
Abstract
Protonic ceramic electrochemical cells, a promising technology for energy conversion and storage, have garnered significant interest in recent years owing to their superior low-temperature (< 600 °C) performance relative to solid oxide electrochemical cells. However, the sluggish kinetics of oxygen electrodes have impeded further advancements. Despite considerable research efforts, the development of practically applicable oxygen electrodes remains challenging. We herein review the recent research focusing on the fundamental understanding and development of oxygen electrode materials. Furthermore, we provide a range of material design strategies for enhancing the catalytic activity of oxygen electrodes along with a concise overview of potential derivative applications. Finally, the perspectives and potential directions for the development of oxygen electrodes for high-performance protonic ceramic electrochemical cells are presented.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Growing energy needs and deteriorating environmental circumstances necessitate the establishment of a sustainable society that relies on renewable energy sources. Hydrogen, in particular, plays a pivotal role in bridging the gap between various contemporary and prospective energy sources. In this regard, solid oxide electrochemical cells (SOECs) have garnered significant attention in recent decades as environmentally friendly electrochemical devices capable of both power generation and hydrogen production through a bidirectional operation [1,2,3,4,5,6]. Depending on the type of ion conducted through the electrolyte, two categories of SOECs have emerged: oxygen ion-conducting SOECs and proton-conducting electrochemical cells (PCECs). Although oxygen ion-conducting SOECs have traditionally been the primary focus of research, the development of promising proton-conducting electrolyte materials has boosted interest in PCECs. Conventional SOECs require high operating temperatures (> 700 °C) because of the low oxygen ion conductivity of the electrolyte materials at low operating temperatures. However, owing to the higher mobility of protons compared to oxygen ions, proton-conducting fuel cells (PCFCs) can address the limitations of SOECs and are therefore deemed more suitable for operation at lower temperatures (below 700 °C) [7]. Operation at lower temperatures can enhance the mechanical and chemical stability of the materials, ultimately improving device reliability and potentially lowering manufacturing costs [8,9,10,11,12]. Moreover, in the fuel cell (FC) mode, PCECs are advantageous over oxygen ion-conducting SOECs because of the absence of fuel dilution at the fuel electrode; this is attributable to the water formation at the oxygen electrode during operation. These advantages have recently motivated intensive research on PCECs.
The development of materials that directly affect the performance and stability of PCECs is a crucial research topic. Among various components, oxygen electrode materials have been widely studied. The reactions occurring at the oxygen electrode determine the rate of the overall electrochemical reaction, thus exerting the most significant influence on the electrochemical performance of PCECs [13]. Consequently, the development of catalytically active oxygen electrode materials is a key challenge to be addressed for the future of PCECs. In conventional SOECs, mixed ion and electron conductors (MIECs) with perovskite structures, capable of simultaneously conducting oxygen ions and electrons (or holes), have been extensively applied as oxygen electrode materials to significantly expand the active sites for the oxygen reduction reaction (ORR) in the FC mode and the oxygen evolution reaction (OER) in the electrolysis (EC) mode. The electrochemical reactions of the oxygen electrode in SOECs are as follows:
However, at the oxygen electrode of the PCECs, the electrochemical reactions would involve further combinations with protons in both FC and EC modes:
The electrochemical reaction in PCEC involves the simultaneous participation of oxygen ions, proton, and electrons. Therefore, the electrochemical reaction, when utilizing a conventional oxygen electrode with only oxygen ion conductivity and electronic conductivity, is confined to the interface with the electrolyte which exhibits proton conductivity [14,15,16,17]. In this regard, studies have increasingly sought to elucidate protonic defects and proton conduction mechanisms in oxygen electrodes to extend the electrochemically active area to the entire surface [18,19,20]. Furthermore, novel approaches have been explored for tailoring PCEC oxygen electrodes.
In this review, we summarize the recent advances in oxygen electrode materials for PCECs. The key properties of the materials are outlined, along with methodologies, for an in-depth analysis. This review also encompasses studies that designed the chemical compositions of materials, developed composite oxygen electrodes with ionic conductors, and explored surface modifications and 3D structural designs. We aimed to provide comprehensive guidelines for the development of future oxygen electrodes and briefly introduce the potential applications stemming from these advancements.
2 Key properties of oxygen electrodes
The perovskite oxide family has been extensively investigated for its application as oxygen electrodes in various PCECs studies. As regards the simple perovskite-based materials (ABO3−δ), the A-sites typically comprise cations with oxidation states of + 2 or + 3, whereas the B-sites are primarily composed of cations with oxidation states of + 3 or + 4. Examples of such materials include BaCeO3 and BaZrO3 (Fig. 1a) [21, 22]. In the case of double perovskite-based materials (AA′B2O5+δ), lanthanides such as Pr, Nd, and Gd are commonly employed for the A cation, whereas cations such as Ba, Sr, and transition metals are used for the A′ cation. Representative instances encompass PrBaCO2O5+δ (PBC) [23], PrBa0.5Sr0.5Co1.5Fe0.5O5+δ (PBSCF) [24], NdBa0.5Sr0.5Co1.5Fe0.5O5+δ (NBSCF) [19], and GdBaCo2O5+δ (GBC) [25] (Fig. 1b). The chemical formula of the Ruddlesden–Popper phases is An+1BnO3n+1, where n denotes the number of octahedral layers within the perovskite layer [26,27,28]. Typically, La is employed for the A-sites, whereas Ni and Co are used for the B-sites (Fig. 1c). Recent studies have shown that nonperovskite oxygen electrode materials exhibit excellent performances. Park et al. reported a misfit-layered compound with the chemical composition LnxCa3−xCo4−yTryO9−δ (LCCTO, where Ln represents Gd, Nd, and La, and Tr denotes Fe, Mn, Ni, and Cu) [29, 30]. Figure 1d shows the crystal structure of Na0.15Ca2.85Co4O9–δ (NCCO) as one of the representatives. The misfit-layered structure exhibits a stacking structure comprising two transition metal oxide subsystems. Among the three nonequivalent oxygen sites, oxygen vacancy formation is the most favorable in the central Co–O layer of rock salt-type Ca2CoO3 layer compared to the side Ca–O layer of rock salt-type Ca2CoO3 layer or the CoO2 layer. NCCO exhibited higher catalytic performance compared to other misfit-layered compounds (Fig. 1e) which is attributed to the lowest oxygen vacancy formation energy in average.

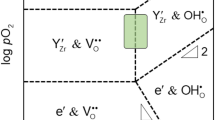
Copyright 2019, Elsevier. d Misfit-layered structure and e comparison of area-specific resistances depending of crystal structures. Reproduced with permission [30]. Copyright 2022, Wiley. f Scheme of effective reaction areas for PCFCs with O2−/e− conducting oxygen electrode, triple-conducting composite oxygen electrode, and triple-conducting single-phase oxygen electrode. Reproduced with permission [16]. Copyright 2022, Wiley. g Transport pathways in the B–O plane for protons, electron holes, and oxygen ions. h Conductivity of holes, protons, and oxygen in Ba0.85K0.15ZrO3−δ at 550 °C (stripped) and 700 °C (solid) under O2 and N2 atmospheres. The conductivity of the dominant carrier is emphasized by a black frame. Reproduced with permission [41]. Copyright 2020, Springer Nature
a Simple perovskite structure, b double perovskite structure, c Ruddlesden–Popper structure. Reproduced with permission [22].
While the primary focus of prior studies on materials for SOECs has been on developing MIECs that simultaneously possess electronic and oxygen ionic conductivities, current research endeavors aim to improve the catalytic performance by further enhancing the proton conductivity within PCECs. Materials capable of conducting electrons, oxygen, and protons within a single framework are referred to as triple-conducting oxides (TCOs), with proton conductivity significantly influencing the catalytic performance. Figure 1f shows the dependence of the number of catalytically active sites on the properties of the electrode material. For materials that conduct both oxygen and electrons, electrochemical reactions primarily occur at the electrolyte interface. When combined with a proton conductor, this occurs at the interface between the two materials. However, in the case of TCOs which facilitate the conductivity of oxygen, proton, and electron, the entire surface of the electrode actively participates in the electrochemical reactions. Therefore, the electrochemically active region is no longer confined to the interface between the oxygen electrode and the electrolyte (Farthest to the right in Fig. 1f). Consequently, ensuring reasonable conductivities for all three components under PCEC operating conditions becomes a critical challenge, although the contribution of each component can be highly sensitive to variations in the temperature or gas atmosphere (Fig. 1g, h).
3 Materials characterization
Intensive research efforts have been devoted to elucidating the critical properties of PCEC oxygen electrodes, namely, proton uptake and proton conduction. Three possible routes for proton uptake were proposed (Fig. 2a) [20].

Copyright 2019, American Chemical Society. b Oxygen nonstoichiometry as a function of temperature and c hydration properties under H2O–air and D2O–air at 400 °C of misfit-layered structures. Reproduced with permission [29]. Copyright 2021, RSC. d Time-resolved Raman analysis of surface functional groups on BaO, during the exposure to wet 50% propane (3%, H2O, 2% H2, 45% Ar) at 450 °C and e integrated peak intensities of Raman band corresponding to –OH and –CO3 groups as a function of gas exposure time. Reproduced with permission [34]. Copyright 2015, American Chemical Society. TOF-SIMS result of PBSCF bulk samples after heat treatments: f under dry air conditions at 500 °C and g under D2O–humidified air conditions at 300 °C. h 3D map of 2D distribution under D2O–humidified air conditions at 300 °C. Reproduced with permission [35]. Copyright 2022, RSC
a Three types of proton uptake mechanisms: hydration mechanism, hydrogenation mechanism, and new-type hydration mechanism. Reproduced with permission [20].
First, a hydration mechanism was suggested, in which water molecules decompose into hydroxide ions and protons (Eq. 5). Then, OH occupies the oxygen vacancy and forms proton bonds with the nearby lattice oxygen \(\left( {{\text{O}}^{2 - } } \right)\).
Second, the hydrogenation mechanism involves the occupation of protons at the expense of holes, releasing oxygen if the oxide materials have an appreciable hole concentration (Eq. 6). This indicates that oxide materials can incorporate protons in the absence of Vo.
Third, a new hydration reaction was proposed, in which the association of \({\text{V}}_{\text{O}}^{ \cdot \cdot }\) and water molecules is coupled with the carrier exchange reaction between the oxygen hole \(\left( {{\text{O}}_{\text{O}}^\cdot } \right)\) and metals (Eq. 7). The incorporation of water molecules into the oxide leads to the hydration and oxidation of adjacent cations, thereby replacing the Mn3+–\({\text{O}}^-\) configuration with Mn4+–\({\text{O}}^{2 - }\). This mechanism indicates that transition metal oxides with large amounts of \({\text{V}}_{\text{O}}^{ \cdot \cdot }\) and \({\text{O}}_{\text{O}}^\cdot\) carriers are promising candidates for TCO development.
For proton conduction, vehicle and Grotthuss mechanisms have been established for oxide materials [31,32,33]. The vehicle mechanism describes the movement of protons in conjunction with O2− ions acting as carriers, requiring relatively high activation energy owing to the lower mobility of O2−. In contrast, the Grotthuss mechanism involves protons repeatedly breaking and forming OH bonds between different O2− ions, resulting in relatively lower activation energy. Therefore, the Grotthuss mechanism is widely accepted in TCOs. Building on this foundational understanding, recent studies have reported efforts toward in-depth analyses of protonic defect formation and proton conduction. Additionally, efforts to characterize the surface reactions and stability in various atmospheres have been reported.
An iodometric titration method was employed to determine the oxygen nonstoichiometry of the oxygen electrode materials. Saqib et al. reported that misfit-layered structures generate numerous oxygen vacancies that play a vital role in their intrinsic OER/ORR activities [29]. Figure 2b shows that the oxygen nonstoichiometry of the misfit-layered structure such as Gd0.3Ca2.7Co3.82Cu0.18O9−δ (GCCCO) was remarkably larger than that of the BSCF, and the high oxygen deficiency of the misfit-layered structure was further confirmed via Raman spectroscopy. Figure 2c elucidates the hydration reaction of the structure using thermogravimetric analysis performed under a fixed H2O and D2O condition at 400 °C (approximately 0.017 atom fraction). Mass relaxation profiles exposed to either H2O or D2O appeared to saturate, with differences close to a mass ratio of D/H = 2, suggesting proton uptake and diffusion (formation of hydroxide defects). Li et al. investigated the coking resistance of BaO to gain insights into its detailed mechanism. Figure 2d shows the transformations of the surface functional groups of BaO during propane exposure, which were studied using in situ Raman spectroscopy [34]. The intensities of the key Raman bands are plotted as a function of the exposure time (Fig. 2e). BaO powders, when heated up initially, showed a strong Raman band at 3580 cm–1 associated with the − OH group vibrations. Following the introduction of propane, the − OH band on BaO slowly decreased in intensity, while the − CO3 band mildly increased in intensity before 1000 s. Between 1300 and 1500 s, abrupt intensity changes simultaneously occur for both the − OH and − CO3 Raman bands, resulting in the appearance of a strong − CO3 band and complete elimination of the − OH band. Secondary ion mass spectroscopy (SIMS) is a well-established technique for the direct detection of hydrogen with high resolution in terms of the amount and spatial distribution. Furthermore, SIMS provides the depth profiles of the species in combination with an ion milling instrument. Im et al. verified the proton solubility of PBSCF by employing two types of SIMS [35]. First, the depth-directional distribution of elements was examined using D-SIMS. Both 2D and 2DO species were simultaneously tracked to verify the presence of 2D in the bulk. In addition, 56Fe was collected to confirm whether the SIMS analyses of the dry air- and D2O-treated samples were carried out under identical conditions. After confirming the proton incorporation and even distribution of proton in PBSCF with D-SIMS, time-of-flight (TOF)-SIMS was additionally used (Fig. 2f, g) for the 3D reconstruction of 2D element with a size of 50 × 50 × 1 μm3 (Fig. 2h), indicating that the proton is not restrictedly distributed in specific regions such as the grain boundary but throughout the bulk.
