
Abstract
Meckel-Gruber syndrome (MKS) is a rare lethal autosomal recessive inherited disorder. Missed diagnosis might happen in clinical works due to an unclear genotype–phenotype correlation. We analyzed two families visiting our center; the parents are normal; each of the family aborted a fetus at 12WG. Following ultrasonography and pathological examination, both were diagnosed as MKS. Whole exome sequencing identified a compound heterozygous of two novel variants of CEP290 and a heterozygous of a novel variant of CC2D2A. Frameshift mutations in ZNF77 were also detected. Western blot analyzing whole-brain tissue showed that the expression of ZNF77, CC2D2A, and CEP290 was enhanced. HEK293T transfected with over-expression wildtype/mutated ZNF77 plasmid showed that SHH was increased in wildtype ZNF77 cells, while SHH and CC2D2A were increased in mutated ZNF77 cells. Our research provided two novel pathogenic variants of CEP290 and CC2D2A and suggested that ZNF77 might promote the expression of CC2D2A and regulate the amount of SHH.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Meckel-Gruber Syndrome (MKS, OMIM *24,900), one of the most serious ciliopathies and the most common neural tube defect (NTDs) [1], is a rare lethal autosomal recessive inherited disorder. Globally, the incidence of MKS is 1/140000–1/13250 [2]. A couple of carriers has a possibility of 25% to produce a MKS fetus, and both two genders gain the same risk. Important systems and organs could be impacted in MKS, leading to a wide phenotypic spectrum, which a “classic triad” (occipital encephalocele, cystic kidney dysplasia, polydactyly) [3] might characterize in the majority of patients. Clinical prenatal diagnosis usually comes from transvaginal ultrasound from 11 to 16 weeks of gestation. However, MKS has a high heterogeneity. Clinical manifestations are different at the general, organic, and even tissue level among different cases. The advancement of assisted reproductive technology, the development of molecular diagnostic technology, and preimplantation genetic testing (PGT) can prevent the occurrence of this disease to a certain extent. However, the accumulation of resources of the nation is a necessary prerequisite for the research of this type of genetic disease.
From the perspective of genetics, variants from at least 14 germlines have been already reported to cause MKS: MKS1, TMEM216, TMEM67, CEP290, RPGRIP1L, CC2D2A, NPHP3, TCTN2, B9D1, B9D2, TMEM231, Kif14, TMEM107, TXNDC15, etc. [4]. MKS follows the pattern of Mendelian inheritance, whereas non-Mendelian inheritance also contributes to some extent [5]. CEP290 (OMIM *610,142) locates on 12q21.32(genomic coordinates: 12:88,049,012–88,142,215;GRch38), with a total length of 92.3 kb and contains 55 exons [6]. Mutations of CEP290 lead to a MKS type 4 (OMIM *611,134). CC2D2A (OMIM *612,013) locates on 4p15.32(genomic coordinates: 4:15,468,659–15,601,556;GRCH38), 131.5 kb, 38 exons on total, encoding 1620 amino acids [7]. Mutations of CC2D2A leads to MKS6 (OMIM *612,284). Furthermore, no clear genotype/phenotype correlations have been clarified nowadays. Some certain inheritance patterns such as basal exon skipping [8], modifier alleles, and stochastic effects might explain this genetic pleiotropy [5], whereas the mutational spectrum still needs to be expanded. From the perspective of histology, MKS is one of the most severe ciliopathies. Primary cilia (PC), a kind of non-motile cilia, play an essential part during fetal growth. Multiple cell signals situate on PCs; one of them is called SHH (sonic hedgehog) pathway; its function consists tightly with cilia [9]. In mammals, SHH is expressed in early embryogenesis and works as a morphogen to pattern the architecture of CNS[10]. In-depth exploration of the relationship between gene mutations and signals will help to clarify the pathogenesis of MKS and is greatly benefit for disease prevention, diagnosis, and treatment.
ZNF77 maps to the 19th chromosome who carries many ZNF loci and other genes with zinc finger encoding motifs [11]. Zinc finger proteins, combining with some DNAs or RNAs, serves as a transcript factor [12]. Bioinformatic analysis via human tissue-specific networks suggests that ZNF77 controls defensins, elastase, and calmodulin expression, which is firstly described by a research of pathogenic lung colonization [13]. Thus, we suppose that frameshift mutation of ZNF77 might influence the amount of CEP290 and CC2D2A downstream, leading to alternation of cilia and SHH signal which locate on its membrane.
