
Abstract
We report an adolescent male who presented with diabetic ketoacidosis (DKA). He was diagnosed to have type 1 diabetes mellitus at the age of 12 years and had been initiated on insulin. On physical examination, he had a distinct senile-looking coarse facies with clinical stigmata of insulin resistance in the form of acanthosis nigricans and hypertrichosis. Additionally, he required more than 3 units/kg/day of insulin during recovery from DKA. The clinical and biochemical profile of the patient led to the suspicion of insulin resistance syndrome which was confirmed by the detection of homozygous missense variation in exon 2 of the insulin receptor gene (INSR) on clinical exome testing. The patient was put on insulin sensitizers along with insulin which led to a marked improvement in glycemic control. The case highlights the importance of a good clinical examination for a correct diagnosis and discusses the challenges in management.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
Insulin resistance is broadly defined as the state in which a greater than normal amount of insulin is required to elicit a quantitatively normal response [8]. Syndromes of insulin resistance include variants of or autoantibodies to the insulin receptor and lipodystrophy [4]. Diabetic ketoacidosis (DKA), although a rare presentation, can occur in these patients due to impaired insulin signaling, and a few cases have been described in the literature [12]. A diagnosis of insulin resistance syndrome (IRS) has important clinical implications. We report a young male who was misdiagnosed as type 1 diabetes mellitus (T1DM) for the initial 5 years and who presented to us with DKA and was later diagnosed with IRS. The case highlights the importance of subtle clinical signs which provide clues to the diagnosis and which, if overlooked, may lead to untoward consequences.
Case presentation
A 17-year-old male presented to our emergency department with osmotic symptoms (polyuria and increased thirst) for 3–5 days, respiratory distress, and altered sensorium for 1 day before presentation. He was diagnosed with T1DM 5 years ago when he had osmotic symptoms and had lost around 5-kg weight in 2–3-month duration. The initial blood glucose was around 300 mg/dl (16.6 mmol/l). There was one hospital admission at the initial diagnosis, although the history given by the parents did not suggest DKA. The patient was initiated on basal-bolus insulin therapy at that time. However, the blood glucose was not well controlled on insulin, and parents stopped insulin after 2 months and switched his treatment to non-allopathic therapy. During the initial 5 years when the patient was not on insulin, the blood glucose control was poor, but there was no history of hospitalization for DKA. His growth was also affected markedly resulting in poor height gain, and there was a lack of pubertal changes.
The patient also had a history of skin changes in the form of darkening of the skin over the nape of the neck and axilla for the last 7 years (starting 2 years before the onset of diabetes), and he consulted a dermatologist for the same. He was diagnosed with acanthosis nigricans, and insulin levels were told to be high. He was not on any treatment at that time. RBS checked at that time was told to be normal. Certain other investigations were advised at that time, but the patient did not follow through.
The patient was first in birth order and was born out of a non-consanguinous marriage.There was no history of gestational diabetes mellitus in the mother or any other perinatal complications. He attained developmental milestones appropriate for his age. There was no family history of diabetes mellitus (DM) in the preceding three generations on the maternal or the paternal side. He has one younger sibling who is 13 years old and is healthy. There was no history of sibling death.
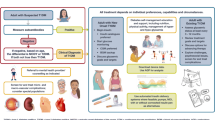
On examination, the patient was thin built with a generalized cachexic look. He was grossly dehydrated with Kussmaul’s breathing. He had tachycardia with a pulse rate of 130 per minute, and his blood pressure was 96/60 mm of Hg. On detailed examination, he had coarse senile-looking facies and thick lips with generalized loss of subcutaneous fat. He had hypertrichosis over the back of the trunk and grade 4 acanthosis nigricans over the nape of his neck, axilla, and popliteal region (Fig. 1). His weight was 30.2 kg (< 3rd centile as per Indian Academy of Pediatrics (IAP) growth charts 5–18 years for boys), and height was 145 cm (< 3rd centile as per IAP growth charts 5–18 years for boys) with a mid parental height of 165.5 cm (10th–25th centile) and a body mass index (BMI) of 14.3 kg/m2 (< 3rd centile as per IAP growth chart). He had a testicular volume of 8–10 cm3 bilaterally with a stretched penile length of 5.5 cm (< 2.5 SD for age) and sparse axillary and pubic hair suggestive of arrested puberty. He did not have organomegaly or any other abnormalities on systemic examination.

