
Abstract
The intracellular signaling pathways that regulate myometrial contractions can be targeted by drugs for tocolysis. The agents, 2-APB, glycyl-H-1152, and HC-067047, have been identified as inhibitors of uterine contractility and may have tocolytic potential. However, the contraction-blocking potency of these novel tocolytics was yet to be comprehensively assessed and compared to agents that have seen greater scrutiny, such as the phosphodiesterase inhibitors, aminophylline and rolipram, or the clinically used tocolytics, nifedipine and indomethacin. We determined the IC50 concentrations (inhibit 50% of baseline contractility) for 2-APB, glycyl-H-1152, HC-067047, aminophylline, rolipram, nifedipine, and indomethacin against spontaneous ex vivo contractions in pregnant human myometrium, and then compared their tocolytic potency. Myometrial strips obtained from term, not-in-labor women, were treated with cumulative concentrations of the contraction-blocking agents. Comprehensive dose–response curves were generated. The IC50 concentrations were 53 µM for 2-APB, 18.2 µM for glycyl-H-1152, 48 µM for HC-067047, 318.5 µM for aminophylline, 4.3 µM for rolipram, 10 nM for nifedipine, and 59.5 µM for indomethacin. A single treatment with each drug at the determined IC50 concentration was confirmed to reduce contraction performance (AUC) by approximately 50%. Of the three novel tocolytics examined, glycyl-H-1152 was the most potent inhibitor. However, of all the drugs examined, the overall order of contraction-blocking potency in decreasing order was nifedipine > rolipram > glycyl-H-1152 > HC-067047 > 2-APB > indomethacin > aminophylline. These data provide greater insight into the contraction-blocking properties of some novel tocolytics, with glycyl-H-1152, in particular, emerging as a potential novel tocolytic for preventing preterm birth.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Preterm birth (PTB), defined as birth before 37 completed weeks of gestation, is a significant determinant of neonatal mortality, disease, and disability in surviving children [1]. Spontaneous premature labor (PTL) is the leading cause of PTB, accounting for approximately 45% of cases [1]. The risk of PTL is increased by prior preterm birth, preterm premature rupture of the membranes (PPROM), uterine overdistension, stress, and immunologically mediated processes [1, 2]. In attempts to prevent PTB, various tocolytics have been trialed for blocking spontaneous PTL, with the goal of extending pregnancy long enough for corticosteroid administration to mature the fetal lungs and improve birth outcomes. Tocolytic compounds examined include betamimetics, such as salbutamol and ritodrine, calcium ion (Ca2+) channel blockers (CCBs), such as nifedipine (NIF), Ca2+ competitors, such as magnesium sulfate, inhibitors of prostaglandin-endoperoxide synthase 2 (PTGS2), such as indomethacin (IND), oxytocin (OT) receptor antagonists (OTRA), such as Atosiban, and nitric oxide (NO) donors, such as nitroglycerine. These tocolytics are now well-characterized and work through either inhibiting pro-contraction signaling pathways, in particular by suppressing elevation of intracellular Ca2+ levels and blocking the availability and actions of uterotonins, or through activating pro-relaxation signaling pathways, in particular by raising intracellular cyclic adenosine monophosphate (cAMP) levels. Several systematic reviews and meta-analyses have been conducted in different parts of the world to assess the relative effectiveness of different tocolytic agents [3,4,5,6,7,8]. As a consequence, the guidelines for tocolytic management differ internationally [3, 9], and tocolytics licensed in one region of the world may or may not be licensed elsewhere. Where certain classes of tocolytics are not licensed, clinicians may utilize them off-label as second-line therapy. Approximately 75% of drugs used in obstetrics for tocolysis are unlicensed [10], this is because there are no specific therapeutic agents explicitly developed for tocolytic management except atosiban. Most tocolytics currently in use were developed for other medical indications, but were found to have tocolytic actions [10]. Moreover, there is no single agent currently available as a first-line tocolytic that is not associated with risks of side-effects [11]. Atosiban is associated with fewer side-effects than other tocolytics, but there is little evidence of efficacy [12]. All currently available tocolytic agents have relatively limited efficacy in postponing PTL and improving neonatal outcomes. As such, there remains a pressing need to evaluate the myometrial contraction blocking capabilities of novel drugs to improve tocolytic therapy.
Here, we report a comprehensive ex vivo analysis of three novel contraction-blocking agents: 2-aminoethoxydiphenyl borate (2-APB), glycyl-H-1152 dihydrochloride (GH), and HC-067047. We examined the potency of these “novel tocolytics” against spontaneous pregnant human myometrial contractions ex vivo and compared them to previously studied tocolytics, including NIF and IND, which have been used clinically for preventing PTB, as well as rolipram (ROL) and aminophylline (AMP), which have been previously investigated for uterine contraction inhibition.
2-APB was originally introduced as an inhibitor of inositol trisphosphate (IP3) receptors (IP3R) [13], which are a family of closely related Ca2+ channels embedded within the sarcoplasmic reticulum (SR). However, subsequent studies in intact cells have revealed that 2-APB also inhibits Ca2+ entry via store-operated channels (SOC); an effect that is independent of IP3R inhibition [14,15,16]. Thus, 2-APB has non-specific inhibitory effects on both IP3R and SOC, as well as on other Ca2+ transporters, e.g., sarcoplasmic Ca2+ ATPase (SRCA) pumps and transient receptor potential (TRP) channels of the TRPC family [17,18,19,20]. In maintaining Ca2+ homeostasis, G-protein coupled receptors, via activation of phospholipase C (PLC), generate intracellular IP3. IP3 then diffuses rapidly within sarcoplasm (cytoplasm) to bind with IP3Rs and release intracellular Ca2+ stores from the SR into the sarcoplasm [21,22,23]. The resulting increase in intracellular Ca2+ triggers the slow activation of SOCs in the sarcolemma (plasma membrane), which mediates store-operated Ca2+ entry (SOCE) into the sarcoplasm. Entry of Ca2+ through the sarcolemma contributes to the refilling of the SR Ca2+ stores and to the total intracellular pool of free Ca2+ in the sarcoplasm. Sarcoplasmic Ca2+ then binds to calmodulin, which activates the canonical signal transduction pathway that culminates in myosin light chain (MLC) phosphorylation and the actin-myosin cross bridge cycling that generates contractility [24, 25]. By inhibiting the activity of both SOC and IP3Rs, as well as other Ca2+ transporters, 2-APB prevents the elevation of sarcoplasmic Ca2+ levels, and in turn, inhibits myometrial contractility. Since its discovery [13], 2-APB has been tested on myometrial strips of rodents in several studies where it was found to inhibit both agonist-stimulated (OT, pennogenin tetraglycoside, Lannea acida plant extract, Ficus deltoidea plant extract) and spontaneous myometrial contractions [26,27,28,29,30,31,32]. However, despite potently inhibiting pregnant rodent uterine contractility, the effects of 2-APB on spontaneous pregnant human myometrial contractions were yet to be examined.
GH is a selective inhibitor of rho-kinase (ROCK). ROCK increases the sensitivity of uterine myocytes to Ca2+ through increased phosphorylation of MLC [33]. ROCK expression (mRNA abundance and protein levels) is upregulated at the end of pregnancy and is likely involved in the processes that underpin the increased myometrial contractility at term [34]. Thus, by inhibiting ROCK, GH reduces the sensitivity of uterine myocytes to Ca2+. GH has been found to inhibit both spontaneous and OT-stimulated pregnant human myometrial contractions ex vivo [35, 36]; however, comprehensive dose–response analyses were yet to be conducted to determine the potency of GH compared to other tocolytics.
HC-067047 is an inhibitor of transient receptor potential subfamily V, member 4 (TRPV4), a nonselective cation channel that is permeable to extracellular Ca2+ [37,38,39,40]. TRPV4 is activated by physiological stimuli, including stretch, swelling, heat, and pressure that may be relevant to human labor [41, 42]. Through inhibiting TRPV4 channels, HC-067047 prevents the influx of extracellular Ca2+ through these channels, thus preventing elevation of intracellular Ca2+ levels and myometrial contractility. TRPV4 is highly expressed in pregnant human myometrium and TRPV4 protein levels increase as gestation progresses [43]. These findings suggest that TRPV4 inhibition could be a potential novel tocolytic strategy [43, 44]; however, the effects of TRPV4 inhibitors, such as HC-067047, on spontaneous pregnant human myometrial contractions were yet to be investigated.
We also examined the non-selective phosphodiesterase (PDE) inhibitor, AMP, and the selective PDE4 inhibitor, ROL. By inhibiting PDEs, which are responsible for the breakdown of cAMP, both AMP and ROL raise intracellular cAMP levels, which promotes uterine relaxation. The tocolytic effects of theophylline, the active ingredient of AMP, and ROL have been reported in pregnant rodent and human myometrium [45,46,47,48,49,50,51]. However, their potency in suppressing spontaneous pregnant human myometrial contractions was yet to be comprehensively assessed. We also determined the potency of NIF and IND, which have well-documented tocolytic effects.
We conducted comprehensive dose–response analyses for 2-APB, GH, HC-067047, AMP, ROL, NIF, and IND using strips of pregnant human myometrium undergoing spontaneous contractions ex vivo. The location at which each of these agents affects uterine myocyte contraction signaling pathways is shown in Fig. 1. We then compared the contraction-blocking potency of the agents to assess the tocolytic potential of 2-APB, GH, and HC-067047 as novel tocolytics.

