
Abstract
Melanoma, a highly prevalent cancer worldwide, exhibits remarkable diversity and plasticity, with the adverse prognosis of advanced melanoma remaining a focal point of investigation. Despite the emergence of novel drugs and combination therapies improving patient outcomes, challenges such as drug resistance and incomplete mechanistic understanding persist. Transcriptional programs play a pivotal role in determining the characteristics of both normal and tumour cells, with their dysregulation of these programs being a hallmark of melanoma. Abnormalities in transcription regulation not only impact the characteristics of melanoma cells but also influence the tumor’s metabolism and immune microenvironment, forming a complex network in tumours. Thus, understanding these changes comprehensively is crucial for unravelling the mechanisms underlying melanoma initiation, progression, response to targeted and immune therapies, and treatment resistance. This review primarily explores the transcriptional features in normal melanocytes and melanoma cells, emphasizing their profound impact on cell metabolism and immune evasion. Furthermore, the plasticity of melanoma cells and its relationship with treatment resistance and metastasis are highlighted, emphasizing the importance of targeting dysregulated transcriptional factors and pathways. Finally, potential clinical implications in targeting transcriptional abnormalities are highlighted, particularly in metastatic or treatment-resistant melanomas. This comprehensive overview aims to contribute to the advancement of melanoma research and the development of precise and effective treatments.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Melanoma is one of the most common tumours worldwide, exhibiting significant diversity and plasticity [1, 2]. Advanced melanoma typically leads to poor prognosis, and recent exciting progress has been made in the research of advanced melanoma and therapy-resistant melanoma. In addition to traditional chemotherapy, the US Food and Drug Administration (FDA) has approved a range of novel anti-melanoma drugs, including targeted therapy against point mutations and immune checkpoint therapy [3]. Simultaneously, a series of combination treatment strategies have entered phase III clinical trials [4, 5].
The study of the gene expression regulatory mechanisms in melanoma is of great significance for understanding the development of the disease. Many papers describe gene variations that affect cell signaling, transcription factors (TFs), epigenetic modifications, chromatin regulatory factors, and chromosomal structures, which may form a complex network [6]. The transcription program operating in normal or tumour cells determines the characteristics of the cells, established through sequential gene expression processes involving major TFs and more signaling molecules [7]. In addition to directly affecting the characteristics of melanoma itself, such as proliferation and metastasis, abnormalities in transcription regulation may indirectly impact the metabolism and immune microenvironment of tumour cells, resulting in poor response to treatment and prognosis [8].
This review provides a summary of the identified changes in dysregulated transcription programs in melanoma. We particularly emphasize the key components mediating transcriptional changes in proliferative or metastatic melanoma [1] which may contribute to controlling advanced melanoma [9]. Delving into the complexity of the melanoma genome and exploring dysregulated transcription programs as key drivers of tumour development aims to provide a comprehensive overview to advance melanoma research and the development of targeted therapeutic strategies.
2 Transcriptional programs in normal melanocytes
2.1 The origin of melanocytes
Melanocytes in the human body originate during neurogenesis in the embryonic stage, typically around 6–8 weeks, known as melanoblasts [6]. These melanoblasts develop from neural crest cells and migrate to various locations, including the eyes, inner ears, heart, adipose tissue, mucosa, brain, and skin [10]. Once within the epidermis, melanoblasts proliferate and differentiate, leading to colonization of the entire embryonic skin. During the 15th-24th weeks of human embryonic development, hair follicles begin to form. During this period, a portion of mature melanocytes differentiate and migrate to the bulb of the hair follicles, while others differentiate into melanocyte stem cells (MSCs), a population of embryonic progenitors with self-renewal capability, which also migrate and settle in the hair follicles [11, 12]. In the subsequent developmental process, melanocyte stem cells provide mature melanocytes to the hair bulb to produce pigment for the newly formed hair follicles, as well as supplying mature melanocytes to the interfollicular epidermis, serving as a reservoir for mature melanocytes in adult skin [13]. Initially existing mature melanocytes in the dermis undergo asymmetric division during the embryonic period to supplement the mature melanocytes in the epidermis and gradually disappear after childhood [14]. However, mature melanocytes persist in the dermis, resulting in blue or gray patches on the skin, known as nevi or Mongolian spots [14]. Therefore, melanin in adult skin is secreted by epidermal melanocytes, and two populations of melanocytes exist in hair follicles: mature melanocytes capable of producing melanin and melanocyte stem cells that serve as a reservoir for mature melanocytes [6].
2.2 Roles of MITF in melanocyte development, survival and proliferation
Microphthalmia-associated transcription factor (MITF) is a transcription factor that regulates the transcriptional expression of downstream genes by binding directly to DNA or in conjunction with cofactors. MITF is a basic helix-loop-helix-zip (bHLHZip) transcription factor encoded on chromosomes 3p12.3–14.115. In the process of melanocyte development, MITF plays a crucial role. MITF may be the first transcription factor to appear in specifying melanocytes during development [15]. In humans, melanoblasts first emerge as epidermal cells expressing MITF in embryos at 6–8 weeks of gestation. During melanoma development in mice, MITF is detected at approximately embryonic days 9–10, along with DCT, marking the establishment of melanocyte specificity [6, 16]. Loss of MITF leads to the complete absence of neurocristic pigment cells, resulting not only in a lack of pigmentation but also in abnormal development of the ears and eyes, as observed in patients with type 2A Waardenburg syndrome (WS) and Tietz syndrome [17].
MITF is the most important transcription factor in melanoma development and the maintenance of melanocyte function. During human embryonic development, mature melanocytes actively proliferate in the epidermis and, under the influence of various transcription factors, including MITF, acquire the enzymes necessary for melanin production, eventually differentiating into mature melanocytes [6]. Under UV irradiation, MITF responds to upregulated α-melanocyte-stimulating hormone (α-MSH) and key melanocortin 1 receptor ligand (MC1R) signaling pathway to induce the expression of pigment-producing enzymes, including tyrosinase (TYR), tyrosinase-related protein 1 (TYRP1), and dopachrome tautomerase (DCT), participating in the regulation of melanin production [18, 19]. Another pathway regulating melanin production, the stem cell factor (SCF)-KIT pathway, also converges on MITF to activate genes involved in melanin production [17, 20].
MITF promotes differentiation-related functions in melanocytes, regulating genes associated with pigmentation such as TYR, TYRP1, DCT, MLANA, SILV, and SLC24A5, as well as cell adhesion-related genes including cancer embryonic antigen-related cell adhesion molecule 1 (CAECAM), and genes involved in melanosome transport such as RAB27a and MYOSIN5a (MYO5a) [15, 21]. Moreover, MITF plays a crucial role in the survival of melanocytes. It regulates survival through BCL-2 (an anti-apoptotic member of the BCL2 family), and decreases in MITF below a threshold level result in reduced BCL2, potentially inducing apoptosis in mature melanocytes [22]. The role of MITF in melanocyte proliferation is multifaceted. An optimal level of MITF promotes melanocyte proliferation, whereas low levels of MITF are associated with dedifferentiation, increased invasiveness, and elevated levels of p27 (CDKN1B), a cyclin-dependent kinase inhibitor protein, leading to reduced proliferation. Conversely, cells with high MITF activity induce cell cycle arrest through increased expression of p21 (CDKN1A) and p16 INK4a (CDKN2A), which negatively impacts cell proliferation [15, 23].
2.3 Roles of other transcription factors in melanocytes
In the differentiation, melanogenesis, survival, and proliferation of normal melanocytes, various transcription factors other than MITF play important roles. Sry Related HMG box 10 (SOX10) is a high-mobility group (HMG) transcription factor involved in melanocyte migration [24]. Concurrently, SOX10 direct activation of MITF transcription.