Clinical picture of the patient showing A hypertrichosis over the back of his trunk, B acanthosis nigricans over the axilla, C acanthosis nigricans at the back of the neck
Initial investigations revealed evidence of severe DKA with a blood glucose value of 476 mg/dl (26.4 mmol/l), pH of 7.0, serum bicarbonate of 6 mmol/l, and 4 + urinary ketones on urine dipstick testing.The biochemical profile of the patient is described in Table 1.
The patient was managed with intravenous fluids and insulin (U40) with frequent monitoring of blood glucose and correction of the electrolyte abnormalities during DKA. He required up to 3.6 U/kg/day of U40 regular insulin intravenously during DKA. After recovery from the acute phase, he was started therapy on basal-bolus insulin regimen. However, he did not achieve glycemic control even with 100 units of insulin per day (3.33 units/kg/day) while on subcutaneous basal bolus regimen.
There was no evidence of diabetic retinopathy or neuropathy. Urinary albumin creatinine ratio was planned on the follow-up visit after adequate glycemic control. Meanwhile, further workup revealed the absence of antibodies against glutamic acid decarboxylase 65-kDa isoform (GAD65), normal liver function tests, normal thyroid profile, and mildly deranged lipid profile. Ultrasonography of the abdomen did not reveal any organomegaly or fatty liver. He also had evidence of hypogonadotropic hypogonadism which was attributed to be of hypothalamic origin due to poor glycemic status for a long duration. A re-evaluation of the hypothalamic-pituitary–gonadal axis was planned for later when the patient achieves target glycemic control. His bone age was appropriate for the chronological age.
Considering the clinical clues, the high insulin requirement, and absent GAD65 antibodies, the possibility of insulin resistance syndrome or lipodystrophic syndromes was entertained, and genetic testing for the same was planned. Dual energy X-ray absorptiometry (DXA) for body composition is a useful investigation for the assessment of body fat distribution and aids in the diagnosis of lipodystrophy but was not done due to nonavailability in the institute. The patient’s clinical exome testing detected a homozygous missense variant in exon 2 of the insulin gene (INSR) resulting in the amino acid substitution of tryptophan for arginine at codon 41 (p.Arg41Trp) consistent with the variant previously reported in a patient affected with Rabson-Mendenhall syndrome (RMS) [3]. The genetic variant detected is depicted in Table 2. The patient and his parents were counseled regarding the disease and its implications.
He was started on low doses of metformin which was later escalated to the maximally tolerated dose. He was discharged on 50 units of U40 regular insulin and 40 units U100 glargine per day along with metformin 2.5 g/day.
Outcome and follow-up
The glycemic status of the patient has markedly improved after adding metformin, although it is still inadequate. Pioglitazone 15 mg/day was added to his treatment regimen, and it is planned to increase the dose depending on the glycemic record. Parents have been counseled regarding the disease implications and plan of management. U-100 regular insulin is being planned if the insulin requirement persists to be high.
Discussion
We have described an adolescent male who was diagnosed with T1DM and who presented with DKA requiring very high doses of insulin and who, on genetic testing, was ultimately found to have an insulin receptor variant. Variants in INSR can cause the insulin-resistance syndromes — Donohue syndrome, RMS, and type A insulin resistance [5, 7]. These syndromes represent a continuum of the clinical spectrum with Donohue syndrome being the most severe type and fatal in the first decade, type A insulin resistance being the mildest form, and RMS intermediate between the two [8]. Diagnosis is suspected based on clinical examination as well as laboratory diagnostic tests with markedly elevated insulin levels being a consistent feature and is confirmed by genetic testing.