Overview of myometrial contraction signaling pathways and tocolytic action. The tocolytic agents examined block myometrial contractility through either inhibiting pro-contraction signaling pathways (2-APB, glycyl-H-1152, HC-067047, nifedipine, and indomethacin) or through activating pro-relaxation signaling pathways (aminophylline, rolipram)
Materials and Methods
Drugs and Reagents
Drugs and reagents were obtained from the following sources: NIF (cat No N7634), IND (cat No I7378), and AMP (cat No A1755, MW-210.21) were purchased from Sigma-Aldrich Pty Ltd (Sydney, Australia); ROL (cat No 0905), GH (cat No 2485, batch-specific MW 458.3), 2-APB (cat No 1224), and HC-067047 (cat No 4100) were purchased from Tocris (Bristol, UK). Other miscellaneous reagents were purchased from Sigma-Aldrich Pty Ltd (Sydney, Australia) and ThermoFisher Scientific Inc (Sydney, Australia).
Human Myometrial Specimens
Biopsy specimens of human myometrial tissue were obtained from women, undergoing elective cesarean section at the John Hunter Hospital, NSW, Australia. Biopsies were collected with the approval of the Hunter and New England Area Human Research Ethics Committees (2019/ETH12330) and all participants gave informed written consent. Myometrial biopsies were obtained from the upper lip of the incision in the lower uterine segment. All myometrial biopsies were obtained from term pregnancies (37–40 weeks of gestation) where the woman was not-in-labor (NIL). The clinical indications for elective cesarean section were breech presentation or a previous cesarean. All women were examined clinically and those with signs of infection, or with diabetes mellitus, or treated with any medication other than prenatal vitamins were excluded from the study. Patient demographic data are shown in Table 1. Upon collection, the myometrial biopsies were placed in pre-chilled phosphate buffered saline (PBS) on ice for transportation and were used within 60 min to commence myometrial dose–response contraction assays.
Myometrial Contraction Assays
Myometrial contraction assays were performed as previously described [52,53,54] using an 8-channel Radnoti Tissue-Organ Bath System (Radnoti Glass Technology Inc., Monrovia, CA, USA) equipped with MLT0201 force transducers (ADInstruments, Bella Vista, NSW, Australia) and eight temperature-controlled organ baths. Human myometrial specimens were dissected into 8 × 1.5 × 1.5 mm tissue strips then connected to the force transducers using nylon thread and stainless-steel tissue clips (ADInstruments). Each strip was lowered into a separate organ bath containing 15 mL modified Krebs–Henseleit buffer solution (KREBS) (no Ca2+, no NaHCO3) (Sigma-Aldrich, cat no K3753-10X1L) supplemented to 2.5 mM CaCl2 and 25 mM NaHCO3. Organ baths were maintained at 37 °C and KREBS continuously gassed with 95% O2 and 5% CO2. The transducer position was adjusted to apply 1 g of tension to each strip. The strips were then equilibrated whereby every 10 min for a total of 30 min, the organ baths were drained then refilled with 15 mL of KREBS (tissue strips washed). Due to tissue creep during stabilization, tissue length was increased to return tension to 1 g. Washing and re-tensioning to 1 g was repeated twice more (each strip tensioned to 1 g a total of 3 times). Thereafter, tension stabilized between 0.5 and 0.9 g and strips were left to develop spontaneous rhythmic contractions ex vivo. Under the described conditions, the myometrial strips took approximately 2 h to establish spontaneous contractions with consistent amplitude and frequency. Testing protocols were then begun, and strips were then maintained under isometric conditions for the remainder of the experimental run. In control experiments, tension was maintained near baseline tension for 6–7 h (Fig. S1). In experiments involving serial drug additions, some treatments resulted in changes of baseline tension. To accommodate these baseline changes, AUC were calculated from the tension between contractions observed immediately prior to drug addition. Contraction data were captured and visualized in real-time using a PowerLab 8/35 data-acquisition system and LabChart software (ADInstruments). Contraction traces were analyzed for key contraction parameters, including amplitude (g) and frequency (contractions/h), and integration of these values to determine the area under the curve (AUC) (g tension × sec). AUC was considered an index of contraction performance and was calculated based on the total area for all contractions generated during each 30 min treatment window (Fig. S2).
Longevity of Spontaneous Contractions in Pregnant Human Myometrium Ex Vivo
To ensure that myometrial strips were able to contract for the duration of the tocolytic studies, strips were allowed to generate spontaneous contractions ex vivo for 7 h. Traces were then analyzed (in 60 min blocks) to confirm that there was no significant change in the resting tension, contraction amplitude, contraction frequency, or AUC across the 7 h period of spontaneous ex vivo contractions.
Dose–Response Study
Before administering drug treatments, a contraction baseline was established for each tissue strip during which 1 h of contractions of consistent amplitude and frequency was recorded. Following the establishment of the baseline, treatments were added to the organ baths and the effects on contractility recorded. For each tissue strip, cumulative concentrations of drugs were administered at 30 min intervals. The effect of each drug was assessed against each strip’s contraction baseline (each strip has an internal control). Seven drugs (2-APB, GH, AMP, HC-067047, ROL, NIF, IND) were analyzed against myometrium from different term pregnant NIL women (replicate numbers as indicated). To control for any effects of the drug vehicles (dimethyl sulfoxide (DMSO), Milli-Q water or KREBS buffer), equivalent cumulative volumes of vehicles were assessed against separate tissue strips during each contraction assay.
Data Analysis
Analysis of AUC was performed using LabChart 8.0 Pro with the dose–response module (ADInstruments). For each strip, the last 30 min of contractions immediately prior to commencing treatments was used as the baseline (100%). Effects of treatments were normalized against the baseline and data expressed as percent (%) of baseline contractility. Dose–response curves for AUC were generated using the non-linear regression model of GraphPad Prism 8.0 (GraphPad Software Inc., San Diego, CA, USA) and fitted through the (log inhibitor vs normalized response-variable slope) equation, Y = 100/(1 + 10^((Log IC50-X)*HillSlope)). The concentration of each drug required to inhibit ex vivo myometrial contractility by 50% (IC50) was determined as being a 50% reduction in total AUC relative to the contraction baseline. An ordinary one-way ANOVA followed by Dunnett’s multiple comparisons test was used to determine significant differences between the baseline and mean AUC of each dose used in the dose–response curve. A probability (P) value of < 0.05 was considered statistically significant.
Confirmation of IC50
We sought to confirm the accuracy of the IC50 values determined for each drug. Baseline contractility for tissue strips was recorded for 1 h. Each drug was then applied to individual contracting strips as a single treatment at the IC50 determined for each drug (with exception of HC-067047, which could not be solubilized at the required concentration predicted to be the IC50). Contractility was recorded for a further 1 h. The effect of administering each drug at the IC50 on AUC was then determined.
Results
Longevity of Spontaneous Contractions and Assessment of Drug Vehicles
We first sought to confirm that term NIL myometrial strips were able to maintain consistent spontaneous rhythmic contractions ex vivo for 7 h, which was the maximum duration of cumulative tocolytic treatments. The strips exhibited spontaneous rhythmic contractions within 2 h of the final equilibration wash/re-tension (Fig. S2, panel A). Contractions remained stable for over 7 h in that comparison of the 60 min periods revealed no significant changes in resting tension, contraction amplitude, frequency, or AUC (n = 5) across the 7 h period (Fig. S2, panels B–E).
During the treatment time courses (2.5–3.5 h), the administration of cumulative doses of DMSO (maximum of 0.42%), Milli-Q water, or KREBS (drug vehicles) had no effect on the contraction amplitude, frequency, or AUC (Fig. S3, panel A–C and Fig. 3, panel A–G).
Dose–Response Analyses
Each of the drugs analyzed dose-dependently inhibited contractions in spontaneously contracting strips of pregnant human myometrium ex vivo. The appropriate dosing regimens for each drug were optimized by prior organ bath contraction studies to determine a dose–response regimen for each drug, whereby the lowest drug concentration had no significant effect, and the highest concentration exerted the maximal inhibitory effect on contractions (ICmax). For all drugs except HC-067047, ICmax was the abolition of contractions (0% of baseline AUC) (data not shown). All drugs, except HC-067047, were therefore equally effective at abolishing spontaneous pregnant human myometrial contractions ex vivo but exhibited different potencies (different IC50 values) (Fig. 2).

Representative traces showing the effect of cumulative drug treatments on contractions. Spontaneously contracting pregnant human myometrial strips were treated with cumulative concentrations of A 2-APB (n = 8), B GH (n = 8), C HC-067047 (n = 7), D AMP (n = 8), E ROL (n = 8), F NIF (n = 10), and G IND (n = 10). Dotted lines indicate the points at which the treatment was added to the bath
The cumulative concentrations of the novel tocolytics, 2-APB (1–120 µM), GH (0.1–80 µM), and HC-067047 (1–300 µM), affected spontaneous contractions similarly in that for each of these drugs, as contraction amplitude was reduced, contraction frequency significantly increased (Fig. 2, panels A–C). In contrast, as AMP (50–800 µM) and ROL (0.001–150 µM) inhibited contraction amplitude, contraction frequency significantly decreased (Fig. 2, panels D–E). This was particularly the case for ROL. The effects of NIF (0.1–50 nM) were consistent with the effects of 2-APB, GH, and HC-067047, in that as NIF reduced contraction amplitude, contraction frequency increased (Fig. 2, panel F), while the effects of IND (1–120 µM) were consistent with AMP and ROL, in that IND reduced contraction frequency as amplitude was inhibited (Fig. 2, panel G).