Additionally, signal transducer and activator of transcription 3 (STAT3) may directly contribute to melanocyte proliferation and may play a crucial role in α-MSH-induced melanin production [25, 26]. cAMP response element binding protein (CREB), as a direct effector of protein kinase A (PKA), plays an important role in the MC1R-induced melanin production pathway [27]. Recent studies have shown that the CNC family transcription factor Nrf3 is expressed in the basal layer of the epidermis, where melanocytes are located, and it coordinates melanin production through phagocytosis and autophagy [28].
UV radiation cause DNA damage and affect the specificity of TFs binding to DNA. Intermittent high-intensity UV radiation during childhood has been identified as a significant modifiable risk factor for melanoma in the white race [29]. But for the colored skin, the association between UV exposure and melanoma was weaker [30]. UV exposure reduces the DNA binding specificity of transcription factors (TFs) and leads to competition between TFs and DNA repair proteins, and as a result, this interference with DNA repair by TFs promotes melanoma development through UV-induced mutations [31].
3 Transcriptional dysregulation and the impacted factors in melanoma
3.1 Key transcription factors
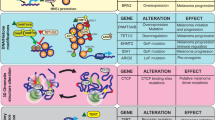
Transcription factor dysregulation, epigenetic modification, and metabolic reprogramming all contribute to melanoma development (Fig. 1). First and foremost, abnormal expression of key transcription factors plays a significant role in the development of melanocytes into melanoma cells [32]. MITF, isolated approximately 30 years ago, has been extensively studied by numerous scientists and found to serve as a key coordinator of various aspects of melanocyte and melanoma biology [15]. In normal melanocytes, MITF regulates melanocyte development, melanin synthesis, and melanocyte survival [6]. In melanoma cells, MITF plays a crucial role in determining the phenotype of melanoma cells, with high MITF levels associated with a more proliferative state and low MITF levels linked to increased invasiveness, but both are capable of tumour formation [21, 33].

Dysregulation of transcription factors, epigenetic modification, and metabolic reprogramming in melanoma development. A During the process of melanocyte development into melanoma cells, transcription factors such as MITF, SOX10, and SOX9 become dysregulated. B Epigenetic modifications of genetic material such as DNA methylation, histone acetylation, chromatin remodeling, and non-coding RNAs. C Metabolic reprogramming of carbohydrates, lipids, and proteins in a relatively hypoxic and nutrient-limiting environment
Early studies on the systematic analysis of the cancer genome revealed that MITF is a target of amplification in melanoma cells [34]. MITF amplification has been identified in approximately 20% of melanoma populations, and studies on melanoma cell lines have shown that superenhancer, BRAF mutations and p16 inactivation can lead to MITF amplification [35]. MITF amplification is more prevalent in metastatic disease and is associated with decreased overall patient survival; however, it is not related to the chemotherapy response in melanoma patients [34, 36]. Recently, single-cell sequencing data has revealed the upregulation of MITF in acral melanoma, promoting lymph node metastasis through the activation of the fatty acid oxidation (FAO) pathway [37]. In addition to gene amplification, mutations in the MITF gene are also associated with melanoma development, where MITF is like an oncogene in melanoma [32]. The E318K mutation in MITF can lead to both familial and sporadic melanoma [38], while the MITF-E87R mutation promotes tumour progression by enhancing invasion through increased expression of S100A4 and reduced adhesion [39]. MITF is also regulated by factors other than genetic factors. High expression of MITF in melanoma is associated with Super enhancers (SE) [35]. SEs are clusters of enhancers bound by a high density of transcription factors and co-factors, often associated with genes that control and define cell identity [40, 41]. SOX10, a known transcriptional regulator of MITF, has been identified as an SE marker in some melanoma cell lines with high MITF expression [42, 43]. Several other transcription factors, such as PAX3, CREB, ZEB2, and LEF1, also promote MITF mRNA transcription in melanoma cells [44, 45]. Moreover, MITF can cooperate with lymphoid-enhancing factor-1 (LEF-1) as a non-DNA-binding co-activator to enhance its self-expression in melanoma [46]. Conversely, various transcription factors, including ATF2, c-MYC, HOXA1, GLI2, BRN2, and NF-κB can inhibit MITF mRNA expression [47]. In addition to various splice isoforms, the MITF protein undergoes extensive post-translational modification. Serine and threonine phosphorylation affect protein stability, ubiquitination possibly regulates target specificity and protein stability, acetylation may decrease MITF DNA-binding affinity and residence time, and tyrosine phosphorylation enhances MITF’s ability to regulate a range of target gene transcription [15, 48]. Therefore, MITF expression and regulation are complex processes.
SOX10, a lineage-specific transcription factor crucial for MITF expression, plays a significant role in melanoma cell growth [42]. It mediates phenotypic switching in melanoma, facilitating transitions between states with distinct proliferative, invasive, and drug-resistant characteristics. High expression of SOX10 positively regulates melanoma cell proliferation, contributing to tumour growth. Conversely, the loss of SOX10 results in a shift toward an invasive phenotype, marked by increased expression of mesenchymal genes [49]. This adaptability allows melanoma cells to thrive in diverse microenvironments. Moreover, SOX10 is linked to the immune phenotype in melanoma, with high SOX10 expression associated with lower PD-L1 levels - a sign of negative regulation of PD-L1 expression [50]. Research indicates that SOX10 serves as a highly specific marker for lymph node melanoma metastasis [51].
Similarly, SOX9, another member of the SOX family characterized by the conserved high-mobility group (HMG) DNA-binding domain, acts similarly in adult skin as SOX10 does during embryogenesis [17]. SOX9 also contributes to NEDD9 expression along with high SOX10, thereby enhancing metastatic properties. Elevated SOX9 expression restores the invasiveness of SOX10 knockdown cells in melanoma, and heightened SOX9 levels promote metastasis within heterogeneous melanoma populations [52].
Paired box 3 (PAX3) is a member of the paired box domain family of transcription factors [53], and LEF-1 is a transcription factor involved in the Wnt signaling pathway [54]. They all contribute to the growth and development of normal melanocytes through their interaction with MITF, tyrosinase-related protein-2 (TRP-2), tyrosinase (Tyr), and cyclin-dependent kinase 2 (Cdk2), but also participate in the malignant melanoma development [55]. Importantly, canonical Wnt signaling has been greatly implicated in the phenotype switching in tumor progression, and LEF1 is a crucial a transcription factor that promotes the canonical Wnt/β-catenin signaling pathway in melanoma [56].
3.2 Epigenetic modifications
Beyond gene mutations and TFs, epigenetics emerges as a pivotal player in the early diagnosis, disease monitoring, and prognosis evaluation of melanoma, offering new insights for targeted therapy [57]. DNA methylation, among the earliest and extensively studied epigenetic modifications, leads to gene silencing in melanoma, particularly in the highly methylated promoters of relevant tumour suppressor genes. This phenomenon may contribute to the initiation and progression of tumour cells [58, 59].
Histone acetylation typically correlates with activation of gene expression. Heightened activity of histone deacetylases (HDAC) can downregulate multiple tumour suppressor genes, fostering melanoma cell progression and inducing resistance to alkylating drugs [58]. Notably, acetylation of lysine 27 on histone H3 (H3K27ac) has recently emerged as a crucial mechanism regulating MITF expression, thereby enhancing the metastatic potential of melanoma cells. Additionally, histone methylation, occurring on lysine and/or arginine residues, may directly influence gene transcription, thus playing a direct role in the transformation of melanocytes into melanoma cells [60, 61].