Our patient was a lean male who presented to us with severe DKA and had the onset of disease in young adolescence. He was diagnosed with T1DM by a local practitioner 5 years back when he had presented with osmotic symptoms and was treated likewise for 5 years before presenting tous. However, certain clinical features were atypical of T1DM. First is the presence of acanthosis nigricans, hypertrichosis, and coarse, senile-looking facies at diagnosis. These are markers of insulin resistance and should have suggested an alternative diagnosis or at least closer follow-up for the clinical course of the disease. It is likely that this finding was missed at diagnosis. This warrants a good and thorough physical examination of patients. Second, there were no symptoms or admission to hospital suggestive of DKA in the past 5 years after diagnosis that is also unlikely to occur in type 1 DM, though history only might not be reliable in this aspect. Thirdly was the requirement of very large doses of insulin during treatment as well as recovery from DKA. The above points and the absence of GAD antibodies suggest insulin resistance syndromes or lipodystrophy as the two differential diagnoses. The absence of dyslipidemia and liver steatosis in our patient favored insulin resistance syndrome over lipodystrophy. At this point, body composition analysis or measurement of serum adiponectin levels could have helped in distinguishing between the two. However, due to the nonavailability of these at our institute and due to cost constraints, we proceeded with genetic testing which confirmed the diagnosis of IRS in this case.
Various INSR variants have been reported previously in the literature. Missense variants are the most common followed by nonsense variants. Rarely, splice site variants, insertions, deletions, or complex gene rearrangements are also found. Most of the variants located in the first 11 exons result in Donohue syndrome, while the variants in the beta subunit are found more frequently in patients with RMS [1]. Our patient was detected to have a homozygous missense variant in exon 2 of the INSR gene resulting in the amino acid substitution of tryptophan for arginine at codon 41 (p.Arg41Trp). This variant was first detected by Grasso et al. in a 16-day-old child who presented with poor feeding and hyperglycemia and had phenotypic characteristics consistent with RMS [3]. DKA, although a rare presentation in IRS, has been reported in the literature [12]. The pathophysiology of DKA in IRS is not clear. A progressive and rapid decline in insulin levels with age has been documented in patients with IRS. The patients are not capable of suppressing hepatic glucose production and release, leading to constant hyperglycemia, and with a further decline in insulin levels, the capacity to suppress fatty acid oxidation is compromised, leading to ketoacidosis [6].
Robinson et al. reported a case series of patients with insulin resistance presenting with DKA [12]. They described three patients — two with antibodies to insulin receptor — type B insulin resistance and one with compound heterozygous variant in insulin receptor. Two of the patients presented with severe DKA that was triggered by an underlying infection while no precipitant infection was found in one patient. These patients required a much higher dosage of insulin (more than 1000 units/day) during DKA treatment as compared to our patient. Based on their experience, they concluded that unusually high doses of insulin are required to achieve the target goal in patients with IRS when compared to patients with type 1 and type 2 DM. This might be due to the fact that at higher doses of insulin, the dose–response curve is extremely shallow.
Treatment of this condition is difficult and involves addressing hyperinsulinemia and insulin resistance. Metformin and pioglitazone are effective in the initial period of management [2]. Insulin requirement is very high resulting in high volumes of subcutaneous injection which can be painful [10]. U-100 and U-500 insulin can be helpful in these cases. However, given the rarity of the condition and scarcity of data, the safety profile of such high doses of insulin is still lacking. Also, again due to the rarity of the condition, it would be difficult to find specialists with experience in handling such high doses of insulin during DKA making it a challenge in management.
Until the last follow-up, our patient has been able to maintain blood glucose values in the range of 150–250 mg/dl (although still suboptimal) with insulin doses of 3 units/kg/day along with metformin 2.5 g/day and pioglitazone 15 mg/day. The patient is on follow-up, and there is a plan to up-titrate insulin further to optimize blood glucose and to consider U-100 insulin if dose volume becomes very high. His family has been counseled regarding further implications of the disease.
Newer treatment modalities like recombinant insulin-like growth factor 1 (IGF-1) and subcutaneous leptin have shown promising results in RMS, but these have been tried only in a few cases [9, 11]. However, they remain unaffordable and unavailable for many patients in resource-limited settings. Hence, there is a need to find new therapeutic options to address this challenging condition.