Tocolytic ICmax
2-APB and GH abolished spontaneous ex vivo contractions at 120 and 80 µM, respectively, whereas HC-067047 failed to completely abolish contractions even at the highest cumulative concentration tested (300 µM). At concentrations of ≥ 100 µM, HC-067047 precipitated out of solution within the organ baths. As such, the contractility recorded at the 100, 200, and 300 µM concentrations does not accurately reflect the effects of HC-067047 against spontaneous pregnant human myometrial contractions ex vivo. AMP and ROL abolished spontaneous contractions (ICmax) at 800 and 150 µM, respectively. The traditional tocolytics, NIF and IND, abolished contractions at 50 nM and 120 µM, respectively.
Determination of the Half-Maximal Inhibitory Concentration (IC50)
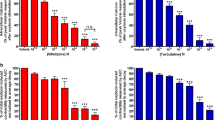
To determine tocolytic potency, dose–response curves were generated (Fig. 3, panels A–G) then the IC50 value determined for each drug. For the novel tocolytics, IC50 values were determined to be 53 µM for 2-APB (n = 8), 18.2 µM for GH (n = 8), and 48 µM for HC-067047 (n = 7). For the PDE inhibitors, IC50 values were 318.5 µM for AMP (n = 8) and 4.3 µM for ROL (n = 8), while for the traditional tocolytics, IC50 values were 10 nM for NIF (n = 10) and 59.5 µM for IND (n = 10). Contractility data are summarized in Table 2.

Dose–response curves. Plotted dose–response curves for the effect of A 2-APB (n = 8), B GH (n = 8), C HC-067047 (n = 7), D AMP (n = 8), E ROL (n = 8), F NIF (n = 10), and G IND (n = 10) on spontaneous pregnant human myometrial contractions ex vivo. Contractility was measured as AUC and expressed relative to the contraction baseline. Data are presented as mean ± SEM. There was a significant reduction in AUC in response to the cumulative doses of drugs. Comparisons were made between baseline and mean AUC of each dose using ordinary one-way ANOVA followed by Dunnett’s multiple comparisons test. A probability (P) value of < 0.05 was considered statistically significant. The asterisks indicate a significant difference, where 4 asterisks indicate P < 0.0001
Confirmation of Half-Maximal Inhibitory Concentration (IC50 )
Having determined the IC50 for each drug via the dose–response analyses, we then applied the IC50 to contracting strips as a single treatment to ascertain the accuracy of the determined IC50 values. Representative contraction traces are shown in Fig. 4, panels A–F. AUC analysis (1 h pre-treatment vs 1 h post-treatment) confirmed that for each drug, AUC was reduced by approximately 50% (2-APB = 49 ± 3% (n = 10); GH = 48 ± 3% (n = 10); AMP = 46 ± 4% (n = 10); ROL = 48 ± 5% (n = 5); NIF = 50 ± 3% (n = 10); IND = 49 ± 3% (n = 10) (Fig. 4, panel G).

Confirmation of the experimentally determined IC50 values. Representative traces showing the extent of contraction inhibition following treatment of pregnant human myometrial strips with the experimentally determined IC50 concentrations for A 2-APB (n = 10), B GH (n = 10), C AMP (n = 10), D ROL (n = 5), E NIF (n = 10), and F IND (n = 10). Black solid lines indicate the boundaries of the 1 h pre- and post-treatment analysis windows, and red dotted lines indicate the points at which the treatment was added to the bath. G The mean percent inhibition (relative to the contraction baselines) induced by each drug when applied at the IC50 for 60 min
The Tocolytic Effect of the Drugs Is Reversible
To validate that the contraction inhibition was mediated by drugs and not due to diminished cellular viability and/or metabolic restriction of the tissue, the myometrial strips were washed with KREBS solution after tocolytic treatment to ascertain whether spontaneous contractions resumed. For all drugs analyzed, the strips resumed contracting after the washout procedure following a brief (< 30 min) recovery period (Fig. 5, panels A–G).

Reversibility of the tocolytics. Representative traces showing that after washout of the drugs, contractions spontaneously resumed in pregnant human myometrial strips treated with A 2-APB (n = 5), B GH (n = 5), C HC-067047 (n = 5), D AMP (n = 5), E ROL (n = 5), F NIF (n = 5), and G IND (n = 5). Red dotted lines indicate the points at which organ baths were drained then refilled with fresh KREBS buffer
Discussion
Evidence suggests that PTL is a syndrome attributable to multiple pathological processes, including infection or inflammation, uterine overdistension, stress, ischemia or hemorrhage, endocrine disorders, immunologically mediated processes [55], and a gene expression pattern distinct from term labor [56]. Thus, PTL is a heterogeneous condition of multiple dysfunctions of preterm tissue that may lead to myometrial contractions, membrane/decidual activation, and/or cervical ripening. These three attributes constitute the common terminal pathway of both preterm and term birth. Hence, it is unclear whether spontaneous PTL results from premature activation of the term labor process, or due to pathological insults initiating uterine transformation from quiescence to overt labor [57]. Although evidence indicates that preterm and term labor are distinct processes [56], the preterm and term labor ultimately converge at the level of the myometrial contractile proteins. In this regard, there are no data available reporting that the levels of contractile proteins relevant to this study (IP3R, TRPV4, L-type Ca2+ channels, and ROCK) change between preterm and term human myometrium. Therefore, in the absence of such data, tocolytic agents that target these proteins should not be dismissed, as they may yet have relevance to tocolysis during PTL.
In this study, we performed comprehensive dose–response analyses to examine the contraction-blocking potency of three potential new tocolytics, 2-APB, GH, and HC-067047. We compared the IC50 of these new tocolytics to the PDE inhibitors, AMP and ROL, and to the clinically deployed tocolytics, NIF and IND. In terms of inhibiting spontaneous pregnant human myometrial contraction ex vivo (measured as AUC), the order of potency of the tocolytics from highest to lowest was NIF > ROL > GH > HC-067047 > 2-APB > IND > AMP (Table 3).
As a non-specific inhibitor of IP3Rs and SOC, 2-APB blocks the Ca2+ entry from both intracellular stores [13] and extracellular space [14,15,16], which prevents the elevation of cytosolic Ca2+ levels that drives contractions. Suppression of myometrial contractions in vitro by 2-APB was first reported by Ascher-Landsberg et al., who showed inhibition of both OT-stimulated and spontaneous contractions in rat myometrium, with contractions ultimately abolished at 100 µM [26]. This generally aligns with other subsequent studies in rodents, where 2-APB inhibited uterine contractions stimulated by different agonists, including OT and pennogenin tetraglycoside [26,27,28,29,30,31,32]. In our study, 2-APB abolished spontaneous pregnant human myometrial contractions at 120 µM, which is largely consistent with Ascher-Landsberg et al. [26], and we identified an IC50 (50% reduction in baseline AUC) for 2-APB of 53 µM. However, unlike Gravina et al., who reported a reduction in mouse myometrial contraction frequency following treatment [29], we observed that in pregnant human myometrium, contraction frequency increased as amplitude decreased in response to cumulative 2-APB treatments. Moreover, existing literature indicates that 2-APB exerts concentration-dependent biphasic effects on both SOC and IP3Rs. Patch-clamp studies on intact cell lines have shown that 2-APB has a stimulatory effect on Ca2+ entry via SOC and the IP3R gating system at lower concentrations (< 10 µM), and thus caused a transient increase in the amplitude of Ca2+ rise, whereas higher concentrations (> 10 µM) of 2-APB inhibits Ca2+ entry [15, 58, 59]. However, a biphasic effect of 2-APB was not observed in our myometrial tissue strip studies. We generated concentration–response curves based on assessment of both AUC (Fig. 3) and amplitude alone (data not shown) and in both cases, we detected no stimulatory effect of 2-APB on myometrial contractility at low concentrations (1 µM). The inconsistency may be attributable to the different experimental models in that the biphasic effect of 2-APB was observed against a non-uterine myocyte cell in monoculture using patch-clamp analysis, whereas our study utilized strips of pregnant human myometrium in a contraction bioassay system [15, 58, 59]. In support of this, existing literature demonstrates that isoform expression of STIM (1–2), a SR membrane protein that induces the opening of the SOCs, and ORAI (1–3), a plasma membrane protein that forms the pore of SOCs, differs between myometrial cells in culture and myometrial tissue [60,61,62]. 2-APB is shown to have differential effects on different STIM and ORAI isoforms [63, 64], which together mediate SOCE. Thus, the absence of a biphasic effect in the present study may be attributable to differences in expression of STIM and/or ORAI isoforms, compared to cell lines.