Chromatin remodelling entails the reorganization of chromatin from a condensed state to a transcriptionally accessible state and relies on the activity of polycomb group (PcG) proteins. This process influences the development of melanoma cells by affecting the expression of relevant genes [58].
Non-coding RNAs are mainly divided into two categories: long non-coding RNAs (lncRNA) and small non-coding RNAs (sncRNA). Among sncRNAs, microRNAs (miRNA) (19–22 bps) primarily disrupt post-transcriptional gene expression by interfering with the 3’UTR region of mRNA [62]. SAMMSON, a lncRNA associated with melanoma, is considered one of the key lncRNAs linked with melanoma occurrence, with expression observed in over 90% of melanomas. SAMMSON regulates tumour metabolism by activating the mitochondrial p32 protein, thereby maintaining oxidative phosphorylation and mitochondrial homeostasis [63].
3.3 Metabolic reprogramming
Metabolic reprogramming stands out as a defining characteristic of cancer. Within tumours, enhanced aerobic glycolysis is a common phenomenon, and in melanoma, the prevalent BRAF mutation provides an additional boost to the glycolytic capacity of melanoma cells, resulting in tumour growth and progression [64]. Tumour-specific nitrite production can inhibit the tricarboxylic acid cycle in tumours, restoring the tumour immune microenvironment and sensitizing tumour cells to immune therapy [65]. Moreover, oxidative phosphorylation in mitochondria, regulated by MITF-coexpressed lncRNA SAMMSON, promotes melanoma growth by directly regulating the mitochondrial master regulator p32 [62]. However, high level of oxidative phosphorylation in mitochondria disturb melanoma occurrence.
Disruption of lipid metabolism emerges as another distinctive metabolic feature of melanoma. Reports indicate overweight/obese (OW/OB) patients with metastatic melanoma show unexpected improvements in prognosis after treatment with immune checkpoint inhibitors (ICIs) and BRAF-targeted therapy [66]. The higher expression of PD-1 on T cells in obese individuals enhancing sensitivity to anti-PD1 may explain this phenomenon. Peroxisomes, crucial players in lipid metabolism, are implicated, and mouse experiments suggest that targeting peroxisome biogenesis can heighten melanoma sensitivity to MAPK pathway inhibition, thereby overcoming MAPKi resistance [67].
Imbalances in amino acid metabolism, particularly involving serine, glutamine, and branched-chain amino acids (BCAA), are intricately linked to melanoma’s pathogenesis. 3-phosphoglycerate dehydrogenase (PHGDH) serves as the rate-limiting enzyme in de novo serine synthesis. Melanoma exhibits sensitivity to serine synthesis restrictions under nutrient-limiting conditions. Dietary supplementation of serine or increased PHGDH expression may promote melanoma progression. Glutamine playsv a vital role in normal cellular metabolism. Regional deficiency leads to excessive histone methylation, inducing cell dedifferentiation and enhancing melanoma cells’ resistance to targeted therapy. Conversely, dietary glutamine supplementation can inhibit melanoma growth in vivo through epigenetic suppression. Furthermore, branched-chain amino acids (BCAA), including leucine (Leu), isoleucine (Ile), and valine (Val), promote tumour cell proliferation by supporting acetyl-CoA production in the tricarboxylic acid cycle [64].
4 The melanoma plasticity and the relationship with treatment resistance
In normal developmental processes, cells undergo specific differentiation programs to generate cells and tissues with distinct functions. However, when exposed to particular environmental stimuli, cells with differentiation potential undergo non-heritable phenotypic changes, continuously adapting to the environment for survival [68]. The transition between epithelial and mesenchymal phenotypes, known as epithelial-mesenchymal transition (EMT), enables tumors to switch between proliferative and invasive phenotypes (Fig. 2A). This phenomenon is especially prevalent in tumour cells, enabling them to promptly respond to microenvironmental stress and treatment interventions, leading to metastasis and drug resistance in clinical practice [69]. Cutaneous malignant melanoma bears a high burden of gene mutations, besides genetic changes, melanoma lineage plasticity also plays an important role in the process of metastasis and drug resistance, which is affected by key factors like MITF, ZEB family, SNAI1, and twist-related protein 1 (TWIST1) & TWIST2, as well as epigenetic modifications such as DNA methylation and histone acetylation (Fig. 2). The multilayered regulation of phenotypic plasticity, including epigenetic and transcriptional control, is essential in determining and maintaining melanoma cell states [68, 70,71,72]. A comprehensive understanding of the dynamic nature of phenotypic plasticity is crucial for addressing two major unresolved clinical challenges – cancer metastasis and treatment resistance.

Mechanisms of lineage plasticity in melanoma. A Melanocytes exhibit two distinct phenotypes: those undergoing EMT transition to an invasive phenotype, and those undergoing MET transition to a proliferative phenotype. B Transcription factors involved in EMT, such as SNAI and ZEB, along with epigenetic modifications of genetic material, influence the phenotypic transition of melanoma cells. C Melanocytes undergoing EMT can form vascular structures and promote the growth of melanoma masses
4.1 Epithelial-mesenchymal transition in melanoma regulated by EMT-TFs
The epithelial–mesenchymal transition (EMT) is characterized by the loss of epithelial phenotype and the acquisition of mesenchymal properties. Specifically, it involves the loss of cell–cell adhesion and apical-basal polarity functions, while cells acquire enhanced migratory and invasive mesenchymal characteristics to varying degrees [8, 73], melanoma cells undergoing EMT transformation lose e-cadherin, a crucial cell-adhesion glycoprotein [72]. The reverse process is described as mesenchymal- epithelial transition (MET). EMT is a common occurrence in various cancer cells, recent studies reveal that melanoma cells undergo EMT or MET, transitioning between proliferative and invasive phenotypes [68, 72, 74]. MITF is recognized as a key transcription factor in EMT [75]. Additionally, EMT is regulated by several other transcription factors (EMT-TFs) [76], including zinc finger E-box binding homeobox 1 and 2 (ZEB1 and ZEB2) [75, 77], snail family transcriptional repressor 1 and 2 (SNAI1 and SNAI2) [78], and TWIST1 [79].
ZEB2 is normally expressed in melanocytes, but its expression gradually decreases during the transition to melanoma, while ZEB1 expression increases [79]. In human melanoma, the upregulation of ZEB2 promotes cell growth and proliferation by activating the expression of MITF, and reduces invasion ability [80]. However, recent analysis using single-cell RNA sequencing has provided evidence that ZEB1 negatively regulates melanocyte proliferation programs and upregulates invasive/stem-like programs [79]. Early studies have demonstrated that the loss of Snai1 has a profound effect on the formation of the mesoderm during embryonic development [81, 82]. Besides, it has been observed that the expression of membrane transport proteins can be induced by members of the SNAI family, contributing to the process of EMT [83]. Studies in non-small cell lung cancer have demonstrated that excessive expression of TWIST1 mediates resistance to MET-dependent TKIs by suppressing p27 expression [84]. The acetylation status of TWIST1 plays a regulatory role in EMT, where diacetylated TWIST1-acK73/76 interacts with BRD8 (a component of the TIP60-Com complex) and histone H4-acK5/8, recruiting the TIP60-Com complex to activate mesenchymal target genes and MYC, thereby promoting target gene expression and cancer metastasis [78].
4.2 Epigenetic regulation in melanoma through interaction with EMT-TFs
Although transcription factors can directly regulate target genes, their binding to genes is ultimately temporary. In this context, epigenetic regulation, mainly involving three levels: DNA methylation, histone modification, and RNA interference, stabilizes cellular plasticity [85,86,87]. EMT-TFs interact synergistically with epigenetic enzymes to regulate downstream effectors of EMT.