Wang et al. introduced machine learning (ML) to efficiently screen the hydrated proton concentrations of 3200 oxides [36]. They also revealed that the cation radius, tolerance factor, and melting point of the ions are essential for guiding the design of TCOs. For the screened material with chemical composition of (La0.7Ca0.3)(Co0.8Ni0.2)O3 (LCCN7382), the hydration energy was calculated from the dehydrated (Fig. 3a) and the hydrated (Fig. 3b) models, revealing that the value of − 0.50 eV was less than or equal to those for other oxides such as BSCF. Furthermore, Fig. 3c shows the calculated barrier energy for proton transport (0.30 eV), which is comparable to those of BaCeO3, SrZrO3, and BaZrO3. Figure 3d shows the process of proton conduction in the LCCN7382, divided into initial state (IS), transition state (TS), and final state (FS). The reaction barriers of oxygen molecule for the three possible sites at Co–Co, Co–Ni, and Ni–Ni were studied (Fig. 3e); the results showed that Co–Co and Co–Ni sites are beneficial for the ORR and that Ni–Ni sites are beneficial for the ORR. Lu et al. studied the proton diffusion along the intraoctahedral direction in perovskite oxides (Fig. 3f) [37]. To investigate the effect of doping Nb5+ and Sc3+, the largest migration barriers based on minimum-energy path calculations were compared. The relatively small energy barrier in the co-doped model compared with that in the single-doped model expedited the performance improvement in the co-doped sample. Ren et al. calculated the effect of fluorine doping on O–H bonding, which plays a crucial role in proton transfer within bulk perovskite oxides [38]. Figure 3g shows the H adsorption energies of the pristine and fluorine-doped models. The fluorine-doped model showed weaker O–H bonding; this can be attributed to the negative inductive effect of F−, resulting in decreased negative charge density. This effect was also observed in the plot of the O–H bonding distance (Fig. 3h); the H site is closer to the MO6 octahedra than to the MO5F octahedra, suggesting a lower energy cost in the fluorinated system to remove protons from M–OH to the adjacent positions. Ciucci et al. provided rational guidelines for designing high-performance TCOs by optimizing their stability and reactivity [39]. Introducing p-type defects via A-site substitution was found to promote the formation of \({\text{V}}_{\text{O}}^{ \cdot \cdot }\), resulting in higher hydration ability but reduced structural stability. They showed that structural destabilization could be alleviated by strengthening the metal–oxygen bond by replacing iron with zirconium, yttrium, or tin in perovskite oxides. Figure 3i displays the formation energy, which is indicative of the material stability against \({\text{V}}_{\text{O}}^{ \cdot \cdot }\) formation and hydration energies. A-site-deficient BaFe0.875Y0.125O3 exhibited good stability and high performance. Figure 3j presents the ORR free energies computed as a function of \({\text{V}}_{\text{O}}^{ \cdot \cdot }\) and \({\text{OH}}_{\text{O}}^\cdot\) concentrations to understand their effects on ORR kinetics. The calculated overpotential suggests that the rate-determining step of the ORR strongly depends on the H+ availability (\({\text{V}}_{\text{O}}^{ \cdot \cdot }\) and \({\text{OH}}_{\text{O}}^\cdot\) concentrations).

Copyright 2022, Wiley. f Proton mobilities of the BCFNS-based perovskite oxides along the proton diffusion pathway (intra-octahedron direction) and minimum-energy paths. Reproduced with permission [37]. Copyright 2023, Elsevier. g H adsorption energy and h distance comparison for pristine and fluorinated system. Reproduced with permission [38]. Copyright 2022, Elsevier. i Plot of computed formation energy against oxygen vacancy formation energy and hydration energy for BaFeO3 and its derivative materials. j Standard ORR free energies computed at the Ba0.875Fe0.875Y0.125O3 surface. Reproduced with permission [39]. Copyright 2022, Springer Nature
Models of a dehydrated and b hydrated LCCN7382. c Proton-conducting path and d the IS, TS, and FS in LCCN7382. e Three reaction states for ORR and OER at Co–Ni sites. Reproduced with permission [36].
4 Materials design
4.1 Chemical doping
Although various crystal structures have been considered for oxygen electrodes, the chemical space has mostly been explored for perovskite oxides. Partial substitution of the A-, B-, and O-sites of the parent perovskite with other ions has been an efficient strategy to improve material properties such as ionic conductivity, electrical conductivity, thermal expansion, and chemical stability. In this section, we discuss the effects of doping and the underlying mechanisms that depend on the doping sites.
4.1.1 A-site doping
The A-site cations typically occupy 12-fold cuboctahedral coordinated positions, with a valence state of + 2 [40]. In most cases, the A-site cations have larger ionic radii than the B-site cations [16]. One promising approach to designing an oxygen electrode by using perovskite materials is to increase the basicity of the oxygen sites, which facilitates proton uptake [41]. This can be realized by incorporating dopant cations with lower electronegativities than the host ions. For example, Zohourian et al. investigated the proton concentration (CH) and oxygen vacancies in (Ba, Sr, La)FeO3−δ with variations of La, Sr contents at A-site (Fig. 4a) [42]. In the series of Ba1−xLaxFeO3−δ, the CH substantially decreases as La replaces Ba; this can be ascribed to a decrease in oxygen vacancy. However, the drastic decrease in CH was attributed to the replacement of Ba (electronegativity = 0.85) with La (electronegativity = 1.1), which decreased the basicity of the oxygen ion. Moreover, in the comparison between the CH of Ba0.95La0.05FeO2.53 and Ba0.5Sr0.5FeO2.5, increasing the Ba-to-Sr (electronegativity = 0.95) ratio contributed to enhanced proton uptake despite having similar amounts of oxygen vacancies, primarily owing to the increased material basicity.

Copyright 2018, Wiley. b Scheme of the hopping and rotating steps for proton migration. c EIS plots of the cell using BSCF and BKSCF oxygen electrodes tested at 600 °C (top) and 700 °C (bottom). Reproduced with permission [43]. Copyright 2019, RSC. d Proton migration features in SFMO derivatives. Energy profiles of the minimum-energy path for O − H···O jumps. Insets: corresponding minima and transition state structures. Turquoise, blue, and orange lines refer to proton transfer in a TM plane framed by two Sr (Sr/Sr), one Ba and one Sr (Ba/Sr), and one K and one Sr (K/Sr) atoms, respectively. Reproduced with permission [45]. Copyright 2016, American Chemical Society. e Schematic illustration of the charge compensation mechanism. f The calculated proton concentration of the BaxCo0.4Fe0.4Zr0.1Y0.1O3−δ (x = 1, 0.95, and 0.9) oxides at different temperatures. Reproduced with permission [46]. Copyright 2019, RSC. Energy barriers associated with g proton rotation and h proton hopping in three systems, BZY (BaZrO3): ideal structure with no A ion vacancies, BZY-1Ba: one A ion vacancy, and BZY-2Ba: two A ion vacancies. Reproduced with permission [47]. Copyright 2020, Elsevier. i EIS of PBSLCC and PBSC in wet air (3% H2O). j Comparisons on the area percentage of the lattice Sr and surface Sr contents of PBSLCC and PBSC samples after the same treatments in wet air for 600 h at 550 °C. k Temperature dependence of the polarization resistance of PBSLCC and other reported excellent electrodes. Reproduced with permission [52]. Copyright 2023, Wiley
a Proton concentration at 250 °C and \(p_{{\text{H}}_2 {\text{O}}} \approx 0.016\) atm for variation of La and Sr content on the perovskite’s A-site. Reproduced with permission [42].
Another approach is based on the fact that dopants with a large ionic radius at the A-site have the potential to enhance ionic conductivity. The mechanism underlying the increase in the ionic conductivity upon doping with larger cations is related to the principle of proton and oxygen ion conduction. The Grotthuss mechanism is widely recognized as the predominant proton transport mechanism in oxide materials [16]. Figure 4b illustrates the proton migration process, which includes both the hopping and rotation steps [43]. Similarly, oxygen ions diffused through thermally activated hopping, moving from one oxygen vacancy site to another [44]. Theoretically, A-site dopants with large ionic radii may increase the lattice parameter of the unit cell, and the enlarged lattice could offer more room for proton rotation, thereby reducing the rotating energy barrier [16]. In addition, the large ionic radii of the A-site dopants could contribute to achieving high symmetry (i.e., cubic) in the perovskite structure [40, 41]. The perovskite structure is known to increase the O–O bond length as it deviates from the cubic phase to other structures, such as orthorhombic and tetragonal. The reduced bond length enables the protons to hop shorter distances, resulting in a lower energy barrier associated with proton hopping. However, the perovskite structure with low symmetry, which deviates from cubic, prevents oxygen sites from having a uniform hydration enthalpy, which may decrease proton mobility [41]. Therefore, the high symmetry of the perovskite structure achieved by introducing A-site dopants with a high ionic radius could enhance the ionic conductivity. Xu et al. analyzed the effect of substituting K+ with Ba2+ in Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) through both experiment and density function theory (DFT) calculation [43]. The lattice parameter of Ba0.4K0.1Sr0.5Co0.8Fe0.2O3−δ (BKSCF) and BSCF was measured to be 3.985 Å and 3.967 Å, respectively, with the observed discrepancy attributed to the different ionic radius of K+ (164 pm) and Ba2+ (161 pm) at the 12-coordination. Furthermore, BKSCF exhibited weaker transition metal–oxygen bond strength than did BSCF, indicating higher oxygen ion activity in BKSCF. Interestingly, the rotation energy barrier for proton migration was lower in BKSCF, whereas the hopping energy barrier was lower in BSCF, as confirmed by DFT calculations. The results indicated that the distance between the oxygen ions may increase because of the expanded unit cell, and this may have an adverse effect on proton conduction. Nevertheless, Fig. 4c shows the large polarization resistance difference between the BSCF and BKSCF symmetric cells at 600 °C and 700 °C, respectively, indicating that dopants with a large ionic radius could increase the overall catalytic activity of oxygen electrode for PCECs. In contrast, Muñoz-García et al. reported that doping large ions into the A-site lowers the energy barrier associated with proton hopping [45]. Figure 4d shows variation of hopping energy barrier for proton migration of the Sr2Fe1.5Mo0.5O6−δ (SFMO), Ba0.25Sr1.75Fe1.5Mo0.5O6−δ (BSFMO), and K0.25Sr1.75Fe1.5Mo0.5O6−δ (KSFMO). BSFMO (0.322 eV) has a lower hopping energy barrier for proton migration compared to that of the SFMO (0.495 eV) because of the larger lattice constants resulting from substituting Sr2+ (144 pm) with Ba2+ (161 pm). Meanwhile, lattice constant of KSFMO (7.917 Å) is lower than BSFMO (7.948 Å) even though the ionic radius of K+ (164 pm) is larger than that of Ba2+ (161 pm). The results showed that the aliovalent substitution of K+ affects the electronic structure of Fe–O, resulting in a decrease in the Fe–O bond length, and, consequently, a reduction in the cell volume. In addition, the substitution of K+ changes the internal distortion pattern along the minimum-energy path allowing the proton in KSFMO to hop without an octahedral rotating motion of TM − O(H) − TM bond (Fig. 4d). Proton hopping without octahedral rotating motion elucidates the underlying reason why KSFMO (0.225 eV) exhibits the lowest energy barrier as compared to BSFMO (0.322 eV) and SFMO (0.495 eV).
Another interesting approach to designing oxygen electrodes for PCECs is based on A-site deficiency. Additional oxygen vacancies can be induced via A-site deficiency, which leads to enhanced proton uptake and oxygen ion conductivity [41]. Ren et al. studied the effects of A-site-deficient BCFZY (BaxCo0.4Fe0.4Zr0.1Y0.1O3−δ) [46]. Figure 4e illustrates two distinct mechanisms through which charge compensation may occur: (1) oxygen vacancy formation and (2) increasing the oxidation state of Co3+/Fe3+. XPS analysis confirmed that Ba deficiency has little effect on the oxidation state of Co and Fe in both Ba0.9Co0.4Fe0.4Zr0.1Y0.1O3−δ (B0.9CFZY) and Ba0.95Co0.4Fe0.4Zr0.1Y0.1O3−δ (B0.95CFZY) and that most of the charge compensation introduces the formation of oxygen vacancies. Moreover, thermogravimetric analysis indicated that Ba0.9CFZY exhibited the highest proton uptake ability (Fig. 4f). However, the A-site deficiency could increase the hopping and rotating energy barriers for proton migration, as shown in Fig. 4g, h [47]. The absence of A-site ions, on one hand, provides more extensive free space conducive to proton rotation. On the other hand, it strengthens the bond between oxygen ions and adjacent A-site ions. This increased bond strength impedes the rotating motion of protons and local structural deformation, thus promoting proton transfer. Therefore, the fact that A-site deficiency can enhance proton uptake but reduce proton mobility should be considered in the rational material design of oxygen electrodes for PCECs.