In this study, we investigated the genealogy of two MKS families. Whole exome sequencing was performed, and mutated locus on CEP290, CC2D2A, and ZNF77 were analyzed. Three novel variants were identified to cause MKS and would contribute to its genotype spectrum. Thus, genetic counseling, prenatal diagnosis, and PGT could be benefited referring to a more complete database. Meanwhile, to research the role of ZNF77, we built ZNF77 over-expressed and mutated model in HEK-293 T cell. Through results using western blot, we confirmed a causality between ZNF77 and CC2D2A and a possibility of over-expression or repressed degrade of SHH when ZNF77 germline is mutated.
Materials and Methods
Patients and Families
We investigated two pedigrees diagnosed as MKS, admitted by the Reproductive Medicine Center of Zhongnan Hospital of Wuhan University, Wuhan, China, in January 2019–March 2019. (Fig. 1) Detailed genetic consulting has been performed, and basic information was collected as follows:

Familial pedigree charts. The arrow indicates the proband, circles indicates female members, square represents males, the slash denotes deceased family member. (a) Case 1, II-7, 24 years old, healthy. II-8, 22 years old, healthy. III-7 was induced labor due to MKS; III-8, the proband, showed MKS symptoms (including occipital encephalocele, cystic kidney). (b) Case 2, II-1, 28, and II-2, 25; III-1, an aborted fetus of unknown gender, induced labor due to MKS; III-2, the proband, he showed MKS symptoms (occipital encephalocele, cystic kidney dysplasia, postaxial polydactyly)
In case 1, the mother was 22 years old, and the father was 26 years old. Their first pregnancy was terminated at 24 weeks of gestation with ultrasound findings of facial deformity, encephalocele, and cystic kidney type I and a diagnosis of MKS. No definite mutations and chromosome abnormality was detected from copy number variants (CNV) test of fetus umbilical cord blood. Samples from aborted fetus could not be found by now.
In case2, the mother was 27 years old, and the father is 29 years old. Their first pregnancy ended at 16 WG because of parietal encephalocele, cystic kidney, and polydactyly (suspected MKS), without genetic testing.
Both two couples were healthy, without markable history of inherited diseases, and denied consanguineous marriage. Encephalocele was found in both families at about 12 WG displayed by ultrasound; the fetuses were aborted at 12 + 5 and 12 + 1 WG, respectively. (Fig. 2) Histological examination, whole exome sequencing, and sanger sequencing were performed.

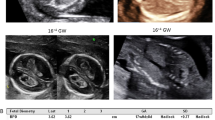
Prenatal ultrasound images. (a) Ultrasonography showed the brain of a normal fetus in sagittal section (a1) and in coronal section (a2). The fetus is of 12 weeks gestation. (b) Ultrasonography at 12th + 3 week of gestation of the brain of the fetal proband in case 1, in sagittal section. An apical hypoechoic bulge about 2.2 × 1.6 cm covered with entire envelope (occipital encephalocele?), incomplete skull, and mixed intracranial construction can be observed (white arrow in panel b1, b2). (c) Ultrasonography at 12th week of gestation of the fetal proband in case 2. An occipital mixed hypoechoic bulge about 1.4 × 0.7 cm in size (occipital encephalocele?) along with incomplete skull can be seen (yellow arrow in panel c1). The midline has disappeared and replaced by multiple anechoic area, one of which is about 0.8 × 0.4 cm
The research was approved by the Ethics Committee of Zhongnan Hospital of Wuhan University [2020053 K]. We obtained written informed consent from all the participant of this study.
Methods
Whole Exome Sequencing and Bioinformatical Analysis
Two milliliters of peripheral blood was taken from parents; EDTA was used for anticoagulation; 1 g of the hip muscle from the fetus were harvested and minced. Genomic DNA was extracted with the method of phenol and chloroform extraction. High-throughput sequencing platform was used to detect the exon regions and flanking intron regions (20 bp) of 20,999 genes in the human whole exome genome. The results were aligned with the reference sequence of the human genome hg38 (GRcH38). Sanger sequencing were used to verify the mutations of family members according to the screening results (Table 1). Primers were designed by the software Primer5 to amplify the exon regions and splicing regions of mutations in CEP290, CC2D2A, and ZNF77 gene. Clinical significance of variants were estimated referring to guideline published by American College of Medical Genetics and Genomics (ACMG).
The referred database and version of predict software are as follows: ClinVar (2020–03-16), DPS6500 (V2), Thousand Genome (Phase3), GnomAD (r2.0.1), ExAC (r0.3.1), BPGD (V3.1), SecondaryFinding_Var (V1.1_2020.3), dbscSNV (1.1), SpliceAI (1.3), dbNSFP (2.9.1), SIFT, MutationTaste, Polyphen2, Phylop, GERP, etc.
