and White American Youth and Young Adults, Am J Hypertens.
Mulukutla SR, et al. Black race is associated with digital artery endothelial dysfunction: results from the Heart SCORE study. Eur Heart J. 2010;31(22):2808–15.
Schnabel RB, et al. Noninvasive vascular function measurement in the community cross-sectional relations and comparison of methods. Circ Cardiovasc Imaging. 2011;4(4):371–80.
Benjamin EJ, et al. Clinical Correlates and Heritability of Flow-Mediated Dilation in the Community: The Framingham Heart Study. Circulation. 2004;109(5):613–9.
Zhao J, et al. Heritability of flow-mediated dilation: a twin study. J Thromb Haemost. 2007;5(12):2386–92.
Larsson E, et al. Hypertension and genetic variation in endothelial-specific genes. PLoS ONE. 2013;8(4):62035.
Liu T, et al. Association between daily cigarette consumption and hypertension moderated by CYP2A6 genotypes in Chinese male current smokers. J Hum Hypertens. 2013;27(1):24–30.
Natarajan P, et al. Multiethnic exome-wide association study of subclinical atherosclerosis. Circ Cardiovasc Genet. 2016;9(6):511–20.
Bis JC, et al. Meta-analysis of genome-wide association studies from the CHARGE consortium identifies common variants associated with carotid intima media thickness and plaque. Nat Genet. 2011;43(10):940–8.
Feitosa MF et al (2018) Novel genetic associations for blood pressure identified via gene-alcohol interaction in up to 570K individuals across multiple ancestries, PLoS One, 13(6).
López-Mejías R et al. (2014) Lack of association between ABO, PPAP2B, ADAMST7, PIK3CG, and EDNRA and carotid intima-media thickness, carotid plaques, and cardiovascular disease in patients with rheumatoid arthritis, Mediators Inflamm 2014.
Carnevale D, Lembo G PI3Kγ in hypertension: A novel therapeutic target controlling vascular myogenic tone and target organ damage, Cardiovasc Res, vol. 95, no. 4. Oxford Academic, pp. 403–408, 01-Sep-2012.
Hoffmann TJ et al. (2017) Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation, Nat Genet
Warren HR, et al. Genome-wide association analysis identifies novel blood pressure loci and offers biological insights into cardiovascular risk. Nat Genet. 2022;49(3):403–15.
van SJ et al. (2013) Genome-wide association study of coronary and aortic calcification implicates risk loci for coronary artery disease and myocardial infarction. Atherosclerosis 228(2): 400–405.
Wirtwein M, et al. Elevated ambulatory systolic-diastolic pressure regression index is genetically determined in hypertensive patients with coronary heart disease. Blood Press. 2017;26(3):174–80.
Jj M, et al. Large scale association analysis for identification of genes underlying premature coronary heart disease: cumulative perspective from analysis of 111 candidate genes. J Med Genet. 2004;41(5):334–41.
Gertow K, et al. Identification of the BCAR1-CFDP1-TMEM170A locus as a determinant of carotid intima-media thickness and coronary artery disease risk. Circ Cardiovasc Genet. 2012;5(6):656–65.
Arya R, et al. A genetic association study of carotid intima-media thickness (CIMT) and plaque in Mexican Americans and European Americans with rheumatoid arthritis. Atherosclerosis. 2018;271:92–101.
Nandakumar P, et al. Analysis of putative cis-regulatory elements regulating blood pressure variation. Hum Mol Genet. 2020;29(11):1922–32.
Vojinovic D et al. (2018) Whole-genome linkage scan combined with exome sequencing identifies novel candidate genes for carotid intima-media thickness, Front Genet 9, no. OCT, pp 1–10
Ali AT, Boehme L, Carbajosa G, Seitan VC, Small KS, Hodgkinson A (2019) Nuclear genetic regulation of the human mitochondrial transcriptome, Elife 8
Paradossi U, Ciofini E, Clerico A, Botto N, Biagini A, Colombo MG. Endothelial function and carotid intima-media thickness in young healthy subjects among endothelial nitric oxide synthase Glu298→Asp and T-786→C polymorphisms. Stroke. 2004;35(6):1305–9.