Recently, high-entropy materials (HEMs), such as Li-based batteries, water electrolysis/fuel cells, and solar cells, have received attention in the fields of energy storage and conversion [48]. HEMs such as high-entropy oxides, alloys, and other compounds are defined as materials comprising a solid solution of more than five components [49, 50]. HEMs, as single-phase materials composed of various elements, can exhibit unique catalytic properties, chemical stability, and phase stability [48]. High-entropy perovskite oxides (HEPOs) can be synthesized by introducing five or more distinct cations at either the A- or B-sites. Unfortunately, previously reported HEPO oxygen electrodes, which enhance the B-site configuration entropy for oxygen ion-conducting solid oxide cells, exhibit limited catalytic activity [51]. In contrast, He et al. synthesized Pr0.2Ba0.2Sr0.2La0.2Ca0.2CoO3−δ (PBSLCC) as HEPO oxygen electrodes for PCECs with increased A-site configuration entropy [52]. In wet air at 550 °C, a symmetric cell with PBSLCC exhibits a lower degradation rate than PrBa0.8Sr0.2Co2O3−δ (PBSC); this is suggested as a low-entropy oxygen electrode (Fig. 4i). Furthermore, Fig. 4j shows that surface Sr content of PBSLCC (27.1%) and PBSC (38.1%) after wet air treatment at 550 °C. The higher surface Sr content of PBSC compared with that of PBSLCC indicated that more Sr segregation occurred on the surface of PBSC. In addition to its high chemical stability, PBSLCC exhibited relatively lower area-specific resistances (ASRs) in BZCYYb-supported symmetric cells than most other advanced oxygen electrodes. (Fig. 4k).
4.1.2 B-site doping
B-site cations occupies sixfold coordinated positions and their typical valence states are + 3 and + 4 [16]. BO6 octahedra are generally acknowledged as active sites for various electrochemical reactions, and B-site cations are considered to be more involved in ionic and electronic conduction than A-site cations [40]. As aforementioned, the strategy for enhancing proton uptake by reducing the electronegativity of the cation is also applicable to B-site ion substitutions. Figure 5a shows that the lower average electronegativities of the A- and B-site cations tend to increase the proton uptake ability of the material [42]. However, reducing the electronegativity of the B-site cations does not always result in positive effects. For instance, Kim et al. investigated the electrochemical properties of BaCo1−xTaxO3−δ by varying the ratio of Ta (electronegativity = 1.5) ions to Co (Electronegativity = 1.88) ions [53, 54]. The Raman spectra in Fig. 5b show that excess Ta doping, as observed in BaCo0.6Ta0.4O3−δ (BCT40), hinders the perovskite breathing mode at approximately 670 cm−1, suggesting a reduction in mobile oxygen vacancies. Concurrently, the corresponding Raman spectra demonstrate the positive effect of doping with high-valence cations. The valence state of tantalum is fixed at + 5, and doping with high-valence cations can create an oxygen vacancy disorder that stabilizes the perovskite structure and contributes to the formation of high crystallographic symmetry in the perovskite [40, 54]. As shown in Fig. 5b, pure perovskite modes at around 530 and 670 cm−1 are observed only when Ta replaced at least 20 mol% of Co. Furthermore, among tested compositions, BCT20 (BaCo0.8Ta0.2O3−δ) exhibited the lowest polarization resistance in a symmetric cell supported by Sm0.2Ce0.8O2−δ (SDC) within the temperature range of 450–650 °C. As a result, BCT20 was regarded as the most catalytically active material among BaCo1−xTaxO3−δ (x = 0, 0.1, 0.2, 0.3, and 0.4). However, Fig. 5c shows that electrical conductivity of BaCo1−xTaxO3−δ (x = 0, 0.1, 0.2, 0.3, and 0.4) decreases as Co is substituted with Ta and electronic conductivity of BCT20 is 1–12 S cm−1 between 350 and 800 °C. Typically, the preference for PCEC oxygen electrodes is high electronic conductivity, approximately 100 S cm−1. However, under well-designed electrode morphology, particularly with a thin oxygen electrode film, a lower conductivity of 1 S cm−1 remains viable. Therefore, the electrical conductivity of most perovskite oxides is suitable for use as oxygen electrodes in PCFCs [40]. Notably, BCT20, with 20 mol% Ta substitution, exhibited the lowest activation energy in a protonic symmetric cell compared to other state-of-the-art oxygen electrodes reported to date (Fig. 5d).

Copyright 2018, Wiley. b Raman spectra of BCO, BCT10, BCT20, BCT30 and BCT40. c Electrical conductivities of BCO, BCT10, BCT20, BCT30, and BCT40 in the temperature range of 300–800 °C. d Performance comparison of BCT20 oxygen electrode relative to several benchmark oxygen electrodes measured in a protonic conductor-based symmetric cell configuration. Reproduced with permission [54]. Copyright 2023, RSI(e) The configure and O–O bond length for the O2 molecular adsorbed LSFMo surface. f EIS plots for the LSF cell and the LSFMo cell measured at 700 °C under the OCV condition. Reproduced with permission [55]. Copyright 2021, Elsevier. g O 1 s in XPS spectra for BCFZY and BCFZYM powders. h Comparison of NH3-TPD profiles for BCFZY and BCFZYM. i Arrhenius plots of the ASRs of BCFZY and BCFZYM electrodes from 450 to 650 °C in 5 vol% H2O air. Reproduced with permission [56]. Copyright 2022, Elsevier. j Schematic of fluorination inductive effect to promote the hydration reaction and the proton mobility in perovskite oxides. k TOF-SIMS depth profile of m/z = 2 (D) signal in D2O-treated BCFZY(F) samples. l Electrocatalytic activity of fluorinated BCFZY electrodes: typical Nyquist plots of symmetric BCFZY(F)|BZCYYb = BCFZY(F) cells at 600 °C. Reproduced with permission [38]. Copyright 2022, Elsevier
a ∆G°hydrat at 700 K versus average “ion electronegativity” of A- and B-site elements (assuming predominant Fe3+ and Co3+ oxidation state). The red dashed line corresponds to the correlation line where electrolyte materials are located. Reproduced with permission [42].
Furthermore, Xu et al. investigated the effects of substituting high-valence cations for the B-sites [55]. They confirmed that Mo ions (Mo5+/Mo6+) were successfully incorporated into the B-sites of La0.5Sr0.5FeO3−δ (LSF). According to DFT calculations, the O–O bond length of the O2 molecule adsorbed on the surface is larger for La0.5Sr0.5Fe0.9Mo0.1O3−δ (LSFMo) (126.2 pm) than LSF (125.1 pm) (Fig. 5e), implying that Mo doping further weakens the O–O bond. In addition, Mo doping influences the electronic structure of Fe such that electrons adjacent to Fe are injected into the adsorbed O2 molecule, reducing the O–O bond strength. Given that the breakdown of O2 molecules is a crucial stage in the ORR, LSFMo demonstrates superior catalytic activity compared to LSF. Moreover, XPS revealed a higher ratio of adsorbed oxygen to lattice oxygen in LSFMo (1.85) than in LSF (1.36), demonstrating the enhanced ORR capability of LSFMo and confirming its superior hydration ability with increased hydroxyl groups compared to LSF. As a result, LSFMO (0.056 Ω cm2) exhibits lower polarization resistance than LSF (0.12 Ω cm2) in half cells with NiO-BCZY anode and BZCY electrolyte at 700 °C (Fig. 5f).
In contrast, doping with low-valence cations such as Mg2+, Al2+/Al3+, and Ni2+/Ni3+ can modify the total metal oxidation state, increasing the number of oxygen vacancies, which may lead to improved ORR activity [56,57,58]. Liang et al. successfully incorporated 5 mol% Mg into the B-site of BCFZY despite the relatively large ionic radius of Mg2+ (0.74 Å), attributed to the small dopant amount and complete substitution of the A-site with the larger cation Ba2+ [56]. For Ba(Co0.4Fe0.4Zr0.1Y0.1)0.95Mg0.05O3−δ (BCFZYM), it was observed that the average oxidation state of Co increased slightly compared to BCFZY, while the average oxidation state of Fe remained at the same level. However, the total metal oxidation state decreased compared to that of BCFZY because of 5 mol% Mg doping. Figure 5g shows the difference in lattice oxygen (O2−) signal between BCFZY and BCFZYM, indicating an increased oxygen vacancy in BCFZYM. Furthermore, BCFZYM exhibited a higher signal of O22−/O− and OH−/O2 than did BCFZY, indicating that BCFZYM has more surface oxygen vacancies to promote the ORR/OER and improve the hydration ability, respectively. Figure 5h shows that BCFZYM exhibits an NH3 desorption peak at a lower temperature compared to BCFZY, indicating an enhanced hydration capability, which is consistent with the XPS results. Consequently, BCFZYM exhibited a lower polarization resistance and activation energy than did BCFZY in a symmetric cell with the BZCYYb electrolyte (Fig. 5i).
4.1.3 O-site doping
The oxygen sites in the perovskite structure can partake in proton, oxygen, electron conduction [40]. Substitution of the oxygen ions by monovalent anion such as Cl− and F− has recently been regarded as a promising approach to enhancing the catalytic activity of perovskite materials [41]. In addition, anion doping can reduce the spin of electrons belonging to transition metal ions, suppressing the thermal expansion of the material [40]. Zhang et al. synthesized BaFe0.6Co0.3Ce0.1O2.95−δCl0.05 (BFCC-Cl) by successfully incorporating 5 mol% Cl− into O-site of BaFe0.6Co0.3Ce0.1O3−δ (BFCC), and BFCC-Cl exhibits better electrochemical performance than BFCC [59]. Furthermore, Ren et al. reported BaCo0.4Fe0.4Zr0.1Y0.1O2.9−δ F0.1 (BCFZYF) with 10 mol% F− doped in BCFZY as an oxygen electrode [38]. Fluorination has several advantages owing to the higher electronegativity of F− than that of O2−. This enhances the polarity of M–\({\text{V}}_{\text{O}}^{ \cdot \cdot }\)–M, promoting H2O adsorption on the oxygen vacancies and concurrently reducing the electron density of the oxygen ions, thus weakening the proton–oxygen ion bonds and facilitating proton diffusion (Fig. 5j). In addition, a decrease in the negative charge of lattice oxygen can decrease the bond energy between the oxygen ions and adjacent cations, leading to increased oxygen mobility [60]. Figure 5k shows TOF-SIMS depth profiles of BCFZY and BCFZYF after D2O treatment at 500 °C. The consistent signal count of element D (m/z = 2) across the probing depths suggests a uniform distribution of protons throughout the bulk. Additionally, the increased D signal in BCFZYF relative to that in BCFZY signifies the enhanced hydration capacity of BCFZYF. As a result, BCFZYF exhibited the lower polarization resistance than BCFZY in BZCYYb-supported symmetric cell at 600 °C. However, monovalent anion dopants may induce cation vacancies and extinction of oxygen vacancies via a charge compensation mechanism, which could impede oxygen transport or hydration [41]. For instance, Belova et al. confirmed that the ionic conductivity decreases as the concentration of F− increases above a certain value because the reduction in protons and oxygen vacancies would dominate the effect of increased charge carrier mobility due to fluorination [61].
4.2 Composites
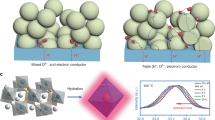
Approaches to developing mixtures of MIEC and proton-conducting oxides (PCO) have also been intensively studied [62,63,64]. As depicted in Fig. 6a, when employing only MIEC as an oxygen electrode for PCECs, the active sites are constrained to the interface between the electrolyte and the oxygen electrode [17]. However, when MIEC was mixed with PCO, the active sites extended inside the oxygen electrode layer (right side of Fig. 6a). Figure 6b shows that Sm0.5Sr0.5CoO3−δ (SSC)-BZCYYb composite oxygen electrode exhibits distinct improvement of cell performance over oxygen electrode using SSC only as MIEC. In the case of TCOs, the entire surface in contact with the gas phase can be an electrochemically active site (Fig. 6c). Therefore, the TCO-PCO composite electrodes seem to have fewer electrochemically active sites compared to the TCO electrodes. As a result, there was no significant enhancement in cell performance of PBSCF-BZCYYb as TCO-PCO composite oxygen electrode from 500 to 650 °C (Fig. 6d). Yang et al. investigated the effects of introducing a PBSCF-BZCYYb functional layer between the electrolyte and the oxygen electrode (Fig. 6e) [65]. Incorporating the functional layer led to a decreased activation energy and polarization resistance, consequently improving the peak power density within the low-temperature range (Fig. 6f, g). These results were attributed to the extension of the electrode–electrolyte interface, which enhanced the charge transfer reactions. In addition to designing a composite electrode by physically mixing TCO and PCO, composite electrodes in which the material phases spontaneously self-assemble into two phases (i.e., MIEC and PCO) at the nanoscale have also been reported. Song et al. successfully fabricated a self-assembled nanocomposite, combining BaCexYyCozO3−δ (P-BCCY) phase for H+/e− conduction with BaCoxCeyYzO3−δ (M-BCCY) and BaCoO3−δ (BC) phases for O2−/e− conduction through high-temperature calcination. Furthermore, they confirmed that the each phase is well mixed and in intimate contact at the nanoscale (Fig. 6h) [66]. Figure 6i shows that the nanocomposite BCCY exhibits higher Dchem and kchem value than BSCF and BCFZY. Moreover, it has lower ASR in BZCYYb-supported symmetric cell than BSCF and BCFZY (Fig. 6j).