DNA methylation is catalyzed by a group of proteins called DNA methyltransferases (DNMTs), and high methylation in gene promoter regions can lead to transcriptional repression [87]. DNA hypermethylation has been associated with the invasive phenotype of malignant melanoma [88]. DNMT interacts with EMT-TFs such as SNAI and ZEBs [89], recruited to the promoter region of the E-cadherin gene, leading to downregulation of E-cadherin expression [90], which may induce melanoma cells to undergo EMT. In other tumours, the expression of EMT-TFs has been shown to be regulated by DNA methylation [76, 91].
Histone acetylation modifications also play a role in melanoma plasticity. Numerous histone-modifying enzymes form complexes with EMT-TFs, affecting chromatin condensation and site accessibility during gene regulation. Classical complexes include the NuRD Complex, PRC1 Complex, PRC2 Complex, and SWI/SNF Chromatin-Remodeling Complex [92]. During the process of EMT triggered by the transforming growth factor-beta (TGF-β) signaling, Histone Deacetylase 1 (HDAC1) mediates global histone deacetylation and the acquisition of specific histone H3 lysine 27 acetylation (H3K27ac) marks on enhancers, thereby promoting the occurrence of this process [89].
Long non-coding RNAs (lncRNAs) have emerged as potential regulators of melanoma lineage plasticity. Additionally, the lncRNA SAMMSON acts as a target of the lineage-specific transcription factor SOX10, and targeting SAMMSON has shown promise in enhancing the sensitivity to MAPK-targeted therapy [92].
4.3 EET and EMT contribute to MAPKi resistance in melanoma therapy
During treatment, melanoma cells undergo phenotypic transitions that contribute to the development of drug resistance [1, 93]. Zhou et al. observed EMT-like phenotypic changes in melanoma cells during MAPKi treatment [74]. Geoffrey Richard et al. confirmed a correlation between high levels of ZEB1 expression and intrinsic resistance to MAPKi in BRAFV600 mutant cell lines and tumours. ZEB1 overexpression is sufficient to drive resistance to MAPKi by promoting a reversible transition to a MITFlow/p75high stem cell-like and tumorigenic phenotype [94]. Therefore, a better understanding of the role of EMT-TFs in melanoma cell plasticity will aid in designing novel combination therapies targeting specific EMT-TFs and the MAPK pathway.
Meanwhile, during tumour growth, epithelial–endothelial transition (EET) occurs (Fig. 2C), where stem cell-like cancer cells differentiate into new blood vessels to supply blood to tumour cells [8]. This proces, also known as angiogenic mimicry (VM), refers to a functional microcirculation pattern associated with endothelium-dependent and mosaic vessels in most malignant tumours. The use of inhibitors of VEGF receptor tyrosine kinase activity may facilitate the occurrence of this process [95]. A recent study reveals that endothelial transformation of metastatic melanoma cells can survive as resting cells and may regain metastatic properties through endothelial interstitial transformation (EndMT) for potential spread [96]. However, the role of EMT-TFs in the process of melanoma cell dormancy and reactivation remains unclear, leaving significant research gaps.
4.4 Impacts of melanoma plasticity on anti-tumour immunotherapy
In addition to regulating transcription factors, EMT also promotes immune suppression in melanoma by upregulating PD-L1 expression, inducing immunosuppressive Treg cells, inhibiting CD8+ cytotoxic cells and NK cells, and promoting antigen presentation, thus affecting tumour immune microenvironment [97,98,99]. These alterations in the tumour immune microenvironment facilitate the development of an invasive phenotype in melanoma cells, increasing their propensity for metastasis. During treatment, melanoma cells undergo phenotypic transitions leading to the development of drug resistance [1, 93]. Zhou et al. observed EMT-like phenotypic changes in melanoma cells during MAPKi treatment [74] This phenotypic plasticity also affects immunotherapy, as evidenced by the innate anti-PD1 resistance signature (IPRES), which shares similarities to MAPKi resistance features, including the expression of AXL, ROR2, and WNT5A [93]. Additionally, highly invasive melanoma cells also exhibit higher intrinsic drug resistance. Therefore, targeting characteristic molecules during the EMT process is emerging as a promising therapeutic approach to address melanoma metastasis and chemotherapy resistance.
5 Clinical implications by targeting dysregulated transcription
Lineage plasticity and dysregulated transcriptional regulation play pivotal roles in melanoma progression, influencing treatment responses and disease outcomes. In this context, targeting specific molecular elements associated with dysregulated transcription holds significant clinical implications. The following outlines potential clinical applications.
5.1 Targeting dysregulated transcriptional factors in melanoma
Key TFs also play pivotal roles in transcriptional dysregulation, which makes the approach to target TFs in melanoma seem to be feasible. For instance, the small molecular inhibitor, ML329, was tested effective to inhibit cell survival in two MITF-dependent melanoma cell lines, SK-MEL-5 and MALME-3 M, rather than in the MITF-independent A375 cell line [100]. However, the translational application by targeting the MITF has not been well progressed, since the role of MITF in mediating melanoma biological properties and responses to targeted therapies have been ambiguous. Moreover, the paired-like homeodomain transcription factor 1 (PITX1) shows certain treatment potential by regulating SOX gene family mRNA transcription, resulting in melanoma suppression with impaired cell proliferation and increased apoptosis [101], although further investigations are still in need.
5.2 Targeting epigenetic regulation-related pathways
Addressing epigenetic regulation-related pathways is another crucial strategy. However, the epigenome-based insights into melanoma regulatory transcription, as well as the implications with targeted therapies are generally limited. Some promising examples with profound clinical benefit include inhibitors targeting EZH2 and DOT1L, LSD1, BET, which not only reverse abnormal differentiation states in melanoma, but also increase the sensitivity to treatment [102]. Besides, the expression of non-coding RNA, SAMMON, which possesses as an informative biomarker of melanoma biology and BRAF inhibitor resistance could be targeted by the above mentioned PITX1 mRNA in a SOX10-dependent manner [101]. Besides, cinobufagin that suppressed Wnt/β-catenin target genes such as Axin-2, cyclin D1, and c-Myc in melanoma via LEF1 inhibition also turns out to be a potential anti-melanoma drug [55].
5.3 Targeting metabolic reprogramming in transcriptional dysregulation
Metabolic reprogramming is a distinctive feature in melanoma, and targeting this pathway holds promise for intervening in melanoma metabolic reprogramming [103]. The adipocyte enhancer-binding protein 1 (AEBP1), which is a transcriptional repressor influencing genes relating to the metabolic state of melanoma cells, was proved as a potential biomarker for cancer prognosis and a therapeutic target [104, 105]. Another effective targets modulating melanoma redox is the nuclear factor erythroid 2-related factor 2 (NRF2) in response to the oxidative stress burden during melanoma development [106].
5.4 Targeting plasticity-related metastasis and drug resistance
Regulating metastasis and drug resistance involves targeting specific transcription factors. Transcription factors like RUNX2, SOX9, and TWIST2, associated with metastasis and phenotype switching, could be targeted to prevent melanoma metastasis [107]. For instance, RUNX2 mediates resistance to BRAF inhibitor Vemurafenib, suggesting the potential benefits of combination therapy to enhance effectiveness [108, 109]. Besides, the metabolic reprogramming-related AEBP1 gene greatly contributes to the metastatic abilities of malignant melanoma, which was reported to upregulate EMT-related genes [105].
Additionally, targeting c-JUN, HIF-1, STAT3, and NF-κB, which regulate PD-L1 expression, may reduce the risk of treatment resistance [110]. c-JUN, involved in inflammation-induced dedifferentiation and the acquisition of pro-inflammatory characteristics, stands out as a potential key regulator of treatment resistance [111, 112]. Besides, STAT1, a regulator of PD-L1 expression, emerges as a promising target for combination immunotherapy [113]. Therefore, a comprehensive understanding and targeted intervention in dysregulated transcriptional control elements present promising avenues for developing precise and effective therapeutic strategies, ultimately improving treatment responses in melanoma patients (Table 1).