Histological Hematoxylin–Eosin Staining Photography
Aborted fetuses were autopsied; their bilateral kidneys, brains, and livers were harvested and stained by hematoxylin–eosin. Tissue blocks were taken to paraffin imbedding and cut into 5 μm sections. The section was placed into xylene to remove the wax, passed through decreasing concentration of alcohol baths (100%, 90%, 80%, 70%) and water, stained in hematoxylin for 3–5 min, and then washed in running tap water until blue. Dip in 1% acid alcohol (1% HCl in 70% alcohol) for a few seconds. Dip in ammonia water until the sections became blue. Stain in 1% Eosin Y for 10 min. The three steps above all follow by tap water washing. Dehydrate in increasing concentration of alcohols. Put slides in two xylene baths for clearing and mount them into DPX. Sections were evaluated with a 200 magnification in the microscope.
Western Blot Analysis of Whole-Brain Tissue of the Probands
Whole brains were harvested from each of the fetus, and a normal fetus of 12 WG aborted from an unmarried pregnancy. RIPA lysis buffer containing HALT Protease and phosphatase inhibitor cocktail solution was used to extract proteins. A bead homogenizer was used for homogenization; afterwards tissue lysates were incubated on ice for 1 h. Then centrifuge proteins at 14,000 g for 30 min at 4℃. The supernatant was added with 2 × sample buffer containing 1 mM dithiothreitol. The Pierce BCA Protein Assay Kit was used to estimate protein. Boil samples for 10 min, and take 30 µg to load on 8–12% SDS-PAGE gels. After all protein were transferred onto PVDF membrane, primary antibodies were added and incubated at 4 °C overnight. Then secondary antibodies were incubated at room temperature for 1 h. Membranes are scanned with gel imager (Peiqing equipment, Shanghai), and proteins are visualized and qualified using Image Labe.
Construction of Recombinant Plasmid, Transfection, and Western Blot Analysis
First, recombinant plasmid PCDNA3.1 should be generated. DNA primers are designed in accordance with human ZNF77 gene and Flag sequencing from GenBank database. Restriction enzyme cutting site are added at the both side of primers. Following the instruction, the primers were used as templates for PCR amplification. Products were separated by 1% agarose gel electrophoresis and recovered by a DNA gel recovery kit. Target genes and PCDNA3.1 vector are both digested by restriction enzymes (EcoRI and XhoI) at 37℃ for 2 h. Mix target genes and vector at a ratio of 3:1 and add DNA ligase T4 at a room temperature for 1 h. E. coli DH5α are employed to plasmid multiplication. Competent E. coli DH5α was transformed with the ligation at 37℃, with 200 r/min and 45 min shaking. The solution of bacteria were cultured at 37℃ in lysogeny broth (LB) medium supplied with 200 μg/ml ampicillin. After overnight culturing, 1% agarose gel electrophoresis and DNA sequencing were performed to verify successfully transformed plasmid.
HEK-293 T cells (ATCC)[14] were cultured in DMEM (11,995,073) supplemented with penicillin (50 μg ml–1) and streptomycin (50 μg ml–1) along with 10% (vol/vol) FBS and placed into 37℃, CO2 5% incubator. The human embryonic kidney (HEK293T) cell line was obtained from China Center for Type Culture Collection (CCTCC, China, http://cctcc.whu.edu.cn/). The medium was replaced every 2 days. When cell fusion reached about 90%, the cells were digested with 0.25% trypsin, collected, and seeded into 96-well flat-bottom cell culture plates for plasmid treatment at 96 cells/well. Cells were randomly divided into three groups: group with over-expressed ZNF77, group with mutated ZNF77, and group with empty vector. Transfections were carried out 18–24 h post-seeding with 30 ng PCDNA3.1 plasmid (on the basis of group) and the lipofection reagent (Neofect transfer reagent) 200 μl and buffer 3 μl per transfection and culture for 24 h.
Evaluation and density qualification were performed by western blot as described by previous step.
Results
Whole Exome Sequencing and Sanger Sequencing Found Variants in CEP290 and CC2D2A and Frameshift Mutation in ZNF77
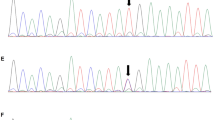
To identify the virulence gene, we performed trio whole exome sequencing and sanger sequencing verification of aborted fetus and their parents. Through WES, the proband of case 1 (III-8) was identified to have a compound heterozygous mutation in CEP290 gene combining a locus c.2340_2341 delGA (p.Asn781*) and a novel locus c.2053-2A > G. Sanger sequencing confirmed that c.2340_2341 delGA (p.Asn781*) came from the father (II-7); c.2053-2A > G came from the mother (II-8). Sanger sequencing verified their heterozygous state, and they all had a normal phenotype (Fig. 3a, b).