Copyright 2022, Elsevier. e Ion conduction and surface/interface charge transfer reaction in BZCYYb-based PCFCs using a PBSCF oxygen electrode with PBSCF/BZCYYb functional layers (left) and cross-sectional SEM image of the PCFC sample with PBSCF/BZCYYb functional layers after cell performance measurements (right). f Comparison of polarization ASRs measured at the OCV from PCFCs with and without functional layers. g PPD comparison of the PCFCs with and without functional layers at 450–600 °C. Reproduced with permission [65]. Copyright 2021, Wiley. h STEM image of a BCCY composite particle. i The fitted values of Dchem and kchem of BCCY composite at temperature range of 500–650 °C from electronic conductivity relaxation curves. j Arrhenius plots of the ASRs of the BZCYYb-supported symmetrical cells with BCCY composite, BSCF and BCFZY oxygen electrodes at 400–650 °C in 5 vol% H2O air. Reproduced with permission [66]. Copyright 2019, Elsevier
a Schematic illustration of the electrochemically active site for oxygen electrode systems: MIEC (left) and MIEC + PCO composite (right). b PPD comparison of noncomposite (SSC) and composite (BZCYYb/SSC) oxygen electrodes based on BZCYYb using humidified (3 vol% H2O) H2 as fuel and dry air as oxidant from 650 to 500 °C. c Schematic illustration of the electrochemically active site for oxygen electrode systems: TIEC (left) and TIEC + PCO composite (right). d PPD comparison of noncomposite (PBSCF) and composite (BZCYYb/PBSCF) oxygen electrodes based on BZCYYb using humidified (3 vol% H2O) H2 as fuel and dry air as oxidant from 650 to 500 °C. Reproduced with permission [17].
4.3 Surface modification
4.3.1 Infiltration
Infiltration is an effective method for improving the catalytic activity of electrodes. During this process, the precursor solution infiltrated the porous scaffold. Subsequent heat treatment causes decomposition of the nitrate solution, leading to a uniform distribution of nanoscale catalysts across the surface of the scaffold [67]. This method creates a hierarchical nanostructure, expands the active triple-phase boundaries (TPBs), and enhances the kinetics of oxygen-related reactions. For example, a hybrid catalyst coating containing PrNi0.5Mn0.5O3 and exsolved PrOx particles on an LSCF oxygen electrode exhibited enhanced tolerance to contaminant poisoning (Sr segregation due to H2O) [68]. Zhou et al. deposited a proton-conducting material BCO onto an LSCF oxygen electrode via a one-step infiltration method (Fig. 7b) [69]. As shown in Fig. 7c, d, the degradation rate of the Rp was reduced from 1.3 × 10−3 for the bare cell to 4.1 × 10−4 Ω cm2 h−1 for the BCO-coated LSCF. During the long-term stability test, the evolution of the intermediate- and low-frequency arcs in the bare LSCF EIS increased significantly, owing to the deteriorated surface reaction activity in the steam environment. This degradation highlights the importance of the BCO catalyst coating, which primarily contributes to facilitating the rate of oxygen surface exchange. Likewise, multiphase (MP) catalyst coating composed of a conformal Pr1−xBaxCoO3−δ thin film and in situ exsolved BaCoO3−δ (BCO) nanoparticles was infiltrated on LSCF oxygen electrodes utilized in PCEC [70]. The MP catalyst coating enhanced the stability of LSCF in a high concentration of H2O and the catalytic activity, with a high peak power density of 1.04 W cm−2 in the fuel cell mode. In addition, the electrolyte can be used as a scaffold for oxygen electrode infiltration [13]. Bi et al. impregnated Sm0.5Sr0.5CoO3−δ (SSC) and PrBaCo2O5+x (PBCO) to the proton-conducting backbone reached to the electrolyte-interfacial area, extending the TPBs by using nanoparticles, providing more sites for ORR [71]. A triple-conductive material, BaCo0.4Fe0.4Zr0.1Y0.4O3–δ, was infiltrated into both the fuel and oxygen electrode sides, extending the reaction sites by enhancing the flow of oxygen ions, electrons, and protons within both electrodes. The infiltration of BCFZY in the electrodes led to a significant increase in power density, reaching 1.06 W cm−2 at 650 °C, nearly tripling the value observed in cells without BCFZY incorporation [72]. Pr6O11 infiltration into the proton-conducting electrolyte scaffold (La0.75Sr0.25)0.95MnO3±δ–BaZr0.85Y0.15O3−δ (LSM-BZY15) improved the electrochemical performance [73]. Analysis of the relaxation time distribution (DRT) confirmed that Pr6O11 significantly enhanced the ORR/OER process and diffusion of oxygen species into the TPB. As a result, the maximum power density reached 188 and 275 mW cm−2 at 550 °C and 600 °C, respectively, improving 141% and 96%, compared to the bare LSM-BZY15 electrode. In addition, NiO nanoparticles, which are neither oxygen electrodes nor proton-conducting materials but exhibit high adsorption capacity and fast dissociation ability for oxygen, were infiltrated into the BCFZY-BaZr0.1Ce0.7Y0.1Yb0.1O3−δ composite oxygen electrode layer to enlarge the active sites for surface exchange reaction kinetics without fading of TPB [74].

Copyright 2022, Wiley. b SEM images of the bare LSCF and BCO-LSCF oxygen electrode. c Time dependence of the electrode polarization resistance (Rp) of the symmetric cells with the bare LSCF and BCO-LSCF electrodes measured in humidified air at 600 °C under open-circuit conditions. d Nyquist plots of EIS of symmetrical cells with the bare LSCF and BCO-LSCF electrodes measured at different times in humidified air (3% H2O) at 600 °C under open-circuit conditions. Reproduced with permission [69]. Copyright 2021, Wiley. e Schematic diagram of water-mediated exsolution on triple-conducting oxides and HR-TEM image of water-mediated exsolved BCFZY-Ag. f Impedance spectra of BCFZY (top) and BCFZ-Ag (bottom) oxygen electrodes before and after the introduction of water (pH2O = 0.03 atm) in the air at 650 °C. g Arrhenius plot of the area-specific resistance of BCFZY and BCFZY-Ag oxygen electrodes at 450–650 °C. Reproduced with permission [77]. Copyright 2022, RSC. h HR-TEM image of BCFZYN-095. i EDX-mapping result of BCFZYN-095. j Dchem and kchem values (fitted) of BCFZYN-100 and BCFZYN-095 from 500 to 700 °C. Reproduced with permission [78]. Copyright 2022, Wiley
a Schematic of surface engineering methods. Reproduced with permission [7].
4.3.2 Exsolution
One solution is the self-precipitation of small, evenly dispersed metal nanoparticles on an oxide support via partial reduction [75]. An in situ exsolution of active metals and alloys on electrode surfaces presents several benefits, including the uniform distribution of nanoparticles and, notably, the generation of oxygen vacancies. These oxygen vacancies create extra active sites and facilitate oxygen ion diffusion, catalyzing the ORR kinetics [23]. Zhu et al. developed a highly active and robust Sr0.95Nb0.1Co0.9O3−δ oxygen electrode for low-temperature SOECs via Ag nanoparticle exsolution, which exhibited high activity for ORR and achieved a meager ASR (~ 0.214 Ω cm2 at 500 °C) [76]. Kim et al. reported an Ag-doped triple-conducting oxide of Ba0.95Ag0.05Co0.4Fe0.4Zr0.1Y0.1O3−δ (BCFZY-Ag) and demonstrated water-mediated exsolution during the electron hole-oriented proton uptake process from water vapor [77]. As shown in Fig. 7e, a schematic of the sequential steps of the water-mediated exsolution process is presented. High-resolution transmission electron microscopy (HR-TEM) showed that the dissolved Ag nanoparticles were anchored to the surface of the BCFZY powder after 1 vol% water treatment with air (pO2 = 0.21 atm, 550 °C). The Ag nanoparticles act as a catalytic core to boost the ORR and reshape the landscape of the triple-conducting oxide surface, leading to rapid surface oxygen exchange. This, in turn, reduced the polarization resistance and enabled a maximum power density of up to 1.20 W cm−2 at 650 °C (Fig. 7f, g). Alternatively, selective cation exsolution can be enabled by manipulating the nonstoichiometry of the A-site cations. Liang et al. suggested Ba0.95(Co0.4Fe0.4Zr0.1Y0.1)0.95Ni0.05O3−δ (BCFZYN-095) with a slight A-site cation deficiency [78]. As shown in Figs. 7h, i, upon calcination, the perovskite-based nanocomposite comprises a major perovskite phase (m-BCFZYN-095) and minor NiO nanoparticles agglomerated on the perovskite surface. The A-site deficiency and Ni cation exsolution significantly improved the oxygen surface exchange, bulk diffusion, and proton bulk conductivity of BCFZYN-095 (Fig. 7j). These enhancements led to superior cathodic performance, resulting in a peak power density of 1.04 W cm−2 achieved at 650 °C. In addition to single-element exsolution, Xu et al. introduced a double perovskite backbone, PrBaCo1.6Fe0.2Nb0.2O5+δ, covered with in situ dissolved nanoparticles of Nb-deficient PrBaCo1.6Fe0.2Nb0.2−xO5+δ. The heterostructured oxygen electrode, induced by steam-containing air, exhibited good durability in the fuel/electrolysis cell dual mode, sustaining ± 0.5 A cm−2 in cycling tests for 200 h, and demonstrated excellent catalytic activity for the ORR/OER, achieving a current density of 2.148 A cm−2 at 1.3 V in the electrolysis cell mode at 650 °C [79].
4.4 3D structure design
The innovative design of three-dimensional (3D) structures, such as fibers and meshes, opens new possibilities for maximizing the active sites of oxygen electrodes with reactants and enhancing their catalytic activity [80,81,82,83,84]. Electrospinning is the most widely used technique for nanofiber fabrication; it uses an electrostatic field formed between a syringe pump and a collector connected to a high-voltage power source [85]. The precursor solution was extruded from the tip of the needle and collected on a metal plate to form randomly oriented nanofiber mats. The as-spun nanofiber mats were heat-treated to obtain stoichiometric amounts of porous and continuous fiber [86]. Designing oxygen electrodes with 3D structures presents several challenges, including the difficulty in maintaining the original microstructure during oxygen electrode preparation because of processes such as mixing and grinding, which can change the morphology of the fibers [80]. It is also challenging to optimize the porosity and surface area during the sintering process to balance charge transfer and gas transport in MIECs [87, 88]. Lee et al. developed a fibrous La0.6Sr0.4Co0.2Fe0.8O3−δ-BaCe0.5Zr0.35Y0.15O3−δ (LSCF-BCZY) composite cathode. The combination of the increased surface area and enhanced ionic conductivity due to the embedded proton-conducting BCZY particles enables efficient charge transfer at the cathode, thereby improving the performance of PCFCs [86]. Umer et al. incorporated NiO nanoparticles onto the surfaces of LSCF nanofibers, resulting in enhanced connectivity within the oxygen electrode layer while maintaining the original microstructure [89]. Figure 8a schematically shows the increased number of contact points on the LSCF nanofiber owing to the uniform distribution of NiO nanoparticles in an optimized ratio. This incorporation created efficient pathways for charge transfer and enhanced the ORR activity of the oxygen electrode. Furthermore, because NiO does not undergo grain growth at the operating temperature owing to its high sintering temperature, the amount of NiO-LSCF-gas TPB remains constant as the LSCF nanofiber thickens, preventing the degradation of the ORR activity (Fig. 8b). A self-structured mesh framework was introduced alongside the fibrous microstructure to enhance mass transport within the porous frame. After immersing the woven fabric in a precursor solution and subjecting it to heat treatment, a self-architectured mesh structure consisting of hollow PNC fibers was created [90]. This mesh-structured PNC fiber electrode exhibited a 10% increase in current density and a 15.7% increase in peak power density at 600 °C, attributed to its ability to enhance gas diffusion. Similarly, Bian et al. prepared PBSCF 3D mesh-structured oxygen electrodes with different self-architecturing temperatures to adjust porosity and fiber morphology [91]. Figure 8c shows the bundle structure of well-knitted 3DECs. The hollow fiber structure and surrounding pores could be tailored by adjusting the calcination temperature. While higher porosity enhances mass transfer, excessive oxygen electrode porosity reduces the contact area between the electrode and electrolyte, thereby increasing the charge transfer resistance at the interface. The EC-800, with a porosity of 52.1%, achieved the lowest charge transfer resistance among the different calcination temperatures. As demonstrated by the significant reduction in polarization resistance in Fig. 8d, the optimized 3DEC’s porosity facilitates faster oxygen transfer, resulting in a 30% enhancement in peak power density at 600 °C operation.

Copyright 2023, American Chemical Society. c Schematic illustration of a full cell testing set-up and SEM image of 3D structured PBSCF. d EIS and PPD comparison among cells with PBSCF conventional and 3D structured oxygen electrode. Reproduced with permission [91]. Copyright 2021, Wiley
a Schematic of the dependences of NiO added in the LSCF nanofiber 3D mesh oxygen electrode. b Current density and power density of the cell with bare LSCF nanofiber and that with 15 wt% NiO-decorated LSCF nanofiber as a function of time. Reproduced with permission [89].
5 Applications in protonic ceramic electrochemical cells
5.1 Recent progress in the FC operation
In this section, we present the experimental results of PCECs employing a range of electrode materials, including single-phase MIECs, single-phase TIECs, and composite oxygen electrodes, which were discussed in previous sections. Comprehensive details of the performance data are shown in Fig. 9a and Table 1. Furthermore, the long-term durability test results, as depicted in Fig. 9c and Table 1, reveal that most PCFCs demonstrate a limited lifespan, typically extending to several hundred hours, highlighting the need for further investigation to enhance their durability under various fuel conditions.