Nassereddine S, et al. The polymorphism G894T of endothelial nitric oxide synthase (eNOS) gene is associated with susceptibility to essential hypertension (EH) in Morocco. BMC Med Genet. 2018;19(1):1–8.
Giri A et al. (2019) Trans-ethnic association study of blood pressure determinants in over 750,000 individuals, Nat Genet.
Dhingra R, et al. Relations of matrix remodeling biomarkers to blood pressure progression and incidence of Hypertension in the community. Circulation. 2009;119(8):1101–7.
Beilby JP, Chapman CML, Palmer LJ, McQuillan BM, Thompson PL, Hung J. Stromelysin-1 (MMP-3) gene 5A/6A promoter polymorphism is associated with blood pressure in a community population. J Hypertens. 2005;23(3):537–42.
Liu X, et al. Associations between polymorphisms of the CXCL12 and CNNM2 gene and hypertension risk: a case-control study. Gene. 2018;675:185–90.
Lambrinoudaki I, et al. Subclinical atherosclerosis and vascular stiffness in premenopausal women: association with NOS3 and CYBA polymorphisms. Heart Vessels. 2018;33(12):1434–44.
Zhang L, et al. Lack of associations of ten candidate coronary heart disease risk genetic variants and subclinical atherosclerosis in four U.S. populations: the population architecture using genomics and epidemiology (PAGE) study. Atherosclerosis. 2013;228(2):390–9.
Zhang X, Liu L, Gu R, Wang X (2019) Association between the ADAMT33 variant and carotid artery intima-media thickness in the Chinese Han population
Heßler N, et al. Linkage and association analysis identifies TRAF1 influencing common carotid intima-media thickness. Stroke. 2016;47:2904–9.
Zekavat SM, et al. Genetic association of finger photoplethysmography-derived arterial stiffness index with blood pressure and coronary artery disease. Arterioscler Thromb Vasc Biol. 2019;39(6):1253–61.
Fung K, et al. Genome-wide association study identifies loci for arterial stiffness index in 127,121 UK Biobank participants OPEN. Nat Sci Rep. 2019;9:9143.
Qi Y, et al. Novel mechanism of blood pressure regulation by forkhead box class O1-mediated transcriptional control of hepatic angiotensinogen. Hypertension. 2014;64(5):1131–40.
Desch M et al. (2010) IRAG determines nitric oxide-and atrial natriuretic peptide-mediated smooth muscle relaxation, Cardiovasc Res.
Tarasov KV, et al. COL4A1 is associated with arterial stiffness by genome-wide association scan. Circ Cardiovasc Genet. 2009;2(2):151–8.
Evangelou E, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. 2018;50(10):1412–25.
Levy D, et al. Framingham heart study 100K project: genome-wide associations for blood pressure and arterial stiffness. BMC Med Genet. 2007;8(SUPPL. 1):S3.
Mitchell GF, et al. Common genetic variation in the 3β-BCL11B gene desert is associated with carotid-femoral pulse wave velocity and excess cardiovascular disease risk the aortagen consortium. Circ Cardiovasc Genet. 2012;5(1):81–90.
Rimpelä JM, et al. Genome-wide association study of nocturnal blood pressure dipping in hypertensive patients. BMC Med Genet. 2018;19(1):1–11.
Gupta RM, et al. A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression. Cell. 2017;170(3):522-533.e15.
Schutte AE, Volpe M, Tocci G, Conti E. Revisiting the relationship between blood pressure and insulin-like growth factor-1. Hypertension. 2014;63(5):1070–7.
Li JP, et al. The association between paired basic amino acid cleaving enzyme 4 gene haplotype and diastolic blood pressure. Chin Med J (Engl). 2004;117(3):382–8.
Ehret GB, et al. Genetic variants in novel pathways influence blood pressure and cardiovascular disease risk. Nature. 2011;478(7367):103–9.
Mitchell GF, et al. Vascular stiffness and genetic variation at the endothelial nitric oxide synthase locus: the framingham heart study. Hypertension. 2007;49(6):1285–90.