By inhibiting the isoenzyme, ROCK [65], GH reduces the Ca2+ sensitivity of uterine myocytes. The myometrial contraction and relaxation cycle depends on the equilibrium between phosphorylation and dephosphorylation of MLC, where myosin light chain kinase (MLCK) phosphorylates MLC to promote contraction and myosin light chain phosphatase (MLCP) dephosphorylates MLC to promote relaxation. MLCK is a Ca2+-dependent enzyme that is activated by the formation of the Ca2+-calmodulin complex in response to an intracellular Ca2+ surge [66,67,68,69]. In contrast, MLCP is negatively regulated by a Ca2+-independent mechanism where a regulatory subunit of MLCP is phosphorylated by ROCK, blocking the action MLCP and potentiating the effect of MLCK. Thus, the mechanism requires less Ca2+ to regulate MLC phosphorylation; a phenomenon called Ca2+ sensitization [33]. It has recently been demonstrated that ROCK-mediated Ca2+ sensitization of contractility can be induced by muscarinic or OT receptor stimulation in rat and human myometrium [70]. Hudson et al. and Aguilar et al. reported that cumulative concentrations and a single concentration (1 µM) of GH inhibited both OT-stimulated and spontaneous contraction of human myometrium ex vivo [35, 36]. Our comprehensive dose–response analyses add to these data as we have shown that in spontaneously contracting pregnant human myometrium ex vivo, the IC50 for GH is 18.2 µM and contractions are abolished at 80 µM. Our analyses also demonstrate that as GH inhibits contraction amplitude, contraction frequency increases, similar to 2-APB.
The third novel tocolytic we examined was HC-067047, a selective inhibitor of TRPV4 channels. TRPV4 plays a role in extracellular Ca2+ influx in response to various stimuli (stretch, swelling, heat, or pressure). There are limited studies examining the tocolytic effect of HC-067047. In mouse and rat uterine tissue strips, a single treatment with 1 µM HC-067047 inhibited contractions stimulated by OT and GSK1016790A [43, 71]. However, when used to treat pregnant human myometrium at the same concentration (1 µM), HC-067047 had only a slight inhibitory effect against OT-stimulated contractions [72].
Interestingly, Villegas et al. reported that activation of TRPV4 channels by the TRPV4 agonists, GSK1016790A and 4αPDD, resulted in the inhibition of OT-stimulated contractions in pregnant human myometrium [73]. This was attributed to an indirect effect whereby Ca2+ influx through TRPV4 channels causes K+ efflux via BKCa-channel activation, which, in turn, causes membrane hyperpolarization that inhibits L-type Ca2+ channels. This contrasts with the findings of Ying et al., who reported that GSK1016790A increased the contractility of mouse uterine tissue, which was then inhibited by HC-067047 [43]. Ying et al. also reported that HC-067047 inhibited OT-stimulated mouse uterine contractions, as well as delayed parturition in both RU486- and inflammation-induced mouse models of PTL [43]. Moreover, Singh et al. reported that HC-067047 inhibited GSK1016790A-induced contractility in murine strips ex vivo [71], while another ex vivo study with rat myometrial strips reported that the TRPV4 antagonist, RN1734, significantly decreased uterine contractility, whereas the TRPV4 agonist, RN1747, increased contractility [74]. The reason for the discrepancy in relation to the effects of the TRPV4 agonism is unclear but may be related to temporal and physical Ca2+ compartmentalization that plays a role in fine tuning contractility [72]. Nonetheless, our findings are consistent with prior studies from the mouse and rat, in that TRPV4 antagonism by HC-067047 unequivocally inhibited ex vivo spontaneous pregnant human myometrial contractions in a concentration-dependent manner (1, 10, 100 µM). Precipitation of HC-067047 at higher concentrations (200, 300 µM) means that the potency of HC-067047 is likely higher than that determined during this study. HC-067047 may therefore be a novel avenue for tocolysis; however, an effective delivery strategy may be required that overcomes the low aqueous solubility, such as delivery via uterine-targeted nanoliposomes [53] or vaginally administered mucus penetrating nanoparticles [75].
As a non-selective PDE inhibitor, AMP leads to the intracellular accumulation of cAMP, which operates through various mechanisms to promote uterine myocyte relaxation (see Fig. 1). Prior studies have demonstrated smooth muscle and myometrial contraction inhibition by AMP in rodents [48, 49, 76] and human [51, 77,78,79,80,81]. In an ex vivo study with pregnant human uterine strips, Bird et al. reported that AMP (40 and 100 µM) produced concentration-dependent inhibition of OT-stimulated contractions [77]. In another study, Verli et al. reported that increasing AMP concentrations (0.01 nM–10 µM) reduced OT-stimulated human myometrial contractions by 25% of baseline contractility [51]. Leroy et al., Berg et al., and Lai et al. demonstrated that AMP inhibited spontaneous pregnant human myometrial contractions in a concentration-dependent manner, where Leroy et al. and Berg et al. determined IC50 values for AMP of ≤ 100 µM [78, 80] and Lai et al. reported at 263 µM [81]. In the present study, AMP also inhibited spontaneous pregnant human myometrial contractions in a concentration-dependent manner; however, a high concentration (800 µM) of AMP was required to abolish contractility. Moreover, our determined IC50 for AMP of 318.5 µM is higher than that reported by Leroy et al., Berg et al. (100 µM), and Lai et al. (263 µM). The reason for these differences is currently unknown.
While AMP is a non-selective PDE inhibitor, ROL selectively inhibits PDE4, which is highly expressed in pregnant human myometrium at term [82]. Verli et al. reported that increasing concentrations of ROL (0.01 nM–10 µM) inhibited OT-stimulated contractions in pregnant human myometrial strips, and at the highest concentration tested of 10 µM, the authors found that ROL inhibited 62% of baseline contractility. Leroy et al., Bardou et al., and Martinez et al. reported that ROL inhibited spontaneous pregnant human myometrial contractions in a concentration-dependent manner, with 50% inhibition of contractility observed at 100 nM, 158 nM, and 22.7 µM, respectively [45, 78, 83]. In our analyses, ROL abolished ex vivo spontaneous pregnant human myometrial contractions at 150 µM and we determined the IC50 to be 4.3 µM. We also noted that for both AMP and ROL treatment, as contraction amplitude decreased, contraction frequency decreased; this contrasts the effects of 2-APB, GH, and HC-067047, where we observed that contraction frequency increased as amplitude was inhibited.
As an inhibitor of voltage-gated L-type Ca2+ channels, NIF blocks the influx of extracellular Ca2+ that underpins uterine myocyte contractility [84, 85]. The effects of NIF are well established [86, 87] and it is currently used clinically for tocolysis in various countries [88]. In the present study, we determined an IC50 for NIF of 10 nM. This is lower than the IC50 values reported by Moynihan et al. [87] (55 nM) and Ballejo et al. [89] (200 nM) against spontaneously contracting pregnant human myometrium, but is largely consistent with the IC50 values reported by Kuc et al. [90] (8.68 nM) and Longo et al. [91] (7.4 nM) against OT-stimulated contractions in pregnant human myometrium. Our determined IC50 of 10 nM, and with contractions abolished at 50 nM, makes NIF orders of magnitude more potent than all the other drugs examined in this study.
Within pregnant myometrium, IND blocks the synthesis of the prostaglandins that promote uterine contractions [92] and has been widely used as a tocolytic for many years. The tocolytic effect of IND was first reported by Vane et al. [93] and has been subsequently examined many times [94,95,96,97,98]. Across these studies, IND was reported to inhibit myometrial contractions at different concentrations and there is no emergent consensus as to an IC50. In our analyses, contractions were abolished by 120 µM IND and we determined the IND IC50 to be 59.5 µM. During prior studies on spontaneously contracting pregnant human myometrium, Arrowsmith et al. [94] and Johnson et al. [96] reported IND IC50 values of 35.4 and 278 µM, respectively, placing our findings within the range of these prior studies.
Strengths and Limitations
This study was the first to conduct a comprehensive dose–response study for novel tocolytics, 2-APB, GH, and HC-067047, to determine IC50 concentrations and compare their potency with clinically used tocolytics. A strength of this study was our confirmation of determined IC50 for each drug, where we confirmed that a ~ 50% reduction in baseline AUC was achieved (with exception of HC-067047 due to solubility limitations) when each drug was applied to contracting myometrial strips as a single treatment. These data support the accuracy of our determined IC50 values within our experimental setting and may provide insight into the relative tocolytic potency that could be expected from the different drugs in a clinical setting.
The study has only examined tocolytic potency against unstimulated (spontaneous) ex vivo contractions in term, NIL pregnant human myometrium. The authors acknowledge that there is increasing evidence that there are distinct differences between preterm and term labor [56], which may call into question the relevance of these data gleaned from term NIL myometrium. However, as previously mentioned, there are no data available indicating that levels of IP3R [99,100,101], TRPV4 [43], and ROCK [34, 102, 103] change between preterm and term pregnant human myometrium. Additionally, changes in expression of L-type Ca2+ channels [104,105,106,107], PTGS1 [108, 109], PTGS2 [109], and PDE4 [51, 110, 111] have been reported between preterm and term myometrium; however, each of these proteins was reported to exhibit higher expression in preterm myometrium than at term, suggesting that tocolytics targeting these proteins may actually have greater relevance during PTL than labor at term.
Lastly, this unstimulated model is standard technique for elucidating contraction pathways; however, further valuable insight would be garnered by examining agonist-stimulated contractions, as well as term IL myometrium and preterm NIL and IL myometrium.