Performance comparison of representative PCEC oxygen electrodes in a FC mode and b EC mode [3, 24, 25, 38, 56, 57, 63, 64, 66, 77, 78, 90, 106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131]. Long-term durability comparison of representative PCEC oxygen electrodes in c FC mode and d EC mode [3, 24, 25, 38, 56, 57, 63, 66, 77, 78, 90, 107,108,109,110,111,112,113,114,115,116,117,118,119,120,121, 123, 125, 126]
5.2 Recent progress in the EC operation
In this section, we summarize the latest findings on PCECs under EC operation and offer a brief overview of their key attributes and advancements in hydrogen generation. The EC performances of PCECs utilizing oxygen electrode materials, including various types of composites, are illustrated in Fig. 9b and Table 2. The long-term durability test results in the electrolysis cell mode are depicted in Fig. 9d and detailed in Table 2.
When comparing the electrochemical performance and long-term durability test results of the different oxygen electrode types, the single-phase TIEC materials consistently outperformed the composites (Fig. 9a–c). Therefore, the future direction of oxygen electrode development should prioritize the development of high-efficiency single-phase TIECs without inhomogeneous mixing of conductors.
6 Versatile gas utilization
PCECs offer significant advantages when utilizing a diverse range of gases [13, 92]. Notably, steam is generated from the oxygen electrode side, enabling high fuel utilization without diluting the fuel gas [93]. This enhances the overall fuel utilization efficiency when gases (e.g., CO2, CO, NH3, and etc.). When using ammonia as fuel, the ammonia decomposition reaction achieves full conversion at 500 °C under atmospheric pressure, making it highly feasible as a fuel source for PCECs at low temperatures [94, 95]. Furthermore, a lower operational temperature offers the potential to achieve the high-efficiency direct production of CH4 via CO2 decomposition [96]. This chapter explores the applications of PCECs utilizing different types of gases, which extend beyond oxygen electrode applications and encompass broader proton-conducting ceramic membranes.
Nanocatalysts have been proposed to improve the catalytic activity and stability of fuel electrode materials for NH3 and CO2 decomposition, utilizing nanocatalysts has been suggested. Zagoraios et al. and Kalaitzidou et al. enhanced the selectivity of CO2 hydrogenation to CH4 by depositing Ru nanoparticles on BZY electrolyte [97, 98]. By tuning of the Ir–O hybridization of the Ir–ceria-based catalysts, high selectivity (> 95%) for either CO or CH4 can be achieved [99]. Liu et al. recently developed an innovative electrocatalyst, Sr2Fe1.4Mo0.5O6−δ-Ni0.175 (SFM-Ni0.175), composed of in situ exsolved Ni–Fe alloyed nanoparticles for efficient CO2 to CO conversion [100]. In Fig. 10a, a unique fuel electrode with an oxide phase and in situ exsolved bimetallic nanoparticles significantly reduced overpotential and improved CO production rates, achieving nearly 100% CO selectivity. Ni–Fe alloys play a vital role in enhancing the electrochemical activity and maintaining structural stability. Figure 10b shows the alternation in NH3 adsorption strength and associative N2 desorption barrier heights attributed to the presence of the Ni–Fe alloy using DFT calculations [101]. The Fe modification accelerated the anodic reactions and reduced the Rp, consequently improving the maximum power densities from 1.398 to 1.609 W cm−2 at 700 °C. Similarly, incorporating a small amount of Pd doping in the electrolyte (Ba(Zr0.1Ce0.7Y0.1Yb0.1)0.95Pd0.05O3−δ) enhanced the catalytic activity of the fuel electrode, resulting in a maximum power density of 724 mW cm–2 at 650 °C operated in NH3, 160% higher than a similar cell without Pd incorporation [102].

Copyright 2022, Elsevier. b Minimum-energy pathways of the NH3 decomposition in Fe modified Ni-BZCYYb calculated by DFT calculations. Those in parentheses denote the NH3 decomposition on Ni(111). Reproduced with permission [101]. Copyright 2022, RSC. c Schematic illustration of PCFC and the mechanism of hydrocarbon reforming, WGS, sulfur, and carbon cleaning. d Current density–voltage–power density curves of PCFCs and their corresponding peak power densities, lifetime, and degradation rate for selected fuels under various conditions: hydrogen (fuel F1), ammonia (F2), methanol (F3), iso-butane (F4), n-butane (F5), simulated desulfurized natural gas (F6), simulated natural gas with 19.5 p.p.m. H2S (F7), propane (F8), methane (steam/carbon ratio S:C = 2, F9), methane (S:C = 2.5, F11), ethanol (F10), and iso-octane (F12) at 600 °C. Reproduced with permission [92]. Copyright 2018, Springer Nature. e Schematic illustration of the PCMR for hydrogen production, where the net endothermic chemical reaction is balanced with the heat evolved from the galvanic operation of the electrochemical cell, and heat transfer is illustrated in red. Reproduced with permission [104]. Copyright 2017, Springer Nature. f Schematic illustration of SMR, WGS, and ammonia synthesis in a PCMR. Reproduced with permission [105]. Copyright 2020, Elsevier
a Schematic illustration of the CO2-PCEC and CO production rate as a function of current density with SFM-Ni0.175 at 400–600 °C. Reproduced with permission [100].
Using a PCEC as a fuel decomposer, Duan et al. operated PCECs on 11 different fuel streams without modifying their composition or architecture [92]. As illustrated in Fig. 10c, the fuel electrode microstructure functioned as a location for carbon and sulfur mitigation. When OH(s) species are generated on the BaZr0.8Y0.2O3−δ (BZY surface, they have the capability to react with carbon that has adhered to the Ni, effectively preventing the formation of coke deposits. As a result, the well-dispersed Ni particles on the BZY support consistently demonstrated stable catalytic performance, even during prolonged operation with various hydrocarbon fuels, as shown in Fig. 10d. Pan et al. performed CO2-H2O co-electrolysis with a 5-cm2 large-area PCEC unit cell stack using BaCe0.4Zr0.4Y0.1Yb0.1O3−δ as electrolyte and BCFZY-BCZYYb composite as oxygen electrode, and achieved 71.2% CH4 yield ratio with H2 recycling [103].
Moreover, among the PCECs’ compartments, proton-conducting electrolytes can also function as fuel reformers, such as protonic ceramic membrane reactors (PCMRs), which handle a range of reactions reminiscent of those in a Haber–Bosch plant. Figure 10e presents a schematic diagram of a PCMR for high-purity hydrogen production from a steam methane reformer (SMR), featuring a single-stage process with minimal energy loss and yielding a nearly pure stream of CO2 as a valuable byproduct for efficient carbon capture [104]. Similarly, Kyriakou et al. successfully demonstrated the integration of these reactions in a single BaZrO3-based PCMR operating at atmospheric pressure [105]. Figure 10f illustrates the Ni-BaZr0.7Ce0.2Y0.1O3−δ (Ni-BZCY72) fuel electrode compartment, where the CH4-H2O mixture undergoes highly selective conversion, resulting in the simultaneous production of CO2 and H+. The latter is then transported to the VN-Fe oxygen electrode through the proton-conducting membrane. In VN, lattice N reacts with H+ to create NH3, whereas the N vacancy is continuously replenished by the dissociated N2 from the gas phase. Furthermore, when coupled with a PCFC, the byproduct hydrogen can be harnessed to produce electricity and purify the reactant nitrogen from the surrounding ambient air.
7 Summary and perspectives
To develop PCECs, research efforts have been directed toward improving the catalytic performance of oxygen electrodes by increasing the proton conductivity. To achieve this objective, various structures have been introduced, accompanied by advanced analytical methodologies for characterizing TCOs. In this context, substantial research effort has been focused on gaining a fundamental understanding of the crystal structure and properties of PCEC oxygen electrodes, particularly those related to proton absorption and conduction. Furthermore, cutting-edge technologies have been employed to effectively assess the hydrated proton concentration in materials, thus revealing crucial factors in the design of TCOs.
Several studies have explored the chemical space of various materials to address the challenges associated with the slow kinetics at the oxygen electrode in PCECs. Among the numerous approaches, an effective method involves the partial substitution of ions at the A-, B-, and O-sites within the parent perovskite, which enhances the intrinsic capability of the material as an oxygen electrode for PCECs. For example, introducing high-valence dopants at the B-site can notably augment both reactivity and stability, particularly at lower temperatures. Moreover, there has been a recent surge in interest in HEPOs as potential materials for oxygen electrodes owing to their enhanced stability. In light of the ongoing research on various dopant elements, to further advance the development of oxygen electrodes, integration with state-of-the-art technologies, such as computational calculations and ML, which can predict physical properties and suggest descriptors for the rational design of new compositions, is required.
Diverse oxygen electrode designs have been investigated using TCOs, MIECs, PCOs, and their variations. Although combining PCOs with MIECs as composite electrodes results in enhanced performance by extending their active sites, the TIEC-PCO composites appear to exhibit a diminished electrochemically active surface area in comparison with single TCO oxygen electrodes. Surface modifications (e.g., infiltration and exsolution) of oxygen electrodes with various types of catalysts have been widely employed to improve catalytic activity and stability. Furthermore, 3D structural oxygen electrodes have been suggested to increase surface exchange, ionic conduction kinetics, and gas diffusion. The 3D nanofiber and mesh-structured electrodes exhibited enhanced current and power densities. In addition, PCECs provide substantial advantages when employing diverse ranges of gases (e.g., NH3, CO2, and CO) because of their lower operating temperature (400–600 °C). Although the majority of PCECs in the literature deliver high performance in both the FC and EC modes (~ 1 W/cm2 of MPD and ~ 1 A/cm2 of current density (at 1.3 V) at 600 °C for FC and EC modes, respectively), their long-term stability needs to be improved for the reliable operation of PCECs.
Data availability
Data sharing not applicable—no new data generated.
References
E.D. Wachsman, K.T. Lee, Lowering the temperature of solid oxide fuel cells. Science 334, 935 (2011). https://doi.org/10.1126/science.1204090
H. An, H.-W. Lee, B.-K. Kim, J.-W. Son, K.J. Yoon, H. Kim, D. Shin, H.-I. Ji, J.-H. Lee, A 5 × 5 cm2 protonic ceramic fuel cell with a power density of 1.3 W cm–2 at 600 °C. Nat. Energy 3, 870 (2018). https://doi.org/10.1038/s41560-018-0230-0
D. Kim, K.T. Bae, K.J. Kim, H.-N. Im, S. Jang, S. Oh, S.W. Lee, T.H. Shin, K.T. Lee, High-performance protonic ceramic electrochemical cells. ACS Energy Lett. 7, 2393 (2022). https://doi.org/10.1021/acsenergylett.2c01370
