
Abstract
In molecular photochemistry, charge-transfer emission is well understood and widely exploited. In contrast, luminescent metal-centered transitions only came into focus in recent years. This gave rise to strongly phosphorescent CrIII complexes with a d3 electronic configuration featuring luminescent metal-centered excited states which are characterized by the flip of a single spin. These so-called spin-flip emitters possess unique properties and require different design strategies than traditional charge-transfer phosphors. In this review, we give a brief introduction to ligand field theory as a framework to understand this phenomenon and outline prerequisites for efficient spin-flip emission including ligand field strength, symmetry, intersystem crossing and common deactivation pathways using CrIII complexes as instructive examples. The recent progress and associated challenges of tuning the energies of emissive excited states and of emerging applications of the unique photophysical properties of spin-flip emitters are discussed. Finally, we summarize the current state-of-the-art and challenges of spin-flip emitters beyond CrIII with d2, d3, d4 and d8 electronic configuration, where we mainly cover pseudooctahedral molecular complexes of V, Mo, W, Mn, Re and Ni, and highlight possible future research opportunities.
Graphical abstract

Similar content being viewed by others

1 Introduction and scope of the review
Photoactive complexes are both fundamentally interesting and highly valuable in many applications, e.g., optical devices, catalysis and biomedicine [1,2,3,4]. Traditionally there is an excessive reliance on compounds containing precious transition metal ions like RuII, IrIII, OsII or PtII due to their favorably high intrinsic ligand field splitting and strong spin–orbit coupling (SOC) [5,6,7,8,9]. In most cases, the emissive states are of charge transfer (CT) character, be it metal-to-ligand (MLCT), ligand-to-metal (LMCT), ligand-to-ligand (LL’CT) or intra-ligand charge transfer (ILCT), while metal-centered (MC) states are often non-emissive and facilitate non-radiative deactivation [10, 11].
Most prominently, this occurs in FeII complexes where efficient relaxation via low-energy MC states had precluded long MLCT lifetimes and phosphorescence for a long time [12,13,14,15,16,17,18,19,20]. An octahedral FeII complex with tridentate N^N^N ligands showed a 3/5MC lifetime of >1.6 ns and sensitized 1O2 [15] and an iron(II) complex with a hexadentate tren(py)3 ligand reduces quinones by photoinduced electron transfer from its 5MC state (tren(py)3 = tris(2-pyridyl-methyliminoethyl)amine) [18]. An excited 3CT state lifetime of 3 ns was achieved by an iron(II) complex with strongly covalent Fe–Namido bonds due to a high barrier for the 3MC/3CT interconversion [21]. Most recently, the first emissive mononuclear FeII complex has been reported [22]. It shows NIR-II luminescence in the range of 1030–1600 nm originating from a 3MLCT state with a lifetime of 1 ns in benzene solution at room temperature.
Rare examples of d6 complexes showing luminescent 3MC states were presented with [CoIII(CN)6]3– and more recently with a hexacarbene CoIII complex [23, 24]. The strongly σ-donating ligands imposed a very high ligand field splitting. This raised the energy of MC states so they can act as long-lived emissive excited states [24].
A fundamentally different type of phosphorescence from MC states appears in octahedral d3–CrIII complexes. Instead of interconfigurational states with occupied antibonding orbitals, associated emissive MC states feature the same electronic configuration as the ground state (t2g)3, but differ by a single flipped electron spin. Hence, this luminescence from intraconfigurational states was named ‘spin-flip emission’. Although CrIII complexes have been known for many years, a conceptual breakthrough toward intense spin-flip emission led to an increased interest in the past six years [11, 25,26,27,28,29].
Beyond the d3 electronic configuration, spin-flip emission is also conceivable in octahedral d2, d4 and d8 complexes, but examples are much less prevalent in the literature than for d3 complexes. In this review, we outline the theoretical frame required to understand spin-flip luminescence with respect to ligand field theory, symmetry, intersystem crossing (ISC) and relaxation pathways using various well-described CrIII complexes. We also show how emission energies can be tuned in these systems over a range of 5800 cm–1, and which applications exploit their unique excited state properties. Finally, we summarize the advances of the field with special emphasis on the often-undervalued central ions VII, VIII, CrIV, MoIII, WIII, MnIV, ReIV and NiII. While spin-flip emission has been observed in many solids doped with suitable transition metal ions [30,31,32,33,34,35,36,37,38,39,40,41,42,43,44,45,46] and many lanthanide complexes show emissive metal-centered ff-transitions [47,48,49], this review focuses on mononuclear molecular systems with d-block transition metal ions.
2 Implications from ligand field theory
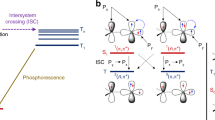
Ligand field theory is a powerful tool to understand and design spin-flip emitters. In an octahedral ligand field the five degenerate d-orbitals split into two sets, three lower t2g and two higher eg* orbitals (Oh notation), which differ in energy by the ligand field splitting ∆O [50, 51]. The ligand field concomitantly leads to splitting of the atomic terms of the central metal forming ligand field terms of different symmetries. The energies of these terms in dependence of ∆O are visualized in Tanabe-Sugano (TS) diagrams and depend on ∆O as well as the Racah parameters B and C (Fig. 1) [52, 53]. While both B and C describe the interelectronic repulsion, the parameter C only affects the energies of states with multiplicities lower than the maximum for a given electronic configuration (for example those of the doublet states in d3 ions) [54]. The relative state energies in a TS diagram depend on the ratio C/B which is often arbitrarily set to 4.0 but in reality varies between complexes [55]. Figure 1 shows the TS diagrams for octahedral d2, d3, d4 and d8 complexes [52, 53]. These electronic configurations feature a set of intraconfigurational states with low energy at high ∆O (e.g., 2E and 2T1 for d3, Oh notation with g/u omitted). These intraconfigurational states possess a nearly unchanged electron distribution compared to the ground state, e.g., (t2g)3 for the 4A2 state in d3. This has two consequences: their energy is essentially ligand field-independent (Eqs. 1 and 2) and they show a geometry close to the ground-state geometry (nested states, 2E/2T1 states in Fig. 2, weak coupling limit). In contrast, the occupation of orbitals of different energies in interconfigurational states like the 4T2 and 4T1 states in d3 ions (Eqs. 3 and 4) or charge-transfer states results in horizontally shifted and broad potential wells (4T2 state in Fig. 2, strong coupling limit). Consequently, this shift can lead to enhanced non-radiative relaxation to the ground state and broader emission bands, while spin-flip emission is typically very sharp [56].

Tanabe-Sugano (TS) diagrams of a d2, b d3, c d4 and d d8 transition metal ions in octahedral fields with C/B = 4 [50, 52, 53, 58]. Important crossing points are marked with circles. Exemplary intraconfigurational microstates relevant for spin-flip emission and detrimental interconfigurational microstates were empirically derived from complete active space self-consistent field (CASSCF) calculations of perfectly octahedral model complexes [MH6]n– (M = VIII, CrIII, MoII, NiII; see Supporting Information for computational details). Dotted lines in the microstates indicate strong mixing

Schematic potential energy diagram of an octahedral d3 transition metal complex with ∆O beyond the first quartet-doublet crossing point 4T2–2E in the TS diagram (Fig. 1b) and energies of the Franck–Condon (FC) states. Radiative (phosphorescence kPh and fluorescence kFl) and non-radiative decay pathways (back-intersystem crossing kbISC, dissociation kdiss and internal conversion kIC) and exemplary microstates of the relevant states are depicted [56]
In general, the transition energies from the ground state to excited MC states with the highest possible multiplicity like 4T2 and 4T1 in the d3 electron configuration can be described with ∆O and the Racah parameter B (Eqs. 3 and 4), while those with lower multiplicity like the 2E and 2T1 states also require the Racah parameter C (Eqs. 1 and 2) [50, 57]. It should be noted that Eqs. 1 and 2 were generated by assuming C/B = 4 for the calculation of the configurational interaction terms as multiples of B2/∆O [50, 57]. For exact solutions, the reader is referred to Ref. [58].
As discussed in more detail below, tuning of the excited MC state energies via the ligand field strength ∆O is well understood and heavily exploited in the design of spin-flip emitters (see Sect. 3.1). Yet designing systems with tailored Racah parameters B and C and thus spin-flip phosphorescence energy is difficult (see Sect. 4). Similarly, to achieve MLCT emission from FeII complexes, many studies focused on imposing a high ligand field splitting ∆O to raise the MC states as potential deactivating states above the MLCT states [10, 19, 59]. Recently, a new design strategy featured increased metal–ligand covalency leading to decreased interelectronic repulsion, which counteracted the lower ∆O and yielded an excited state lifetime of 3 ns of a pseudo-octahedral iron(II) complex [21].
While ligand field theory and the derived TS diagrams are useful to identify certain trends, they come with some limitations: (1) The diagrams refer to a perfectly octahedral coordination geometry. (2) Spin–orbit coupling (SOC) and hence mixing of states with different multiplicity is neglected. (3) Both a lower symmetry of the ground state and SOC lead to splitting of degenerate ligand field terms. (4) TS diagrams reflect the state energies at the ground-state geometry (Franck–Condon state) and neglect excited state energy lowering by excited state distortion. (5) CT states are not considered but can sometimes play an important role in the photodynamics of spin-flip emitters as discussed below.
3 Prerequisites for strong spin-flip emission
In this section, parameters influencing spin-flip emission and excited state relaxation pathways are discussed using d3–CrIII complexes as instructive and well-explored examples.
3.1 Strong ligand field splitting to avoid relaxation via MC states
Spin-flip phosphorescence is favored when the spin-flip states are the lowest energy excited states, even though it is a spin-forbidden process. As discussed in the previous section, this energy level ordering requires a high ligand field splitting ∆O. In case of CrIII this necessitates strongly σ-donating ligands, since the 3d orbitals possess a rather contracted radial distribution function (primogenic effect) [60, 61]. This limits overlap with ligand orbitals, which is referred to as a low intrinsic ligand field strength [62]. For d3 ions in an octahedral ligand field, the 4T2 state rises above the 2E and 2T1 states with increasing ∆O (Fig. 1b). For CrIII ions this could not be fully achieved using traditional ligands like en or tpy with their homoleptic complexes showing only weak spin-flip phosphorescence ([CrIII(en)3]3+ Cr13+: \(\Phi\) = 0.0062%, [CrIII(tpy)2]3+ Cr23+: \(\Phi\) < 0.001%; en = 1,2-ethylenediamine, tpy = 2,2’;6’,2”-terpyridine; Scheme 1, Table 1) [63, 64]. These ligands form five-membered chelate rings with the metal ion which leads to substantial deviation of 7°–11° from perfectly octahedral geometry (∠(N–Cr–N) = 90° or 180°) and a weak σ-orbital overlap [56, 65]. As a result, Cr13+ and Cr23+ only reach the first 4T2–2E crossing point in the TS diagram (Fig. 1b). Weakly luminescent CrIII(acac)3 Cr3 serves to discuss the effects of a small ∆O (acac– = acetylacetonato, Scheme 1, Table 1).

CrIII complexes discussed in this review
For Cr3, quantum chemical calculations placed the 4T2 state close to the 2T1 state in the FC region, i.e., close to the first quartet-doublet crossing point 4T2–2E in the TS diagram (Fig. 1b), leading to a high density of states. Furthermore, large SOC constants of 100–170 cm–1 were calculated between the 4T2 and 2T1 states [66]. This is in agreement with El Sayed’ rule stating that SOC between two states is large when a change in spin multiplicity is accompanied by a change in orbital angular momentum. The orbital character changes during ISC from the 4T2 state with (t2g)2(eg*)1 to the 2T1 state with (t2g)3(eg*)0 electron configuration [67]. Consequently, wavepacket simulations predicted an ultrafast 4T2 → 2T1 ISC for Cr3 [66]. In fact, fs-transient absorption studies with ligand field excitation revealed ISC with \(\tau\)ISC < 100 fs, that competes with vibrational cooling (VC) in the 4T2 state. An experimental time constant of \(\tau\) = 1.1(1) ps was assigned to VC in the doublet states state [68].
Apart from facilitating ISC, a small energy separation between 4T2 and 2E/2T1 states enables back-intersystem crossing (bISC) from the doublet manifold to the 4T2 state [69]. This results in low phosphorescence quantum yields and a low photostability of the complexes, since the 4T2 state with its (t2g)2(eg*)1 electronic configuration is Jahn–Teller distorted and potentially dissociative [25, 70, 71].
A conceptual breakthrough was achieved with [CrIII(ddpd)2]3+ Cr43+ (ddpd = N,N’-dimethyl-N,N’-dipyridin-2-ylpyridine-2,6-diamine, Scheme 1, Table 1) showing a very strong and long-lived dual emission in the near-infrared (NIR, 738 and 775 nm, \(\Phi\) = 11%, \(\tau\) = 899 µs) after 4A2 → 4LMCT or 4A2 → 4T2 excitation at 435 nm [25]. In Cr43+, the tridentate tpy-like ligand was formally expanded by NMe bridges leading to six-membered chelate rings with almost perfectly octahedral coordination with respect to the [CrN6] coordination polyhedron. The resulting very strong ligand field raised the 4T2 state to the level of the 2T2 state, close to the second quartet-doublet crossing point 4T2–2T2 in the TS diagram (Fig. 1b) [72]. At this crossing point with roughly degenerate 4T2 and 2T2 states at the FC geometry, ISC from the 4T2 state to the 2T2 state might be facilitated by a high density of doublet states [67] as well as a large SOC constant of 42 cm–1 between the 4T2(1) and 2T2(2) states as calculated using multi-reference methods [73]. Furthermore, the internal conversion (IC) 2T2 → 2E/2T1 might be faster than the bISC process 2T2 → 4T2 resulting in efficient population of the emissive 2E/2T1 states. In fact, after excitation to the 4T2 states, fast ISC and vibrational cooling (VC) populate the thermalized doublet states 2E/2T1 within \(\tau\) = 3.5 ps [74]. The significant 2E–4T2 energy gap of the relaxed excited states of 7100 cm–1 effectively prevents bISC and enables radiative relaxation (kPh) to the ground state [25]. Cr43+ is called ‘Molecular Ruby’ because of its optical properties reminiscent of the gemstone ruby (Al2O3:Cr3+) [25]. The nickname was recently adapted for the emerging class of strongly luminescent CrIII complexes [25, 28, 73].
3.2 Relaxation via CT states
Aside from interconfigurational MC states, CT states need to be considered as relaxation pathways in spin-flip emitters as well. Complexes of VII, CrIII and MnIV all feature a d3 electronic configuration. However, due to the different oxidation state of the central metal ions, low-energy MLCT and LMCT states can arise in VII and MnIV complexes, respectively (see below for more details) [42, 89]. For CrIII, CT states are of relatively high energy (e.g., the superposition of 4LMCT and 4T2 absorption bands at 435 nm in Cr43+ [25]) or they can be avoided altogether in the region of the 4T2 absorption as demonstrated with [CrIII(bpmp)2]3+ Cr53+ and [CrIII(tpe)2]3+ Cr63+ (bpmp = 2,6-bis(2-pyridylmethyl)pyridine, tpe = 1,1,1-tris(pyrid-2-yl)ethane; Scheme 1, Table 1) [73, 78]. Since low-energy CT states or their admixture to spin-flip states as in [VII(bpy)3]2+ (bpy = 2,2’-bipyridine) may act as relaxation pathways for long-lived spin-flip states [89], the relative energies of ligand and central metal orbitals need to be taken into account when designing ligands for spin-flip emitters.