Final Remarks
This study represents a comprehensive analysis of the myometrial contraction-blocking potency of the novel tocolytics, 2-APB, glycyl-H-1152, and HC-067047, and their potency comparison against the traditional tocolytics, nifedipine and indomethacin, as well as other potential candidates, rolipram and aminophylline (Table 2). Among the novel tocolytics, glycyl-H-1152 was the most potent followed by HC-067047 and 2-APB. Glycyl-H-1152 was also found to be a more potent inhibitor of ex vivo myometrial contractions than indomethacin and aminophylline, but less potent than nifedipine and rolipram, making glycyl-H-1152 the third most potent contraction blocker assessed (Table 3). These data provide us with greater insight into the contraction blocking potency of these drugs, with glycyl-H-1152 in particular emerging as a potential novel tocolytic due to its substantial potency. Glycyl-H-1152 may be an excellent candidate for encapsulation into uterine-targeted nanoliposomes [53] or vaginally administered mucus penetrating nanoparticles [75] as novel tocolytic strategies for preventing preterm birth. Such platforms may also facilitate the administration of hydrophobic drugs, such as HC-067047. Further studies are warranted to assess the tocolytic efficacy and safety of these agents in vivo using preterm birth models.
Data Availability
Not applicable.
Code Availability
Not applicable.
References
Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;371(9606):75–84. https://doi.org/10.1016/S0140-6736(08)60074-4.
Di Renzo GC, Roura LC, Facchinetti F, Antsaklis A, Breborowicz G, Gratacos E, Husslein P, Lamont R, Mikhailov A, Montenegro N, Radunovic N, Robson M, Robson SC, Sen C, Shennan A, Stamatian F, Ville Y. Guidelines for the management of spontaneous preterm labor: identification of spontaneous preterm labor, diagnosis of preterm premature rupture of membranes, and preventive tools for preterm birth. J Matern Fetal Neonatal Med. 2011;24(5):659–67. https://doi.org/10.3109/14767058.2011.553694.
Haas DM, Caldwell DM, Kirkpatrick P, McIntosh JJ, Welton NJ. Tocolytic therapy for preterm delivery: systematic review and network meta-analysis. BMJ. 2012;345:e6226. https://doi.org/10.1136/bmj.e6226.
Bain ES, Middleton PF, Crowther CA. Maternal adverse effects of different antenatal magnesium sulphate regimens for improving maternal and infant outcomes: a systematic review. BMC Pregnancy Childbirth. 2013;13(1):195. https://doi.org/10.1186/1471-2393-13-195.
Duckitt K, Thornton S, O'Donovan OP, Dowswell T. Nitric oxide donors for treating preterm labour. Cochrane Database Syst Rev. 2014;2014:5:CD002860. https://doi.org/10.1002/14651858.CD002860.pub2.
Neilson JP, West HM, Dowswell T. Betamimetics for inhibiting preterm labour. Cochrane Database Syst Rev. 2014:2:Cd004352. https://doi.org/10.1002/14651858.CD004352.pub3.
Conde-Agudelo A, Romero R, Kusanovic JP. Nifedipine in the management of preterm labor: a systematic review and metaanalysis. Am J Obstet Gynecol. 2011;204:2:134 e1–20. https://doi.org/10.1016/j.ajog.2010.11.038.
Hammers AL, Sanchez-Ramos L, Kaunitz AM. Antenatal exposure to indomethacin increases the risk of severe intraventricular hemorrhage, necrotizing enterocolitis, and periventricular leukomalacia: a systematic review with metaanalysis. Am J Obstet Gynecol. 2015;212:4:505 e1–13. https://doi.org/10.1016/j.ajog.2014.10.1091.
Alfirevic Z. Tocolytics: do they actually work? BMJ. 2012;345:e6531. https://doi.org/10.1136/bmj.e6531.
Lamont CD, Jorgensen JS, Lamont RF. The safety of tocolytics used for the inhibition of preterm labour. Expert Opin Drug Saf. 2016;15(9):1163–73. https://doi.org/10.1080/14740338.2016.1187128.
American College of O, Gynecologists. Committee on Practice B-O. ACOG practice bulletin no. 127: management of preterm labor. Obstet Gynecol. 2012;119:6:1308–17. https://doi.org/10.1097/AOG.0b013e31825af2f0.
Flenady V, Reinebrant HE, Liley HG, Tambimuttu EG, Papatsonis DN. Oxytocin receptor antagonists for inhibiting preterm labour. Cochrane Database Syst Rev. 2014:6:Cd004452. https://doi.org/10.1002/14651858.CD004452.pub3.
Maruyama T, Kanaji T, Nakade S, Kanno T, Mikoshiba K. 2APB, 2-aminoethoxydiphenyl borate, a membrane-penetrable modulator of Ins(1,4,5)P3-induced Ca2+ release. J Biochem. 1997;122(3):498–505. https://doi.org/10.1093/oxfordjournals.jbchem.a021780.
Djillani A, Nusse O, Dellis O. Characterization of novel store-operated calcium entry effectors. Biochim Biophys Acta. 2014;1843(10):2341–7. https://doi.org/10.1016/j.bbamcr.2014.03.012.
Prakriya M, Lewis RS. Potentiation and inhibition of Ca(2+) release-activated Ca(2+) channels by 2-aminoethyldiphenyl borate (2-APB) occurs independently of IP(3) receptors. J Physiol. 2001;536(Pt 1):3–19. https://doi.org/10.1111/j.1469-7793.2001.t01-1-00003.x.
Bootman MD, Collins TJ, Mackenzie L, Roderick HL, Berridge MJ, Peppiatt CM. 2-aminoethoxydiphenyl borate (2-APB) is a reliable blocker of store-operated Ca2+ entry but an inconsistent inhibitor of InsP3-induced Ca2+ release. FASEB J. 2002;16(10):1145–50. https://doi.org/10.1096/fj.02-0037rev.
Bilmen JG, Wootton LL, Godfrey RE, Smart OS, Michelangeli F. Inhibition of SERCA Ca2+ pumps by 2-aminoethoxydiphenyl borate (2-APB). 2-APB reduces both Ca2+ binding and phosphoryl transfer from ATP, by interfering with the pathway leading to the Ca2+-binding sites. Eur J Biochem. 2002;269:15:3678–87. https://doi.org/10.1046/j.1432-1033.2002.03060.x.
Ma HT, Venkatachalam K, Parys JB, Gill DL. Modification of store-operated channel coupling and inositol trisphosphate receptor function by 2-aminoethoxydiphenyl borate in DT40 lymphocytes. J Biol Chem. 2002;277(9):6915–22. https://doi.org/10.1074/jbc.M107755200.
Missiaen L, Callewaert G, De Smedt H, Parys JB. 2-Aminoethoxydiphenyl borate affects the inositol 1,4,5-trisphosphate receptor, the intracellular Ca2+ pump and the non-specific Ca2+ leak from the non-mitochondrial Ca2+ stores in permeabilized A7r5 cells. Cell Calcium. 2001;29(2):111–6. https://doi.org/10.1054/ceca.2000.0163.
Lievremont JP, Bird GS, Putney JW Jr. Mechanism of inhibition of TRPC cation channels by 2-aminoethoxydiphenylborane. Mol Pharmacol. 2005;68(3):758–62. https://doi.org/10.1124/mol.105.012856.
Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361(6410):315–25. https://doi.org/10.1038/361315a0.
Joseph SK. The inositol triphosphate receptor family. Cell Signal. 1996;8(1):1–7. https://doi.org/10.1016/0898-6568(95)02012-8.
Putney JW Jr. A model for receptor-regulated calcium entry. Cell Calcium. 1986;7(1):1–12. https://doi.org/10.1016/0143-4160(86)90026-6.
Arthur P, Taggart MJ, Mitchell BF. Oxytocin and parturition: a role for increased myometrial calcium and calcium sensitization? Front Biosci. 2007;12:1093–9946 (Print):619–33. https://doi.org/10.2741/2087.
Smith R. Parturition. N Engl J Med. 2007;356(3):271–83. https://doi.org/10.1056/NEJMra061360.
Ascher-Landsberg J, Saunders T, Elovitz M, Phillippe M. The effects of 2-aminoethoxydiphenyl borate, a novel inositol 1,4, 5-trisphosphate receptor modulator on myometrial contractions. Biochem Biophys Res Commun. 1999;264(3):979–82. https://doi.org/10.1006/bbrc.1999.1602.
Wang L, Jia C, Yu Z, Liu X, Kang L, Cong Y, Shan Y, Zhao Z, Ma B, Cong Y. Pennogenin tetraglycoside induces rat myometrial contraction and MLC20 phosphorylation via PLC-IP(3) and rhoA/rho kinase signaling pathways. PLoS ONE. 2012;7(12):e51536. https://doi.org/10.1371/journal.pone.0051536.
Ngadjui E, Kouam JY, Fozin GRB, Momo ACT, Deeh PBD, Wankeu-Nya M, Nguelefack TB, Watcho P. Uterotonic effects of aqueous and methanolic extracts of Lannea acida in Wistar rats: an in vitro study. Reprod Sci. 2021;28(9):2448–57. https://doi.org/10.1007/s43032-021-00465-x.