J.H. Kim, D. Kim, S. Ahn, K.J. Kim, S. Jeon, D.-K. Lim, J.K. Kim, U. Kim, H.-N. Im, B. Koo, An universal oxygen electrode for reversible solid oxide electrochemical cells at reduced temperatures. Energy Environ. Sci. (2023). https://doi.org/10.1039/D2EE04108A
L. Mathur, Y. Namgung, H. Kim, S.-J. Song, Recent progress in electrolyte-supported solid oxide fuel cells: a review. J. Korean Ceram. Soc. 60, 614 (2023). https://doi.org/10.1007/s43207-023-00296-3
H. Bae, Y. Shin, L. Mathur, D. Lee, S.-J. Song, Defect chemistry of p-type perovskite oxide La0.2Sr0.8FeO3−δ: a combined experimental and computational study. J. Korean Ceram. Soc. 59, 876 (2022). https://doi.org/10.1007/s43207-022-00237-6
N. Tsvetkov, D. Kim, I. Jeong, J.H. Kim, S. Ahn, K.T. Lee, W. Jung, Advances in materials and interface understanding in protonic ceramic fuel cells. Adv. Mater. Technol. (2022). https://doi.org/10.1002/admt.202201075
J.H. Park, H.-N. Im, K.T. Lee, Understanding redox cycling behavior of Ni–YSZ anodes at 500 °C in solid oxide fuel cells by electrochemical impedance analysis. J. Korean Ceram. Soc. 58, 606 (2021). https://doi.org/10.1007/s43207-021-00136-2
K.T. Bae, I. Jeong, D. Kim, H. Yu, H.-N. Im, A. Akromjon, C.-W. Lee, K.T. Lee, Highly active cobalt-free perovskites with Bi doping as bifunctional oxygen electrodes for solid oxide cells. Chem. Eng. J. 461, 142051 (2023). https://doi.org/10.1016/j.cej.2023.142051
I. Jeong, S.J. Jeong, B.-H. Yun, J.-W. Lee, C.-W. Lee, W. Jung, K.T. Lee, Physically driven enhancement of the stability of Bi2O3-based ionic conductors via grain boundary engineering. NPG Asia Mater. 14, 53 (2022). https://doi.org/10.1038/s41427-022-00402-7
I. Jeong, C. Yeon, C.-W. Lee, K.T. Lee, Accurate and efficient prediction of highly disordered Bi2O3 with optimum structure pool: combined approach of the special quasirandom structure method and structure sampling. J. Phys. Chem. C 126, 18885 (2022). https://doi.org/10.1021/acs.jpcc.2c05757
H. Yu, I. Jeong, S. Jang, D. Kim, H.N. Im, C.W. Lee, E.D. Wachsman, K.T. Lee, Lowering the temperature of solid oxide electrochemical cells using triple-doped bismuth oxides. Adv. Mater. (2023). https://doi.org/10.1002/adma.202306205
C. Duan, J. Tong, M. Shang, S. Nikodemski, M. Sanders, S. Ricote, A. Almansoori, R. O’Hayre, Readily processed protonic ceramic fuel cells with high performance at low temperatures. Science 349, 1321 (2015). https://doi.org/10.1126/science.aab3987
Y. Bu, S. Joo, Y. Zhang, Y. Wang, D. Meng, X. Ge, G. Kim, A highly efficient composite cathode for proton-conducting solid oxide fuel cells. J. Power. Sources 451, 227812 (2020). https://doi.org/10.1016/j.jpowsour.2020.227812
H.-I. Ji, J.-H. Lee, J.-W. Son, K.J. Yoon, S. Yang, B.-K. Kim, Protonic ceramic electrolysis cells for fuel production: a brief review. J. Korean Ceram. Soc. 57, 480 (2020). https://doi.org/10.1007/s43207-020-00059-4
N. Wang, C. Tang, L. Du, R. Zhu, L. Xing, Z. Song, B. Yuan, L. Zhao, Y. Aoki, S. Ye, Advanced cathode materials for protonic ceramic fuel cells: recent progress and future perspectives. Adv. Energy Mater. 12, 2201882 (2022). https://doi.org/10.1002/aenm.202201882
A. Seong, D. Jeong, M. Kim, S. Choi, G. Kim, Performance comparison of composite cathode: Mixed ionic and electronic conductor and triple ionic and electronic conductor with BaZr0.1Ce0.7Y0.1Yb0.1O3−δ for highly efficient protonic ceramic fuel cells. J. Power. Sources 530, 231241 (2022). https://doi.org/10.1016/j.jpowsour.2022.231241
S. Im, J.-H. Lee, H.-I. Ji, PrBa0.5Sr0.5Co1.5Fe0.5O5+δ composite cathode in protonic ceramic fuel cells. J. Korean Ceram. Soc. 58, 351 (2021). https://doi.org/10.1007/s43207-021-00109-5
J.-I. Lee, K.-Y. Park, H. Park, H. Bae, M. Saqib, K. Park, J.-S. Shin, M. Jo, J. Kim, S.-J. Song, Triple perovskite structured Nd1.5Ba1.5CoFeMnO9−δ oxygen electrode materials for highly efficient and stable reversible protonic ceramic cells. J. Power. Sources 510, 230409 (2021). https://doi.org/10.1016/j.jpowsour.2021.230409
N. Wang, S. Hinokuma, T. Ina, H. Toriumi, M. Katayama, Y. Inada, C. Zhu, H. Habazaki, Y. Aoki, Incorporation of bulk proton carriers in cubic perovskite manganite driven by interplays of oxygen and manganese redox. Chem. Mater. 31, 8383 (2019). https://doi.org/10.1021/acs.chemmater.9b02131
D. Kim, I. Jeong, K.J. Kim, K.T. Bae, D. Kim, J. Koo, H. Yu, K.T. Lee, A brief review of heterostructure electrolytes for high-performance solid oxide fuel cells at reduced temperatures. J. Korean Ceram. Soc. (2022). https://doi.org/10.1007/s43207-021-00175-9
J. Kim, S. Sengodan, S. Kim, O. Kwon, Y. Bu, G. Kim, Proton conducting oxides: a review of materials and applications for renewable energy conversion and storage. Renew. Sustain. Energy Rev. 109, 606 (2019). https://doi.org/10.1016/j.rser.2019.04.042
K.T. Bae, I. Jeong, A. Akromjon, H.-N. Im, K.T. Lee, Robust and efficient Fe/Mn bimetal doped Pr4/3Ba2/3Co2/3Fe2/3Mn2/3O5+δ double perovskite catalysts for direct CO2 electrolysis. Chem. Eng. J. 472, 145015 (2023). https://doi.org/10.1016/j.cej.2023.145015
S. Choi, C.J. Kucharczyk, Y. Liang, X. Zhang, I. Takeuchi, H.-I. Ji, S.M. Haile, Exceptional power density and stability at intermediate temperatures in protonic ceramic fuel cells. Nat. Energy 3, 202 (2018). https://doi.org/10.1038/s41560-017-0085-9
J. Kim, S. Sengodan, G. Kwon, D. Ding, J. Shin, M. Liu, G. Kim, Triple-conducting layered perovskites as cathode materials for proton-conducting solid oxide fuel cells. Chemsuschem 7, 2811 (2014). https://doi.org/10.1002/cssc.201402351
K.J. Kim, M.K. Rath, H.H. Kwak, H.J. Kim, J.W. Han, S.-T. Hong, K.T. Lee, A highly active and redox-stable SrGdNi0.2Mn0.8O4±δ anode with in situ exsolution of nanocatalysts. ACS Catal. 9, 1172 (2019). https://doi.org/10.1021/acscatal.8b03669
D. Kim, K.T. Lee, Effect of lanthanide (Ln = La, Nd, and Pr) doping on electrochemical performance of Ln2NiO4+δ−YSZ composite cathodes for solid oxide fuel cells. Ceram. Int. 47, 2493 (2021). https://doi.org/10.1016/j.ceramint.2020.09.092
K.J. Kim, C. Lim, K.T. Bae, J.J. Lee, M.Y. Oh, H.J. Kim, H. Kim, G. Kim, T.H. Shin, J.W. Han, Concurrent promotion of phase transition and bimetallic nanocatalyst exsolution in perovskite oxides driven by Pd doping to achieve highly active bifunctional fuel electrodes for reversible solid oxide electrochemical cells. Appl. Catal. B 314, 121517 (2022). https://doi.org/10.1016/j.apcatb.2022.121517
M. Saqib, I.-G. Choi, H. Bae, K. Park, J.-S. Shin, Y.-D. Kim, J.-I. Lee, M. Jo, Y.-C. Kim, K.-S. Lee, Transition from perovskite to misfit-layered structure materials: a highly oxygen deficient and stable oxygen electrode catalyst. Energy Environ. Sci. 14, 2472 (2021). https://doi.org/10.1039/D0EE02799E
K. Park, H. Bae, H.K. Kim, I.G. Choi, M. Jo, G.M. Park, M. Asif, A. Bhardwaj, K.S. Lee, Y.C. Kim, Understanding the highly electrocatalytic active mixed triple conducting NaxCa3–xCo4O9–δ oxygen electrode materials. Adv. Energy Mater. 13, 2202999 (2023). https://doi.org/10.1002/aenm.202202999
K.D. Kreuer, A. Rabenau, W. Weppner, Vehicle mechanism, a new model for the interpretation of the conductivity of fast proton conductors. Angew. Chem. Int. Ed. Engl. 21, 208 (1982). https://doi.org/10.1002/anie.198202082
T. Ueki, M. Watanabe, Macromolecules in ionic liquids: progress, challenges, and opportunities. Macromolecules 41, 3739 (2008). https://doi.org/10.1021/ma800171k
K. Kreuer, Aspects of the formation and mobility of protonic charge carriers and the stability of perovskite-type oxides. Solid State Ionics 125, 285 (1999). https://doi.org/10.1016/S0167-2738(99)00188-5
X. Li, M. Liu, S.Y. Lai, D. Ding, M. Gong, J.-P. Lee, K.S. Blinn, Y. Bu, Z. Wang, L.A. Bottomley, In situ probing of the mechanisms of coking resistance on catalyst-modified anodes for solid oxide fuel cells. Chem. Mater. 27, 822 (2015). https://doi.org/10.1021/cm503852v
S. Im, M.A. Berk, S. Yang, B.-K. Kim, K.J. Yoon, J.-W. Son, J.-H. Lee, H.-I. Ji, The proton uptake process in double perovskite triple ionic–electronic conducting oxides for protonic ceramic cells. J. Mater. Chem. A 10, 16127 (2022). https://doi.org/10.1039/D2TA03522G
N. Wang, B. Yuan, C. Tang, L. Du, R. Zhu, Y. Aoki, W. Wang, L. Xing, S. Ye, Machine-learning-accelerated development of efficient mixed protonic-electronic conducting oxides as the air electrodes for protonic ceramic cells. Adv. Mater. 34, 2203446 (2022). https://doi.org/10.1002/adma.202203446
C. Lu, R. Ren, Z. Zhu, G. Pan, G. Wang, C. Xu, J. Qiao, W. Sun, Q. Huang, H. Liang, BaCo0.4Fe0.4Nb0.1Sc0.1O3−δ perovskite oxide with super hydration capacity for a high-activity proton ceramic electrolytic cell oxygen electrode. Chem. Eng. J. 472, 14878 (2023). https://doi.org/10.1016/j.cej.2023.144878
R. Ren, X. Yu, Z. Wang, C. Xu, T. Song, W. Sun, J. Qiao, K. Sun, Fluorination inductive effect enables rapid bulk proton diffusion in BaCo0.4Fe0.4Zr0.1Y0.1O3−δ perovskite oxide for high-activity protonic ceramic fuel cell cathode. Appl. Catal. B Environ. 317, 121759 (2022). https://doi.org/10.1016/j.apcatb.2022.121759
Z. Wang, Y. Wang, J. Wang, Y. Song, M.J. Robson, A. Seong, M. Yang, Z. Zhang, A. Belotti, J. Liu, Rational design of perovskite ferrites as high-performance proton-conducting fuel cell cathodes. Nat. Catal. 5, 777 (2022). https://doi.org/10.1038/s41929-022-00829-9
J. Cao, Y. Ji, Z. Shao, Perovskites for protonic ceramic fuel cells: a review. Energy Environ. Sci. 15, 2200 (2022). https://doi.org/10.1039/D2EE00132B
M. Papac, V. Stevanović, A. Zakutayev, R. O’Hayre, Triple ionic–electronic conducting oxides for next-generation electrochemical devices. Nat. Mater. 20, 301 (2021). https://doi.org/10.1038/s41563-020-00854-8
R. Zohourian, R. Merkle, G. Raimondi, J. Maier, Mixed-conducting perovskites as cathode materials for protonic ceramic fuel cells: understanding the trends in proton uptake. Adv. Func. Mater. 28, 1801241 (2018). https://doi.org/10.1002/adfm.201801241
X. Xu, H. Wang, M. Fronzi, X. Wang, L. Bi, E. Traversa, Tailoring cations in a perovskite cathode for proton-conducting solid oxide fuel cells with high performance. J. Mater. Chem. A 7, 20624 (2019). https://doi.org/10.1039/C9TA05300J
S.J. Skinner, J.A. Kilner, Oxygen ion conductors. Mater. Today 6, 30 (2003). https://doi.org/10.1016/S1369-7021(03)00332-8
A.B. Munoz-Garcia, M. Pavone, First-principles design of new electrodes for proton-conducting solid-oxide electrochemical cells: A-site doped Sr2Fe1.5Mo0.5O6−δ perovskite. Chem. Mater. 28, 490 (2016). https://doi.org/10.1021/acs.chemmater.5b03262
R. Ren, Z. Wang, C. Xu, W. Sun, J. Qiao, D.W. Rooney, K. Sun, Tuning the defects of the triple conducting oxide BaCo0.4Fe0.4Zr0.1Y0.1O3−δ perovskite toward enhanced cathode activity of protonic ceramic fuel cells. J. Mater. Chem. A 7, 18365 (2019). https://doi.org/10.1039/C9TA04335G
Y. Jing, N.R. Aluru, The role of A-site ion on proton diffusion in perovskite oxides (ABO3). J. Power. Sources 445, 227327 (2020). https://doi.org/10.1016/j.jpowsour.2019.227327
A. Amiri, R. Shahbazian-Yassar, Recent progress of high-entropy materials for energy storage and conversion. J. Mater. Chem. A. 9, 782 (2021). https://doi.org/10.1039/D0TA09578H
M. Fu, X. Ma, K. Zhao, X. Li, D. Su, High-entropy materials for energy-related applications. Iscience (2021). https://doi.org/10.1016/j.isci.2021.102177