Learning points/take-home messages
-
Patients with IRS can, rarely, present with diabetic ketoacidosis which is otherwise a common presentation in patients with T1DM especially in young age groups.
-
A thorough clinical examination and biochemical testing can guide us in reaching the correct diagnosis with confirmation on genetic testing.
-
The absence of GAD antibodies, the presence of acanthosis nigricans, high insulin requirement, and genetic testing can differentiate T1DM from IRS.
-
Insulin sensitizers like metformin and pioglitazone are important part of treatment armamentarium in patients with IRS.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Ardon O, Procter M, Tvrdik T, Longo N, Mao R. Sequencing analysis of insulin receptor defects and detection of two novel variants in INSR gene. Mol Genet Metab Rep. 2014;1:71–84.
Fowlkes JL, Bunn RC, Coleman HN, Hall B, Reid MC, Thrailkill KM. Severe deficiencies of IGF-I, IGF-II, IGFBP-3, ALS and paradoxically high-normal bone mass in a child with insulin-resistance syndrome (Rabson-Mendenhall type). Growth Horm IGF Res. 2007;17:399–407.
Grasso V, Colombo C, Favalli V, Galderisi A, Rabbone I, Gombos S, et al. Six cases with severe insulin resistance (SIR) associated with variants of insulin receptor: is a Bartter-like syndrome a feature of congenital SIR? Acta Diabetol. 2013;50:951–7.
Kahn CR, Flier JS, Bar RS, Archer JA, Gorden P, Martin MM, et al. The syndromes of insulin resistance and acanthosis nigricans. Insulin-receptor disorders in man. N Engl J Med. 1976;294:739–745.
Longo N, Singh R, Griffin LD, Langley SD, Parks JS, Elsas LJ. Impaired growth in Rabson-Mendenhall syndrome: lack of effect of growth hormone and insulin-like growth factor-I. J Clin Endocrinol Metab. 1994;79:799–805.
Longo N, Wang Y, Pasquali M. Progressive decline in insulin levels in Rabson-Mendenhall syndrome. J Clin Endocrinol Metab. 1999;84:2623–9.
Longo N, Wang Y, Smith SA, Langley SD, DiMeglio LA, Giannella-Neto D. Genotype-phenotype correlation in inherited severe insulin resistance. Hum Mol Genet. 2002;11:1465–75.
Mantzoros CS, Flier JS. Insulin resistance: the clinical spectrum. Adv Endocrinol Metab. 1995;6:193–232.
McDonald A, Williams RM, Regan FM, Semple RK, Dunger DB. IGF-I treatment of insulin resistance. Eur J Endocrinol. 2007;157(Suppl 1):S51-56.
Mehta AE. Androgen excess disorders in women. Trends Endocrinol Metab. 1998;9:129–30.
Okawa MC, Cochran E, Lightbourne M, Brown RJ. Long-Term Effects of Metreleptin in Rabson-Mendenhall Syndrome on Glycemia, Growth, and Kidney Function. J Clin Endocrinol Metab. 2022;107(3):e1032–e1046. https://doi.org/10.1210/clinem/dgab782.
Robinson C, Cochran E, Gorden P, Brown RJ. Management of diabetic ketoacidosis in severe insulin resistance. Diabetes Care. 2016;39:e116–8.
Acknowledgements
None.
Funding
None.
Author information
Authors and Affiliations
Contributions
NB and KK was involved in preparing the manuscript. NB and KK helped in data collection, tables, and figures. NB, KK, KK, and KS were involved in case management and critical review of manuscript. The corresponding author reviewed the final manuscript. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Ethics approval was taken from the Institute Ethics Committee of AIIMS Rishikesh vide letter no AIIMS/IEC/23/304 dated 25 August 2023. A written informed consent was taken from the mother of the patient, and assent was also taken from the patient.
Consent for publication
A written informed consent was taken from the mother of the patient, and assent was also taken from the patient.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Batra, N., Kaur, K., Kadian, K. et al. Insulin resistance syndrome presenting with diabetic ketoacidosis — a rare presentation. J Rare Dis 2, 15 (2023). https://doi.org/10.1007/s44162-023-00018-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s44162-023-00018-7