Alvim RO, Santos PCJL, Ferreira NE, Mill JG, Krieger JE, Pereira AC. Thioredoxin interacting protein (TXNIP) rs7212 polymorphism is associated with arterial stiffness in the Brazilian general population. J Hum Hypertens. 2012;26(5):340–2.
Brull DJ, et al. Effect of a COL1A1 Sp1 binding site polymorphism on arterial pulse wave velocity: an index of compliance. Hypertension. 2001;38(3):444–8.
Lajemi M et al. (2001) Endothelin gene variants and aortic and cardiac structure in never-treated hypertensives, Am J Hypertens 14(8) I, pp. 755–760.
Mattace-Raso FUS, et al. Angiotensin-converting enzyme gene polymorphism and common carotid stiffness: The Rotterdam study. Atherosclerosis. 2004;174(1):121–6.
Benetos A et al. Influence of angiotensin-converting enzyme and angiotensin II type 1 receptor gene polymorphisms on aortic stiffness in normotensive and hypertensive patients, Circulation 1996.
Dima I, et al. Association of arterial stiffness with the angiotensin-converting enzyme gene polymorphism in healthy individuals. Am J Hypertens. 2008;21(12):1354–8.
Lajemi M (2001) et al. Angiotensin II type 1 receptor-153A/G and 1166A/C gene polymorphisms and increase in aortic stiffness with age in hypertensive subjects, J Hypertens.
Benetos A et al. (1995) Influence of angiotensin II type 1 receptor polymorphism on aortic stiffness in never-treated hypertensive patients, in Hypertension.
Gardier S, Vincent M, Lantelme P, Rial MO, Bricca G, Milon H (2004) A1166C polymorphism of angiotensin II type 1 receptor, blood pressure and arterial stiffness in hypertension, J Hypertens
Mayer O, Filipovský J, Pešta M, Cífková R, Dolejšová M, Šimon J (2008) Synergistic effect of angiotensin II type 1 receptor and endothelial nitric oxide synthase gene polymorphisms on arterial stiffness, J Hum Hypertens.
Taniwaki H, et al. Association of ACE gene polymorphism with arterial stiffness in patients with type 2 diabetes. Diabetes Care. 1999;22(11):1858–64.
Sie MPS et al. (2009) Genetic variation in the renin-angiotensin system and arterial stiffness. the rotterdam study, Clin Exp Hypertens.
Bozec E et al. Arterial stiffness and angiotensinogen gene in hypertensive patients and mutant mice, J Hypertens.
Bonnardeaux A et al. (1994) Angiotensin II type 1 receptor gene polymorphisms in human essential hypertension, Hypertension.
Ehret GB et al. (2016) The genetics of blood pressure regulation and its target organs from association studies in 342,415 individuals, Nat Genet.
Åström Malm I, Alehagen U, Blomstrand P, Dahlström U, De Basso R (2020) Higher blood pressure in elderly hypertensive females, with increased arterial stiffness and blood pressure in females with the Fibrillin-1 2/3 genotype, BMC Cardiovasc Disord 20(1).
Medley TL, Cole TJ, Gatzka CD, Wang WYS, Dart AM, Kingwell BA. Fibrillin-1 genotype is associated with aortic stiffness and disease severity in patients with coronary artery disease. Circulation. 2002;105(7):810–5.
Hong KW, et al. Genetic variations in ATP2B1, CSK, ARSG and CSMD1 loci are related to blood pressure and/or hypertension in two Korean cohorts. J Hum Hypertens. 2010;24(6):367–72.
Burghardt KJ, Grove TB, Ellingrod VL. Endothelial nitric oxide synthetase genetic variants, metabolic syndrome and endothelial function in schizophrenia. J Psychopharmacol. 2014;28(4):349–56.
Korsakova N et al. Genetic polymorphisms as risk factors of endothelial dysfunction in pregnant women, in European Hematology Association, 2018, PB2508.
Montrezol FT, et al. ACE gene plays a key role in reducing blood pressure in the hyperintensive elderly after resistance training. J Strength Cond Res. 2019;33(4):1119–29.