3.3 Symmetry
The intraconfigurational spin-flip transition is governed by two selection rules: it is a spin-forbidden process and additionally Laporte’s rule applies, which forbids electronic transitions between wave functions of the same parity [90]. In centrosymmetric [CrIII(CN)6]3– Cr73– the combination of Laporte and spin selection rules leads to very long-lived emission (\(\tau\) = 3.45 ms) in frozen solution at 77 K with a low radiative rate constant of kPh = 25 s–1 (Scheme 1, Table 1) [80, 91]. Irradiation of Cr73– in aqueous solution can lead to ligand substitution [70]. The tripodal chelating ligand tpe imposes inversion symmetry on [CrIII(tpe)2]3+ Cr63+ (Scheme 1, Table 1) resulting in a record lifetime of 4.5 ms in DClO4/D2O at room temperature while retaining a high phosphorescence quantum yield of 8.2% [78]. Due to the inversion symmetry, the extinction coefficient for the 4A2 → 4T2 transition is very low (\(\varepsilon\) = 30 M–1 cm–1, Laporte forbidden), as is the radiative rate constant of the NIR phosphorescence (kPh = 18 s–1, Laporte and spin-forbidden) [78]. On the other hand, the coordinating nitrogen atoms in the ddpd complex Cr43+ are arranged around a center of symmetry [CrN6] but the overall symmetry is lower due to the orientation of the pyridine rings (point group D2). This allows for a faster radiative decay kPh and thus leads to a shorter lifetime of 899 µs [25]. Lower symmetry also lifts the degeneracy of the E and T states, which influences band shape and transition energy of the spin-flip luminescence [27, 72]. The radiative rate kPh increases by removing the center of inversion from Cr63+ to Cr43+ lifting Laporte’s rule [78].
In addition, symmetry can also influence non-radiative relaxation pathways opened by geometric distortions. In tris(bidentate)chromium(III) complexes with D3 or D3h symmetry like [CrIII(bpy)3]3+ Cr83+, a trigonal distortion of the coordination sphere in the long-lived excited states can lead to surface crossing of the excited doublet states with the ground state [84, 92]. This relaxation pathway limits phosphorescence quantum yields with 0.15% obtained for [CrIII(phen)3]3+ Cr93+ in water (phen = 1,10-phenanthroline, Scheme 1, Table 1) [93]. In 1 M aqueous HCl, the quantum yield is reported as 1.2% with \(\tau\) = 304 µs [82]. The pair [CrIII(en)3]3+ Cr13+ and the trigonally distorted cage complex [CrIII(sen)]3+ Cr103+ displays an even stronger effect with a reduction of the excited state lifetimes from \(\tau\) = 1.2–1.85 µs to \(\tau\) = 0.0001 µs (sen = 4, 4’, 4”-ethylidenetris(3-azabutane-1-amine); Scheme 1, Table 1) [63, 75, 84]. The hexadentate ligand in Cr103+ apparently enables efficient non-radiative relaxation pathways due to trigonal distortion which are unavailable in Cr13+.
3.4 Multi-phonon relaxation
The low energy of the doublet states in CrIII-based spin-flip emitters (typically 12,800–15,000 cm–1) [94] enables non-radiative decay via energy transfer to vibrational overtones of nearby X–H oscillators (X = C, N, O) [95]. This constitutes a major obstacle for efficient molecular emitters with organic ligands but not for oxidic materials such as ruby. By almost quantitative deuteration of the ligand in [CrIII([D9]-ddpd)2]3+ [D18]-Cr43+, a record quantum yield of 30% could be achieved (Table 1) [79]. This effect is due to the lower energy of the C–D fundamental mode (≈2200 cm–1) and its overtones compared to C–H vibrations (≈3000 cm–1). To deactivate the excited doublet state in the deuterated Molecular Ruby, energy transfer to a higher vibrational overtone (\(\nu\)6) with a lower \(\nu\)0 → \(\nu\)6 extinction coefficient is necessary than with C–H oscillators (\(\nu\)4 + \(\nu\)5). In case of acetonitrile, solvent deuteration had a negligible effect, whereas \(\tau\) and \(\Phi\) of Cr43+ significantly increased from 898 to 1164 µs and from 11.0 to 14.2%, respectively, in D2O instead of H2O [79].
In [CrIII(bpmp)2]3+ Cr53+ (Scheme 1, Table 1) with an emission maximum at 709 nm, selective α-deuteration of the ligand yielding [CrIII([D2]-bpmp)2]3+ [D4]-Cr53+ increased the quantum yield and lifetime from 20 to 25% and from 1.8 to 2.5 ms, respectively (Table 1) [73]. Clearly, the C–H oscillators closest to the CrIII center (d ≈ 3.0 Å) affect the multiphonon energy transfer the most, while the more distant oscillators play only a minor role due to the d–6 dependence of the corresponding rate constant [95].
Due to the higher energy of the N–H vibrations (3400 cm–1) and their different anharmonicity, multiphonon quenching is very pronounced in complexes like Cr13+, Cr103+ and [CrIII(H2tpda)2]3+ Cr113+ (H2tpda = 2,6-bis(2-pyridylamino)pyridine; Scheme 1, Table 1). N–H/N–D exchange on the ligands increased quantum yields by factors of 2.2–25 [84, 85]. Again, multiphonon quenching strongly depends on the Cr⋯(H–X) distance d with N–H bonds in Cr13+ (d = 2.48 Å) being closer to the metal center than in Cr113+ (d = 3.1–3.8 Å) [65, 85].
3.5 Solvent effects and counter ions
Apart from deuteration effects discussed in the previous section, solvents, salt additives and the counter ions of the complexes can influence their photophysical properties.
In Cr63+, the phosphorescence quantum yield increased from 3.2% in H2O to 4.2% and 5.4% in 0.1 M NaClO4(aq) and 0.1 M HClO4(aq), respectively. It was suggested that the perchlorate ions and the acid protect the charged complex from solvent molecules [78]. A similar effect was found for Cr83+, which possesses increased lifetimes in the presence of salts (e.g., NaClO4) or acids in high concentrations (> 1 M). Here, the effect was rationalized by perchlorate ions filling the pockets between the bpy ligands in Cr83+ leading to a rigidification and thus inhibiting distortional non-radiative relaxation (see Sect. 3.3). The influence of the solvent is much less pronounced for Cr93+ (\(\tau\)HClO4/\(\tau\)H2O ≈ 2) than for Cr83+ (\(\tau\)HClO4/\(\tau\)H2O ≈ 11), probably since the phen ligand scaffold of Cr93+ is more rigid on its own [96]. In general, relatively high phosphorescence quantum yields were found for tris(bidentate)chromium(III) polypyridyl complexes like Cr83+ (\(\Phi\) = 0.25%), Cr93+ (\(\Phi\) = 1.2%) and [CrIII(Ph2phen)3]3+ Cr123+ (\(\Phi\) = 3.0%) in 1 M HCl(aq) (Table 1, Scheme 1, Ph2phen = 4,7-diphenyl-1,10-phenanthroline) [82]. For Cr43+, there was no effect on lifetime or quantum yield when using dilute HClO4(aq) instead of water [25, 78].
Absorption/emission profiles and lifetimes of the bpy complex Cr83+ remained unchanged in non-aqueous solvents like MeOH, MeCN, dimethylformamide (DMF) and ethylene glycol [96]. Yet, the 4T2→2E ISC efficiency decreased in these solvents [97]. The emission lifetime of the cyanido complex Cr73– correlated with solvent polarity [91], while the emission of the en complex Cr13+ remained unaffected by the presence of MgCl2 up to 5.2 M [98].
Cr4[BF4]3 forms a contact ion pair with one [BF4]– anion on average in acetonitrile solution, due to the high charge of the complex cation [99]. In fact, the counter ions of Cr43+ affect the phosphorescence lifetime and quantum yield [100]. In acetonitrile solution, \(\Phi\) increased from 5.2% with chloride anions to 13.6% with tetrakis(3,5-bis(trifluoromethyl)phenyl)borate [BArF24]–. This change was attributed to reduced self-quenching when employing bulky anions. However, tetraphenylborate [BPh4]– led to lower quantum yields of 9.2% likely due to the introduction of additional C–H oscillators close to the CrIII center in contact ion pairs enabling multi-phonon relaxation (see above). The counter ions affected lifetime and quantum yield in parallel. Thus, only the non-radiative relaxation pathways from the long-lived 2E/2T1 states were influenced by the anions, while the fast evolution from initially excited 4T2 or 4LMCT states to the doublet manifold (ISC, VC, IC) remained unaffected [100].
The Molecular Ruby Cr53+ features acidic methylene bridges in the ligand due to the high positive charge. Deprotonation of Cr53+ is reversible and forms a non-emissive complex. Therefore, addition of an acid is required to prevent deprotonation and to harvest the full luminescence potential of Cr53+. The acidic protons also lead to a stronger interaction with solvent molecules and counter ions via hydrogen bonding. As a result, in deaerated D2O/DClO4 quantum yields of 13.4, 15.7 and 20.0% were obtained for the [BF4]–, [ClO4]– and [PF6]– salts of Cr53+, respectively [73].
4 Tuning emission energy in Molecular Rubies
In charge-transfer emitters (LMCT, MLCT, LL’CT), it is straightforward to tune excited state energies for example by introducing electron-donating or -withdrawing substituents on the ligand [101,102,103]. Methods for tuning the energy of metal-centered spin-flip states are not so obvious [27, 73, 88]. Their energies depend on the nephelauxetic effect, i.e., the covalency of the metal–ligand bond and the Racah parameters B and C [50, 57]. The archetypical Molecular Ruby Cr43+ and many of its congeners emit in the NIR-I spectral region between 720 and 780 nm [25, 27, 28, 64, 78, 85]. By increasing the metal–ligand covalency with a monoanionic carbazolato ligand in [CrIII(dpc)2]+ Cr13+ (dpc– = 3,6-di-tert-butyl-1,8-di(pyridine-2-yl)-carbazolato; Scheme 1, Table 1), the emission band shifted to the NIR-II peaking at 1067 nm in frozen solution at 77 K [87]. The high covalency of the bond between CrIII and the anionic ligand (B = 470–550 cm–1) decreased the repulsion of the d electrons and led to an unprecedentedly low energy of the spin-flip emission. However, an admixed 2LMCT state in Cr13+ increased the excited state distortion which facilitated non-radiative relaxation to the ground state leading to a low quantum yield of \(\Phi\) < 0.00089% and a relatively short lifetime of \(\tau\)1 = 1.4 µs (88%) and \(\tau\)2 = 6.3 µs (12%) at 77 K. In addition, it is plausible to assume that multi-phonon relaxation of the excited state (see above) plays a significant role because the NIR-II emission band might have a large spectral overlap with the absorption bands of the ligands’ aromatic C–H overtones [87]. Similarly a low-energy phosphorescence at 910 nm was found for fac-CrIII(ppy)3 Cr14 (ppy = anion of 2-phenylpyridine; Scheme 1, Table 1), an isostructural analog of the famous CT emitter IrIII(ppy)3, with a lifetime of 9.5 µs in 2-MeTHF (2-MeTHF = 2-methyltetrahydrofuran) at room temperature. At 77 K, the maximum shifted to 890 nm with shoulders at 910 and 1020 nm and a lifetime of 48 µs. The low quantum yield of 0.03% at room temperature in dichloromethane solution was rationalized with multiphonon quenching via C–H oscillators of the ligands, trigonal distortion in the excited state reminiscent of [CrIII(bpy)3]3+ Cr83+ and self-quenching enabled by intermolecular \(\pi\)–\(\pi\) and CH–\(\pi\) interactions of this neutral complex Cr14 [88].
In CrIII complexes with amine ligands like Cr13+, Cr103+ or [CrIII(NH3)6]3+ Cr153+, a weak red emission between 657 and 690 nm was observed, but lifetimes and quantum yields were poor (Scheme 1, Table 1) [63, 84, 104]. Recently, the emission maximum of a highly emissive Molecular Ruby was successfully blue-shifted to 709 nm by employing a methylene-bridged tripyridine ligand in Cr53+ [73]. Compared to Cr43+, this marks an increase of 1200 cm–1 in doublet state energy. This trend was correctly predicted by complete active space self-consistent field calculations with N-electron valence perturbation correction (CASSCF(7,12)-NEVPT2) [73].
A theoretical upper limit of the emission energy can be derived from ligand field theory using the Racah parameter B of the central metal ion. Assuming C/B = 4.0, a 2E energy of 19.5 B is predicted for an octahedral d3 complex [58]. With B = 918 cm–1 for the free Cr3+ ion [50], this corresponds to 17,900 cm–1 (559 nm). The doublet state energies are also determined by the ratio C/B. This is nicely demonstrated by ruby (Al2O3:Cr3+) which features a highly ionic metal ligand bond with B = 822 cm–1 but due to the low ratio C/B = 3.2 merely shows red emission at 694 nm (14,400 cm–1) [105].
An alternative strategy to ligand design for the tuning of the emission energy is changing the central metal ion. A lower charge and/or extended d-orbitals as in 4d/5d transition metals result in a lower interelectronic repulsion. Consequently, highly charged central ions like MnIV (B = 1064 cm–1) [50] should in principle lead to high spin-flip energies. However, in the end the covalence of the metal–ligand bond is the decisive factor, which needs to be considered for each complex individually. A more detailed discussion can be found in Sect. 6.
5 Applications
The phosphorescence of CrIII emitters is quenched by triplet dioxygen 3O2 via doublet-singlet Dexter-type energy transfer forming 1O2 with 61% quantum yield in the case of Cr43+. This excited state reactivity allows utilizing Cr43+ as an optical oxygen sensor and as photosensitizer for the α-cyanation of aliphatic amines via 1O2/trimethylsilylcyanide [25, 74].
The complex Cr123+ (Scheme 1, Table 1) was successfully employed as a photoredox catalyst in radical cationic [4 + 2] cycloaddition reactions [106]. Photoexcited Cr123+ is reductively quenched by the substrate (e.g., trans-anethol). The resulting radical cation reacts with a diene like isoprene. Interestingly, the catalysis requires the presence of O2 likely functioning as an electron shuttle. Oxygen can oxidize Cr122+ to regenerate the photocatalyst Cr123+ and to form superoxide. Finally, superoxide reduces the cationic intermediate after reaction of the oxidized alkene and the diene yielding the [4 + 2] cycloaddition product [86, 107]. This catalytic scheme strongly benefits from the very long excited state lifetime of 13 µs of Cr123+ in CH3NO2 even under aerobic conditions [86].
Cr43+ shows dual phosphorescence from its doublet states at 738 and 775 nm at room temperature because these two states are in thermal equilibrium with an energy difference of 650–700 cm–1 [108]. Thus, the complex was employed as a self-referenced ratiometric optical temperature sensor [108, 109].