a–c: Sanger sequencing peak figure. d, e: Sequence of ZNF77. Variants are noted by red square frame (ps. Frameshift mutation started at chr19:2,939,037. Compared with normal negative chain of chromosome 19.). (a) CEP290 c.2340_2341 delGA:p.Asn781*,exon 22. (b) CEP290 c.2053-2A > G, IVS20. (c) CC2D2A c.4333C > T, exon 35. (d) Frameshift mutation of ZNF77 in case 1. (e) Frameshift mutation of ZNF77 in case 2
c.2340_2341 delGA(p.Asn781*) locates in exon 22 of CEP290, with an estimation as pathologenic (PVS1 + PM2 + PM3_Strong + PP4) on the basis of guideline from The American College of Medical Genetics and Genomics (ACMG)[15]: The variant c.2340_2341delGA (p.Asn781*) located in exon 20 was a frameshift mutation, which is relatively rare. It is predicted that this mutation may lead to the premature amino acid stop code of protein synthesis (PVS1). So far, this mutation has not been reported in the ExAC database and the Thousand People Database (PM2). Morbid variants exist in recessive genetic diseases (PM3). The phenotype and family genetic characteristics of the proband are highly consistent with MKS syndrome (PP4).
c.2053-2A > G is located in the region of intron of CEP290. According to ACMG [15], the locus is evaluated as pathogenic (PVS1 + PM2 + PM3): This variant is located in the classical mRNA splicing region, where the nucleotide sequence is highly conserved. It is predicted that abnormal mRNA splicing and editing might happen, and thus, correct mRNA molecule cannot be formed (PVS1). The variant has not been reported in the ExAC database and the Thousand People Database by now (PM2). In recessive genetic diseases, pathogenic variants (PM3) are detected in the transposition.
In addition, we found a novel variant c.4333C > T(p.R1445X)in exon 35 of CC2D2A. Sanger sequencing confirmed the proband (III-2) to be homozygous nonsense mutation and his parents (II-1, II-2) to be in a heterozygous state (Fig. 3c). Likewise, the parents are with normal phenotype. According to ACMG [15], the locus has been considered as pathogenic (PVS2 + PM2): The variant locates on exon 35 and leads to a nonsense mutation and thus a premature stop codon. Nonsense-mediated decay might account for the lethal phenotype. In addition, it is known that certain loss-of-function (LOF) mutation have already been reported (PVS1). The locus has not been reported in any related literature nor was it included in the gnomAD database (PM2).
In addition, frameshift mutations were detected in exon 2 of ZN77 in both probands (Fig. 3d, e).
Basic Conditions of Patients, Ultrasound, Gross Specimen, and Pathological Examination
In case 1, specimen of aborted body showed that the fetus was 9.5 cm in length and 21.1 g in weight. The head circumference was about 7 cm, and a 1.5 × 1 cm defect was located on cranial bone, from which part of the brain tissue weighing 1.3 g bulge out. Other brain tissue weighs 1.3 g which remain in the skull, with a mixture of structure and vaguely recognizable cerebrum and cerebellum (Fig. 4a). Hematoxylin–eosin staining pathological images of the brain demonstrated local edema, and neuronal degeneration within neurons, glial cells, and primitive neural tubes can be seen as well. Those kidneys demonstrated primary glomerulus and renal tubules especially the later undergoing cystic dilatation (Fig. 5).

Clinical photographs. (a) Proband in case 1, fetus aborted at 12th + 5 week of gestation. The skull is defect, and the brain tissue can be seen (black arrow). (b) Proband in case 2, fetus aborted at 12th + 1 week of gestation. The picture showed encephalocele and polydactyly (black arrow)

Histopathological examination under microscope of essential organs. Normal tissues have been obtained from fetus of 12 weeks gestation and without relative disease. (a) Fetus proband in case 1. Histopathological images showed structure of primary glomerulus and renal tubules in both bilateral kidneys, part of those tubules undergoing cystic dilatation (black arrow in panels 1, 2). Local brain tissue edema and neuronal degeneration within neurons, glial cells, and primitive neural tubes (black arrow in panel 3). Hepatic cells are normal (panel 4) HE staining. (b) Fetus proband in case 2. Extensive cystic renal tissues can be observed, without obvious cortex and medulla, on both side of the kidneys (black arrow in panels 1,2). Image of encephalocele tissues could not find stratification (black arrow in panel 3). Hepatic cells are normal (panel 4). HE staining
In case 2, the gross specimen appeared 9 cm in length and 18.7 g in weight; the head circumference was 6.0 cm, biparietal diameter is 2.0 cm. The fetus had facial and skull hypoplasia, and part of the brain bulged from the cranial notch, about 3.0 × 2.0 × 0.3 cm in volume and 7.0 g in weight. The size of left kidney is about 2.0 × 1.0 × 1.8 cm, and the right is 2.0 × 1.0 × 1.8 cm; altogether the kidneys weigh 1.8 g; extra fingers and toes could be seen on his limbs (Fig. 4b). HE staining pathological images demonstrated concentrated neurons and inapparent stratification; cystic dilatations without obvious cortex and medulla, adrenal could not be found (Fig. 5).