6 Future challenges
Melanoma plasticity is a process wherein cells alter their phenotype, presenting different molecular and/or histological characteristics, playing a crucial role in cancer progression and treatment resistance [93]. While potential genetic changes within tumours can enhance lineage plasticity, it is primarily a dynamically controlled process by transcriptional and epigenetic dysregulation [1]. This review extensively explores the categories of genetic mutations, transcription, and epigenetic regulatory factors influencing lineage amplification and plasticity in melanoma. Additionally, it delves into their interactions with other malignant features such as energy metabolism, metastasis, and drug resistance. Strategies for detecting and treating highly plastic tumours are also discussed.
The future of this field holds significant promise through the advancement of high-throughput single-cell and spatial analysis techniques. These developments, coupled with tools for tracing individual cells, are anticipated to drive significant progress. Similarly, accurately recapitulating various manifestations of plasticity in tumour models, sampled at relevant time points, will be crucial for advancing our understanding. Effectively deciphering and utilizing the cell atlases generated by these comprehensive large-scale approaches will be a major challenge, and high-throughput genetic perturbation strategies are expected to assist in this respect. Overall, future research is poised to unveil the molecular mechanisms driving melanoma plasticity and leverage this knowledge to formulate more precise and effective therapeutic strategies, ultimately enhancing treatment responses and prognostic outcomes for melanoma patients.
Availability of data and materials
Not applicable.
References
Arozarena I, Wellbrock C. Phenotype plasticity as enabler of melanoma progression and therapy resistance. Nat Rev Cancer. 2019;19(7):377–91.
Long GV, Swetter SM, Menzies AM, Gershenwald JE, Scolyer RA. Cutaneous melanoma. Lancet Lond Engl. 2023;402(10400):485–502.
Parra LM, Webster RM. The malignant melanoma market. Nat Rev Drug Discov. 2022;21(7):489–90.
Simon AC, Elder CT, Gyori DJ. Metastatic cutaneous melanoma: navigating the evolving treatment landscape. J Hematol Oncol Pharm. 2024;14(1):34–41.
Zhu S, Zhang T, Zheng L, Liu H, Song W, Liu D, et al. Combination strategies to maximize the benefits of cancer immunotherapy. J Hematol OncolJ Hematol Oncol. 2021;14(1):156.
Centeno PP, Pavet V, Marais R. The journey from melanocytes to melanoma. Nat Rev Cancer. 2023;23(6):372–90.
Morgan MP, Finnegan E, Das S. The role of transcription factors in the acquisition of the four latest proposed hallmarks of cancer and corresponding enabling characteristics. Semin Cancer Biol. 2022;86(Pt 3):1203–15.
Davies A, Zoubeidi A, Beltran H, Selth LA. The transcriptional and epigenetic landscape of cancer cell lineage plasticity. Cancer Discov. 2023;13(8):1771–88.
Senft D. The hierarchy of melanoma. Nat Rev Cancer. 2022;22(12):658.
Renauld JM, Davis W, Cai T, Cabrera C, Basch ML. Transcriptomic analysis and ednrb expression in cochlear intermediate cells reveal developmental differences between inner ear and skin melanocytes. Pigment Cell Melanoma Res. 2021;34(3):585–97.
Ge W, Tan SJ, Wang SH, Li L, Sun XF, Shen W, et al. Single-cell transcriptome profiling reveals dermal and epithelial cell fate decisions during embryonic hair follicle development. Theranostics. 2020;10(17):7581–98.
Morita R, Sanzen N, Sasaki H, Hayashi T, Umeda M, Yoshimura M, et al. Tracing the origin of hair follicle stem cells. Nature. 2021;594(7864):547–52.
Ji S, Zhu Z, Sun X, Fu X. Functional hair follicle regeneration: an updated review. Signal Transduct Target Ther. 2021;6(1):66.
Baykal C, Yılmaz Z, Sun GP, Büyükbabani N. The spectrum of benign dermal dendritic melanocytic proliferations. J Eur Acad Dermatol Venereol. 2019;33(6):1029–41.
Goding CR, Arnheiter H. MITF—the first 25 years. Genes Dev. 2019;33(15–16):983–1007.
Vandamme N, Berx G. From neural crest cells to melanocytes: cellular plasticity during development and beyond. Cell Mol Life Sci CMLS. 2019;76(10):1919–34.
D’Mello SAN, Finlay GJ, Baguley BC, Askarian-Amiri ME. Signaling pathways in melanogenesis. Int J Mol Sci. 2016;17(7):1144.
Nguyen NT, Fisher DE. MITF and UV responses in skin: From pigmentation to addiction. Pigment Cell Melanoma Res. 2019;32(2):224–36.
Estrada C, Mirabal-Ortega L, Méry L, Dingli F, Besse L, Messaoudi C, et al. MITF activity is regulated by a direct interaction with RAF proteins in melanoma cells. Commun Biol. 2022;5(1):1–13.
Ostojić J, Yoon YS, Sonntag T, Nguyen B, Vaughan JM, Shokhirev M, et al. Transcriptional co-activator regulates melanocyte differentiation and oncogenesis by integrating cAMP and MAPK/ERK pathways. Cell Rep. 2021;35(7):109136.
Gelmi MC, Houtzagers LE, Strub T, Krossa I, Jager MJ. MITF in normal melanocytes, cutaneous and uveal melanoma: a delicate balance. Int J Mol Sci. 2022;23(11):6001.
McGill GG, Horstmann M, Widlund HR, Du J, Motyckova G, Nishimura EK, et al. Bcl2 regulation by the melanocyte master regulator Mitf modulates lineage survival and melanoma cell viability. Cell. 2002;109(6):707–18.
Kim N, Kim S, Lee MW, Jeon HJ, Ryu H, Kim JM, et al. MITF promotes cell growth, migration and invasion in clear cell renal cell carcinoma by activating the RhoA/YAP signal pathway. Cancers. 2021;13(12):2920.
Qi J, Ma L, Guo W. Recent advances in the regulation mechanism of SOX10. J Otol. 2022;17(4):247–52.
Shin SY, Choi JH, Jung E, Gil HN, Lim Y, Lee YH. The EGR1–STAT3 transcription factor axis regulates α-melanocyte–stimulating hormone-induced tyrosinase gene transcription in melanocytes. J Invest Dermatol. 2019;139(7):1616–9.
Montaudié H, Sormani L, Dadone-Montaudié B, Heim M, Cardot-Leccia N, Tulic MK, et al. CLEC12B decreases melanoma proliferation by repressing signal transducer and activator of transcription 3. J Invest Dermatol. 2022;142(2):425–34.
Hseu YC, Vudhya Gowrisankar Y, Wang LW, Zhang YZ, Chen XZ, Huang PJ, et al. The in vitro and in vivo depigmenting activity of pterostilbene through induction of autophagy in melanocytes and inhibition of UVA-irradiated α-MSH in keratinocytes via Nrf2-mediated antioxidant pathways. Redox Biol. 2021;44:102007.
Waku T, Nakada S, Masuda H, Sumi H, Wada A, Hirose S, et al. The CNC-family transcription factor Nrf3 coordinates the melanogenesis cascade through macropinocytosis and autophagy regulation. Cell Rep. 2023;42(1):111906.
Brunsgaard EK, Jensen J, Grossman D. Melanoma in skin of color: Part II. Racial disparities, role of UV, and interventions for earlier detection. J Am Acad Dermatol. 2023;89(3):459–68.