Hydrostatic pressures for example in diamond anvil cells are usually measured optically via the shift in emission energies of ruby’s R1/R2 lines (approx. –0.77(3) and –0.84(3) cm–1 kbar–1) [110, 111]. Interestingly, for Cr4[BF4]3 much larger shifts of –14.8 and –9.5 cm–1 kbar–1 were found for the low- and high-energy emission, respectively, in aqueous solution, in methanol and in the solid state. The large barochromic effect is explained by subtle changes in the coordination geometry of the complex induced by high pressures [72, 112].
The high doublet energy and long excited state lifetime of the spin-flip state of Cr53+ allowed for a Dexter-type doublet-triplet energy transfer to 9,10-diphenylanthracene followed by efficient green-to-blue triplet–triplet annihilation upconversion (2 × 532 nm → 432 nm) with a high quantum yield of 12.0% (maximum value is 50%) [113]. The long-lived excited states in CrIII complexes can also be used to increase the excited state lifetime of lanthanide ions via CrIII → LnIII energy transfer as exemplified by binuclear [CrIIILnIII(L1)3]6+ (LnIII = NdIII: CrNd, LnIII = YbIII: CrYb, Fig. 3a and 3b) and trinuclear [CrIIILn’IIICrIII(L2)3]9+ helicate complexes (Ln’III = NdIII: CrNdCr, Ln’III = ErIII: CrErCr, Ln’III = YbIII: CrYbCr, Fig. 3a, c). Using the 4A2 → 2E excitation of the CrIII centers at 750 nm, lifetimes in the millisecond region were reached for the lanthanide emissions between 1000 and 1670 nm [114, 115]. For CrNdCr and CrYbCr quantum yields of 2.7(1) and 3.0(3) % were determined, respectively [115]. Furthermore, CrErCr (Fig. 3c) yielded green upconverted ErIII emission (4S3/2 → 4I15/2) with NIR irradiation via a sequential energy transfer upconversion (ETU) process (2 × 750 nm → 543 nm) in the solid state at 10 K and in frozen CH3CN solution at 30 K. The efficiency of the CrIII → ErIII energy transfer amounts to 50% [116].

Structures of a ligands L1 and L2, b binuclear CrIII–LnIII complexes and c trinuclear CrIII–LnIII–CrIII complexes [114,115,116] with red, green and blue colors used for carbon atoms on different ligands; chromium colored in yellow; lanthanide in violet; nitrogen colored in gray; oxygen colored in orange; hydrogen atoms were omitted for clarity
Alternatively, the Molecular Ruby Cr43+ operated as energy acceptor in a cooperative upconversion process from the 2F7/2 → 2F5/2 transition of Yb3+ in the [Cr4][YbIII(dpa)3] double salt yielding NIR-to-NIR upconverted photons (2 × 980 nm → 775 nm; dpa = 2,6-pyridine-dicarboxylate) [117].
A particularly promising application of spin-flip emission is circularly polarized luminescence (CPL) with potential applications like biosensing, telecommunication and security inks [118,119,120,121]. Unlike tpy, ligands like ddpd or dqpOMe (dqpOMe = 2,2´-(4-methoxypyridine-2,6-diyl)diquinoline) employed in Molecular Rubies form six-membered chelate rings with boat conformations. Two enantiomers (P,P) and (M,M) arise from the resulting double helix of the ligands around the central ion (Fig. 4). In several instances, a separation was possible using HPLC with chiral stationary phases. Apart from rich electronic circular dichroism, the separated enantiomers also showed strong CPL [28, 29, 122,123,124]. The key figure for quantification is the dissymmetry factor glum representing the excess of left-handed polarized over right-handed polarized light intensity IL and IR (Eq. 5). The physical description of glum (Eq. 5) includes the electronic and magnetic transition dipole moments |µab| and |mba|, respectively, as well as the angle \(\tau\)ab between the two vectors. Equation 5 shows that a high dissymmetry factor glum is expected for transitions a → b which are spin-forbidden (low |µab|) and magnetic dipole allowed (high |mba|) [125]. Both conditions are met by the 2E → 4A2 transition in Molecular Rubies yielding outstanding |glum| values of 0.09 for Cr43+ and 0.20 for [CrIII(dqpOMe)2]3+ Cr163+ (Scheme 1, Table 1) [29, 122]. Dissymmetry factors as high as this are rarely achieved with CT emitters and usually necessitate the use of lanthanide complexes exploiting metal-centered ff-transitions [123, 125, 126].

Molecular structures of enantiomers of [CrIII(ddpd)2]3+ Cr43+ with red and blue colors used for carbon atoms on different ddpd ligands to clarify each ligand’s helicity; chromium colored in green; nitrogen colored in gray; hydrogen atoms were omitted for clarity [122]
Intraconfigurational spin-flip transitions can potentially be exploited in optically addressable qubits. Complexes of CrIII, CrIV, VIII and NiII like Cr43+, Cr17–Cr22, V4, Ni12+ and Ni22+ (Tables 1 and 2, Schemes 1, 2 and 5) were proposed as suitable candidates [127,128,129,130,131,132]. Their properties are discussed in more detail below.
6 Spin-flip emitters based on other transition metals and electronic configurations
This section highlights spin-flip emissive complexes of 3d, 4d and 5d metal ions with suitable d electron configurations.
6.1 d 2 – TiII, VIII, CrIV, MoIV, TcV, ReV
The relevant excited states for spin-flip emission in the d2 configuration in an octahedral field are the 3T1 ground state, the interconfigurational 3T2 and intraconfigurational 1T2/1E excited states (Fig. 1a).
Divalent group 4 metal ions are potential candidates for spin-flip emissive d2 complexes [37, 38]. However, stable complexes of, e.g., TiII are quite rare and their investigation so far focused on electrochemical properties and reactivity [133,134,135,136,137,138,139]. Thus, no spin-flip luminescence with TiII complexes has been reported to date, while examples for luminescent solid-state materials containing Ti2+, such as MgCl2:Ti2+ and NaCl:Ti2+, exist [37, 38].
A weak, structured emission was found for [VIII(urea)6]3+ V13+ at 77 K in the solid state peaking at 992, 1010, 1011 and 1187 nm (Table 2) [140,141,142]. Trigonal Jahn–Teller distortion splits the 3T1 ground state by 1400 cm–1 to 3A1 and 3E states. This ground-state splitting of octahedral d2 metal complexes is a key difference to d3 spin-flip emitters with their orbitally non-degenerate 4A2 ground state (Fig. 1a,b). In VIII complexes, the total luminescence intensity is distributed to a large number of possible spin-flip transitions 1T2/1E → 3T1 with differing energies [143].
[VIII(ddpd)2]3+ V23+ is the first VIII complex showing NIR luminescence at room temperature in solution (Scheme 2, Table 2). This was achieved by using the strong \(\sigma\)-donor ddpd that has been previously employed in the first Molecular Ruby [25, 62]. The ligand field splitting is so large that the complex is located well above the first crossing point (1T2, 1E)/3T2, placing the spin-flip states below the interconfigurational 3T2 states in the TS diagram (Fig. 1a). When excited at 306 nm, V23+ shows a weak NIR-II phosphorescence peaking at 982, 1088 and 1109 nm in solution at 298 K. The bands were assigned to the spin-flip transitions from 1T2/1E to the split 3T1 ground state. A quantum yield of 0.00018% was found for this NIR-II emission in CD3CN at room temperature. Low energy excitation at 620 nm is less efficient in populating the metal-centered 1T2/1E states. In butyronitrile at 77 K, the luminescence decayed biexponentially with lifetimes of \(\tau\)1 = 790 ns (93%) and \(\tau\)2 = 8800 ns (7%). In addition to the NIR-spin-flip emission, blue fluorescence possibly originating from a 3LMCT state was detected at 396 nm in CD3CN at 298 K with a high quantum yield of 2.1%. This dual emissive behavior was rationalized with an inefficient ISC and fast spin-allowed IC and fluorescence. In contrast to Cr43+, non-radiative deactivation of the low-energy 1T2/1E spin-flip states via multiphonon relaxation (electronic-to-vibrational energy transfer to vibrational overtones of C–H oscillators) unexpectedly does not play a significant role as evidenced by the very similar lifetimes and quantum yields of the non-deuterated and perdeuterated vanadium(III) complexes [62, 79]. The low quantum yield can then be attributed to a poor ISC efficiency and efficient non-radiative decay pathways beyond multiphonon relaxation. Possibly the inefficient ISC may be caused by the low density of acceptor states in the singlet manifold as \(\Delta\)O in V23+ lies below the second triplet-singlet crossing point 1A1/3T2 in the TS diagram (Fig. 1a) [62].

NIR-II spin-flip emission was also detected for the heteroleptic complex mer-VIIICl3(ddpd) V3 (Scheme 2, Table 2) in the solid state at room temperature with bands at 1102, 1219 and 1256 nm and a phosphorescence lifetime of 0.5 µs [144]. Ligand deuteration significantly increased the phosphorescence lifetime to 3.4 µs. Transient absorption spectroscopy showed that the long-lived singlet states are populated after \(\tau\) = 1.4 ps, which is an upper limit for the time constant of the ISC. Trajectory surface hopping simulations within a linear vibronic coupling model arrived at a similar value of 1.7 ± 0.3 ps [145]. Interestingly, under hydrostatic pressure V3 showed a hypsochromic shift by + 10 cm–1 kbar–1 in contrast to the bathochromic shifts found for Cr43+. This positive shift was rationalized by the combined effect of changes in singlet energy and ground state splitting under pressure [144].
Spin-flip emission occurs also in five-coordinate VIII{(C6F5)3tren}(CNtBu) V4 ({(C6F5)3tren}3– = 2,2’,2’’-tris[(pentafluorphenyl)amido]trimethylamine; Scheme 2, Table 2) which was proposed as an optically addressable molecular quantum bit candidate [128]. In this coordination geometry, a 3A ground state and 3E and 1E MC excited states arise (Fig. 5a). Excitation at 640 nm assigned as a spin-allowed 3A → 3E transition yielded a 1E → 3A emission around 1240 nm in 2-MeTHF at 77 K and in single crystals. No emission was detected at room temperature in fluid solution. Long lifetimes of 11.1 and 3.0 µs were measured of single crystals of V4 at 4 K and at room temperature, respectively, substantiating the assignment of the emission as phosphorescence. ISC was found to occur within < 4.2 ps followed by VR with a time constant of 26 ps [128]. The rather slow ISC compared to V3 [144] or CrIII(acac)3 Cr3 (Scheme 1) [68, 146] was attributed to restrictions of vibrational modes along the ISC reaction coordinate imposed by the rigid substituted tren ligand [128].

The TS diagram of tetrahedral d2 complexes is analogous to the octahedral d8 case (Figs. 1d and 5b). Hence spin-flip emission from a 1E → 3A2 transition could be achieved with a high ligand field splitting in tetrahedral d2 complexes [147]. In fact, tetrahedral CrIV complexes with anionic alkyl or aryl ligands Cr17–Cr22 emit between 897 and 1025 nm at 4 K and were proposed as optically addressable qubit candidates (Scheme 2, Table 2) [130, 131]. For the investigation, the complexes Cr18–Cr22 were diluted in isostructural SnIV host lattices while a SnIV(2,4-dimethylphenyl)4 lattice was used for CrIV(CH2CPh3)4 Cr17 (CH2CPh3– = 2,2,2-triphenyleth-1-yl). A resulting incompatibility with this host lattice in Cr17 served to explain its broad emission compared to the extremely narrow bandwidths obtained for Cr18–Cr22. Because of the stronger nephelauxetic effect in the aryl complexes Cr20–Cr22, their emission is of lower energy than in the alkyl derivatives Cr17–Cr19 [131]. The phosphorescence lifetimes of Cr20–Cr22 in the host lattices at 4 K were determined as 3.3, 5.7 and 6.9 µs, respectively [130].
Long-lived phosphorescence from oxido and nitrido complexes featuring M=O, O=M’ = O or M’≡N (M = MoIV; M’ = TcV, ReV, OsVI) moieties is well known [148,149,150,151,152,153,154]. Although all of these complexes feature a d2 electronic configuration, their emission does not arise from an intraconfigurational spin-flip state but rather from an interconfigurational MC states. The complexes’ D4h symmetry and strong \(\pi\)-bonds to the nitrido or oxido ligands give rise to a 1A1 ground state with (dxy)2 configuration and an emissive 3E state with (dxy)1(dyz, dxz)1 configuration [148,149,150,151,152,153,154], which is distinctly different from the 3T1 ground state and 1T2/1E spin-flip states expected for octahedral d2 complexes (Fig. 1a) [50].
6.2 d 3 – MoIII, WIII, VII, MnIV and ReIV
Luminescent molecular CrIII complexes have been known for a long time with many reviews covering this substance class [56, 63, 143, 155, 156, 158]. In contrast to CrIII, luminescent complexes of MoIII and WIII were hardly investigated. To our knowledge, ten emissive molecular MoIII complexes and only one emissive WIII complex were reported in three publications [159,160,161]. This is probably due to the fact that MoIII and WIII complexes are less stable than CrIII complexes despite their (t2g)3 electronic configuration [162]. It was suggested that the higher ionic radii facilitate decomposition via seven-coordinate intermediates or that decomposition may be catalyzed by byproducts with different oxidation states [162]. In general, intermediate oxidation states like + III are more difficult to stabilize in 5d and 6d transition metal complexes resulting in complexes sensitive to oxidation [163, 164].
MoIII and WIII ions provide a very high intrinsic ligand field splitting due to better overlap of the large 4d and 5d orbitals with orbitals of the coordinating ligands. In addition, the heavier elements enable strong SOC, which is expected to enhance ISC rates [161, 165,166,167]. Absorption spectroscopy revealed that even with weak \(\pi\)-donating ligands like chloride in [MoIIICl6]3– Mo13– the 4T2 state is located well above the 2E/2T1/2T2 states and thus \(\Delta\)O is past the first and second crossing points in the TS diagram (Fig. 1b) [160]. The emission bands of the known MoIII complexes Mo13–, [MoIII(NCS)6]3– Mo23–, mer-MoIIICl3(L)3 Mo3–Mo5 (L = py, urea, tu; py = pyridine, tu = thiourea), mer-MoIIIBr3(urea)3 Mo6, fac-MoIII(Me3[9]aneN3)X3 Mo7–Mo9 (Me3[9]aneN3 = 1,4,7-trimethyl-1,4,7-triazacyclononane; X = Cl, Br, I) and fac-[MoIII{HB(Me2Pz)3}Cl3]– Mo10– ({HB(Me2Pz)3}– = tris(3,5-dimethyl-1H-pyrazol-1-yl)hydroborate) appear between 1090 and 1400 nm with emission lifetimes of several hundred nanoseconds and poor quantum yields of 0.0061–0.012% (Scheme 3, Table 3) [160, 161]. Multiphonon relaxation via C–H oscillators of the ligands or solvent molecules might play an important role in the deactivation of the excited states. Another reason for the poor quantum yields measured for Mo7–Mo9 might be that despite the high SOC in MoIII [165], ISC could be slow due to a low density of doublet states in the region of the initially excited 4T2 state at the FC geometry. For WIIICl3(Me3[9]aneN3) W1 (Scheme 3, Table 3) the emission peaked at 1400 nm, but the complex was too unstable for a more detailed investigation [161]. The trend of the energies going from CrIII to MoIII and WIII can be explained with respect to the weaker interelectronic repulsion in this series (Cr3+: B = 918 cm–1, Mo3+: B = 610 cm–1) [50, 161].