Expression CEP290, CC2D2A, and ZNF77 Were Enhanced in MKS Fetus
We performed western blot utilizing whole-brain tissue from two fetus. In comparison with normal brain, both patients (lane 3 and lane 4) had higher density of CC2D2A an ZNF77, suggesting that the expressions of CC2D2A and ZNF77 were increased (Fig. 6a).

Western blotting. Numbers on the right indicate molecular weight. (a) Western blot analysis of whole-brain tissue from a normal fetus and probands in case 1 and case 2. Compared with control, the density of ZNF77.CEP290 and CC2D2A appeared to decreased. (b, c) Western blot analysis of Flag-tagged ZNF77 and Flag-tagged mutated ZNF77 transfected HEK293cell cell. Lanes 1–3 demonstrate empty plasmid as control, with lanes 4–6 demonstrating ZNF77-transfected cells and lanes 7–9 mutated ZNF77-transfected. Density of the proteins is directly shown by relative intensity in histogram. Remarkable increase is noted by asterisk above cross line; the more asterisks the stronger increase. SHH has increased when ZNF77 was transfected and enhanced more significantly when mutated ZNF77. CC2D2A showed notable increased when mutated ZNF77 was transfected. CEP290 has no obvious change
ZNF77 Plasmid Transfected HEK293T Cells to Convince Correlations Between ZNF77 and CC2D2A, SHH
A histogram was used to quantify the density; remarkable increasing was noted by asterisk above cross line; the more asterisks the stronger increased. Compared to cells transfected by empty vector, SHH had increased in ZNF77 over-expressed cells; nevertheless, CC2D2A and CEP290 did not alter significantly; in ZNF77 mutated cells, SHH had a higher enhancement, along with a notable enhancement of CC2D2A and stable CEP290 (Fig. 6b, c). In other words, over-expressed ZNF77 might promote the expression of SHH; mutated ZNF77 might enhance SHH as well as CC2D2A.
Discussion
MKS is categorized as ciliopathies and represents the most serious conditions. Ciliopathies present a general term of diverse monogenetic disorders displaying abnormal developmental and degenerative symptoms, caused by a hair-like cellular organelle, cilium [16]. German physicist Johann Friedrich Meckel had firstly reported the disease in 1882; he described two siblings with typical features: occipital encephalocele, cystic kidneys, and polydactyly [17]. Later an investigator Gruber noted the same condition [18]. MKS has been reported more frequently since 1960s, and then, gradually up-to-date diagnostic criteria has been refined and abstracted. We referred to a review [19] to make a diagnosis of MKS. Nowadays we acknowledge a classical triad (occipital encephalocele, polydactyly, cystic kidney) as recognizable character of MKS [19], yet it is often controversial whether a diagnosis of MKS should be provided with all of the followings. After all, penetrance rates are not consistent in different case reports, classical triad do not always exist in one case. In our case 1, polydactyly does not appear. Thus, iconography is considered not to be the unique method to detect a MKS fetus; the importance of molecular DNA testing has been emphasized. Meanwhile, since MKS is a rare inherited disease, we could not get plenty of samples. Correspondingly, every sample is important for the research of the disease.
In cilium, a region at the base that links axoneme microtubules and surrounding membranes is called “transition zone” (TZ). Multiple proteins situated in TZ and formed functional complexes, consisting MKS module and NPHP module [20]. Centrosomal protein of 290 kDa (CEP290) locates in TZ, centrosome/basal bodies, and pericentriolar satellites [21, 22]; interacts with other components in TZ; and together serves as barriers at ciliary gate [23].
Mutations of CEP290 lead to a continual phenotypic spectrum, from Leber congenital amaurosis (LCA) which is least severe, Joubert syndrome (JBTS) which is middle severe, to MKS type4, the most severe. Few cases have been reported about CEP290-related MKS. The first variation c.2144 T > G(p.Leu715*) homozygous mutation that causes MKS4 in Chinese population is identified in 2019 [24]. In their case, the proband displayed occipital encephalocele and cystic dilatation, thus diagnosed as MKS and aborted.
In our case 1, the variant c.2340_2341 delGA (p.Asn781*) is a frameshift mutation. Yan Xu et al. had reported correspondence with LCA in 2016 [25]. Lushan LI et al. had reported in two compound heterozygous fetus. Both cases contained the variant; the probands had manifestations of enlarged ventricles, bilateral polycystic kidney dysplasia, and oligohydramnios. They did not show encephalocele; thus, they had not been diagnosed as MKS [26]. In brief, in our case, this variant firstly appeared in a MKS patient. (Fig. 7a).