Gravina FS, Parkington HC, Kerr KP, de Oliveira RB, Jobling P, Coleman HA, Sandow SL, Davies MM, Imtiaz MS, van Helden DF. Role of mitochondria in contraction and pacemaking in the mouse uterus. Br J Pharmacol. 2010;161(6):1375–90. https://doi.org/10.1111/j.1476-5381.2010.00949.x.
Salleh N, Ahmad VN. In-VITRo effect of Ficus deltoidea on the contraction of isolated rat’s uteri is mediated via multiple receptors binding and is dependent on extracellular calcium. BMC Complement Altern Med. 2013;13(1):359. https://doi.org/10.1186/1472-6882-13-359.
Chung S, Kim YH, Joeng JH, Ahn DS. Transient receptor potential c4/5 like channel is involved in stretch-induced spontaneous uterine contraction of pregnant rat. Korean J Physiol Pharmacol. 2014;18(6):503–8. https://doi.org/10.4196/kjpp.2014.18.6.503.
Guo L, Su J, Deng BW, Yu ZY, Kang LP, Zhao ZH, Shan YJ, Chen JP, Ma BP, and Cong YW. Active pharmaceutical ingredients and mechanisms underlying phasic myometrial contractions stimulated with the saponin extract from Paris polyphylla Sm. var. yunnanensis used for abnormal uterine bleeding. Hum Reprod. 2008;23:4:964–71. https://doi.org/10.1093/humrep/den001.
Somlyo AP, Somlyo AV. Ca2+ sensitivity of smooth muscle and nonmuscle myosin II: modulated by G proteins, kinases, and myosin phosphatase. Physiol Rev. 2003;83(4):1325–58. https://doi.org/10.1152/physrev.00023.2003.
Niiro N, Nishimura J, Sakihara C, Nakano H, Kanaide H. Up-regulation of rho A and rho-kinase mRNAs in the rat myometrium during pregnancy. Biochem Biophys Res Commun. 1997;230(2):356–9. https://doi.org/10.1006/bbrc.1996.5960.
Hudson CA, Heesom KJ, Lopez BA. Phasic contractions of isolated human myometrium are associated with rho-kinase (ROCK)-dependent phosphorylation of myosin phosphatase-targeting subunit (MYPT1). Mol Hum Reprod. 2012;18(5):265–79. https://doi.org/10.1093/molehr/gar078.
Aguilar HN, Tracey CN, Zielnik B, Mitchell BF. Rho-kinase mediates diphosphorylation of myosin regulatory light chain in cultured uterine, but not vascular smooth muscle cells. J Cell Mol Med. 2012;16(12):2978–89. https://doi.org/10.1111/j.1582-4934.2012.01625.x.
Sanborn BM. Relationship of ion channel activity to control of myometrial calcium. J Soc Gynecol Investig. 2000;7(1):4–11. https://doi.org/10.1016/s1071-5576(99)00051-9.
Birnbaumer L, Zhu X, Jiang M, Boulay G, Peyton M, Vannier B, Brown D, Platano D, Sadeghi H, Stefani E, Birnbaumer M. On the molecular basis and regulation of cellular capacitative calcium entry: roles for Trp proteins. Proc Natl Acad Sci U S A. 1996;93(26):15195–202. https://doi.org/10.1073/pnas.93.26.15195.
Liedtke W, Kim C. Functionality of the TRPV subfamily of TRP ion channels: add mechano-TRP and osmo-TRP to the lexicon! Cell Mol Life Sci. 2005;62(24):2985–3001. https://doi.org/10.1007/s00018-005-5181-5.
Nilius B, Vriens J, Prenen J, Droogmans G, Voets T. TRPV4 calcium entry channel: a paradigm for gating diversity. Am J Physiol Cell Physiol. 2004;286(2):C195-205. https://doi.org/10.1152/ajpcell.00365.2003.
Becker D, Blase C, Bereiter-Hahn J, Jendrach M. TRPV4 exhibits a functional role in cell-volume regulation. J Cell Sci. 2005;118(Pt 11):2435–40. https://doi.org/10.1242/jcs.02372.
Benfenati V, Caprini M, Dovizio M, Mylonakou MN, Ferroni S, Ottersen OP, Amiry-Moghaddam M. An aquaporin-4/transient receptor potential vanilloid 4 (AQP4/TRPV4) complex is essential for cell-volume control in astrocytes. Proc Natl Acad Sci U S A. 2011;108(6):2563–8. https://doi.org/10.1073/pnas.1012867108.
Ying L, Becard M, Lyell D, Han X, Shortliffe L, Husted CI, Alvira CM, Cornfield DN. The transient receptor potential vanilloid 4 channel modulates uterine tone during pregnancy. Sci Transl Med. 2015;7:319:319ra204. https://doi.org/10.1126/scitranslmed.aad0376.
Smith R. Reapplying the uterine brake in preterm labor. Science Translational Medicine. 2015;7:319:319fs51–319fs51. https://doi.org/10.1126/scitranslmed.aad9788.
Fernandez-Martinez E, Ponce-Monter H, Soria-Jasso LE, Ortiz MI, Arias-Montano JA, Barragan-Ramirez G, Mayen-Garcia C. Inhibition of uterine contractility by thalidomide analogs via phosphodiesterase-4 inhibition and calcium entry blockade. Molecules. 2016;21(10):1332. https://doi.org/10.3390/molecules21101332.
Tyson EK, Smith R, Read M. Evidence that corticotropin-releasing hormone modulates myometrial contractility during human pregnancy. Endocrinology. 2009;150(12):5617–25.
Coutinho EM, Vieira Lopes AC. Inhibition of uterine motility by aminophylline. Am J Obstet Gynecol. 1971;110(5):726–9. https://doi.org/10.1016/0002-9378(71)90261-4.
Laifer SA, Ghodgaonkar RB, Zacur HA, Dubin NH. The effect of aminophylline on uterine smooth muscle contractility and prostaglandin production in the pregnant rat uterus in vitro. Am J Obstet Gynecol. 1986;155(1):212–5. https://doi.org/10.1016/0002-9378(86)90113-4.
Buckle JW, Nathanielsz PW. Modification of myometrial activity in vivo by administration of cyclic nucleotides and theophylline to the pregnant rat. J Endocrinol. 1975;66(3):339–47. https://doi.org/10.1677/joe.0.0660339.
Lipshitz J. Uterine and cardiovascular effects of aminophylline. Am J Obstet Gynecol. 1978;131(7):716–8. https://doi.org/10.1016/0002-9378(78)90232-6.
Verli J, Klukovits A, Kormanyos Z, Hajagos-Toth J, Ducza E, Seres AB, Falkay G, Gaspar R. Uterus-relaxing effect of beta2-agonists in combination with phosphodiesterase inhibitors: studies on pregnant rat in vivo and on pregnant human myometrium in vitro. J Obstet Gynaecol Res. 2013;39(1):31–9. https://doi.org/10.1111/j.1447-0756.2012.01929.x.
Paul J, Maiti K, Read M, Hure A, Smith J, Chan EC, Smith R. Phasic phosphorylation of caldesmon and ERK 1/2 during contractions in human myometrium. PLoS ONE. 2011;6(6): e21542. https://doi.org/10.1371/journal.pone.0021542.
Paul JW, Hua S, Ilicic M, Tolosa JM, Butler T, Robertson S, Smith R. Drug delivery to the human and mouse uterus using immunoliposomes targeted to the oxytocin receptor. Am J Obstet Gynecol. 2017;216:3:283 e1–283 e14. https://doi.org/10.1016/j.ajog.2016.08.027.
Paul JW, Kemsley JO, Butler TA, Tolosa JM, Thompson MB, Smith R, Whittington CM. A comparison of uterine contractile responsiveness to arginine vasopressin in oviparous and viviparous lizards. J Comp Physiol B. 2020;190(1):49–62. https://doi.org/10.1007/s00360-019-01254-4.
Romero R, Dey SK, Fisher SJ. Preterm labor: one syndrome, many causes. Science. 2014;345(6198):760–5. https://doi.org/10.1126/science.1251816.
Phung J, Wang CA, Reeders J, Chan E-C, Riveros C, Zakar T, Paul JW, Pennell CE, Smith R. Preterm labor is a distinct process from term labor following computational analysis of human myometrium. Am J Obstet Gynecol. 2022;226(1):106.e1-106.e16. https://doi.org/10.1016/j.ajog.2021.07.002.
de Laat MWM, Pieper PG, Oudijk MA, Mulder BJM, Christoffels VM, Afink GB, Postma AV, Ris-Stalpers C. The clinical and molecular relations between idiopathic preterm labor and maternal congenital heart defects. Reprod Sci. 2013;20(2):190–201. https://doi.org/10.1177/1933719112446083.
Wu J, Kamimura N, Takeo T, Suga S, Wakui M, Maruyama T, Mikoshiba K. 2-Aminoethoxydiphenyl borate modulates kinetics of intracellular Ca(2+) signals mediated by inositol 1,4,5-trisphosphate-sensitive Ca(2+) stores in single pancreatic acinar cells of mouse. Mol Pharmacol. 2000;58(6):1368–74. https://doi.org/10.1124/mol.58.6.1368.