Investigations of novel MoIII and WIII emitters may offer insights on the effects that very high ligand field splittings \(\Delta_\text{O}\) have on the efficiency of the ISC processes and the radiative and non-radiative rates for the spin-flip state relaxation. Applying the lessons learned from the Molecular Rubies might help in the design and synthesis of stable MoIII and WIII complexes with strong emission in the NIR-II spectral region.
Vanadium(II) complexes also feature a d3 electronic configuration and are thus potential candidates for spin-flip emission similar to CrIII. The Racah parameter B(V2+) = 766 cm–1 of the free ion is lower compared to B(Cr3+) = 918 cm–1 [50] indicating lower doublet energies for VII complexes [89]. However, the lower oxidation state also leads to a lower intrinsic ligand field splitting \(\Delta_{\text{O}}\) [171]. In addition, a relatively facile oxidation of V2+ to V3+ may generate low-energy MLCT states. In fact, early studies on tris(bidentate)vanadium(II) complexes using bpy, bpy derivatives and phen V52+–V82+ (Scheme 4) concluded that their lowest excited states have 4MLCT character. These complexes featured excited state lifetimes in the low nanosecond region and no luminescence [172]. The excited state assignment was recently called into question and a mixed 2MC/2MLCT state was proposed instead based on electrochemical, quantum chemical and transient absorption studies [89]. The partial 2MLCT character leads to geometric distortion facilitating non-radiative decay compared to CrIII complexes with their nested doublet excited states [25]. Crystal structures of [VII(bpy)3]2+ V52+ and [VII(phen)3]2+ V82+ (Scheme 4) further revealed a significant trigonal distortion in the ground state. According to quantum chemical calculations, ISC pathways differ between V52+/V82+ and their CrIII homologues Cr83+/Cr93+. Both findings were rationalized with a stronger metal–ligand \(\pi\)-interactions in the VII complexes [173].

Structures of VII complexes discussed in the manuscript
Complexes of the heavier homologues NbII and TaII are rare and no spin-flip emission has been reported to date [137, 174,175,176,177,178].
For the d3-ion MnIV, a very early report stated that K2[MnIVCl6] K2Mn1 emits at 820 nm in solution (Scheme 5, Table 3) [168, 169]. Apart from this, the only reported emissive complex is [MnIV{PhB(MeIm)3}2](OTf)2 Mn2(OTf)2 ({PhB(MeIm)3}– = phenyltris(3-methylimidazol-2-yl)borate, Scheme 5, Table 3) [42, 43]. This hexacarbene complex shows a long-lived 2E → 4A2 spin-flip phosphorescence at 814 nm and a weak 4LMCT fluorescence between 600 and 750 nm in the solid state at room temperature. When considering the higher Racah parameter of free Mn4+ (1064 cm–1) compared to Cr3+ (918 cm–1) [50], higher doublet state energies should be accessible in MnIV complexes. In Mn2(OTf)2, however, the emission energy is lower than in most CrIII complexes due to a high covalency of the bonds between the MnIV ion and the anionic tricarbene ligands (nephelauxetic effect) [42, 43, 57].

To the best of our knowledge, [nBu4N]2[ReIVX6] (X = Cl, Br) [nBu4N]2Re1 and [nBu4N]2Re2 (Scheme 5, Table 3) present the only emissive molecular ReIV complexes in solution reported so far [160, 170]. Excitation at 360 and 414 nm in MeCN leads to NIR-II phosphorescence at 1340 and 1380 nm, respectively. For Re12–, lifetimes of 80 and 140 ns and an estimated upper limit of 0.02% for the quantum yield were reported [160, 170]. Re22– showed a phosphorescence lifetime of 40 ns and photosolvation upon LMCT excitation with UV light [179]. Analogous to the MoIII and WIII cases, the extended 5d orbitals and low interelectronic repulsion in ReIV lead to high-energy 4T2 states and very low lying doublet states. The challenges accompanied with the heavier group 6 metal ions outlined above apply for ReIV as well.
6.3 d 4 – CrII, MoII, WII, MnIII and ReIII
The d4 complexes are a special case among the electronic configurations discussed in this section. Depending on \(\Delta\)O, they can be high-spin or low-spin, resulting in dramatically different energy-level ordering (Fig. 1c). States of three different multiplicities (singlet, triplet, quintet) become relevant. Spin-flip emission is only conceivable in low-spin d4 complexes. A very high \(\Delta_{\text{O}}\) (≫ 38 B for C/B = 4) is required to establish the spin-flip states 1T2 and 1E as the lowest excited states below the high-spin state 5E (Fig. 1c) [58]. The d4 configuration is different from the others since deactivation of the potential spin-flip state via a 1T1 → 5E transition would entail a change of the total spin of \(\Delta\)S = 2 instead of just 1. However, while efficient ISC processes with \(\Delta\)S = 2 are relatively rare, they cannot be excluded [180, 181]. Another challenge in this electronic configuration might be that the 5E high-spin state always lies below the lowest triplet MC state 3E. A relaxation cascade 3E → 1T2/1E after 3T1 → 3E excitation is thus probably impeded by non-radiative deactivation of the 3E state via the 5E state similar to certain d6-FeII complexes [19]. Therefore, in a potential d4 spin-flip emitter, the 1T2 and 1E states need to be populated via other routes, e.g., CT states. Finally, similar to the d2 electron configuration, the 3T1 ground state of the low-spin d4 electron configuration is orbitally degenerate giving rise to Jahn–Teller distortions (Fig. 1a,c). Overall, spin-flip emission from d4 transition metal complexes has not been achieved yet for molecular systems. In principle complexes of group 6, ions in the oxidation state + II are potential candidates. However, there are only few examples of low-spin octahedral CrII complexes requiring exceptionally strong ligands like CN– [182, 183]. The reduced Molecular Ruby [CrII(ddpd)2]2+ Cr42+ shows spin-crossover at room temperature and dark excited states with microsecond lifetimes [184]. Further preparative and handling challenges for divalent group 6 metal ions include their sensitivity to oxidation, dimerization or cluster formation [185,186,187,188,189,190].
Other candidates for d4 spin-flip emission are, e.g., MnIII, TcIII and ReIII. However, known emissive MnIII complexes only show luminescence from ligand-centered transitions [191,192,193,194]. While octahedral TcIII and ReIII complexes are quite common [195, 196], to the best of our knowledge no spin-flip emission has been reported so far.
Due to the very high \(\Delta\)O required for spin-flip emission in the d4 case, complexes of 4d and 5d transition metal ions seem to be promising candidates.
6.4 d 8 – NiII, PdII and PtII
In an octahedral ligand field, d8 ions like NiII possess a 3A2 ground state, a 3T2 excited state and a 1E spin-flip state (Fig. 1d). Compared to the other d electron configurations, the 1E state in d8 is unique because the spin-flip occurs in the eg* orbitals with \(\sigma\)-instead of \(\pi\)-symmetry. Interestingly, the 1E state consists of an unpaired and a spin-paired microstate (Fig. 1d). Population of the spin-paired microstate might lead to excited state Jahn–Teller distortion facilitating non-radiative decay to the ground state.
In principle, a strong ligand field could raise the 3T2 state above the 1E state and enable spin-flip phosphorescence. Octahedral NiII complexes with strong donor ligands like phen, tpm, bpy, tpy and ddpd (Ni12+–Ni52+; tpm = tris(pyrid-2-yl)methane; Scheme 6) show ligand field splittings of 17–18B which are close to the 3T2/1E crossing point in the TS diagram (Fig. 1d) [129, 197,198,199,200,201], while homo- and heteroleptic complexes with poly(pyrazolyl)methane ligands showed a lower \(\Delta\)O of 11–15B [202]. Absorption spectroscopy revealed that the 3T2 and 1E states are not sufficiently separated with 1E transitions detected as shoulders on the 3T2 bands. In this case, the spin-forbidden 3A2 → 1E absorptions are enhanced by intensity borrowing from the nearby 3T2 band [201]. The lowest energy adiabatic state is strongly anharmonic due to coupling with components of the 3T2 state via SOC which reduces the spin-flip character [203]. In summary, a ligand field splitting \(\Delta\)O ≫ 18B is necessary to bring spin-flip luminescence within reach [200]. However, as the classical example of [NiII(CN)4]2– illustrates, a square-planar coordination geometry with a singlet ground state is favored with very strong ligands [204]. Thus, a balance between these two extremes is necessary for spin-flip emission.

Structures of NiII complexes discussed in the manuscript
It was reported that Ni12+ and Ni22+ are emissive in the solid state at 150 K [129]. However, these findings have been called into question since the reported emission bands for Ni12+ and Ni22+ are almost superimposable, which is unlikely considering their different symmetry [143].
The heavier homologues PdII and PtII prefer a square-planar coordination geometry due to their high intrinsic ligand field splitting [185]. Pseudooctahedral complexes of PdII and PtII are very rare and often require sophisticated ligands to avoid the formation of a square-planar geometry [205,206,207,208,209,210]. No spin-flip phosphorescence in these types of complexes was reported to date.
7 Conclusion
The numerous examples of spin-flip luminescent complexes in this review substantiate that metal-centered states can be more than just non-radiative relaxation pathways for charge-transfer states, but that spin-flip chromophores constitute a useful class of phosphorescent complexes complementary to charge-transfer chromophores. A deeper understanding of the requirements for efficient spin-flip emission has given rise to the emerging class of highly luminescent CrIII complexes (Molecular Rubies) and the first luminescent VIII complexes. This marks substantial progress in the ongoing endeavor to establish photoactive complexes based on earth-abundant metals as sustainable alternatives to precious or rare earth metal complexes [11, 19, 158]. In this context, circularly polarized luminescence is a promising application of enantiopure chiral spin-flip emitters, which makes full use of their unique excited state properties [123].
This review also highlighted many open venues for more fundamental research. It is still a challenge to tune the relative energies of the relevant states to achieve, e.g., high-energy spin-flip emission. In addition, the role of the density of high-energy 2T2 doublet states for efficient intersystem crossing in excited CrIII complexes remains unclear to this date. 4d and 5d complexes of MoIII, WIII or ReIV offer ideal conditions to study d3 spin-flip emission beyond the second quartet-doublet crossing point in the Tanabe-Sugano diagram. For some metal ions like NiII, reports on spin-flip emission are limited to doped solids [46], while convincing evidence in molecular systems is lacking. Here, it remains a challenge to establish a sufficiently high ligand field splitting that separates the initially excited interconfigurational states and the intraconfigurational spin-flip states without resulting in a square-planar coordination geometry.
Availability of data and material
Supporting Information for quantum chemical calculations.
Code availability
Not applicable.