Siedlinski M, et al. Vascular transcriptome profiling identifies Sphingosine kinase 1 as a modulator of angiotensin II-induced vascular dysfunction. Sci Rep. 2017;7(1):1–13.
Di Pietro D, Oliveti M, Sommella E, Al E Sortilin evokes endothelial dysfunction and arterial hypertension through the dysregulation of sphingolipid metabolism and oxidative stress, in ESC Congress 2020—The Digital Experience—Programme, 2020.
Yoshino S et al (2016) Sex-specific genetic variants are associated with coronary endothelial dysfunction, J Am Heart Assoc 5(4).
Newton-Cheh C, et al. Genome-wide association study identifies eight loci associated with blood pressure. Nat Genet. 2009;41(6):666–76.
McMaster W, Saleh M, Kirabo M, Itani H, Harrison D, Madhur M (2022) Deficiency of LNK/SH2B3 promotes hypertension, endothelial dysfunction, and aortic dissection, Angiogenesis, vol. 17, no. Developmental Vascular Biology and Genetics Workshop VI and Vascular Inflammation Workshop organized by NAVBO: ABSTRACTS, pp. 935–984, 2014.
Dale BL, Madhur MS Linking Inflammation and Hypertension via LNK/SH2B3.
Ference BA, Julius S, Mahajan N, Levy PD, Williams KA, Flack JM. Clinical effect of naturally random allocation to lower systolic blood pressure beginning before the development of hypertension. Hypertension. 2014;63(6):1182–8.
Smyth SS, Cheng HY, Miriyala S, Panchatcharam M, Morris AJ Roles of lysophosphatidic acid in cardiovascular physiology and disease, Biochimica et Biophysica Acta—Molecular and Cell Biology of Lipids, 1781(9). NIH Public Access, pp. 563–570, Sep-2008.
Gu K, Zhang Y, Sun K, Jiang X (2020) Associations between PHACTR1 gene polymorphisms and pulse pressure in Chinese Han population, Biosci Rep. 40(6).
Zhang D, et al. Genome-wide linkage and association scans for pulse pressure in Chinese twins. Hypertens Res. 2012;35(11):1051–7.
Ingelsson E, Syvänen AC, Lind L. Endothelium-dependent vasodilation in conduit and resistance vessels in relation to the endothelial nitric oxide synthase gene. J Hum Hypertens. 2008;22(8):569–78.
Shibao CA, et al. A Common CD36 Variant Influences Endothelial Function and Response to Treatment with Phosphodiesterase 5 Inhibition. J Clin Endocrinol Metab. 2016;101(7):2751–8.
Liu X, Meng F, Yang P. Association study of CD36 single nucleotide polymorphisms with essential hypertension in the Northeastern Han Chinese. Gene. 2013;527(1):410–5.
Akpinar TS, et al. Endothelial constitutive nitric oxide synthase, angiotensin converting enzyme, angiotensin II type 1 receptor gene polymorphisms and endothelial functions in healthy individuals. Eur Rev Med Pharmacol Sci. 2014;18(1):39–45.
Fan M, et al. CYBA C242T gene polymorphism and flow-mediated vasodilation in a population of young adults: The cardiovascular risk in young finns study. J Hypertens. 2007;25(7):1381–7.
Rafiq A, Aslam K, Malik R, Afroze D. C242T polymorphism of the NADPH oxidase p22PHOX gene and its association with endothelial dysfunction in asymptomatic individuals with essential systemic hypertension. Mol Med Rep. 2014;9(5):1857–62.
Guangda X, Yuhua W. Apolipoprotein e4 allele and endothelium-dependent arterial dilation in Type 2 diabetes mellitus without angiopathy. Diabetologia. 2003;46(4):514–9.
Traylor M, et al. Influence of genetic variation in PDE3A on endothelial function and stroke. Hypertension. 2020;75(2):365–71.
Maass PG, et al. PDE3A mutations cause autosomal dominant hypertension with brachydactyly. Nat Genet. 2015;47(6):647–53.
Luft FC et al. (2019) Abstract 039: The PDE3A gene regulates blood pressure, Hypertension 74(Suppl_1).