A map of reported pathogenic variants of MKS. Domains are shown by the strip above and exons are shown below. Orange spots in the middle stand for variants we have counted. We denoted our novel variants by red square box at the position corresponding its location in real amino sequence. (One of them is in intron region so it hasn’t been marked.) Surrounding variants are written out to prove that our variants are novel. (A) CEP290. (B) CC2D2A. Variant information were downloaded from NCBI ClinVar database, and domain information were referred to NCBI protein database, Pfam database, and SMART database
Coiled-coil and C2 domain-containing 2A (CC2D2A) is a member of MKS/B9 module. It contains 3 possible coil domains, a C-terminal C2 domain, a potential CaMKII recognition site, and a PKC phosphorylation site, and 2 possible nuclear localization signals [27]. CC2D2A is a component of the basal body protein complex, forming a ring structure that functions in the transition zone at the base of the cilia. This complex serves as a barrier that restricts the diffusion of proteins between the plasma and the ciliary membrane [28]. In our case 2, the novel variant c.4333C > T (p.R1445X) was located on the 35 exon. A premature stop codon that initiated nonsense-mediated decay might account for the lethal phenotype [29]. The variant we have found in CC2D2A is novel as well as the variants in CEP290 (Fig. 7b).
Western blot analysis of the brain tissue from both probands showed enhanced CEP290, CC2D2A, and ZNF77 in case 1 and case 2 (lane 2 and lane 3). However, as a 100% mortality rare inherited disease, with an early gestational age, we could not get more tissues. Thus, we could not perform statistical analysis.
In addition to three novel variants, our study brings up an effect of ZNF77 in certain MKS-related gene expression and signal pathway. ZNF77 maps to 19 chromosome which carries many ZNF loci and other genes with zinc finger encoding motifs [11]. With a role of transcript factor, proteins of zinc-finger families could combine with DNAs and RNAs [12]. Bioinformatic analysis via human tissue-specific networks suggests ZNF77 controls defensins, elastase, and calmodulin expression, which is firstly described by a research of pathogenic lung colonization [13]. Other literatures brought forward its contributions to early-onset dyslipidemia, myocardial infarction and chronic kidney diseases, etc. [30]. Our research firstly discovered mutations in ZNF77 might have effects on embryogenesis. HEK-293 T cell serves as a widely used cell model; SHH plays a role of a signal molecule from the start of SHH pathway. Its enhanced expression might represent an activated pathway. To further investigate the correlation between ZNF77 and proteins above, we combined ZNF77 and mutated ZNF77 with PCDNA3.1 plasmid to build over-expressed ZNF77-PCDNA3.1 plasmid and mutated ZNF77-PCDNA3.1 plasmid. Afterwards we transfected HEK293T cells with those two plasmids and empty vector to construct over-expressed ZNF77 model and mutated ZNF77 model. Then we proceeded western blot of lysates of HEK293T cells to estimate altered expression of target proteins. Targeted proteins are tagged by Flag; a histogram is used to quantify the density; remarkable increase is noted by asterisk above cross line; the more asterisks the stronger increase. Western blot of transfected HEK293T cells revealed that mutated ZNF77 instead of normal or over-expressed ZNF77 could enhance expression of CC2D2A. We suppose that active ZNF77 regulates the amount of CC2D2A to remain stable, possibly inhibits it from over-expressing. Unaltered CC2D2A in ZNF77-over-expressed cells reveals that the inhibition has nothing to do with normal amount of active ZNF77, as well as an excess. CEP290 staying changeless interprets no markable correlations thereof.
In mammals, Hedgehog is classified into 3 types, desert hedgehog (DHH), Indian hedgehog (IHH), and sonic hedgehog (SHH) [31]. During neurogenesis, SHH plays a role of morphogen, mitogen, and guidance molecule [32]. Lacking of SHH might result in neural tube dorsalization and polydactyly [9]. From embryo period to postnatal growth, SHH constantly takes effects in cerebellar pattering and maturation. Its abnormal expression not only leads to embryo neural defects but contributes to serious neural diseases, such as Parkinson’s disease, autism spectrum disorder, depression, dementia, and stroke as Sita et al. has reviewed [33]. SHH works as the start of SHH signal, primarily being translated into 45 kDa precursor who shortly afterwards auto-proteolytically cleaved into signaling active N-terminus and inactive C-terminus [34]. SHH diffuses from localized source to distant cells to elicit transcriptional responds through ligand that lying on ciliary membrane [35]. Some certain cancers including medulloblastoma (MB) [36] and carcinoma basal cell [37] are mediated by overactive SHH pathway; thus, in neurons, activation of SHH might induce neural recovery. Shh plays a role in the proliferation of cerebellar granule neuron precursor (CGNPs) and regulates its cell cycle by upregulating or maintaining cyclin gene and cyclin-Rb axis in G1 [38]. SHH binds to its specific receptor Ptch1 [39] on ciliary membrane and co-receptors Cdo/Boc and then undergoes endocytosis and lysosomal degradation mediated by Megalin to negatively feedback its concentration [40]. In our study, over-expression of ZNF77 caused a corresponding increase in SHH expression and inactivated ZNF77 protein even greater. We suggest that the enhancement might attribute to a promoted expression or an inhibited degradation, probably not CC2D2A. To be sure, ZNF77 is a factor who effects on SHH.