References
Bizzarri, C., Spuling, E., Knoll, D. M., Volz, D., & Bräse, S. (2018). Sustainable metal complexes for organic light-emitting diodes (OLEDs). Coordination Chemistry Reviews, 373, 49–82. https://doi.org/10.1016/j.ccr.2017.09.011
Li, X., Xie, Y., & Li, Z. (2021). Diversity of luminescent metal complexes in OLEDs: Beyond traditional precious metals. Chemistry - An Asian Journal, 16(19), 2817–2829. https://doi.org/10.1002/asia.202100784
Twilton, J., Le, C., Zhang, P., Shaw, M. H., Evans, R. W., & MacMillan, D. W. C. (2017). The merger of transition metal and photocatalysis. Nature Reviews Chemistry. https://doi.org/10.1038/s41570-017-0052
Imran, M., Ramzan, M., Qureshi, A. K., Khan, M. A., & Tariq, M. (2018). Emerging applications of porphyrins and metalloporphyrins in biomedicine and diagnostic magnetic resonance imaging. Biosensors, 8(4), 95. https://doi.org/10.3390/bios8040095
Chou, P.-T., & Chi, Y. (2006). Osmium- and ruthenium-based phosphorescent materials: Design, photophysics, and utilization in OLED fabrication. European Journal of Inorganic Chemistry, 2006(17), 3319–3332. https://doi.org/10.1002/ejic.200600364
Liu, Z., Bian, Z., & Huang, C. (2010). Luminescent iridium complexes and their applications. In H. Bozec & V. Guerchais (Eds.), Molecular organometallic materials for optics (pp. 113–142). Springer. https://doi.org/10.1007/978-3-642-01866-4_4
Mauro, M., Aliprandi, A., Septiadi, D., Kehr, N. S., & de Cola, L. (2014). When self-assembly meets biology: Luminescent platinum complexes for imaging applications. Chemical Society Reviews, 43(12), 4144–4166. https://doi.org/10.1039/c3cs60453e
Parker, D., Fradgley, J. D., & Wong, K.-L. (2021). The design of responsive luminescent lanthanide probes and sensors. Chemical Society Reviews, 50(14), 8193–8213. https://doi.org/10.1039/d1cs00310k
Shum, J., Leung, P.K.-K., & Lo, K.K.-W. (2019). Luminescent ruthenium(II) polypyridine complexes for a wide variety of biomolecular and cellular applications. Inorganic Chemistry, 58(4), 2231–2247. https://doi.org/10.1021/acs.inorgchem.8b02979
Wenger, O. S. (2019). Is iron the new ruthenium? Chemistry--A European Journal, 25(24), 6043–6052. https://doi.org/10.1002/chem.201806148
Förster, C., & Heinze, K. (2020). Photophysics and photochemistry with Earth-abundant metals—fundamentals and concepts. Chemical Society Reviews, 49(4), 1057–1070. https://doi.org/10.1039/c9cs00573k
Braun, J. D., Lozada, I. B., Kolodziej, C., Burda, C., Newman, K. M. E., van Lierop, J., Davis, R. L., & Herbert, D. E. (2019). Iron(II) coordination complexes with panchromatic absorption and nanosecond charge-transfer excited state lifetimes. Nature Chemistry, 11(12), 1144–1150. https://doi.org/10.1038/s41557-019-0357-z
Carey, M. C., Adelman, S. L., & McCusker, J. K. (2019). Insights into the excited state dynamics of Fe(II) polypyridyl complexes from variable-temperature ultrafast spectroscopy. Chemical Science, 10(1), 134–144. https://doi.org/10.1039/c8sc04025g
Fredin, L. A., Pápai, M., Rozsályi, E., Vankó, G., Wärnmark, K., Sundström, V., & Persson, P. (2014). Exceptional excited-state lifetime of an iron(II)-N-heterocyclic carbene complex explained. Journal of Physical Chemistry Letters, 5(12), 2066–2071. https://doi.org/10.1021/jz500829w
Dierks, P., Kruse, A., Bokareva, O. S., Al-Marri, M. J., Kalmbach, J., Baltrun, M., Neuba, A., Schoch, R., Hohloch, S., Heinze, K., Seitz, M., Kühn, O., Lochbrunner, S., & Bauer, M. (2021). Distinct photodynamics of κ-N and κ-C pseudoisomeric iron(II) complexes. Chemical Communications, 57(54), 6640–6643. https://doi.org/10.1039/d1cc01716k
Dierks, P., Päpcke, A., Bokareva, O. S., Altenburger, B., Reuter, T., Heinze, K., Kühn, O., Lochbrunner, S., & Bauer, M. (2020). Ground- and excited-state properties of iron(II) complexes linked to organic chromophores. Inorganic Chemistry, 59(20), 14746–14761. https://doi.org/10.1021/acs.inorgchem.0c02039
Reuter, T., Kruse, A., Schoch, R., Lochbrunner, S., Bauer, M., & Heinze, K. (2021). Higher MLCT lifetime of carbene iron(II) complexes by chelate ring expansion. Chemical Communications, 57(61), 7541–7544. https://doi.org/10.1039/d1cc02173g
Woodhouse, M. D., & McCusker, J. K. (2020). Mechanistic origin of photoredox catalysis involving iron(II) polypyridyl chromophores. Journal of the American Chemical Society, 142(38), 16229–16233. https://doi.org/10.1021/jacs.0c08389
Dierks, P., Vukadinovic, Y., & Bauer, M. (2022). Photoactive iron complexes: more sustainable, but still a challenge. Inorganic Chemistry Frontiers. https://doi.org/10.1039/D1QI01112J
Chábera, P., Kjær, K. S., Prakash, O., Honarfar, A., Liu, Y., Fredin, L. A., Harlang, T. C. B., Lidin, S., Uhlig, J., Sundström, V., Lomoth, R., Persson, P., & Wärnmark, K. (2018). FeII hexa N-heterocyclic carbene complex with a 528 ps metal-to-ligand charge-transfer excited-state lifetime. Journal of Physical Chemistry Letters, 9(3), 459–463. https://doi.org/10.1021/acs.jpclett.7b02962
Larsen, C. B., Braun, J. D., Lozada, I. B., Kunnus, K., Biasin, E., Kolodziej, C., Burda, C., Cordones, A. A., Gaffney, K. J., & Herbert, D. E. (2021). Reduction of electron repulsion in highly covalent Fe-amido complexes counteracts the impact of a weak ligand field on excited-state ordering. Journal of the Chemical Society. https://doi.org/10.1021/jacs.1c06429
Leis, W., Argüello Cordero, M. A., Lochbrunner, S., Schubert, H., & Berkefeld, A. (2022). A photoreactive iron(II) complex luminophore. Journal of the Chemical Society. https://doi.org/10.1021/jacs.1c13083
Viaene, L., & D’Olieslager, J. (1987). Luminescence from and absorption by the 3T1g level of the hexacyanocobaltate(III) ion. Inorganic Chemistry, 26(6), 960–962. https://doi.org/10.1021/ic00253a039
Kaufhold, S., Rosemann, N. W., Chábera, P., Lindh, L., Bolaño Losada, I., Uhlig, J., Pascher, T., Strand, D., Wärnmark, K., Yartsev, A., & Persson, P. (2021). Microsecond photoluminescence and photoreactivity of a metal-centered excited state in a hexacarbene-co(III) complex. Journal of the American Chemical Society, 143(3), 1307–1312. https://doi.org/10.1021/jacs.0c12151
Otto, S., Grabolle, M., Förster, C., Kreitner, C., Resch-Genger, U., & Heinze, K. (2015). [Cr(ddpd)2]3+: A molecular, water-soluble, highly NIR-emissive ruby analogue. Angewandte Chemie International Edition, 54(39), 11572–11576. https://doi.org/10.1002/anie.201504894
Förster, C., Dorn, M., Reuter, T., Otto, S., Davarci, G., Reich, T., Carrella, L., Rentschler, E., & Heinze, K. (2018). Ddpd as expanded terpyridine dramatic effects of symmetry and electronic properties in first row transition metal complexes. Inorganics, 6(3), 86. https://doi.org/10.3390/inorganics6030086
Doistau, B., Collet, G., Bolomey, E. A., Sadat-Noorbakhsh, V., Besnard, C., & Piguet, C. (2018). Heteroleptic ter-bidentate Cr(III) complexes as tunable optical sensitizers. Inorganic Chemistry, 57(22), 14362–14373. https://doi.org/10.1021/acs.inorgchem.8b02530
Jiménez, J.-R., Doistau, B., Cruz, C. M., Besnard, C., Cuerva, J. M., Campaña, A. G., & Piguet, C. (2019). Chiral molecular ruby Cr(dqp)23+ with long-lived circularly polarized luminescence. Journal of the American Chemical Society, 141(33), 13244–13252. https://doi.org/10.1021/jacs.9b06524
Jiménez, J.-R., Poncet, M., Míguez-Lago, S., Grass, S., Lacour, J., Besnard, C., Cuerva, J. M., Campaña, A. G., & Piguet, C. (2021). Bright long-lived circularly polarized luminescence in chiral chromium(III) complexes. Angewandte Chemie International Edition, 60(18), 10095–10102. https://doi.org/10.1002/anie.202101158
Flint, C. (1971). A vibronic analysis of the transition in solid Cs2MnF6. Journal of Molecular Spectroscopy, 37(3), 414–422. https://doi.org/10.1016/0022-2852(71)90173-1
Chodos, S. L., Black, A. M., & Flint, C. D. (1976). Vibronic spectra and lattice dynamics of Cs2MnF6 and A12MIVF6:MnF62−. The Journal of Chemical Physics, 65(11), 4816–4824. https://doi.org/10.1063/1.432952
Flint, C. D., & Lang, P. F. (1987). Substitution reactions and luminescence spectra of the mixed complexes [ReClxBr 6–x]2–. Journal of the Chemical Society, Dalton Transactions, 8, 1929–1932. https://doi.org/10.1039/DT9870001929
Flint, C. D., & Lang, P. F. (1986). Luminescence spectra of the mixed complexes [TcClxBr 6–x]2–. Journal of the Chemical Society, Dalton Transactions, 5, 921–923. https://doi.org/10.1039/DT9860000921
Preetz, W., Peters, G., & Bublitz, D. (1996). Preparation and spectroscopic investigations of mixed octahedral complexes and clusters. Chemical Reviews, 96(3), 977–1026. https://doi.org/10.1021/cr940393i
Suyver, J. F., Aebischer, A., Biner, D., Gerner, P., Grimm, J., Heer, S., Krämer, K. W., Reinhard, C., & Güdel, H. U. (2005). Novel materials doped with trivalent lanthanides and transition metal ions showing near-infrared to visible photon upconversion. Optical Materials, 27(6), 1111–1130. https://doi.org/10.1016/j.optmat.2004.10.021
Reber, C., Guedel, H. U., Meyer, G., Schleid, T., & Daul, C. A. (1989). Optical spectroscopic and structural properties of V3+-doped fluoride, chloride, and bromide elpasolite lattices. Inorganic Chemistry, 28(16), 3249–3258. https://doi.org/10.1021/ic00315a034
Jacobsen, S. M., & Güdel, H. U. (1989). Higher excited state luminescence in Ti2+:MgCl2 Dynamics of radiative and nonradiative processes. Journal of Luminescence, 43(3), 125–137. https://doi.org/10.1016/0022-2313(89)90009-4
Wenger, O. S., & Güdel, H. U. (2001). Dual luminescence and excited-state dynamics in Ti2+ doped NaCl. The Journal of Physical Chemistry B, 105(19), 4181–4187. https://doi.org/10.1021/jp004183n
Flint, C. D., & Paulusz, A. G. (1981). Infrared and visible luminescence spectra of MoCl63- and MoBr 63- in cubic elpasolite crystals. Molecular Physics, 44(4), 925–938. https://doi.org/10.1080/00268978100102891
Flint, C. D., & Paulusz, A. G. (1980). High resolution infrared and visible luminescence spectra of OsCl62- and OsBr 62- in cubic crystals. Molecular Physics, 41(4), 907–923. https://doi.org/10.1080/00268978000103241
Ji, H., Hou, X., Molokeev, M. S., Ueda, J., Tanabe, S., Brik, M. G., Zhang, Z., Wang, Y., & Chen, D. (2020). Ultrabroadband red luminescence of Mn4+ in MgAl2O4 peaking at 651 nm. Dalton Transactions, 49(17), 5711–5721. https://doi.org/10.1039/D0DT00931H
Harris, J. P., Reber, C., Colmer, H. E., Jackson, T. A., Forshaw, A. P., Smith, J. M., Kinney, R. A., & Telser, J. (2017). Near-infrared 2Eg→4A2g and visible LMCT luminescence from a molecular bis-(tris(carbene)borate) manganese(IV) complex. Canadian Journal of Chemistry, 95(5), 547–552. https://doi.org/10.1139/cjc-2016-0607
Harris, J. P., Reber, C., Colmer, H. E., Jackson, T. A., Forshaw, A. P., Smith, J. M., Kinney, R. A., & Telser, J. (2020). Correction: Near-infrared 2Eg→4A2g and visible LMCT luminescence from a molecular bis-(tris(carbene)borate) manganese(IV) complex. Canadian Journal of Chemistry, 98(5), 250. https://doi.org/10.1139/cjc-2020-0119
Bussiére, G., Beaulac, R., Cardinal-David, B., & Reber, C. (2001). Coupled electronic states in trans-MCl2(H2O)4n+ complexes (M: Ni2+, Co2+, V3+, Cr3+) probed by absorption and luminescence spectroscopy. Coordination Chemistry Reviews, 219–221, 509–543. https://doi.org/10.1016/S0010-8545(01)00349-6
Atanasov, M., Andreici Eftimie, E.-L., Avram, N. M., Brik, M. G., & Neese, F. (2021). First-principles study of optical absorption energies, ligand field and spin-hamiltonian parameters of Cr3+ ions in emeralds. Inorganic Chemistry. https://doi.org/10.1021/acs.inorgchem.1c02650
Wenger, O. S., Bénard, S., & Güdel, H. U. (2002). Crystal field effects on the optical absorption and luminescence properties of Ni2+-doped chlorides and bromides: Crossover in the emitting higher excited state. Inorganic Chemistry, 41(23), 5968–5977. https://doi.org/10.1021/ic020347y
Monteiro, J. H. S. K. (2020). Recent advances in luminescence imaging of biological systems using lanthanide(III) luminescent complexes. Molecules. https://doi.org/10.3390/molecules25092089
Yuan, J., & Wang, G. (2006). Lanthanide-based luminescence probes and time-resolved luminescence bioassays. Trends Analyt. Chem., 25(5), 490–500. https://doi.org/10.1016/j.trac.2005.11.013
Wei, C., Ma, L., Wei, H., Liu, Z., Bian, Z., & Huang, C. (2018). Advances in luminescent lanthanide complexes and applications. Science China Technological Science, 61(9), 1265–1285. https://doi.org/10.1007/s11431-017-9212-7
Lever, A. B. P. (1968). Inorganic electronic spectroscopy (1st ed.). Elsevier.
Ballhausen, C. J. (1962). Introduction to ligand field theory. McGraw-Hill Book Company.
Tanabe, Y., & Sugano, S. (1954). On the absorption spectra of complex ions I. Journal of the Physical Society of Japan, 9(5), 753–766. https://doi.org/10.1143/jpsj.9.753
Tanabe, Y., & Sugano, S. (1954). On the absorption spectra of complex ions II. Journal of the Physical Society of Japan, 9(5), 766–779. https://doi.org/10.1143/JPSJ.9.766
Figgis, B. N., & Hitchman, M. A. (2000). Ligand field theory and its applications. Wiley-VCH.
Zare, D., Doistau, B., Nozary, H., Besnard, C., Guénée, L., Suffren, Y., Pelé, A.-L., Hauser, A., & Piguet, C. (2017). CrIII as an alternative to RuII in metallo-supramolecular chemistry. Dalton Transactions, 46(28), 8992–9009. https://doi.org/10.1039/c7dt01747b
Otto, S., Dorn, M., Förster, C., Bauer, M., Seitz, M., & Heinze, K. (2018). Understanding and exploiting long-lived near-infrared emission of a molecular ruby. Coordination Chemistry Reviews, 359, 102–111. https://doi.org/10.1016/j.ccr.2018.01.004
Jørgensen, C. K. (1963). Spectroscopy of transition-group complexes. In: Advances in chemical physics, (pp. 33–146). Wiley. https://doi.org/10.1002/9780470143513.ch2.
Oppenheim, J. & Miller, J. (2021), Tanabe-Sugano for Mathematica. Retrieved December 07, 2021, from https://github.com/JulesOpp/Tanabe-Sugano. Accessed 07 Dec 2021
Kaufhold, S., & Wärnmark, K. (2020). Design and synthesis of photoactive iron N-heterocyclic carbene complexes. Catalysts, 10(1), 132. https://doi.org/10.3390/catal10010132
Tang, Y., Zhao, S., Long, B., Liu, J.-C., & Li, J. (2016). On the nature of support effects of metal dioxides MO2 (M = Ti, Zr, Hf, Ce, Th) in single-atom gold catalysts: importance of quantum primogenic effect. Journal of Physical Chemistry C, 120(31), 17514–17526. https://doi.org/10.1021/acs.jpcc.6b05338
McCusker, J. K. (2019). Electronic structure in the transition metal block and its implications for light harvesting. Science, 363(6426), 484–488. https://doi.org/10.1126/science.aav9104
Dorn, M., Kalmbach, J., Boden, P., Päpcke, A., Gómez, S., Förster, C., Kuczelinis, F., Carrella, L. M., Büldt, L. A., Bings, N. H., Rentschler, E., Lochbrunner, S., González, L., Gerhards, M., Seitz, M., & Heinze, K. (2020). A vanadium(III) complex with blue and NIR-II spin-flip luminescence in solution. Journal of the American Chemical Society, 142(17), 7947–7955. https://doi.org/10.1021/jacs.0c02122
Kirk, A. D., & Porter, G. B. (1980). Luminescence of chromium(III) complexes. Journal of Physical Chemistry, 84(8), 887–891. https://doi.org/10.1021/j100445a020
Jiménez, J.-R., Doistau, B., Besnard, C., & Piguet, C. (2018). Versatile heteroleptic bis-terdentate Cr(III) chromophores displaying room temperature millisecond excited state lifetimes. Chemical Communications, 54(94), 13228–13231. https://doi.org/10.1039/c8cc07671e
Whuler, A., Brouty, C., Spinat, P. & Herpin, P. (1977). Structure du complexe actif hydraté (+)-Cr(en)3Cl3⋅2H2O. Etude de la configuration absolue et du désordre conformationnel, Acta Crystallogr. B Struct. Sci., 33(9), 2877–2885. https://doi.org/10.1107/S0567740877009674.