Lopes FCPS, Sleiman MG, Sebastian K, Bogucka R, Jacobs EA, Adamson AS. UV Exposure and the risk of cutaneous melanoma in skin of color: a systematic review. JAMA Dermatol. 2021;157(2):213–9.
Mielko Z, Zhang Y, Sahay H, Liu Y, Schaich MA, Schnable B, et al. UV irradiation remodels the specificity landscape of transcription factors. Proc Natl Acad Sci U S A. 2023;120(11):e2217422120.
Gupta R, Janostiak R, Wajapeyee N. Transcriptional regulators and alterations that drive melanoma initiation and progression. Oncogene. 2020;39(48):7093–105.
Sánchez-del-Campo L, Martí-Díaz R, Montenegro MF, González-Guerrero R, Hernández-Caselles T, Martínez-Barba E, et al. MITF induces escape from innate immunity in melanoma. J Exp Clin Cancer Res CR. 2021;40:117.
Garraway LA, Widlund HR, Rubin MA, Getz G, Berger AJ, Ramaswamy S, et al. Integrative genomic analyses identify MITF as a lineage survival oncogene amplified in malignant melanoma. Nature. 2005;436(7047):117–22.
Eliades P, Abraham BJ, Ji Z, Miller DM, Christensen CL, Kwiatkowski N, et al. High MITF expression is associated with super-enhancers and suppressed by CDK7 inhibition in melanoma. J Invest Dermatol. 2018;138(7):1582–90.
Ugurel S, Houben R, Schrama D, Voigt H, Zapatka M, Schadendorf D, et al. Microphthalmia-associated transcription factor gene amplification in metastatic melanoma is a prognostic marker for patient survival, but not a predictive marker for chemosensitivity and chemotherapy response. Clin Cancer Res Off J Am Assoc Cancer Res. 2007;13(21):6344–50.
Wei C, Sun W, Shen K, Zhong J, Liu W, Gao Z, et al. Delineating the early dissemination mechanisms of acral melanoma by integrating single-cell and spatial transcriptomic analyses. Nat Commun. 2023;14(1):8119.
Vergani E, Frigerio S, Dugo M, Devecchi A, Feltrin E, De Cecco L, et al. Genetic variants and somatic alterations associated with MITF-E318K germline mutation in melanoma patients. Genes. 2021;12(9):1440.
Nordlinger A, Dror S, Elkahloun A, Del Rio J, Stubbs E, Golan T, et al. Mutated MITF-E87R in melanoma enhances tumor progression via S100A4. J Invest Dermatol. 2018;138(10):2216–23.
Bradner JE, Hnisz D, Young RA. Transcriptional addiction in cancer. Cell. 2017;168(4):629–43.
Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, et al. Transcriptional super-enhancers connected to cell identity and disease. Cell. 2013;155(4):934–47.
Tudrej KB, Czepielewska E, Kozłowska-Wojciechowska M. SOX10-MITF pathway activity in melanoma cells. Arch Med Sci. 2017;13(6):1493–503.
Yu L, Peng F, Dong X, Chen Y, Sun D, Jiang S, et al. Sex-determining region Y chromosome-related high-mobility-group box 10 in cancer: a potential therapeutic target. Front Cell Dev Biol. 2020;8:564740.
Hartman ML, Czyz M. MITF in melanoma: mechanisms behind its expression and activity. Cell Mol Life Sci. 2015;72(7):1249–60.
Kawakami A, Fisher DE. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab Invest. 2017;97(6):649–56.
Infarinato NR, Stewart KS, Yang Y, Gomez NC, Pasolli HA, Hidalgo L, et al. BMP signaling: at the gate between activated melanocyte stem cells and differentiation. Genes Dev. 2020;34(23–24):1713–34.
Darabi S, Elliott A, Braxton DR, Zeng J, Hodges K, Poorman K, et al. Transcriptional profiling of malignant melanoma reveals novel and potentially targetable gene fusions. Cancers. 2022;14(6):1505.
Louphrasitthiphol P, Loffreda A, Pogenberg V, Picaud S, Schepsky A, Friedrichsen H, et al. MITF-acetylation reprograms MITF target selectivity and residence time. Nat Commun. 2023;14(1):6051.
Capparelli C, Purwin TJ, Glasheen M, Caksa S, Tiago M, Wilski N, et al. Targeting SOX10-deficient cells to reduce the dormant-invasive phenotype state in melanoma. Nat Commun. 2022;13(1):1381.
Yokoyama S, Takahashi A, Kikuchi R, Nishibu S, Lo J, Hejna M, et al. SOX10 regulates melanoma immunogenicity through an IRF4-IRF1 axis. Cancer Res. 2021;81(24):6131–41.
Szumera-Ciećkiewicz A, Bosisio F, Teterycz P, Antoranz A, Delogu F, Koljenović S, et al. SOX10 is as specific as S100 protein in detecting metastases of melanoma in lymph nodes and is recommended for sentinel lymph node assessment. Eur J Cancer Oxf Engl 1990. 2020;137:175–82.
Yang X, Liang R, Liu C, Liu JA, Cheung MPL, Liu X, et al. SOX9 is a dose-dependent metastatic fate determinant in melanoma. J Exp Clin Cancer Res. 2019;38(1):17.
Huang L, Zhai Y, La J, Lui JW, Moore SPG, Little EC, et al. Targeting pan-ETS factors inhibits melanoma progression. Cancer Res. 2021;81(8):2071–85.
Kluge V, Kappelmann-Fenzl M, Fischer S, Zimmermann T, Pommer M, Kuphal S, et al. Alternative Wnt-signaling axis leads to a break of oncogene-induced senescence. Cell Death Dis. 2024;15(2):166.
Kim GH, Fang XQ, Lim WJ, Park J, Kang TB, Kim JH, et al. Cinobufagin suppresses melanoma cell growth by inhibiting LEF1. Int J Mol Sci. 2020;21(18):6706.
Eichhoff OM, Weeraratna A, Zipser MC, Denat L, Widmer DS, Xu M, et al. Differential LEF1 and TCF4 expression is involved in melanoma cell phenotype switching. Pigment Cell Melanoma Res. 2011;24(4):631–42.
Zob DL, Augustin I, Caba L, Panzaru MC, Popa S, Popa AD, et al. Genomics and epigenomics in the molecular biology of melanoma—a prerequisite for biomarkers studies. Int J Mol Sci. 2022;24(1):716.
Mannavola F, D’Oronzo S, Cives M, Stucci LS, Ranieri G, Silvestris F, et al. Extracellular vesicles and epigenetic modifications are hallmarks of melanoma progression. Int J Mol Sci. 2020;21(1):52.
Chen Y, Yi X, Sun N, Guo W, Li C. Epigenetics regulates antitumor immunity in melanoma. Front Immunol. 2022;13:868786.
Azevedo H, Pessoa GC, de Luna Vitorino FN, Nsengimana J, Newton-Bishop J, Reis EM, et al. Gene co-expression and histone modification signatures are associated with melanoma progression, epithelial-to-mesenchymal transition, and metastasis. Clin Epigenetics. 2020;12:127.
Sutopo NC, Kim JH, Cho JY. Role of histone methylation in skin cancers: histone methylation–modifying enzymes as a new class of targets for skin cancer treatment. Biochim Biophys Acta BBA - Rev Cancer. 2023;1878(3):188865.
Grafanaki K, Grammatikakis I, Ghosh A, Gopalan V, Olgun G, Liu H, et al. Noncoding RNA circuitry in melanoma onset, plasticity, and therapeutic response. Pharmacol Ther. 2023;248:108466.