Taken together, mutated ZNF77 can enhance the expression of CC2D2A as well as SHH, whereas over-expressed ZNF77 simply increases that of SHH. We could come up with a hypothesis of causality between ZNF77 and CC2D2A/SHH, especially a role of impacting factor in SHH pathway. Yet there is other approaches excluding CC2D2A for ZNF77 to effect on SHH pathway. Glioblastoma (Gli) proteins are downstream effectors which contains zinc-finger domains[41], aligning with the same function of ZNF77. More in-depth researches of gene expression context and whether there are some feedback mechanisms can be done in the future.
In conclusion, MKS is a severe inheritance disease which cannot be ignored due to its low incidence. Recently reports had gradually increased along with the development of molecular genetics. Our research also brings about several reasons serving as evidence for prenatal diagnosis of MKS. By now, single gene testing has low sensitivity because of the absence of clear genotype–phenotype correlations for MKS. However, once a morbid variant in a family has been identified, prenatal genetic diagnosis by placental chorionic sampling can be prioritized during subsequent pregnancies. PGT might also be used to optimize embryos, then decrease the risk of induction of labor, and elevate the health quality of population. In addition, the discovery of new mutation sites has enriched the MKS pathogenic genotype spectrum. We have also found the potential effect of ZNF77 on CC2D2A and SHH pathways. All of above have laid a good foundation for the follow-up research on the pathogenic mechanism of MKS.
Availability of Data and Material
All data generated or analyzed during this study are included in this published article.
Code Availability
Not applicable.
References
Logan CV, Abdel-Hamed Z, Johnson CA. Molecular genetics and pathogenic mechanisms for the severe ciliopathies: insights into neurodevelopment and pathogenesis of neural tube defects. Mol Neurobiol. 2011;43(1):12–26. https://doi.org/10.1007/s12035-010-8154-0.
Jha T, Bardhan J, Das B, Patra KK, Dhali B, Seth S. Meckel-Gruber syndrome: a rare clinical entity. J Indian Med Assoc. 2010;108(9):611–2.
Radhakrishnan P, Nayak SS, Shukla A, Lindstrand A, Girisha KM. Meckel syndrome: clinical and mutation profile in six fetuses. Clin Genet. 2019;96(6):560–5. https://doi.org/10.1111/cge.13623.
Parisi MA. The molecular genetics of Joubert syndrome and related ciliopathies: the challenges of genetic and phenotypic heterogeneity. Transl Sci Rare Dis. 2019;4(1–2):25–49. https://doi.org/10.3233/TRD-190041.
Shaheen R, Szymanska K, Basu B, Patel N, Ewida N, Faqeih E, Al Hashem A, Derar N, Alsharif H, Aldahmesh MA, Alazami AM, Hashem M, Ibrahim N, Abdulwahab FM, Sonbul R, Alkuraya H, Alnemer M, Al Tala S, Al-Husain M, Morsy H, Seidahmed MZ, Meriki N, Al-Owain M, AlShahwan S, Tabarki B, Salih MA, Faquih T, El-Kalioby M, Ueffing M, Boldt K, Logan CV, Parry DA, Al Tassan N, Monies D, Megarbane A, Abouelhoda M, Halees A, Johnson CA, Alkuraya FS. Characterizing the morbid genome of ciliopathies. Genome Biol. 2016;17(1):242.
Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett BV, Utsch B, Khanna H, Liu Y, Drummond I, Kawakami I, Kusakabe T, Tsuda M, Ma L, Lee H, Larson RG, Allen SJ, Wilkinson CJ, Nigg EA, Shou C, Lillo C, Williams DS, Hoppe B, Kemper MJ, Neuhaus T, Parisi MA, Glass IA, Petry M, Kispert A, Gloy J, Ganner A, Walz G, Zhu X, Goldman D, Nurnberg P, Swaroop A, Leroux MR, Hildebrandt F. The centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4. Nat Genet. 2006;38(6):674–81.