Ando, H., Iuchi, S., & Sato, H. (2012). Theoretical study on ultrafast intersystem crossing of chromium(III) acetylacetonate. Chemical Physics Letters, 535, 177–181. https://doi.org/10.1016/j.cplett.2012.03.043
Penfold, T. J., Gindensperger, E., Daniel, C., & Marian, C. M. (2018). Spin-vibronic mechanism for intersystem crossing. Chemical Reviews, 118(15), 6975–7025. https://doi.org/10.1021/acs.chemrev.7b00617
Juban, E. A., & McCusker, J. K. (2005). Ultrafast dynamics of 2E state formation in Cr(acac)3. Journal of the American Chemical Society, 127(18), 6857–6865. https://doi.org/10.1021/ja042153i
Ballardini, R., Varani, G., Wasgestian, H. F., Moggi, L., & Balzani, V. (1973). Role of the excited states in the photochemical and photophysical behavior of tris(ethylenediamine)chromium(III) in aqueous solutions. Journal of Physical Chemistry, 77(25), 2947–2951. https://doi.org/10.1021/j100643a004
Adamson, A. W. (1967). Photochemistry of complex ions. IV. Role of quartet excited states in the photochemistry of chromium(III) complexes. The Journal of Physical Chemistry, 71(4), 798–808. https://doi.org/10.1021/j100863a003
Knochenmuss, R., Reber, C., Rajasekharan, M. V., & Güdel, H. U. (1986). Broadband near-infrared luminescence of Cr+3 in the elpasolite lattices Cs2NaInCl6, Cs2NaYCl6, and Cs2NaYBr 6. The Journal of Chemical Physics, 85(8), 4280–4289. https://doi.org/10.1063/1.451801
Otto, S., Harris, J. P., Heinze, K., & Reber, C. (2018). Molecular ruby under pressure. Angewandte Chemie International Edition, 57(34), 11069–11073. https://doi.org/10.1002/anie.201806755
Reichenauer, F., Wang, C., Förster, C., Boden, P., Ugur, N., Báez-Cruz, R., Kalmbach, J., Carrella, L. M., Rentschler, E., Ramanan, C., Niedner-Schatteburg, G., Gerhards, M., Seitz, M., Resch-Genger, U., & Heinze, K. (2021). Strongly red-emissive molecular ruby Cr(bpmp)23+ surpasses Ru(bpy)32+. Journal of the American Chemical Society, 143(30), 11843–11855. https://doi.org/10.1021/jacs.1c05971
Otto, S., Nauth, A. M., Ermilov, E., Scholz, N., Friedrich, A., Resch-Genger, U., Lochbrunner, S., Opatz, T., & Heinze, K. (2017). Photo-chromium: sensitizer for visible-light-induced oxidative C−H bond functionalization-electron or energy transfer? ChemPhotoChem, 1(8), 344–349. https://doi.org/10.1002/cptc.201700077
Fukuda, R., Walters, R. T., Macke, H., & Adamson, A. W. (1979). Rate of primary photoproduct formation for aqueous tris(ethylenediamine)chromium(3+) and chloropentaamminechromium(2+). Journal of Physical Chemistry, 83(16), 2097–2103. https://doi.org/10.1021/j100479a009
Barbour, J. C., Kim, A. J. I., deVries, E., Shaner, S. E., & Lovaasen, B. M. (2017). Chromium(III) bis-arylterpyridyl complexes with enhanced visible absorption via incorporation of intraligand charge-transfer transitions. Inorganic Chemistry, 56(14), 8212–8222. https://doi.org/10.1021/acs.inorgchem.7b00953
Yardley, J. T., & Beattie, J. K. (1972). Lifetime of the 4T2g state of tris(2,4-pentanedionato)chromium(III) reexamined. Journal of the American Chemical Society, 94(25), 8925–8926. https://doi.org/10.1021/ja00780a057
Treiling, S., Wang, C., Förster, C., Reichenauer, F., Kalmbach, J., Boden, P., Harris, J. P., Carrella, L. M., Rentschler, E., Resch-Genger, U., Reber, C., Seitz, M., Gerhards, M., & Heinze, K. (2019). Luminescence and light-driven energy and electron transfer from an exceptionally long-lived excited state of a non-innocent chromium(III) complex. Angewandte Chemie International Edition, 58(50), 18075–18085. https://doi.org/10.1002/anie.201909325
Wang, C., Otto, S., Dorn, M., Kreidt, E., Lebon, J., Sršan, L., Di Martino-Fumo, P., Gerhards, M., Resch-Genger, U., Seitz, M., & Heinze, K. (2018). Deuterated molecular ruby with record luminescence quantum yield. Angewandte Chemie International Edition, 57(4), 1112–1116. https://doi.org/10.1002/anie.201711350
Chen, S.-N., & Porter, G. B. (1970). Lifetime of the 4T2g state of chromium(III) complexes. Journal of the American Chemical Society, 92(7), 2189–2190. https://doi.org/10.1021/ja00710a096
Barker, K. D., Barnett, K. A., Connell, S. M., Glaeser, J. W., Wallace, A. J., Wildsmith, J., Herbert, B. J., Wheeler, J. F., & Kane-Maguire, N. A. P. (2001). Synthesis and characterization of heteroleptic Cr(diimine)33+ complexes. Inorganica Chimica Acta, 316(1–2), 41–49. https://doi.org/10.1016/S0020-1693(01)00377-2
McDaniel, A. M., Tseng, H.-W., Damrauer, N. H., & Shores, M. P. (2010). Synthesis and solution phase characterization of strongly photooxidizing heteroleptic Cr(III) tris-dipyridyl complexes. Inorganic Chemistry, 49(17), 7981–7991. https://doi.org/10.1021/ic1009972
Serpone, N., Jamieson, M. A., Henry, M. S., Hoffman, M. Z., Bolletta, F., & Maestri, M. (1979). Excited-state behavior of polypyridyl complexes of chromium(III). Journal of the American Chemical Society, 101, 2907–2916. https://doi.org/10.1021/ja00505a019
Perkovic, M. W., Heeg, M. J., & Endicott, J. F. (1991). Stereochemical perturbations of the relaxation behavior of (2E)chromium(III). Ground-state x-ray crystal structure, photophysics, and molecular mechanics simulations of the quasi-cage complex [4,4’,4”-ethylidynetris(3-azabutan-1-amine)]chromium tribromide. Inorganic Chemistry, 30(16), 3140–3147. https://doi.org/10.1021/ic00016a009
Otto, S., Förster, C., Wang, C., Resch-Genger, U., & Heinze, K. (2018). A strongly luminescent chromium(III) complex acid. Chemistry--A European Journal, 24(48), 12555–12563. https://doi.org/10.1002/chem.201802797
Higgins, R. F., Fatur, S. M., Shepard, S. G., Stevenson, S. M., Boston, D. J., Ferreira, E. M., Damrauer, N. H., Rappé, A. K., & Shores, M. P. (2016). Uncovering the roles of oxygen in Cr(III) photoredox catalysis. Journal of the American Chemical Society, 138(16), 5451–5464. https://doi.org/10.1021/jacs.6b02723
Sinha, N., Jiménez, J.-R., Pfund, B., Prescimone, A., Piguet, C., & Wenger, O. S. (2021). A near-infrared-II emissive chromium(III) complex. Angewandte Chemie (International ed. in English), 60(44), 23722–23728. https://doi.org/10.1002/anie.202106398
Stein, L., Boden, P., Naumann, R., Förster, C., Niedner Schatteburg, G., & Heinze, K. (2022). The overlooked NIR luminescence of the Ir(ppy)3 analogue Cr(ppy)3. Chemical Communications. https://doi.org/10.1039/D2CC00680D
Dill, R. D., Portillo, R. I., Shepard, S. G., Shores, M. P., Rappé, A. K., & Damrauer, N. H. (2020). Long-lived mixed 2MLCT/MC states in antiferromagnetically coupled d3 vanadium(II) bipyridine and phenanthroline complexes. Inorganic Chemistry, 59(20), 14706–14715. https://doi.org/10.1021/acs.inorgchem.0c01950
Laporte, O., & Meggers, W. F. (1925). Some rules of spectral structure. Journal of the Optical Society of America, 11(5), 459. https://doi.org/10.1364/JOSA.11.000459
Dannöhl-Fickler, R., Kelm, H., & Wasgestian, F. (1975). Solvent and temperature dependence of the phosphorescence decay time of hexacyanochromate(III). Journal of Luminescence, 10(2), 103–112. https://doi.org/10.1016/0022-2313(75)90038-1
McDaniel, A. M., Tseng, H.-W., Hill, E. A., Damrauer, N. H., Rappé, A. K., & Shores, M. P. (2013). Syntheses and photophysical investigations of Cr(III) hexadentate iminopyridine complexes and their tris(bidentate) analogues. Inorganic Chemistry, 52(3), 1368–1378. https://doi.org/10.1021/ic302055r
Xiang, H., Cheng, J., Ma, X., Zhou, X., & Chruma, J. J. (2013). Near-infrared phosphorescence: Materials and applications. Chemical Society Reviews, 42(14), 6128–6185. https://doi.org/10.1039/c3cs60029g
Scattergood, P. A. (2020). Recent advances in chromium coordination chemistry: luminescent materials and photocatalysis. In N. J. Patmore & P. I. P. Elliott (Eds.), Organometallic chemistry (pp. 1–34). Royal Society of Chemistry. https://doi.org/10.1039/9781788017077-00001
Kreidt, E., Kruck, C., & Seitz, M. (2018). Nonradiative deactivation of lanthanoid luminescence by multiphonon relaxation in molecular complexes. In J.-C.G. Bünzli, V. K. Pecharsky, G.-Y. Adachi, K. A. Gschneidner, & E. LeRoy (Eds.), Handbook on the physics and chemistry of rare earths (pp. 35–79). Amsterdam, Oxford: North-Holland. https://doi.org/10.1016/bs.hpcre.2018.04.001
Henry, M. S. (1977). Prolongation of the lifetime of the 2E state of tris(2,2’-bipyridine)chromium(III) ion by anions in aqueous solution. Journal of the American Chemical Society, 99(18), 6138–6139. https://doi.org/10.1021/ja00460a068
Henry, M. S., & Hoffman, M. Z. (1978). Solution medium effects on the photophysics and photochemistry of polypyridyl complexes of chromium(III). In M. S. Wrighton (Ed.), Inorganic and organometallic photochemistry (pp. 91–114). American Chemical Society. https://doi.org/10.1021/ba-1978-0168.ch006
Wasgestian, H. F., Ballardini, R., Varani, G., Moggi, L., & Balzani, V. (1973). Quenching of the tris(ethylenediamine)chromium(III) phosphorescence by some transition metal ions in aqueous solutions. Journal of Physical Chemistry, 77(22), 2614–2617. https://doi.org/10.1021/j100640a007
Basu, U., Otto, S., Heinze, K., & Gasser, G. (2019). Biological evaluation of the NIR-emissive ruby analogue [Cr(ddpd)2][BF4]3 as a photodynamic therapy photosensitizer. European Journal of Inorganic Chemistry, 2019(1), 37–41. https://doi.org/10.1002/ejic.201801023
Wang, C., Kitzmann, W. R., Weigert, F., Förster, C., Wang, X., Heinze, K., & Resch-Genger, U. (2022). Matrix effects on photoluminescence and oxygen sensitivity of a molecular ruby. ChemPhotoChem. https://doi.org/10.1002/cptc.202100296
Luo, L.-J., Su, Q.-Q., Cheng, S.-C., Xiang, J., Man, W.-L., Shu, W.-M., Zeng, M.-H., Yiu, S.-M., Ko, C.-C., & Lau, T.-C. (2020). Tunable luminescent properties of tricyanoosmium nitrido complexes bearing a chelating O^N ligand. Inorganic Chemistry, 59(7), 4406–4413. https://doi.org/10.1021/acs.inorgchem.9b03554
Coppo, P., Plummer, E. A., & de Cola, L. (2004). Tuning iridium(III) phenylpyridine complexes in the “almost blue” region. Chemical Communications, 15, 1774–1775. https://doi.org/10.1039/b406851c
Duvenhage, M. M., Visser, H. G., Ntwaeaborwa, O. M., & Swart, H. C. (2014). The effect of electron donating and withdrawing groups on the morphology and optical properties of Alq3. Physica B: Condens Matter, 439, 46–49. https://doi.org/10.1016/j.physb.2013.11.049
Chatterjee, K. K., & Forster, L. S. (1964). Electronic transitions in chromium(III) complexes—IV radiative and non-radiative transition probabilities. Spectrochimica Acta, 20(10), 1603–1609. https://doi.org/10.1016/0371-1951(64)80139-9
Witzke, H. (1971). Semi-empirical evaluations of the Racah B and C parameters from the crystal field spectra of chromium(III) complexes. Theoretica Chimica Acta, 20(2), 171–185. https://doi.org/10.1007/BF00569263
Stevenson, S. M., Shores, M. P., & Ferreira, E. M. (2015). Photooxidizing chromium catalysts for promoting radical cation cycloadditions. Angewandte Chemie International Edition, 54(22), 6506–6510. https://doi.org/10.1002/anie.201501220
Yang, Y., Liu, Q., Zhang, L., Yu, H., & Dang, Z. (2017). Mechanistic investigation on oxygen-mediated photoredox diels-alder reactions with chromium catalysts. Organometallics, 36(3), 687–698. https://doi.org/10.1021/acs.organomet.6b00886
Otto, S., Scholz, N., Behnke, T., Resch-Genger, U., & Heinze, K. (2017). Thermo-chromium: A contactless optical molecular thermometer. Chemistry--A European Journal, 23(50), 12131–12135. https://doi.org/10.1002/chem.201701726
Wang, C., Otto, S., Dorn, M., Heinze, K., & Resch-Genger, U. (2019). Luminescent TOP nanosensors for simultaneously measuring temperature, oxygen, and pH at a single excitation wavelength. Analytical Chemistry, 91(3), 2337–2344. https://doi.org/10.1021/acs.analchem.8b05060
Forman, R. A., Piermarini, G. J., Barnett, J. D., & Block, S. (1972). Pressure measurement made by the utilization of ruby sharp-line luminescence. Science, 176(4032), 284–285. https://doi.org/10.1126/science.176.4032.284
Chijioke, A. D., Nellis, W. J., Soldatov, A., & Silvera, I. F. (2005). The ruby pressure standard to 150GPa. Journal of Applied Physics, 98(11), 114905. https://doi.org/10.1063/1.2135877
Reber, C., Grey, J. K., Lanthier, E., & Frantzen, K. A. (2011). Pressure-induced change of d-d luminescence energies, vibronic structure and band intensities in transition metal complexes. In J. P. Fackler & L. Falvello (Eds.), Techniques in inorganic chemistry (pp. 233–254). Taylor & Francis. https://doi.org/10.1080/02603590500403909
Wang, C., Reichenauer, F., Kitzmann, W. R., Kerzig, C., Heinze, K. & Resch-Genger, U. (2021). Efficient Triplet-Triplet Annihilation Upconversion Sensitized by a Second-Generation Molecular Ruby via an Underexplored Energy Transfer Mechanism, submitted.