S. Dai, Across the board: sheng Dai on catalyst design by entropic factors. Chemsuschem 13, 1915 (2020). https://doi.org/10.1002/cssc.202000448
Z. Liu, Z. Tang, Y. Song, G. Yang, W. Qian, M. Yang, Y. Zhu, R. Ran, W. Wang, W. Zhou, High-entropy perovskite oxide: a new opportunity for developing highly active and durable air electrode for reversible protonic ceramic electrochemical cells. Nanomicro Lett. 14, 217 (2022). https://doi.org/10.1007/s40820-022-00967-6
F. He, Y. Zhou, T. Hu, Y. Xu, M. Hou, F. Zhu, D. Liu, H. Zhang, K. Xu, M. Liu, An efficient high-entropy perovskite-type air electrode for reversible oxygen reduction and water splitting in protonic ceramic cells. Adv. Mater. 35, 2209469 (2023). https://doi.org/10.1002/adma.202209469
C. Sun, Y. Kong, L. Shao, Q. Zhang, X. Wu, N. Zhang, K. Sun, Significant zirconium substitution effect on the oxygen reduction activity of the cathode material NdBaCo2O5+δ for solid oxide fuel cells. ACS Sustain. Chem. Eng. 7, 11603 (2019). https://doi.org/10.1021/acssuschemeng.9b01486
J.H. Kim, D. Kim, S. Ahn, K.J. Kim, S. Jeon, D.-K. Lim, J.K. Kim, U. Kim, H.-N. Im, B. Koo, K.T. Lee, W. Jung, An universal oxygen electrode for reversible solid oxide electrochemical cells at reduced temperatures. Energy Environ. Sci. (2023). https://doi.org/10.1039/D2EE04108A
X. Xu, Y. Xu, J. Ma, Y. Yin, M. Fronzi, X. Wang, L. Bi, Tailoring electronic structure of perovskite cathode for proton-conducting solid oxide fuel cells with high performance. J. Power. Sources 489, 229486 (2021). https://doi.org/10.1016/j.jpowsour.2021.229486
M. Liang, Y. Song, D. Liu, L. Xu, M. Xu, G. Yang, W. Wang, W. Zhou, R. Ran, Z. Shao, Magnesium tuned triple conductivity and bifunctionality of BaCo0.4Fe0.4Zr0.1Y0.1O3−δ perovskite towards reversible protonic ceramic electrochemical cells. Appl. Catal. B Environ. 318, 121868 (2022). https://doi.org/10.1016/j.apcatb.2022.121868
M. Liang, F. He, C. Zhou, Y. Chen, R. Ran, G. Yang, W. Zhou, Z. Shao, Nickel-doped BaCo0.4Fe0.4Zr0.1Y0.1O3−δ as a new high-performance cathode for both oxygen-ion and proton conducting fuel cells. Chem. Eng. J. 420, 127717 (2021). https://doi.org/10.1016/j.cej.2020.127717
Y. Lu, M.Y. Shah, B.S. Almutairi, N. Mushtaq, M. Yousaf, N. Akbar, N. Arshad, M.S. Irshad, Y. Dong, Designing highly active core/shell cathode materials for low-temperature PCFCs. J. Alloys Compd. (2023). https://doi.org/10.1016/j.jallcom.2023.170861
L. Zhang, S. Yang, S. Zhang, A novel perovskite oxychloride as a high performance cathode for protonic ceramic fuel cells. J. Power. Sources 440, 227125 (2019). https://doi.org/10.1016/j.jpowsour.2019.227125
Z. Zhang, Y. Zhu, Y. Zhong, W. Zhou, Z. Shao, Anion doping: a new strategy for developing high-performance perovskite-type cathode materials of solid oxide fuel cells. Adv. Energy Mater. 7, 1700242 (2017). https://doi.org/10.1002/aenm.201700242
K. Belova, S. Baskakova, C. Argirusis, I. Animitsa, The effect of F−-doping on the conductivity of proton conductor Ba4Ca2Nb2O11. Electrochim. Acta 193, 63 (2016). https://doi.org/10.1016/j.electacta.2016.02.052
J. Seo, H.-W. Kim, J.H. Yu, H.J. Park, Electrochemical properties of Ba0.5Sr0.5Co0.8Fe0.2O3 and BaZr0.65Ce0.20Y0.15O3 composite cathodes on Y-doped barium–cerium–zirconium oxide solid electrolyte. J. Korean Ceram. Soc. 59, 217 (2022). https://doi.org/10.1007/s43207-021-00169-7
Z. Liu, Y. Chen, G. Yang, M. Yang, R. Ji, Y. Song, R. Ran, W. Zhou, Z. Shao, One-pot derived thermodynamically quasi-stable triple conducting nanocomposite as robust bifunctional air electrode for reversible protonic ceramic cells. Appl. Catal. B 319, 121929 (2022). https://doi.org/10.1016/j.apcatb.2022.121929
L. Bi, S.P. Shafi, E. Traversa, Y-doped BaZrO3 as a chemically stable electrolyte for proton-conducting solid oxide electrolysis cells (SOECs). J. Mater. Chem. A. 3, 5815 (2015). https://doi.org/10.1039/C4TA07202B
S.J. Yang, W. Chang, H.J. Jeong, D.H. Kim, J.H. Shim, High-performance protonic ceramic fuel cells with electrode–electrolyte composite cathode functional layers. Int. J. Energy Res. 46, 6553 (2022). https://doi.org/10.1002/er.7591
Y. Song, Y. Chen, W. Wang, C. Zhou, Y. Zhong, G. Yang, W. Zhou, M. Liu, Z. Shao, Self-assembled triple-conducting nanocomposite as a superior protonic ceramic fuel cell cathode. Joule 3, 2842 (2019). https://doi.org/10.1016/j.joule.2019.07.004
K.J. Kim, I. Thaheem, I. Jeong, H. Yu, J.H. Park, K.T. Lee, Nanostructured spinel Mn1.3Co1.3Cu0.4O4 as a bifunctional electrocatalyst for high-performance solid oxide electrochemical cells at intermediate temperatures. J. Power. Sources 539, 231611 (2022). https://doi.org/10.1016/j.jpowsour.2022.231611
Y. Chen, S. Yoo, X. Li, D. Ding, K. Pei, D. Chen, Y. Ding, B. Zhao, R. Murphy, B. DeGlee, J. Liu, M. Liu, An effective strategy to enhancing tolerance to contaminants poisoning of solid oxide fuel cell cathodes. Nano Energy 47, 474 (2018). https://doi.org/10.1016/j.nanoen.2018.03.043
Y. Zhou, W. Zhang, N. Kane, Z. Luo, K. Pei, K. Sasaki, Y. Choi, Y. Chen, D. Ding, M. Liu, An efficient bifunctional air electrode for reversible protonic ceramic electrochemical cells. Adv. Func. Mater. 31, 2105386 (2021). https://doi.org/10.1002/adfm.202105386
Y. Niu, Y. Zhou, W. Zhang, Y. Zhang, C. Evans, Z. Luo, N. Kane, Y. Ding, Y. Chen, X. Guo, W. Lv, M. Liu, Highly active and durable air electrodes for reversible protonic ceramic electrochemical cells enabled by an efficient bifunctional catalyst. Adv. Energy Mater. 12, 2103783 (2022). https://doi.org/10.1002/aenm.202103783
L. Bi, S.P. Shafi, E.H. Da’as, E. Traversa, Tailoring the cathode-electrolyte interface with nanoparticles for boosting the solid oxide fuel cell performance of chemically stable proton-conducting electrolytes. Small 14, 1801231 (2018). https://doi.org/10.1002/smll.201801231
C. Kim, H. Lee, I. Jang, S. Kim, H. Jung, M. Ryu, J. Kim, D. Lee, H. Yoon, U. Paik, T. Song, BaCo0.4Fe0.4Zr0.1Y0.1O3−δ triple conductor for boosting electrode efficiency for proton conducting fuel cells. Int. J. Hydrog. Energy 47, 5499 (2022). https://doi.org/10.1016/j.ijhydene.2021.11.140
K. Sun, Z. Yu, Q. Ni, Y. Li, D. Xu, Y. Gu, Y. Zheng, H. Chen, L. Ge, L. Guo, Highly durable Sr-doped LaMnO3-based cathode modified with Pr6O11 nano-catalyst for protonic ceramic fuel cells based on Y-doped BaZrO3 electrolyte. J. Eur. Ceram. Soc. 42, 4266 (2022). https://doi.org/10.1016/j.jeurceramsoc.2022.04.008
H. Lee, H. Jung, C. Kim, S. Kim, I. Jang, H. Yoon, U. Paik, T. Song, Enhanced electrochemical performance and durability of the BaCo0.4Fe0.4Zr0.1Y0.1O3−δ composite cathode of protonic ceramic fuel cells via forming nickel oxide nanoparticles. ACS Appl. Energy Mater. 4, 11564 (2021). https://doi.org/10.1021/acsaem.1c02311
D.-H. Kim, J.K. Kim, D. Oh, S. Park, Y.B. Kim, J. Ko, W. Jung, I.-D. Kim, Ex-solution hybrids functionalized on oxide nanofibers for highly active and durable catalytic materials. ACS Nano 17, 5842 (2023). https://doi.org/10.1021/acsnano.2c12580
Y. Zhu, W. Zhou, R. Ran, Y. Chen, Z. Shao, M. Liu, Promotion of oxygen reduction by exsolved silver nanoparticles on a perovskite scaffold for low-temperature solid oxide fuel cells. Nano Lett. 16, 512 (2016). https://doi.org/10.1021/acs.nanolett.5b04160
J.H. Kim, J. Hong, D.-K. Lim, S. Ahn, J. Kim, J.K. Kim, D. Oh, S. Jeon, S.-J. Song, W. Jung, Water as a hole-predatory instrument to create metal nanoparticles on triple-conducting oxides. Energy Environ. Sci. 15, 1097 (2022). https://doi.org/10.1039/D1EE03046A
M. Liang, Y. Zhu, Y. Song, D. Guan, Z. Luo, G. Yang, S.P. Jiang, W. Zhou, R. Ran, Z. Shao, A new durable surface nanoparticles-modified perovskite cathode for protonic ceramic fuel cells from selective cation exsolution under oxidizing atmosphere. Adv. Mater. 34, 2106379 (2022). https://doi.org/10.1002/adma.202106379
K. Xu, H. Zhang, Y. Xu, F. He, Y. Zhou, Y. Pan, J. Ma, B. Zhao, W. Yuan, Y. Chen, M. Liu, An efficient steam-induced heterostructured air electrode for protonic ceramic electrochemical cells. Adv. Func. Mater. 32, 2110998 (2022). https://doi.org/10.1002/adfm.202110998
Y. Chen, Y. Bu, Y. Zhang, R. Yan, D. Ding, B. Zhao, S. Yoo, D. Dang, R. Hu, C. Yang, M. Liu, A highly efficient and robust nanofiber cathode for solid oxide fuel cells. Adv. Energy Mater. 7, 1601890 (2017). https://doi.org/10.1002/aenm.201601890
W. Zhang, H. Wang, K. Guan, Z. Wei, X. Zhang, J. Meng, X. Liu, J. Meng, La0.6Sr0.4Co0.2Fe0.8O3−δ/CeO2 heterostructured composite nanofibers as a highly active and robust cathode catalyst for solid oxide fuel cells. ACS Appl. Mater. Interfaces 11, 26830 (2019). https://doi.org/10.1021/acsami.9b06668
M. Zhi, S. Lee, N. Miller, N.H. Menzler, N. Wu, An intermediate-temperature solid oxide fuel cell with electrospun nanofiber cathode. Energy Environ. Sci. 5, 7066 (2012). https://doi.org/10.1039/C2EE02619H
J.C. Ruiz-Morales, D. Marrero-López, J. Peña-Martínez, J. Canales-Vázquez, J.J. Roa, M. Segarra, S.N. Savvin, P. Núñez, Performance of a novel type of electrolyte-supported solid oxide fuel cell with honeycomb structure. J. Power. Sources 195, 516 (2010). https://doi.org/10.1016/j.jpowsour.2009.08.017
J.C. Ruiz-Morales, J. Peña-Martínez, J. Canales-Vázquez, D. Marrero-López, C. Savaniu, P. Núñez, Cost-effective microstructural engineering of solid oxide fuel cell components for planar and tubular designs. J. Am. Ceram. Soc. 92, 276 (2009). https://doi.org/10.1111/j.1551-2916.2008.02857.x
H. Tang, Z. Jin, Y. Wu, W. Liu, L. Bi, Cobalt-free nanofiber cathodes for proton conducting solid oxide fuel cells. Electrochem. Commun. 100, 108 (2019). https://doi.org/10.1016/j.elecom.2019.01.022
S. Lee, S. Park, S. Wee, H.W. Baek, D. Shin, One-dimensional structured La0.6Sr0.4Co0.2Fe0.8O3−δ–BaCe0.5Zr0.35Y0.15O3−δ composite cathode for protonic ceramic fuel cells. Solid State Ionics 320, 347 (2018). https://doi.org/10.1016/j.ssi.2018.03.010
W. Pan, Z. Lü, K. Chen, X. Huang, B. Wei, W. Li, Z. Wang, W. Su, Novel polymer fibers prepared by electrospinning for use as the pore-former for the anode of solid oxide fuel cell. Electrochim. Acta 55, 5538 (2010). https://doi.org/10.1016/j.electacta.2010.04.037
H. Jeong, B. Sharma, S. Jo, Y.H. Kim, J.H. Myung, Electrochemical characteristics of La0.8Sr0.2MnO3 (LSM)–scandia-stabilized zirconia (ScSZ) composite cathode. J. Korean Ceram. Soc. 59, 473 (2022). https://doi.org/10.1007/s43207-022-00200-5
M.A. Umer, Y.-C. Yu, K.-R. Lee, C.-J. Tseng, S.-Y. Chen, S.-W. Lee, Three-dimensional NiO-decorated La0.6Sr0.4Co0.2Fe0.8O3−δ nanofibrous mesh as high-performance cathode for protonic ceramic electrochemical cells. ACS Appl. Energy Mater. 6, 1621 (2023). https://doi.org/10.1021/acsaem.2c03498
H. Ding, W. Wu, C. Jiang, Y. Ding, W. Bian, B. Hu, P. Singh, C.J. Orme, L. Wang, Y. Zhang, D. Ding, Self-sustainable protonic ceramic electrochemical cells using a triple conducting electrode for hydrogen and power production. Nat. Commun. 11, 1907 (2020). https://doi.org/10.1038/s41467-020-15677-z
W. Bian, W. Wu, Y. Gao, J.Y. Gomez, H. Ding, W. Tang, M. Zhou, D. Ding, Regulation of cathode mass and charge transfer by structural 3D engineering for protonic ceramic fuel cell at 400 °C. Adv. Func. Mater. 31, 2102907 (2021). https://doi.org/10.1002/adfm.202102907