Chorna-Ornan I, Joel-Almagor T, Ben-Ami HC, Frechter S, Gillo B, Selinger Z, Gill DL, Minke B. A common mechanism underlies vertebrate calcium signaling and Drosophila phototransduction. J Neurosci. 2001;21(8):2622–9. https://doi.org/10.1523/jneurosci.21-08-02622.2001.
Murtazina DA, Chung D, Ulloa A, Bryan E, Galan HL, Sanborn BM. TRPC1, STIM1, and ORAI influence signal-regulated intracellular and endoplasmic reticulum calcium dynamics in human myometrial cells. Biol Reprod. 2011;85(2):315–26. https://doi.org/10.1095/biolreprod.111.091082.
Chin-Smith EC, Slater DM, Johnson MR, Tribe RM. STIM and Orai isoform expression in pregnant human myometrium: a potential role in calcium signaling during pregnancy. Front Physiol. 2014;5:169. https://doi.org/10.3389/fphys.2014.00169.
Sutovska M, Kocmalova M, Sadlonova V, Dokus K, Adamkov M, Luptak J, Franova S. Orai1 protein expression and the role of calcium release-activated calcium channels in the contraction of human term-pregnant and non-pregnant myometrium. J Obstet Gynaecol Res. 2015;41(5):704–11. https://doi.org/10.1111/jog.12626.
Peinelt C, Lis A, Beck A, Fleig A, Penner R. 2-Aminoethoxydiphenyl borate directly facilitates and indirectly inhibits STIM1-dependent gating of CRAC channels. J Physiol. 2008;586(13):3061–73. https://doi.org/10.1113/jphysiol.2008.151365.
DeHaven WI, Smyth JT, Boyles RR, Bird GS, Putney JW Jr. Complex actions of 2-aminoethyldiphenyl borate on store-operated calcium entry. J Biol Chem. 2008;283(28):19265–73. https://doi.org/10.1074/jbc.M801535200.
Matsui K, Higashi K, Fukunaga K, Miyazaki K, Maeyama M, Miyamoto E. Hormone treatments and pregnancy alter myosin light chain kinase and calmodulin levels in rabbit myometrium. J Endocrinol. 1983;97(1):11–9. https://doi.org/10.1677/joe.0.0970011.
Schoenwaelder SM, Burridge K. Bidirectional signaling between the cytoskeleton and integrins. Curr Opin Cell Biol. 1999;11(2):274–86. https://doi.org/10.1016/s0955-0674(99)80037-4.
Zhong C, Chrzanowska-Wodnicka M, Brown J, Shaub A, Belkin AM, Burridge K. Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly. J Cell Biol. 1998;141(2):539–51. https://doi.org/10.1083/jcb.141.2.539.
Gong MC, Iizuka K, Nixon G, Browne JP, Hall A, Eccleston JF, Sugai M, Kobayashi S, Somlyo AV, Somlyo AP. Role of guanine nucleotide-binding proteins–ras-family or trimeric proteins or both–in Ca2+ sensitization of smooth muscle. Proc Natl Acad Sci U S A. 1996;93(3):1340–5. https://doi.org/10.1073/pnas.93.3.1340.
Togashi H, Emala CW, Hall IP, Hirshman CA. Carbachol-induced actin reorganization involves Gi activation of rho in human airway smooth muscle cells. Am J Physiol. 1998;274(5):L803–9. https://doi.org/10.1152/ajplung.1998.274.5.L803.
Taggart MJ, Lee YH, Morgan KG. Cellular redistribution of PKCalpha, rhoA, and ROKalpha following smooth muscle agonist stimulation. Exp Cell Res. 1999;251(1):92–101. https://doi.org/10.1006/excr.1999.4565.
Singh V, Ram M, Kandasamy K, Thangamalai R, Choudhary S, Dash JR, Kumar D, Parida S, Singh TU, Mishra SK. Molecular and functional characterization of TRPV4 channels in pregnant and nonpregnant mouse uterus. Life Sci. 2015;122:51–8. https://doi.org/10.1016/j.lfs.2014.12.010.
Rousseau É, Labelle K, Massenavette L. Involvement of alternative calcium conductance on human myometrial contractile and pharmacological properties. Current Opinion in Gynecology and Obstetrics. 2018;1:1:100–110. https://doi.org/10.18314/cogo.v1i1.1288.
Villegas D, Giard O, Brochu-Gaudreau K, Rousseau É. Activation of TRPV4 channels leads to a consistent tocolytic effect on human myometrial tissues. Eur J Obstet Gynecol Reprod Biol: X. 2021;10:100124. https://doi.org/10.1016/j.eurox.2021.100124.
Ducza E, Csányi A, Szőke É, Pohóczky K, Hajagos-Tóth J, Kothencz A, Tiszai Z, Gáspár R. Significance of transient receptor potential vanilloid 4 and aquaporin 5 co-expression in the rat uterus at term. Heliyon. 2019;5:10. https://doi.org/10.1016/j.heliyon.2019.e02697.
Ensign LM, Tang BC, Wang YY, Tse TA, Hoen T, Cone R, and Hanes J. Mucus-penetrating nanoparticles for vaginal drug delivery protect against herpes simplex virus. Sci Transl Med. 2012;4:138:138ra79. https://doi.org/10.1126/scitranslmed.3003453.
Mitznegg P, Schubert E, Carl H. The influence of theophylline on the motility and the cyclic 3’,5’-AMP content in the pregnant and non-pregnant rat uterus in vitro. Arzneimittelforschung. 1976;26(9):1684–5.
Bird LM, Anderson NC Jr, Chandler ML, Young RC. The effects of aminophylline and nifedipine on contractility of isolated pregnant human myometrium. Am J Obstet Gynecol. 1987;157(1):171–7. https://doi.org/10.1016/s0002-9378(87)80373-3.
Leroy MJ, Cedrin I, Breuiller M, Giovagrandi Y, Ferre F. Correlation between selective inhibition of the cyclic nucleotide phosphodiesterases and the contractile activity in human pregnant myometrium near term. Biochem Pharmacol. 1989;38(1):9–15. https://doi.org/10.1016/0006-2952(89)90142-1.
Berg G, Andersson RG, Ryden G. Effects of different phosphodiesterase-inhibiting drugs on human pregnant myometrium: an in vitro study. Arch Int Pharmacodyn Ther. 1987;290(2):288–92.
Berg G, Andersson RG, Ryden G. In vitro study of phosphodiesterase-inhibiting drugs: a complement to beta-sympathomimetic drug therapy in premature labor? Am J Obstet Gynecol. 1983;145(7):802–6. https://doi.org/10.1016/0002-9378(83)90682-8.
Lai PF, Young RC, Tribe RM, Johnson MR. Evaluating aminophylline and progesterone combination treatment to modulate contractility and labor-related proteins in pregnant human myometrial tissues. Pharmacol Res Perspect. 2021;9(4):e00818. https://doi.org/10.1002/prp2.818.
Leroy MJ, Lugnier C, Merezak J, Tanguy G, Olivier S, Le Bec A, Ferre F. Isolation and characterization of the rolipram-sensitive cyclic AMP-specific phosphodiesterase (type IV PDE) in human term myometrium. Cell Signal. 1994;6(4):405–12. https://doi.org/10.1016/0898-6568(94)90087-6.
Bardou M, Cortijo J, Loustalot C, Taylor S, Perales-Marin A, Mercier FJ, Dumas M, Deneux-Tharaux C, Frydman R, Morcillo EJ, Advenier C. Pharmacological and biochemical study on the effects of selective phosphodiesterase inhibitors on human term myometrium. Naunyn Schmiedebergs Arch Pharmacol. 1999;360(4):457–63.
Fleckenstein A. Specific pharmacology of calcium in myocardium, cardiac pacemakers, and vascular smooth muscle. Annu Rev Pharmacol Toxicol. 1977;17(1):149–66. https://doi.org/10.1146/annurev.pa.17.040177.001053.
Wray S, Jones K, Kupittayanant S, Li Y, Matthew A, Monir-Bishty E, Noble K, Pierce SJ, Quenby S, Shmygol AV. Calcium signaling and uterine contractility. J Soc Gynecol Investig. 2003;10(5):252–64. https://doi.org/10.1016/s1071-5576(03)00089-3.
Forman A, Andersson KE, Persson CG, Ulmsten U. Relaxant effects of nifedipine on isolated, human myometrium. Acta Pharmacol Toxicol (Copenh). 1979;45(2):81–6.
Moynihan AT, Smith TJ, Morrison JJ. The relaxant effect of nifedipine in human uterine smooth muscle and the BK(Ca) channel. Am J Obstet Gynecol. 2008;198:2:237 e1–8. https://doi.org/10.1016/j.ajog.2007.08.074.
Hoh JK, Lappas M, Liu C, Qiao C, Pallavi K, Takeda J, Kim YJ. Preterm birth rate and dilemma of preterm labor treatment in Asia. Placenta. 2019;79:68–71. https://doi.org/10.1016/j.placenta.2019.01.005.
Ballejo G, Calixto JB, Medeiros YS. In vitro effects of calcium entry blockers, chlorpromazine and fenoterol upon human pregnant myometrium contractility. Br J Pharmacol. 1986;89(3):515–23. https://doi.org/10.1111/j.1476-5381.1986.tb11151.x.