Leucci E, Vendramin R, Spinazzi M, Laurette P, Fiers M, Wouters J, et al. Melanoma addiction to the long non-coding RNA SAMMSON. Nature. 2016;531(7595):518–22.
Guo W, Wang H, Li C. Signal pathways of melanoma and targeted therapy. Signal Transduct Target Ther. 2021;6(1):424.
Yang L, Wang D, Jia H, Yang C, Zhang Y, Li H, et al. Tumor-specific peroxynitrite overproduction disrupts metabolic homeostasis for sensitizing melanoma immunotherapy. Adv Mater. 2023;35(29):2301455.
Hahn AW, Menk AV, Rivadeneira DB, Augustin RC, Xu M, Li J, et al. metabolism-obesity is associated with altered tumor metabolism in metastatic melanoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2023;29(1):154–64.
Huang F, Cai F, Dahabieh MS, Gunawardena K, Talebi A, Dehairs J, et al. Peroxisome disruption alters lipid metabolism and potentiates antitumor response with MAPK-targeted therapy in melanoma. J Clin Invest. 2023;133(20):e166644.
Singh M, Yelle N, Venugopal C, Singh SK. EMT: Mechanisms and therapeutic implications. Pharmacol Ther. 2018;182:80–94.
Wouters J, Kalender-Atak Z, Minnoye L, Spanier KI, De Waegeneer M, Bravo González-Blas C, et al. Robust gene expression programs underlie recurrent cell states and phenotype switching in melanoma. Nat Cell Biol. 2020;22(8):986–98.
Jain P, Pillai M, Duddu AS, Somarelli JA, Goyal Y, Jolly MK. Dynamical hallmarks of cancer: phenotypic switching in melanoma and epithelial-mesenchymal plasticity. Semin Cancer Biol. 2023;96:48–63.
Singh D, Siddique HR. Epithelial-to-mesenchymal transition in cancer progression: unraveling the immunosuppressive module driving therapy resistance. Cancer Metastasis Rev. 2024;43(1):155–73.
Pedri D, Karras P, Landeloos E, Marine JC, Rambow F. Epithelial-to-mesenchymal-like transition events in melanoma. FEBS J. 2022;289(5):1352–68.
Manfioletti G, Fedele M. Epithelial-Mesenchymal Transition (EMT). Int J Mol Sci. 2023;24(14):11386.
Zhou Q, Wang J, Zhang Z, Wuethrich A, Lobb RJ, Trau M. Tracking the EMT-like phenotype switching during targeted therapy in melanoma by analyzing extracellular vesicle phenotypes. Biosens Bioelectron. 2024;244:115819.
Vandamme N, Denecker G, Bruneel K, Blancke G, Akay Ö, Taminau J, et al. The EMT transcription factor ZEB2 promotes proliferation of primary and metastatic melanoma while suppressing an invasive, mesenchymal-like phenotype. Cancer Res. 2020;80(14):2983–95.
Nieto MA, Huang RYJ, Jackson RA, Thiery JP. EMT: 2016. Cell. 2016;166(1):21–45.
Richard G, Dalle S, Monet MA, Ligier M, Boespflug A, Pommier RM, et al. ZEB1-mediated melanoma cell plasticity enhances resistance to MAPK inhibitors. EMBO Mol Med. 2016;8(10):1143–61.
Guo R, Wang P, Zheng X, Cui W, Shang J, Zhao Z. SGLT2 inhibitors suppress epithelial-mesenchymal transition in podocytes under diabetic conditions via downregulating the IGF1R/PI3K pathway. Front Pharmacol. 2022;13:897167.
Yu X, He T, Tong Z, Liao L, Huang S, Fakhouri WD, et al. Molecular mechanisms of TWIST1-regulated transcription in EMT and cancer metastasis. EMBO Rep. 2023;24(11):e56902.
Durand S, Tang Y, Pommier RM, Benboubker V, Grimont M, Boivin F, et al. ZEB1 controls a lineage-specific transcriptional program essential for melanoma cell state transitions. Oncogene. 2024;22:1–17.
Carver EA, Jiang R, Lan Y, Oram KF, Gridley T. The mouse snail gene encodes a key regulator of the epithelial-mesenchymal transition. Mol Cell Biol. 2001;21(23):8184–8.
Baulida J, Díaz VM, García de Herreros A. Snail1: a transcriptional factor controlled at multiple levels. J Clin Med. 2019;8(6):757.
Ogihara T, Mizoi K, Kamioka H, Yano K. Physiological roles of ERM proteins and transcriptional regulators in supporting membrane expression of efflux transporters as factors of drug resistance in cancer. Cancers. 2020;12(11):3352.
Kumar V, Yochum ZA, Devadassan P, Huang EHB, Miller E, Baruwal R, et al. TWIST1 is a critical downstream target of the HGF/MET pathway and is required for MET driven acquired resistance in oncogene driven lung cancer. Oncogene. 2024;43:1431–44.
Sun L, Fang J. Epigenetic regulation of epithelia-mesenchymal transition. Cell Mol Life Sci CMLS. 2016;73(23):4493–515.
Andrews MC, Oba J, Wu CJ, Zhu H, Karpinets T, Creasy CA, et al. Multi-modal molecular programs regulate melanoma cell state. Nat Commun. 2022;13(1):4000.
Serrano-Gomez SJ, Maziveyi M, Alahari SK. Regulation of epithelial-mesenchymal transition through epigenetic and post-translational modifications. Mol Cancer. 2016;15:18.
Meevassana J, Varophas S, Prabsattru P, Kamolratanakul S, Ruangritchankul K, Kitkumthorn N. 5-Methylcytosine immunohistochemistry for predicting cutaneous melanoma prognosis. Sci Rep. 2024;14(1):7554.
Skrypek N, Goossens S, De Smedt E, Vandamme N, Berx G. Epithelial-to-mesenchymal transition: epigenetic reprogramming driving cellular plasticity. Trends Genet TIG. 2017;33(12):943–59.
Cui H, Hu Y, Guo D, Zhang A, Gu Y, Zhang S, et al. DNA methyltransferase 3A isoform b contributes to repressing E-cadherin through cooperation of DNA methylation and H3K27/H3K9 methylation in EMT-related metastasis of gastric cancer. Oncogene. 2018;37(32):4358–71.
Bücker L, Lehmann U. CDH1 (E-cadherin) gene methylation in human breast cancer: critical appraisal of a long and twisted story. Cancers. 2022;14(18):4377.
Bracken AP, Brien GL, Verrijzer CP. Dangerous liaisons: interplay between SWI/SNF, NuRD, and Polycomb in chromatin regulation and cancer. Genes Dev. 2019;33(15–16):936–59.
Rambow F, Marine JC, Goding CR. Melanoma plasticity and phenotypic diversity: therapeutic barriers and opportunities. Genes Dev. 2019;33(19–20):1295–318.
Cheli Y, Giuliano S, Botton T, Rocchi S, Hofman V, Hofman P, et al. Mitf is the key molecular switch between mouse or human melanoma initiating cells and their differentiated progeny. Oncogene. 2011;30(20):2307–18.
Sun B, Zhang D, Zhao N, Zhao X. Epithelial-to-endothelial transition and cancer stem cells: two cornerstones of vasculogenic mimicry in malignant tumors. Oncotarget. 2016;8(18):30502–10.
Li X, Karras P, Torres R, Rambow F, van den Oord J, Marine JC, et al. Disseminated melanoma cells transdifferentiate into endothelial cells in intravascular niches at metastatic sites. Cell Rep. 2020;31(11):107765.