Imbert, D., Cantuel, M., Bünzli, J.-C.G., Bernardinelli, G., & Piguet, C. (2003). Extending lifetimes of lanthanide-based near-infrared emitters (Nd, Yb) in the millisecond range through Cr(III) sensitization in discrete bimetallic edifices. Journal of the American Chemical Society, 125(51), 15698–15699. https://doi.org/10.1021/ja0386501
Aboshyan-Sorgho, L., Nozary, H., Aebischer, A., Bünzli, J.-C.G., Morgantini, P.-Y., Kittilstved, K. R., Hauser, A., Eliseeva, S. V., Petoud, S., & Piguet, C. (2012). Optimizing millisecond time scale near-infrared emission in polynuclear chrome(III)-lanthanide(III) complexes. Journal of the American Chemical Society, 134(30), 12675–12684. https://doi.org/10.1021/ja304009b
Aboshyan-Sorgho, L., Besnard, C., Pattison, P., Kittilstved, K. R., Aebischer, A., Bünzli, J.-C.G., Hauser, A., & Piguet, C. (2011). Near-infrared→visible light upconversion in a molecular trinuclear d-f-d complex. Angewandte Chemie International Edition, 50(18), 4108–4112. https://doi.org/10.1002/anie.201100095
Kalmbach, J., Wang, C., You, Y., Förster, C., Schubert, H., Heinze, K., Resch-Genger, U., & Seitz, M. (2020). Near-IR to near-IR upconversion luminescence in molecular chromium ytterbium salts. Angewandte Chemie International Edition, 59(42), 18804–18808. https://doi.org/10.1002/anie.202007200
Bünzli, J.-C.G., & Eliseeva, S. V. (2010). Lanthanide NIR luminescence for telecommunications, bioanalyses and solar energy conversion. Journal of Rare Earths, 28(6), 824–842. https://doi.org/10.1016/S1002-0721(09)60208-8
MacKenzie, L. E., & Pal, R. (2021). Circularly polarized lanthanide luminescence for advanced security inks. Nature Reviews Chemistry, 5(2), 109–124. https://doi.org/10.1038/s41570-020-00235-4
Hao, C., Xu, L., Sun, M., Zhang, H., Kuang, H., & Xu, C. (2019). Circularly polarized light triggers biosensing based on chiral assemblies. Chemistry--A European Journal, 25(53), 12235–12240. https://doi.org/10.1002/chem.201901721
Zinna, F., & Di Bari, L. (2015). Lanthanide circularly polarized luminescence: Bases and applications. Chirality, 27(1), 1–13. https://doi.org/10.1002/chir.22382
Dee, C., Zinna, F., Kitzmann, W. R., Pescitelli, G., Heinze, K., Di Bari, L., & Seitz, M. (2019). Strong circularly polarized luminescence of an octahedral chromium(III) complex. Chemical Communications, 55(87), 13078–13081. https://doi.org/10.1039/c9cc06909g
Poncet, M., Benchohra, A., Jiménez, J.-R., & Piguet, C. (2021). Chiral chromium(III) complexes as promising candidates for circularly polarized luminescence. ChemPhotoChem, 5(10), 880–892. https://doi.org/10.1002/cptc.202100146
Doistau, B., Jiménez, J.-R., & Piguet, C. (2020). Beyond chiral organic (p-Block) chromophores for circularly polarized luminescence: The success of d-block and f-block chiral complexes. Frontiers in Chemistry, 8, 555. https://doi.org/10.3389/fchem.2020.00555
Kreidt, E., Arrico, L., Zinna, F., Di Bari, L., & Seitz, M. (2018). Circularly polarised luminescence in enantiopure samarium and europium cryptates. Chemistry--A European Journal, 24(51), 13556–13564. https://doi.org/10.1002/chem.201802196
Arrico, L., Di Bari, L., & Zinna, F. (2021). Quantifying the overall efficiency of circularly polarized emitters. Chemistry--A European Journal, 27(9), 2920–2934. https://doi.org/10.1002/chem.202002791
Fataftah, M. S., Zadrozny, J. M., Coste, S. C., Graham, M. J., Rogers, D. M., & Freedman, D. E. (2016). Employing forbidden transitions as qubits in a nuclear spin-free chromium complex. Journal of the American Chemical Society, 138(4), 1344–1348. https://doi.org/10.1021/jacs.5b11802
Fataftah, M. S., Bayliss, S. L., Laorenza, D. W., Wang, X., Phelan, B. T., Wilson, C. B., Mintun, P. J., Kovos, B. D., Wasielewski, M. R., Han, S., Sherwin, M. S., Awschalom, D. D., & Freedman, D. E. (2020). Trigonal bipyramidal V3+ complex as an optically addressable molecular qubit candidate. Journal of the American Chemical Society, 142, 20400–20408. https://doi.org/10.1021/jacs.0c08986
Wojnar, M. K., Laorenza, D. W., Schaller, R. D., & Freedman, D. E. (2020). Nickel(II) metal complexes as optically addressable qubit candidates. Journal of the American Chemical Society, 142(35), 14826–14830. https://doi.org/10.1021/jacs.0c06909
Bayliss, S. L., Laorenza, D. W., Mintun, P. J., Kovos, B. D., Freedman, D. E., & Awschalom, D. D. (2020). Optically addressable molecular spins for quantum information processing. Science, 370(6522), 1309–1312. https://doi.org/10.1126/science.abb9352
Laorenza, D. W., Kairalapova, A., Bayliss, S. L., Goldzak, T., Greene, S. M., Weiss, L. R., Deb, P., Mintun, P. J., Collins, K. A., Awschalom, D. D., Berkelbach, T. C., & Freedman, D. E. (2021). Tunable Cr4+ molecular color centers. Journal of the American Chemical Society. https://doi.org/10.1021/jacs.1c10145
Lenz, S., Bamberger, H., Hallmen, P. P., Thiebes, Y., Otto, S., Heinze, K., & van Slageren, J. (2019). Chromium(III)-based potential molecular quantum bits with long coherence times. Physical Chemistry Chemical Physics: PCCP, 21(13), 6976–6983. https://doi.org/10.1039/C9CP00745H
Wijeratne, G. B., Zolnhofer, E. M., Fortier, S., Grant, L. N., Carroll, P. J., Chen, C.-H., Meyer, K., Krzystek, J., Ozarowski, A., Jackson, T. A., Mindiola, D. J., & Telser, J. (2015). Electronic structure and reactivity of a well-defined mononuclear complex of Ti(II). Inorganic Chemistry, 54(21), 10380–10397. https://doi.org/10.1021/acs.inorgchem.5b01796
Reinholdt, A., Pividori, D., Laughlin, A. L., DiMucci, I. M., MacMillan, S. N., Jafari, M. G., Gau, M. R., Carroll, P. J., Krzystek, J., Ozarowski, A., Telser, J., Lancaster, K. M., Meyer, K., & Mindiola, D. J. (2020). A mononuclear and high-spin tetrahedral TiII complex. Inorganic Chemistry, 59(24), 17834–17850. https://doi.org/10.1021/acs.inorgchem.0c02586
Jensen, J. A., Wilson, S. R., Schultz, A. J., & Girolami, G. S. (1987). Divalent titanium chemistry. Synthesis, reactivity, and x-ray and neutron diffraction studies of Ti(BH4)2(dmpe)2 and Ti(CH3)2(dmpe)2. Journal of the American Chemical Society, 109(26), 8094–8096. https://doi.org/10.1021/ja00260a029
Beaumier, E. P., Pearce, A. J., See, X. Y., & Tonks, I. A. (2019). Modern applications of low-valent early transition metals in synthesis and catalysis. Nature Reviews Chemistry, 3(1), 15–34. https://doi.org/10.1038/s41570-018-0059-x
Araya, M. A., Cotton, F. A., Matonic, J. H., & Murillo, C. A. (1995). An efficient reduction process leading to titanium(II) and niobium(II): Preparation and structural characterization of trans-MCl2(py)4 compounds, M = Ti, Nb, and Mn. Inorganic Chemistry, 34(22), 5424–5428. https://doi.org/10.1021/ic00126a009
Fortier, S., & Gomez-Torres, A. (2021). Redox chemistry of discrete low-valent titanium complexes and low-valent titanium synthons. Chemical Communications, 57(80), 10292–10316. https://doi.org/10.1039/D1CC02772G
Kayal, A., Kuncheria, J., & Lee, S. C. (2001). Bishydrotris(pyrazol-1-yl)boratotitanium(II): A stable Tp2M complex of singular reactivity. Chemical Communications, 23, 2482–2483. https://doi.org/10.1039/B108115B
Dingle, R., McCarthy, P. J., & Ballhausen, C. J. (1969). Crystal spectra of hexaurea complexes. III. Optical and magnetic properties of crystals containing the hexaurea vanadium (III) Ion. The Journal of Chemical Physics, 50(5), 1957–1962. https://doi.org/10.1063/1.1671314
Flint, C. D., & Greenough, P. (1972). Luminescence spectrum of the V(urea)63+ ion. Chemical Physics Letters, 16(2), 369–370. https://doi.org/10.1016/0009-2614(72)80294-X
Beaulac, R., Tregenna-Piggott, P. L. W., Barra, A.-L., Weihe, H., Luneau, D., & Reber, C. (2006). The electronic ground state of V(urea)63+ probed by NIR luminescence, electronic Raman, and high-field EPR spectroscopies. Inorganic Chemistry, 45(8), 3399–3407. https://doi.org/10.1021/ic051709f
Dorn, M., East, N. R., Förster, C., Kitzmann, W. R., Moll, J., Reichenauer, F., Reuter, T., Stein, L., & Heinze, K. (2022). d-d and charge transfer photochemistry of 3d metal complexes. In J. Reedijk (Ed.), Comprehensive inorganic chemistry III. Elsevier.
Dorn, M., Kalmbach, J., Boden, P., Kruse, A., Dab, C., Reber, C., Niedner-Schatteburg, G., Lochbrunner, S., Gerhards, M., Seitz, M., & Heinze, K. (2021). Ultrafast and long-time excited state kinetics of an NIR-emissive vanadium(III) complex I: Synthesis, spectroscopy and static quantum chemistry. Chemical Science, 12(32), 10780–10790. https://doi.org/10.1039/d1sc02137k
Zobel, J. P., Knoll, T., & González, L. (2021). Ultrafast and long-time excited state kinetics of an NIR-emissive vanadium(III) complex II. Elucidating triplet-to-singlet excited-state dynamics. Chemica Science, 12(32), 10791–10801. https://doi.org/10.1039/d1sc02149d
Juban, E. A., Smeigh, A. L., Monat, J. E., & McCusker, J. K. (2006). Ultrafast dynamics of ligand-field excited states. Coordination Chemistry Reviews, 250, 1783–1791. https://doi.org/10.1016/J.CCR.2006.02.010
Brik, M., Avram, N., & Avram, C. (2005). Crystal field analysis of the ground and excited state absorption of a Cr4+ ion in LiAlO2 and LiGaO2 crystals. Central European Journal of Physics, 3(4), 508–524. https://doi.org/10.2478/BF02475609
Neyhart, G. A., Bakir, M., Boaz, J., Vining, W. J., & Patrick Sullivan, B. (1991). Photophysics and photochemistry of rhenium(V)-nitrogen triple bonds. Coordination Chemistry Reviews, 111, 27–32. https://doi.org/10.1016/0010-8545(91)84007-R
Savoie, C., & Reber, C. (1998). Emitting state energies and vibronic structure in the luminescence spectra of trans-dioxorhenium(V) complexes. Coordination Chemistry Reviews, 171, 387–398. https://doi.org/10.1016/S0010-8545(98)90059-5
Yam, V.W.-W., & Che, C.-M. (1990). Photochemistry and photophysics of trans-d2-dioxo complexes of osmium(VI). Coordination Chemistry Reviews, 97, 93–104. https://doi.org/10.1016/0010-8545(90)80082-5
Ikeda, H., Ito, A., Sakuda, E., Kitamura, N., Takayama, T., Sekine, T., Shinohara, A., & Yoshimura, T. (2013). Excited-state characteristics of tetracyanidonitridorhenium(V) and -technetium(V) complexes with N-heteroaromatic ligands. Inorganic Chemistry, 52(11), 6319–6327. https://doi.org/10.1021/ic302463v
Sasaki, K., Yamate, H., Yoshino, H., Miura, H., Shimoda, Y., Miyata, K., Onda, K., Ohtani, R., & Ohba, M. (2020). Vapor switching of the luminescence mechanism in a Re(V) complex. Chemical Communications, 56(85), 12961–12964. https://doi.org/10.1039/D0CC05462C
Lanthier, E., Bendix, J., & Reber, C. (2010). Pressure-dependent luminescence spectroscopy of molybdenum(IV) oxo complexes. Dalton Transactions, 39(15), 3695–3705. https://doi.org/10.1039/b924242b
Isovitsch, R. A., Beadle, A. S., Fronczek, F. R., & Maverick, A. W. (1998). Electronic absorption spectra and phosphorescence of oxygen-containing molybdenum(IV) complexes. Inorganic Chemistry, 37(17), 4258–4264. https://doi.org/10.1021/ic971186e
Kirk, A. D. (1981). Chromium(III) photochemistry and photophysics. Coordination Chemistry Reviews, 39(1–2), 225–263. https://doi.org/10.1016/s0010-8545(00)80515-9
Forster, L. S. (1990). The photophysics of chromium(III) complexes. Chemical Reviews, 90(2), 331–353. https://doi.org/10.1021/cr00100a001
Kjær, K. S., van Driel, T. B., Harlang, T. C. B., Kunnus, K., Biasin, E., Ledbetter, K., Hartsock, R. W., Reinhard, M. E., Koroidov, S., Li, L., Laursen, M. G., Hansen, F. B., Vester, P., Christensen, M., Haldrup, K., Nielsen, M. M., Dohn, A. O., Pápai, M. I., Møller, K. B., … Gaffney, K. J. (2019). Finding intersections between electronic excited state potential energy surfaces with simultaneous ultrafast X-ray scattering and spectroscopy. Chemical Science, 10(22), 5749–5760. https://doi.org/10.1039/c8sc04023k
Wegeberg, C., & Wenger, O. S. (2021). Luminescent first-row transition metal complexes. JACS Au. https://doi.org/10.1021/jacsau.1c00353
Schläfer, H., Gausmann, H., & Witzke, H. (1966). Phosphorescence of molybdenum (III) complexes. Journal of Molecular Spectroscopy, 21(1–4), 125–129. https://doi.org/10.1016/0022-2852(66)90131-7
Yao, Q., & Maverick, A. W. (1988). Near-infrared luminescence of octahedral molybdenum(III) and rhenium(IV) complexes in solution. Inorganic Chemistry, 27(10), 1669–1670. https://doi.org/10.1021/ic00283a001
Mohammed, A. K., Isovitsch, R. A., & Maverick, A. W. (1998). Solution photophysics, one-electron photooxidation, and photoinitiated two-electron oxidation of molybdenum(III) complexes. Inorganic Chemistry, 37(11), 2779–2785. https://doi.org/10.1021/ic970955r
Cornioley-Deuschel, C., & von Zelewsky, A. (1987). Stability of d3 diimine complexes: molybdenum(III) vs chromium(III). Inorganic Chemistry, 26(6), 962–963. https://doi.org/10.1021/ic00253a040
Stiefel, E. I. (1977). The coordination and bioinorganic chemistry of molybdenum. In S. J. Lippard (Ed.), Progress in inorganic chemistry (pp. 1–223). Wiley. https://doi.org/10.1002/9780470166239.ch1
Young, C. G. (2004). Molybdenum. In J. A. McCleverty & T. J. Meyer (Eds.), Comprehensive coordination chemistry II (pp. 415–527). Elsevier/Pergamon. https://doi.org/10.1016/B0-08-043748-6/03033-4
Dunn, T. M. (1961). Spin-orbit coupling in the first and second transition series. Transactions of the Faraday Society, 57, 1441. https://doi.org/10.1039/TF9615701441
Koseki, S., Matsunaga, N., Asada, T., Schmidt, M. W., & Gordon, M. S. (2019). Spin-orbit coupling constants in atoms and ions of transition elements: comparison of effective core potentials, model core potentials, and all-electron methods. Journal of Physical Chemistry A, 123(12), 2325–2339. https://doi.org/10.1021/acs.jpca.8b09218
Morrison, C. A. (1992). Crystal fields for transition-metal ions in laser host materials. Springer.