C. Duan, R.J. Kee, H. Zhu, C. Karakaya, Y. Chen, S. Ricote, A. Jarry, E.J. Crumlin, D. Hook, R. Braun, N.P. Sullivan, R. O’Hayre, Highly durable, coking and sulfur tolerant, fuel-flexible protonic ceramic fuel cells. Nature 557, 217 (2018). https://doi.org/10.1038/s41586-018-0082-6
Y. Zhang, R. Knibbe, J. Sunarso, Y. Zhong, W. Zhou, Z. Shao, Z. Zhu, Recent progress on advanced materials for solid-oxide fuel cells operating below 500 °C. Adv. Mater. 29, 1700132 (2017). https://doi.org/10.1002/adma.201700132
F. Ishak, I. Dincer, C. Zamfirescu, Energy and exergy analyses of direct ammonia solid oxide fuel cell integrated with gas turbine power cycle. J. Power. Sources 212, 73 (2012). https://doi.org/10.1016/j.jpowsour.2012.03.083
Y. Li, H. Wang, C. Priest, S. Li, P. Xu, G. Wu, Advanced electrocatalysis for energy and environmental sustainability via water and nitrogen reactions. Adv. Mater. 33, 2000381 (2021). https://doi.org/10.1002/adma.202000381
C. Vogt, M. Monai, G.J. Kramer, B.M. Weckhuysen, The renaissance of the Sabatier reaction and its applications on Earth and in space. Nat. Catal. 2, 188 (2019). https://doi.org/10.1038/s41929-019-0244-4
D. Zagoraios, C. Panaritis, A. Krassakopoulou, E.A. Baranova, A. Katsaounis, C.G. Vayenas, Electrochemical promotion of Ru nanoparticles deposited on a proton conductor electrolyte during CO2 hydrogenation. Appl. Catal. B 276, 119148 (2020). https://doi.org/10.1016/j.apcatb.2020.119148
I. Kalaitzidou, A. Katsaounis, T. Norby, C.G. Vayenas, Electrochemical promotion of the hydrogenation of CO2 on Ru deposited on a BZY proton conductor. J. Catal. 331, 98 (2015). https://doi.org/10.1016/j.jcat.2015.08.023
M. Li, B. Hua, L.-C. Wang, J.D. Sugar, W. Wu, Y. Ding, J. Li, D. Ding, Switching of metal–oxygen hybridization for selective CO2 electrohydrogenation under mild temperature and pressure. Nat. Catal. 4, 274 (2021). https://doi.org/10.1038/s41929-021-00590-5
F. Liu, L. Fang, D. Diercks, P. Kazempoor, C. Duan, Rationally designed negative electrode for selective CO2-to-CO conversion in protonic ceramic electrochemical cells. Nano Energy 102, 107722 (2022). https://doi.org/10.1016/j.nanoen.2022.107722
H. Zhang, Y. Zhou, K. Pei, Y. Pan, K. Xu, Y. Ding, B. Zhao, K. Sasaki, Y. Choi, Y. Chen, M. Liu, An efficient and durable anode for ammonia protonic ceramic fuel cells. Energy Environ. Sci. 15, 287 (2022). https://doi.org/10.1039/D1EE02158C
F. He, Q. Gao, Z. Liu, M. Yang, R. Ran, G. Yang, W. Wang, W. Zhou, Z. Shao, A new Pd doped proton conducting perovskite oxide with multiple functionalities for efficient and stable power generation from ammonia at reduced temperatures. Adv. Energy Mater. 11, 2003916 (2021). https://doi.org/10.1002/aenm.202003916
Z. Pan, C. Duan, T. Pritchard, A. Thatte, E. White, R. Braun, R. O’Hayre, N.P. Sullivan, High-yield electrochemical upgrading of CO2 into CH4 using large-area protonic ceramic electrolysis cells. Appl. Catal. B 307, 121196 (2022). https://doi.org/10.1016/j.apcatb.2022.121196
H. Malerød-Fjeld, D. Clark, I. Yuste-Tirados, R. Zanón, D. Catalán-Martinez, D. Beeaff, S.H. Morejudo, P.K. Vestre, T. Norby, R. Haugsrud, J.M. Serra, C. Kjølseth, Thermo-electrochemical production of compressed hydrogen from methane with near-zero energy loss. Nat. Energy 2, 923 (2017). https://doi.org/10.1038/s41560-017-0029-4
V. Kyriakou, I. Garagounis, A. Vourros, E. Vasileiou, M. Stoukides, An electrochemical Haber–Bosch process. Joule 4, 142 (2020). https://doi.org/10.1016/j.joule.2019.10.006
B. Lin, H. Ding, Y. Dong, S. Wang, X. Zhang, D. Fang, G. Meng, Intermediate-to-low temperature protonic ceramic membrane fuel cells with Ba0.5Sr0.5Co0.8Fe0.2O3−δ–BaZr0.1Ce0.7Y0.2O3−δ composite cathode. J. Power. Sources 186, 58 (2009). https://doi.org/10.1016/j.jpowsour.2008.09.041
G. Li, Y. Zhang, Y. Ling, B. He, J. Xu, L. Zhao, Probing novel triple phase conducting composite cathode for high performance protonic ceramic fuel cells. Int. J. Hydrog. Energy 41, 5074 (2016). https://doi.org/10.1016/j.ijhydene.2016.01.068
Z. Wang, W. Yang, S.P. Shafi, L. Bi, Z. Wang, R. Peng, C. Xia, W. Liu, Y. Lu, A high performance cathode for proton conducting solid oxide fuel cells. J. Mater. Chem. A 3, 8405 (2015). https://doi.org/10.1039/C5TA00391A
L. Zhao, B. He, Y. Ling, Z. Xun, R. Peng, G. Meng, X. Liu, Cobalt-free oxide Ba0.5Sr0.5Fe0.8Cu0.2O3−δ for proton-conducting solid oxide fuel cell cathode. Int. J. Hydrog. Energy 35, 3769 (2010). https://doi.org/10.1016/j.ijhydene.2010.01.039
M. Liu, J. Gao, X. Liu, G. Meng High performance of anode supported Ba(Zr0.1Ce0.7Y0.2)O3−δ (BZCY) electrolyte cell for IT-SOFC. Int. J. Hydrog. Energy 36, 13741 (2011). https://doi.org/10.1016/j.ijhydene.2011.07.087
L. Yang, Z. Liu, S. Wang, Y. Choi, C. Zuo, M. Liu, A mixed proton, oxygen ion, and electron conducting cathode for SOFCs based on oxide proton conductors. J. Power. Sources 195, 471 (2010). https://doi.org/10.1016/j.jpowsour.2009.07.057
Y. Zhou, E. Liu, Y. Chen, Y. Liu, L. Zhang, W. Zhang, Z. Luo, N. Kane, B. Zhao, L. Soule, Y. Niu, Y. Ding, H. Ding, D. Ding, M. Liu, An active and robust air electrode for reversible protonic ceramic electrochemical cells. ACS Energy Lett. 6, 1511 (2021). https://doi.org/10.1021/acsenergylett.1c00432
Z. Luo, Y. Zhou, X. Hu, W. Wang, Y. Ding, W. Zhang, T. Li, N. Kane, Z. Liu, M. Liu, A new class of proton conductors with dramatically enhanced stability and high conductivity for reversible solid oxide cells. Small 19, 2208064 (2023). https://doi.org/10.1002/smll.202208064
J.-I. Lee, K.-Y. Park, H. Park, H. Bae, M. Saqib, K. Park, J.-S. Shin, M. Jo, J. Kim, S.-J. Song, E.D. Wachsman, J.-Y. Park, Triple perovskite structured Nd1.5Ba1.5CoFeMnO9−δ oxygen electrode materials for highly efficient and stable reversible protonic ceramic cells. J. Power. Sources 510, 230409 (2021). https://doi.org/10.1016/j.jpowsour.2021.230409
J.H. Kim, D. Kim, S. Ahn, K.J. Kim, S. Jeon, D.-K. Lim, J.K. Kim, U. Kim, H.-N. Im, B. Koo, K.T. Lee, W. Jung, An universal oxygen electrode for reversible solid oxide electrochemical cells at reduced temperatures. Energy Environ. Sci. 16, 3803 (2023). https://doi.org/10.1039/D2EE04108A
J.H. Kim, J.K. Kim, H.G. Seo, D.-K. Lim, S.J. Jeong, J. Seo, J. Kim, W. Jung, Ex-solved Ag nanocatalysts on a Sr-free parent scaffold authorize a highly efficient route of oxygen reduction. Adv. Func. Mater. 30, 2001326 (2020). https://doi.org/10.1002/adfm.202001326
K. Bae, D.Y. Jang, H.J. Choi, D. Kim, J. Hong, B.-K. Kim, J.-H. Lee, J.-W. Son, J.H. Shim, Demonstrating the potential of yttrium-doped barium zirconate electrolyte for high-performance fuel cells. Nat. Commun. 8, 14553 (2017). https://doi.org/10.1038/ncomms14553
K. Park, H. Bae, H.-K. Kim, I.-G. Choi, M. Jo, G.-M. Park, M. Asif, A. Bhardwaj, K.-S. Lee, Y.-C. Kim, S.-J. Song, E.D. Wachsman, J.-Y. Park, Understanding the highly electrocatalytic active mixed triple conducting NaxCa3–xCo4O9–δ oxygen electrode materials. Adv. Energy Mater. 13, 2202999 (2023). https://doi.org/10.1002/aenm.202202999
M. Saqib, I.-G. Choi, H. Bae, K. Park, J.-S. Shin, Y.-D. Kim, J.-I. Lee, M. Jo, Y.-C. Kim, K.-S. Lee, S.-J. Song, E.D. Wachsman, J.-Y. Park, Transition from perovskite to misfit-layered structure materials: a highly oxygen deficient and stable oxygen electrode catalyst. Energy Environ. Sci. 14, 2472 (2021). https://doi.org/10.1039/D0EE02799E
J. Kim, A. Jun, O. Gwon, S. Yoo, M. Liu, J. Shin, T.-H. Lim, G. Kim, Hybrid-solid oxide electrolysis cell: a new strategy for efficient hydrogen production. Nano Energy 44, 121 (2018). https://doi.org/10.1016/j.nanoen.2017.11.074
H. Bai, Y. Zhang, J. Chu, Q. Zhou, H. Lan, J. Zhou, Oxygen electrode PrBa0.5Sr0.5Co1.5Fe0.5O5+δ–BaZr0.1Ce0.7Y0.1Yb0.1O3−δ with different composite proportions for proton-conducting solid oxide electrolysis cells. ACS Appl. Mater. Interfaces 15, 38581 (2023). https://doi.org/10.1021/acsami.3c07638
E. Vøllestad, R. Strandbakke, M. Tarach, D. Catalán-Martínez, M.-L. Fontaine, D. Beeaff, D.R. Clark, J.M. Serra, T. Norby, Mixed proton and electron conducting double perovskite anodes for stable and efficient tubular proton ceramic electrolysers. Nat. Mater. 18, 752 (2019). https://doi.org/10.1038/s41563-019-0388-2
S. Yang, S. Zhang, C. Sun, X. Ye, Z. Wen, Lattice incorporation of Cu2+ into the BaCe0.7Zr0.1Y0.1Yb0.1O3−δ electrolyte on boosting its sintering and proton-conducting abilities for reversible solid oxide cells. ACS Appl. Mater. Interfaces 10, 42387 (2018). https://doi.org/10.1021/acsami.8b15402
W. Li, B. Guan, L. Ma, S. Hu, N. Zhang, X. Liu, High performing triple-conductive Pr2NiO4+δ anode for proton-conducting steam solid oxide electrolysis cell. J. Mater. Chem. A 6, 18057 (2018). https://doi.org/10.1039/C8TA04018D
D. Huan, N. Shi, L. Zhang, W. Tan, Y. Xie, W. Wang, C. Xia, R. Peng, Y. Lu, New, efficient, and reliable air electrode material for proton-conducting reversible solid oxide cells. ACS Appl. Mater. Interfaces 10, 1761 (2018). https://doi.org/10.1021/acsami.7b16703
L. Lei, Z. Tao, X. Wang, J.P. Lemmon, F. Chen, Intermediate-temperature solid oxide electrolysis cells with thin proton-conducting electrolyte and a robust air electrode. J. Mater. Chem. A 5, 22945 (2017). https://doi.org/10.1039/C7TA05841A
J. Lyagaeva, N. Danilov, G. Vdovin, J. Bu, D. Medvedev, A. Demin, P. Tsiakaras, A new Dy-doped BaCeO3–BaZrO3 proton-conducting material as a promising electrolyte for reversible solid oxide fuel cells. J. Mater. Chem. A 4, 15390 (2016). https://doi.org/10.1039/C6TA06414K
C. Zuo, S. Zha, M. Liu, M. Hatano, M. Uchiyama, Ba(Zr0.1Ce0.7Y0.2)O3–δ as an electrolyte for low-temperature solid-oxide fuel cells. Adv. Mater. 18, 3318 (2006). https://doi.org/10.1002/adma.200601366
M.A. Azimova, S. McIntosh, On the reversibility of anode supported proton conducting solid oxide cells. Solid State Ionics 203, 57 (2011). https://doi.org/10.1016/j.ssi.2011.09.008
F. He, D. Song, R. Peng, G. Meng, S. Yang, Electrode performance and analysis of reversible solid oxide fuel cells with proton conducting electrolyte of BaCe0.5Zr0.3Y02O3−δ. J. Power. Sources 195, 3359 (2010). https://doi.org/10.1016/j.jpowsour.2009.12.079
S. Choi, T.C. Davenport, S.M. Haile, Protonic ceramic electrochemical cells for hydrogen production and electricity generation: exceptional reversibility, stability, and demonstrated faradaic efficiency. Energy Environ. Sci. 12, 206 (2019). https://doi.org/10.1039/C8EE02865F
Acknowledgements
This research was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (2020M1A2A2080864, RS-2023-00247245). This work was also funded and conducted under the “Competency Development Program for Industry Specialists” of the Korean Ministry of Trade, Industry, and Energy (MOTIE) (P0017120). This research was supported by the National Research Council of Science & Technology (NST) Grant by the Korea government (MSIT) (No. CAP22072-000).
Funding
Open Access funding enabled and organized by KAIST.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interest to declare that are relevant to the content of this article.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Oh, S., Kim, H., Jeong, I. et al. Recent progress in oxygen electrodes for protonic ceramic electrochemical cells. J. Korean Ceram. Soc. 61, 224–249 (2024). https://doi.org/10.1007/s43207-023-00360-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43207-023-00360-y