Kuc P, Laudanski P, Pierzynski P, Laudanski T. The effect of combined tocolysis on in vitro uterine contractility in preterm labour. Adv Med Sci. 2011;56(1):88–94. https://doi.org/10.2478/v10039-011-0019-x.
Longo M, Jain V, Vedernikov YP, Hankins GD, Garfield RE, Saade GR. Effects of L-type Ca(2+)-channel blockade, K(+)(ATP)-channel opening and nitric oxide on human uterine contractility in relation to gestational age and labour. Mol Hum Reprod. 2003;9(3):159–64. https://doi.org/10.1093/molehr/gag023.
Landen and C. Mechanisms of indomethacin and nimesulide on inhibition of calcium rises in human uterine myocytes. Obstetrics & Gynecology. 2001;97:5:S6. https://doi.org/10.1016/s0029-7844(01)01143-7.
Vane JR, Williams KI. The contribution of prostaglandin production to contractions of the isolated uterus of the rat. Br J Pharmacol. 1973;48(4):629–39. https://doi.org/10.1111/j.1476-5381.1973.tb08250.x.
Arrowsmith S, Neilson J, Bricker L, Wray S. Differing in vitro potencies of tocolytics and progesterone in myometrium from singleton and twin pregnancies. Reprod Sci. 2016;23(1):98–111. https://doi.org/10.1177/1933719115597788.
Sawdy R, Knock GA, Bennett PR, Poston L, Aaronson PI. Effect of nimesulide and indomethacin on contractility and the Ca2+ channel current in myometrial smooth muscle from pregnant women. Br J Pharmacol. 1998;125(6):1212–7. https://doi.org/10.1038/sj.bjp.0702211.
Johnson WL, Harbert GM, Martin CB. Pharmacologic control of uterine contractility. In vitro human and in vivo monkey studies. Am J Obstet Gynecol. 1975;123:4:364–75. https://doi.org/10.1016/s0002-9378(16)33437-8.
Crankshaw DJ, Dyal R. Effects of some naturally occurring prostanoids and some cyclooxygenase inhibitors on the contractility of the human lower uterine segment in vitro. Can J Physiol Pharmacol. 1994;72(8):870–4. https://doi.org/10.1139/y94-123%M7834575.
Garrioch DB. The effect of indomethacin on spontaneous activity in the isolated human myometrium and on the response to oxytocin and prostaglandin. Br J Obstet Gynaecol. 1978;85(1):47–52. https://doi.org/10.1111/j.1471-0528.1978.tb15825.x.
Morgan JM, De Smedt H, Gillespie JI. Identification of three isoforms of the InsP3 receptor in human myometrial smooth muscle. Pflugers Arch. 1996;431(5):697–705. https://doi.org/10.1007/BF02253832.
Martin C, Chapman KE, Thornton S, Ashley RH. Changes in the expression of myometrial ryanodine receptor mRNAs during human pregnancy. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 1999;1451:2:343–352. https://doi.org/10.1016/S0167-4889(99)00104-4.
Mesonero JE, Tanfin Z, Hilly M, Colosetti P, Mauger J-P, Harbon S. Differential expression of inositol 1,4,5-trisphosphate receptor types 1, 2, and 3 in rat myometrium and endometrium during gestation1. Biol Reprod. 2000;63(2):532–7. https://doi.org/10.1095/biolreprod63.2.532.
Moran CJ, Friel AM, Smith TJ, Cairns M, Morrison JJ. Expression and modulation of rho kinase in human pregnant myometrium. Mol Hum Reprod. 2002;8(2):196–200. https://doi.org/10.1093/molehr/8.2.196.
Friel AM, Curley M, Ravikumar N, Smith TJ, Morrison JJ. Rho A/rho kinase mRNA and protein levels in human myometrium during pregnancy and labor. J Soc Gynecol Investig. 2005;12(1):20–7. https://doi.org/10.1016/j.jsgi.2004.07.002.
Banciu A, Banciu DD, Mustaciosu CC, Radu M, Cretoiu D, Xiao J, Cretoiu SM, Suciu N, Radu BM. Beta-estradiol regulates voltage-gated calcium channels and estrogen receptors in telocytes from human myometrium. Int J Mol Sci. 2018;19(5):1413.
Longo M, Jain V, Vedernikov YP, Hankins GDV, Garfield RE, Saade GR. Effects of L-type Ca2+-channel blockade, K+ATP-channel opening and nitric oxide on human uterine contractility in relation to gestational age and labour. Mol Hum Reprod. 2003;9(3):159–64. https://doi.org/10.1093/molehr/gag023.
Ohkubo T, Kawarabayashi T, Inoue Y, Kitamura K. Differential expression of L- and T-type calcium channels between longitudinal and circular muscles of the rat myometrium during pregnancy. Gynecol Obstet Invest. 2005;59(2):80–5. https://doi.org/10.1159/000082333.
Collins PL, Moore JJ, Lundgren DW, Choobineh E, Chang SM, Chang AS. Gestational changes in uterine L-type calcium channel function and expression in guinea pig1. Biol Reprod. 2000;63(5):1262–70. https://doi.org/10.1095/biolreprod63.5.1262.
Zuo J, Lei ZM, Rao CV, Pietrantoni M, Cook VD. Differential cyclooxygenase-1 and -2 gene expression in human myometria from preterm and term deliveries. J Clin Endocrinol Metab. 1994;79(3):894–9. https://doi.org/10.1210/jcem.79.3.8077379.
Slater DM, Dennes WJB, Campa JS, Poston L, Bennett PR. Expression of cyclo-oxygenase types-1 and -2 in human myometrium throughout pregnancy. Mol Hum Reprod. 1999;5(9):880–4. https://doi.org/10.1093/molehr/5.9.880.
Yulia A, Varley AJ, Singh N, Lei K, Tribe R, Johnson MR. Changes in cAMP effector predominance are associated with increased oxytocin receptor expression in twin but not infection-associated or idiopathic preterm labour. PLoS ONE. 2020;15(11):e0240325. https://doi.org/10.1371/journal.pone.0240325.
Méhats C, Schmitz T, Oger S, Hervé R, Cabrol D, Leroy M-J. PDE4 as a target in preterm labour. BMC Pregnancy Childbirth. 2007;7(1):S12. https://doi.org/10.1186/1471-2393-7-S1-S12.
Acknowledgements
We are grateful to the pregnant women who donated myometrial samples, to our research midwife, Ms. Anne Wright, and acknowledge the help of Obstetrics, Midwifery, and Surgical staff at the John Hunter Hospital, Newcastle, Australia.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions This study was supported by two Hunter Medical Research Institute grants awarded to JWP and RS, and a University of Newcastle International Postgraduate Research Scholarship awarded to MRH. The funding providers had no involvement in the study.
Author information
Authors and Affiliations
Contributions
All authors contributed to the manuscript. MRH and JWP wrote the manuscript. JMT, RCY, and RS edited the manuscript. Figures by MRH. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
Human tissue samples were collected with the approval of the Hunter and New England Area Human Research Ethics Committees (2019/ETH12330).
Consent to Participate
All patients informed written consent.
Consent for Publication
All patients informed written consent.
Conflict of Interests
The authors declare no competing interests.
Supplementary Information
Below is the link to the electronic supplementary material.
43032_2022_1000_MOESM1_ESM.jpg
Supplementary file2 Figure S1. Longevity of spontaneous myometrial contractions ex vivo. (A) A representative trace showing spontaneous pregnant human myometrial contractions recorded ex vivo for > 7 h (n = 5). Panels (B), (C), (D) and (E) show the mean contraction resting tension, amplitude, frequency, and AUC calculated during each 1 h period (expressed as a percentage of baseline contractility). There was no significant change in resting tension, amplitude, frequency, or AUC over the > 7 h recording period. Comparisons were made between baseline and the individual 60 min periods using ordinary one-way ANOVA followed by Dunnett’s multiple comparisons test. A probability (P) value of < 0.05 was considered as statistically significant (JPG 3.35 MB)
43032_2022_1000_MOESM2_ESM.jpg
Supplementary file1 Figure S2. Assessment of contraction parameters. Illustration showing how contraction amplitude and frequency were measured and AUC determined. The horizontal blue dotted line indicates the baseline that was used as lower border for calculating AUC (JPG 976 KB)
43032_2022_1000_MOESM3_ESM.jpg
Supplementary file3 Figure S3. Representative traces showing the effect of cumulative doses of drug vehicles on contractions. Spontaneously contracting pregnant human myometrial strips were treated with cumulative doses (10 μL each) of (A) DMSO(0, 0.06, 0.12, 0.18, 0,24, 0.30, 0.36, 0.42% v/v), (B) KREBS buffer (0, 0.06, 0.12, 0.18, 0,24, 0.30, 0.36, 0.42% v/v) and (C) Milli-Q water(0, 0.06, 0.12, 0.18, 0,24, 0.30% v/v). Dotted lines indicate the points at which the treatment was added to the bath(JPG 3723 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
Cite this article
Hossain, M.R., Tolosa, J.M., Young, R.C. et al. Assessing the Potency of the Novel Tocolytics 2-APB, Glycyl-H-1152, and HC-067047 in Pregnant Human Myometrium. Reprod. Sci. 30, 203–220 (2023). https://doi.org/10.1007/s43032-022-01000-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43032-022-01000-2