Levy D, et al. Genome-wide association study of blood pressure and hypertension. Nat Genet. 2009;41(6):677–87.
Celermajer DS, Sorensen KE, Barley J, Jeffrey S, Carter N, Deanfield J. Angiotensin-converting enzyme genotype is not associated with endothelial dysfunction in subjects without other coronary risk factors. Atherosclerosis. 1994;111(1):121–6.
Logan JG, Engler MB, Kim H. Genetic Determinants of Arterial Stiffness. Adv Data Anal Classif. Feb. 2015;9(1):23–43.
Lacolley P, Challande P, Osborne-Pellegrin M, Regnault V. Genetics and pathophysiology of arterial stiffness. Cardiovasc Res. 2008;81(4):637–48.
Salvi P et al. (2008) Comparative study of methodologies for pulse wave velocity estimation, J Hum Hypertens.
Van Bortel LM, et al. Expert consensus document on the measurement of aortic stiffness in daily practice using carotid-femoral pulse wave velocity. J Hypertens. 2012;30(3):445–8.
Gaeta G, et al. Arterial abnormalities in the offspring of patients with premature myocardial infarction. N Engl J Med. 2000;343(12):840–6.
Clarkson P, Celermajer DS, Powe AJ, Donald AE, Henry RMA, Deanfield JE. Endothelium-dependent dilatation is impaired in young healthy subjects with a family history of premature coronary disease. Circulation. 1997;96(10):3378–83.
MacGregor AJ, Snieder H, Schork NJ, Spector TD Twins—novel uses to study complex traits and genetic diseases, Trends in Genetics 16(3). Trends Genet, pp. 131–134, 01-Mar-2000.
Persu A, et al. Modifier effect of ENOS in autosomal dominant polycystic kidney disease. Hum Mol Genet. 2002;11(3):229–41.
Jones LC, Hingorani AD. Genetic regulation of endothelial function. Heart. 2005;91(10):1275–7.
Casas JP, Bautista LE, Humphries SE, Hingorani AD. Endothelial nitric oxide synthase genotype and ischemic heart disease: meta-analysis of 26 studies involving 23 028 subjects. Circulation. 2004;109(11):1359–65.
Park JY, et al. NFKB1 promoter variation implicates shear-induced NOS3 gene expression and endothelial function in prehypertensives and stage I hypertensives. Am J Physiol—Hear Circ Physiol. 2007;293(4):H2320.
Hopper JL, Mathews JD. A multivariate normal model for pedigree and longitudinal data and the software ‘fisher.’ Aust J Stat. 1994;36(2):153–76.
Rafiq K, Nishiyama A (2014) Is antioxidant therapy effective for advanced hypertension and renal injury?, J Hypertens.
S. ˇ eda O et al. Systematic, genome-wide, sex-specific linkage of cardiovascular traits in french canadians,” 2008.
Mattace-Raso FUS, et al. Determinants of pulse wave velocity in healthy people and in the presence of cardiovascular risk factors: ‘establishing normal and reference values.’ Eur Heart J. 2010;31(19):2338–50.
Alexander Y et al. (2021) Endothelial function in cardiovascular medicine: a consensus paper of the European society of cardiology working groups on atherosclerosis and vascular biology, aorta and peripheral vascular diseases, coronary pathophysiology and microcirculation, and Thr, Cardiovasc. Res..
Acknowledgements
We additionally wish to acknowledge Prof T. Guzik, University of Glasgow, for a supervisory role.
Funding
E. Murray received European Research Council funding to undertake an MD as part of the Inflammatension Study (Grant number ERC-2016-COG awarded to Prof T Guzik).
Author information
Authors and Affiliations
Contributions
All authors contributed to study design and manuscript writing, and approved the final version. ECM and AC additionally contributed to the literature search, data gathering and analysis.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to disclose.
Ethics Approval
As a review of published literature, ethics approval was not required.
Consent for Publication
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Craig, A., Delles, C. & Murray, E.C. A Review of Vascular Traits and Assessment Techniques, and Their Heritability. Artery Res 28, 61–78 (2022). https://doi.org/10.1007/s44200-022-00016-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s44200-022-00016-y