Fleischauer, P. D., & Fleischauer, P. (1970). Photoluminescence of transition metal coordination compounds. Chemical Reviews, 70(2), 199–230. https://doi.org/10.1021/cr60264a002
Sartori, G., Cervone, E., & Cancellieri, P. (1963). The electronic structure of d3 complexes using ligand field theory. Atti della Accademia Nazionale dei Lincei, Classe di Scienze Fisiche, Matematiche e Naturali, 35, 226.
Maverick, A. W., Lord, M. D., Yao, Q., & Henderson, L. J. (1991). Photophysics and photochemistry of hexachlororhenate(IV) and hexabromorhenate(IV). Inorganic Chemistry, 30(3), 553–558. https://doi.org/10.1021/ic00003a040
Holleman, A. F., & Wiberg, N. (2017). Hollemann Wiberg, Grundlagen und Hauptgruppenelemente, Band 1 (103rd ed.). De Gruyter.
Fujita, I., Yazaki, T., Torii, Y., & Kobayashi, H. (1972). Electronic absorption spectra of tris(2,2′-bipyridine)- and Tris(1,10-phenanthroline) complexes of Vanadium(II) and Chromium(II). Bulletin Chemical Society of Japan, 45(7), 2156–2161. https://doi.org/10.1246/bcsj.45.2156
Joyce, J. P., Portillo, R. I., Nite, C. M., Nite, J. M., Nguyen, M. P., Rappé, A. K., & Shores, M. P. (2021). Electronic structures of Cr(III) and V(II) polypyridyl systems: Undertones in an isoelectronic analogy. Inorganic Chemistry, 60(17), 12823–12834. https://doi.org/10.1021/acs.inorgchem.1c01129
Kee, T. (1998). Niobium and tantalum 1995. Coordination Chemistry Reviews, 169(1), 129–152. https://doi.org/10.1016/S0010-8545(98)00004-6
Wexler, P. A., Wigley, D. E., Koerner, J. B., & Albright, T. A. (1991). Preparation, properties, and bonding analysis of tantalum(II) η6-arene complexes. Organometallics, 10(7), 2319–2327. https://doi.org/10.1021/om00053a039
Thiyagarajan, B., Michalczyk, L., Bollinger, J. C., & Bruno, J. W. (1996). Generation of organoniobium(II) radicals and synthesis of heterometallic niobium−mercury compounds. Organometallics, 15(11), 2588–2590. https://doi.org/10.1021/om960152w
Tayebani, M., Gambarotta, S., & Yap, G. (1998). C−H versus C−N bond cleavage promoted by niobium(II) amide. Organometallics, 17(17), 3639–3641. https://doi.org/10.1021/om980298q
Noh, W., & Girolami, G. S. (2008). Mono(cycloheptatrienyl) tantalum chemistry: Synthesis and characterization of new tantalum halide, hydride, and alkyl species. Inorganic Chemistry, 47(22), 10682–10691. https://doi.org/10.1021/ic8015035
Cohen, J. L., & Hoggard, P. E. (2009). Photosolvation reactions of hexabromorhenate(IV). Inorganica Chimica Acta, 362(2), 580–582. https://doi.org/10.1016/j.ica.2008.03.119
McCusker, J. K., Walda, K. N., Dunn, R. C., Simon, J. D., Magde, D., & Hendrickson, D. N. (1993). Subpicosecond 1MLCT -> 5T2 intersystem crossing of low-spin polypyridyl ferrous complexes. Journal of the American Chemical Society, 115(1), 298–307. https://doi.org/10.1021/ja00054a043
Shari‘atiVura-Weis, Y. J. (2021). Ballistic ΔS = 2 intersystem crossing in a cobalt cubane following ligand-field excitation probed by extreme ultraviolet spectroscopy. Physical Chemistry Chemical Physics: PCCP. https://doi.org/10.1039/D1CP04136C
Scarborough, C. C., Sproules, S., Doonan, C. J., Hagen, K. S., Weyhermüller, T., & Wieghardt, K. (2012). Scrutinizing low-spin Cr(II) complexes. Inorganic Chemistry, 51(12), 6969–6982. https://doi.org/10.1021/ic300882r
Eaton, J. P., & Nicholls, D. (1981). The complex cyanides of chromium(II) and chromium(0). Transition Metal Chemistry, 6(4), 203–206. https://doi.org/10.1007/BF00618223
Becker, P. M., Förster, C., Carrella, L. M., Boden, P., Hunger, D., van Slageren, J., Gerhards, M., Rentschler, E., & Heinze, K. (2020). Spin crossover and long-lived excited states in a reduced molecular ruby. Chemistry--A European Journal, 26(32), 7199–7204. https://doi.org/10.1002/chem.202001237
Holleman, A. F., & Wiberg, N. (2017). Hollemann Wiberg, Nebengruppenelemente, Lanthanoide, Actinoide, Transactinoide, Band 2 (103rd ed.). De Gruyter.
Cotton, F. A., Eglin, J. L., & Wiesinger, K. J. (1992). Synthesis and characterization of molybdenum species: Dinuclear and mononuclear species of the molecular formulas [Mo2(O2CCH3)2(LL)2][BF4]2 and [Mo(O)(F)(LL)2][BF4] where LL=bis-phosphine. The use of [Mo(NCCH3)10][BF4]4 as a source for the [Mo2]4+ core. Inorganica Chimica Acta, 195, 11–23.
Cotton, F. A., & Wiesinger, K. J. (1992). Decakis(Acetonitrile)Dimolybdenum(II) Tetrafluoroborate(1-). Inorganic Syntheses, 29, 134–137.
Holste, G. (1975). Mo2(CH3COO)4, Mo2(C6H5COO)4 und [Mo6Cl8]Cl2(CH3COO)2. Zeitschrift für Anorganische und Allgemeine Chemie, 414(1), 81–90. https://doi.org/10.1002/zaac.19754140111
Johnson, K. D., & Powell, G. L. (2008). Microwave-assisted synthesis of dimolybdenum tetracarboxylates and a decanuclear osmium cluster. Journal of Organometallic Chemistry, 693(8–9), 1712–1715. https://doi.org/10.1016/j.jorganchem.2007.11.036
Maverick, A. W., Najdzionek, J. S., MacKenzie, D., Nocera, D. G., & Gray, H. B. (1983). Spectroscopic, electrochemical, and photochemical properties of molybdenum(II) and tungsten(II) halide clusters. Journal of the American Chemical Society, 105(7), 1878–1882. https://doi.org/10.1021/ja00345a034
Eltayeb, N. E., Teoh, S. G., Kusrini, E., Adnan, R., & Fun, H. K. (2010). The manganese(III) complex with chelating Schiff base ligand: X-ray structure, spectroscopic and computational studies. Spectrochimica Acta Part A: Molecular and Biomolecular Spectroscopy, 75(1), 453–457. https://doi.org/10.1016/j.saa.2009.11.006
Majumder, A., Goswami, S., Batten, S. R., Salah El Fallah, M., Ribas, J., & Mitra, S. (2006). Catalytic oxidation of 3,5-di-tert-butylcatechol by a manganese(III) 18-azametallacrown-6 compound: Synthesis, crystal structure, fluorescence, magnetic and kinetic investigation. Inorganica Chimica Acta, 359(8), 2375–2382. https://doi.org/10.1016/j.ica.2006.01.045
Makarska-Bialokoz, M., Pratviel, G., & St. Radzki. (2008). The influence of solvent polarity on spectroscopic properties of 5-[4-(5-carboxy-1-butoxy)-phenyl]-10,15,20-tris(4-N-methylpyridiniumyl)porphyrin and its complexes with Fe(III) and Mn(III) ions. Journal of Molecular Structure, 875(1–3), 468–477. https://doi.org/10.1016/j.molstruc.2007.05.026
Sun, E., Shi, Y., Zhang, P., Zhou, M., Zhang, Y., Tang, X., & Shi, T. (2008). Spectroscopic properties and cyclic voltammetry on a series of meso-tetra(p-alkylamidophenyl)porphyrin liquid crystals and their Mn complexes. Journal of Molecular Structure, 889(1–3), 28–34. https://doi.org/10.1016/j.molstruc.2008.01.014
Abram, U. (2004). Rhenium. In J. A. McCleverty & T. J. Meyer (Eds.), Comprehensive coordination chemistry II (pp. 271–402). Elsevier/Pergamon. https://doi.org/10.1016/B0-08-043748-6/04177-3
Alberto, R. (2004). Technetium. In J. A. McCleverty & T. J. Meyer (Eds.), Comprehensive coordination chemistry II (pp. 127–270). Elsevier/Pergamon. https://doi.org/10.1016/B0-08-043748-6/04022-6
Hancock, R. D., & McDougall, G. J. (1977). The role of electronic delocalization over the chelate ring in stabilizing complexes of 2,2′-bipyridyl. Journal of the Chemical Society Dalton Transactions, 1, 67–70. https://doi.org/10.1039/dt9770000067
Robinson, M. A., Curry, J. D., & Busch, D. H. (1963). Complexes derived from strong field ligands. XVII. Electronic spectra of octahedral Nickel(II) complexes with ligands of the α-diimine and closely related classes. Inorganic Chemistry, 2(6), 1178–1181. https://doi.org/10.1021/ic50010a021
Henke, W., & Reinen, D. (1977). Spektroskopische Untersuchungen zum Jahn-Teller-Effekt des Cu2+-Ions in Terpyridin-Komplexen Cu(terpy)2X2·nH2O [X = NO, ClO, Br−]. Zeitschrift für Anorganische und Allgemeine Chemie, 436(1), 187–200. https://doi.org/10.1002/zaac.19774360123
Dorn, M., Mack, K., Carrella, L. M., Rentschler, E., Förster, C., & Heinze, K. (2018). Structure and electronic properties of an expanded terpyridine complex of Nickel(II) [Ni(ddpd)2](BF4)2. Zeitschrift für Anorganische und Allgemeine Chemie, 644(14), 706–712. https://doi.org/10.1002/zaac.201800101
González, E., Rodrigue-Witchel, A., & Reber, C. (2007). Absorption spectroscopy of octahedral nickel(II) complexes: A case study of interactions between multiple electronic excited states. Coordination Chemistry Reviews, 251(3–4), 351–363. https://doi.org/10.1016/j.ccr.2006.08.011
Nolet, M.-C., Michaud, A., Bain, C., Zargarian, D., & Reber, C. (2006). Allowed and forbidden d-d transitions in poly(3,5-dimethylpyrazolyl)methane complexes of nickel(II). Photochemistry and Photobiology, 82(1), 57–63. https://doi.org/10.1562/2005-06-28-RA-593
Dab, C., East, N. R., Förster, C., Heinze, K. & Reber, C. (2022). to be submitted.
Oppenheim, J. J., McNicholas, B. J., Miller, J., & Gray, H. B. (2019). Electronic Structure of Tetracyanonickelate(II). Inorganic Chemistry, 58(22), 15202–15206. https://doi.org/10.1021/acs.inorgchem.9b02135
Giordano, T. J., Butler, W. M., & Rasmussen, P. G. (1978). Platinum and palladium complexes of thienylpyridine. 2. Quasi-octahedral divalent-metal compounds. Inorganic Chemistry, 17, 1917–1922.
Takeda, N., Shimizu, D., & Tokitoh, N. (2005). Synthesis and structure of a distorted octahedral palladium(II) complex coordinated with a tetrathioether ligand tethered with bulky substituents. Inorganic Chemistry, 44(23), 8561–8568. https://doi.org/10.1021/ic050944v
Pointillart, F., Train, C., Villain, F., Moulin, C. C. D., Gredin, P., Chamoreau, L.-M., Gruselle, M., Aullon, G., Alvarez, S., & Verdaguer, M. (2007). Six-fold oxygen-coordinated triplet (S = 1) palladium(II) moieties templated by tris(bipyridine)ruthenium(II) ions. Journal of the American Chemical Society, 129(5), 1327–1334. https://doi.org/10.1021/ja066817v
Takeda, N., Isobe, T., & Tokitoh, N. (2007). Synthesis of an acyclic diselenodithioether ligand tethered with bulky substituents and its application to the synthesis of a distorted octahedral palladium(II) complex. Heteroatom Chemistry, 18(5), 549–556. https://doi.org/10.1002/hc.20339
Das, D., Vats, B. G., Kannan, S., Maity, D. K., & Drew, M. G. (2013). Steric effects in pyrazole palladium(II) compounds: Synthetic, structural, and theoretical studies of a highly distorted octahedral palladium(II) pyrazole compound. Polyhedron, 54, 104–109. https://doi.org/10.1016/j.poly.2013.01.051
Safin, D. A., Babashkina, M. G., & Garcia, Y. (2013). First paramagnetic Pd(II) complex with a PdN4S2 coordination core. Dalton Transactions, 42(4), 902–905. https://doi.org/10.1039/C2DT32505E
Acknowledgements
The authors would like to thank Prof. Dr. Christian Reber, Dr. Christoph Förster and Nathan Roy East for constructive criticism of the manuscript. Parts of this research were conducted using the supercomputer ELWETRITSCH and advisory services offered by the Technical University of Kaiserslautern (https://elwe.rhrk.uni-kl.de/), which is a member of the AHRP.
Funding
Open Access funding enabled and organized by Projekt DEAL. Financial support from the Deutsche Forschungsgemeinschaft [DFG, Priority Program SPP 2102 “Light-controlled reactivity of metal complexes” (HE 2778/13–1)] is gratefully acknowledged. W. R. K. is grateful to the Chemical Industry Funds for a Kekulé Fellowship.
Author information
Authors and Affiliations
Contributions
WRK conceptualized the article, performed the literature search and wrote the article. JM provided support in writing of the article. KH provided the idea and general concept for the article and critically revised the work. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Conflicts of interest
The authors declare no conflict of interest.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kitzmann, W.R., Moll, J. & Heinze, K. Spin-flip luminescence. Photochem Photobiol Sci 21, 1309–1331 (2022). https://doi.org/10.1007/s43630-022-00186-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43630-022-00186-3