Shen S, Liu X, Guo Q, Liang Q, Wu J, Guan G, et al. Tumor microenvironment remodeling plus immunotherapy could be used in mesenchymal-like tumor with high tumor residual and drug resistant rate. Commun Biol. 2023;6(1):1–17.
Jiang Y, Zhan H. Communication between EMT and PD-L1 signaling: new insights into tumor immune evasion. Cancer Lett. 2020;1(468):72–81.
Hossain SM, Eccles MR. Phenotype switching and the melanoma microenvironment; impact on immunotherapy and drug resistance. Int J Mol Sci. 2023;24(2):1601.
Faloon PW, Bennion M, Weiner WS, Smith RA, Wurst J, Weiwer M, et al. A Small Molecule Inhibitor of the MITF Molecular Pathway. 2012 Dec 13 [updated 2014 Sep 18]. In: Probe Reports from the NIH Molecular Libraries Program [Internet]. Bethesda (MD): National Center for Biotechnology Information (US); 2010–. PMID: 24027801.
Ohira T, Nakagawa S, Takeshita J, Aburatani H, Kugoh H. PITX1 inhibits the growth and proliferation of melanoma cells through regulation of SOX family genes. Sci Rep. 2021;11(1):18405.
Huang F, Santinon F, Flores González RE, del Rincón SV. Melanoma plasticity: promoter of metastasis and resistance to therapy. Front Oncol. 2021;16(11):756001.
Alkaraki A, McArthur GA, Sheppard KE, Smith LK. Metabolic plasticity in melanoma progression and response to oncogene targeted therapies. Cancers. 2021;13(22):5810.
Vivas-García Y, Falletta P, Liebing J, Louphrasitthiphol P, Feng Y, Chauhan J, et al. Lineage-restricted regulation of SCD and fatty acid saturation by MITF controls melanoma phenotypic plasticity. Mol Cell. 2020;77(1):120–137.e9.
Majdalawieh AF, Massri M, Ro HS. AEBP1 is a novel oncogene: mechanisms of action and signaling pathways. J Oncol. 2020;2020:8097872.
Carpenter EL, Becker AL, Indra AK. NRF2 and key transcriptional targets in melanoma redox manipulation. Cancers. 2022;14(6):1531.
Suresh S, Rabbie R, Garg M, Lumaquin D, Huang TH, Montal E, et al. Identifying the transcriptional drivers of metastasis embedded within localized melanoma. Cancer Discov. 2023;13(1):194–215.
Pulica R, Solal KC, Lasfar A, Pulica R, Solal KC, Lasfar A. Role of RUNX2 in melanoma: a new player in tumor progression and resistance to therapy. In: Melanoma. IntechOpen; 2021. https://doi.org/10.5772/intechopen.97105.
Proietti I, Skroza N, Bernardini N, Tolino E, Balduzzi V, Marchesiello A, et al. Mechanisms of acquired BRAF inhibitor resistance in melanoma: a systematic review. Cancers. 2020;12(10):2801.
Tang Y, Durand S, Dalle S, Caramel J. EMT-inducing transcription factors, drivers of melanoma phenotype switching, and resistance to treatment. Cancers. 2020;12(8):2154.
Riesenberg S, Groetchen A, Siddaway R, Bald T, Reinhardt J, Smorra D, et al. MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat Commun. 2015;6:8755.
Hwang SG, Yu SS, Poo H, Chun JS. c-Jun/activator protein-1 mediates interleukin-1β-induced dedifferentiation but not cyclooxygenase-2 expression in articular chondrocytes*. J Biol Chem. 2005;280(33):29780–7.
Yi M, Niu M, Xu L, Luo S, Wu K. Regulation of PD-L1 expression in the tumor microenvironment. J Hematol OncolJ Hematol Oncol. 2021;14(1):10.
SuryoRahmanto A, Swartling FJ, Sangfelt O. Targeting SOX9 for degradation to inhibit chemoresistance, metastatic spread, and recurrence. Mol Cell Oncol. 2016;4(1):e1252871.
Tripathi SK, Sahoo RK, Biswal BK. SOX9 as an emerging target for anticancer drugs and a prognostic biomarker for cancer drug resistance. Drug Discov Today. 2022;27(9):2541–50.
Zeng J, Zhang J, Sun Y, Wang J, Ren C, Banerjee S, et al. Targeting EZH2 for cancer therapy: from current progress to novel strategies. Eur J Med Chem. 2022;238:114419.
Duan R, Du W, Guo W. EZH2: a novel target for cancer treatment. J Hematol OncolJ Hematol Oncol. 2020;13(1):104.
Alexandrova E, Salvati A, Pecoraro G, Lamberti J, Melone V, Sellitto A, et al. Histone methyltransferase DOT1L as a promising epigenetic target for treatment of solid tumors. Front Genet. 2022;13:864612.
Yi Y, Ge S. Targeting the histone H3 lysine 79 methyltransferase DOT1L in MLL-rearranged leukemias. J Hematol OncolJ Hematol Oncol. 2022;15(1):35.
Noce B, Di Bello E, Fioravanti R, Mai A. LSD1 inhibitors for cancer treatment: focus on multi-target agents and compounds in clinical trials. Front Pharmacol. 2023;2(14):1120911.
Fang Y, Liao G, Yu B. LSD1/KDM1A inhibitors in clinical trials: advances and prospects. J Hematol OncolJ Hematol Oncol. 2019;12(1):129.
Chen J, Tang P, Wang Y, Wang J, Yang C, Li Y, et al. Targeting bromodomain-selective inhibitors of BET proteins in drug discovery and development. J Med Chem. 2022;65(7):5184–211.
Jung M, Gelato KA, Fernández-Montalván A, Siegel S, Haendler B. Targeting BET bromodomains for cancer treatment. Epigenomics. 2015;7(3):487–501.
Boregowda RK, Medina DJ, Markert E, Bryan MA, Chen W, Chen S, et al. The transcription factor RUNX2 regulates receptor tyrosine kinase expression in melanoma. Oncotarget. 2016;7(20):29689–707.
Zhao W, Yang H, Chai J, Xing L. RUNX2 as a promising therapeutic target for malignant tumors. Cancer Manag Res. 2021;13:2539–48.
Brennan A, Leech JT, Kad NM, Mason JM. Selective antagonism of cJun for cancer therapy. J Exp Clin Cancer Res. 2020;39(1):184.
Bui BP, Nguyen PL, Lee K, Cho J. Hypoxia-inducible factor-1: a novel therapeutic target for the management of cancer, drug resistance, and cancer-related pain. Cancers. 2022;14(24):6054.
Zou S, Tong Q, Liu B, Huang W, Tian Y, Fu X. Targeting STAT3 in cancer immunotherapy. Mol Cancer. 2020;19(1):145.
Cerezo M, Guemiri R, Druillennec S, Girault I, Malka-Mahieu H, Shen S, et al. Translational control of tumor immune escape via the eIF4F-STAT1-PD-L1 axis in melanoma. Nat Med. 2018;24(12):1877–86.
Yu H, Lin L, Zhang Z, Zhang H, Hu H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther. 2020;5(1):1–23.
Acknowledgements
We thank Dr. Raymond Mao for critically reading the manuscript.
Funding
This work was supported by grants from the Hubei Provincial Natural Science Foundation (2023AFB1024), the Major Program of Wuhan Municipal Health Commission (WX21M01) and the National Natural Science Foundation of China (82130089).
Author information
Authors and Affiliations
Contributions
CS, MC, XN: first draft writing; JL: revision; JT: supervision.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
None.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shen, C., Chen, M., Nian, X. et al. Transcriptional dysregulation and insights into clinical implications in melanoma. Holist Integ Oncol 3, 28 (2024). https://doi.org/10.1007/s44178-024-00091-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44178-024-00091-y