Noor A, Windpassinger C, Patel M, Stachowiak B, Mikhailov A, Azam M, Irfan M, Siddiqui ZK, Naeem F, Paterson AD, Lutfullah M, Vincent JB, Ayub M. CC2D2A, encoding a coiled-coil and C2 domain protein, causes autosomal-recessive mental retardation with retinitis pigmentosa. Am J Hum Genet. 2008;82(4):1011–8. https://doi.org/10.1016/j.ajhg.2008.01.021.
Drivas TG, Wojno AP, Tucker BA, Stone EM, Bennett J. Basal exon skipping and genetic pleiotropy: A predictive model of disease pathogenesis. Sci Transl Med. 2015;7(291):291ra97. https://doi.org/10.1126/scitranslmed.aaa5370.
Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11(5):331–44. https://doi.org/10.1038/nrg2774.
Machold R, Fishell G. Hedgehog patterns midbrain ARChitecture. Trends Neurosci. 2002;25(1):10–1.
Huebner K, Druck T, LaForgia S, Lasota J, Croce CM, Lanfrancone L, Donti E, Pengue G, La Mantia G, Pelicci PG. Chromosomal localization of four human zinc finger cDNAs. Hum Genet. 1993;91(3):217–22.
Laity JH, Lee BM, Wright PE. Zinc finger proteins: new insights into structural and functional diversity. Curr Opin Struct Biol. 2001;11(1):39–46.
Gago S, Overton NLD, Ben-Ghazzi N, Novak-Frazer L, Read ND, Denning DW, Bowyer P. Lung colonization by Aspergillus fumigatus is controlled by ZNF77. Nat Commun. 2018;9(1):3835. https://doi.org/10.1038/s41467-018-06148-7.
Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–24. https://doi.org/10.1038/gim.2015.30.
Hildebrandt F, Benzing T, Katsanis N. Ciliopathies. N Engl J Med. 2011;364(16):1533–43. https://doi.org/10.1056/NEJMra1010172.
Meckel JF. Beschreibung zweier, durch sehr ähnliche Bildungsabweichungen entstellter Geschwister. Dtsch Arch Physiol. 1822;7:99–172.
Gruber GB. Beiträge zur Frage “gekoppelter” Mißbildungen. Beitr Pathol Anat. 1934;93:459–76.
Salonen R, Paavola P. Meckel syndrome. J Med Genet. 1998;35(6):497–501.
Williams CL, Li C, Kida K, Inglis PN, Mohan S, Semenec L, Bialas NJ, Stupay RM, Chen N, Blacque OE, Yoder BK, Leroux MR. MKS and NPHP modules cooperate to establish basal body/transition zone membrane associations and ciliary gate function during ciliogenesis. J Cell Biol. 2011;192(6):1023–41. https://doi.org/10.1083/jcb.201012116.
Craige B, Tsao C-C, Diener DR, Hou Y, Lechtreck K-F, Rosenbaum JL, Witman GB. CEP290 tethers flagellar transition zone microtubules to the membrane and regulates flagellar protein content. J Cell Biol. 2010;190(5):927–40. https://doi.org/10.1083/jcb.201006105.
Kim J, Krishnaswami SR, Gleeson JG. CEP290 interacts with the centriolar satellite component PCM-1 and is required for Rab8 localization to the primary cilium. Hum Mol Genet. 2008;17(23):3796–805. https://doi.org/10.1093/hmg/ddn277.
Jana SC, Mendonça S, Machado P, Werner S, Rocha J, Pereira A, Maiato H, Bettencourt-Dias M. Differential regulation of transition zone and centriole proteins contributes to ciliary base diversity. Nat Cell Biol. 2018;20(8):928–41. https://doi.org/10.1038/s41556-018-0132-1.
Zhang R, Chen S, Han P, Chen F, Kuang S, Meng Z, Liu J, Sun R, Wang Z, He X, Li Y, Guan Y, Yue Z, Li C, Kumar Dey S, Zhu Y, Banerjee S. Whole exome sequencing identified a homozygous novel variant in CEP290 gene causes Meckel syndrome. J Cell Mol Med. 2020;24(2):1906–16. https://doi.org/10.1111/jcmm.14887.
Xu Y, Xiao X, Li S, Jia X, Xin W, Wang P, Sun W, Huang L, Guo X, Zhang Q. Molecular genetics of Leber congenital amaurosis in Chinese: new data from 66 probands and mutation overview of 159 probands. Exp Eye Res. 2016;149:93–9. https://doi.org/10.1016/j.exer.2016.06.019.
Li L, Fu F, Li R, Xiao W, Yu Q, Wang D, Jing X, Zhang Y, Yang X, Pan M, Liu Z, Liao C. Genetic tests aid in counseling of fetuses with cerebellar vermis defects. Prenatal Diagnosis. 2020;1–11. https://doi.org/10.1002/pd.5732.
Tallila J, Jakkula E, Peltonen L, Salonen R, Kestilä M. Identification of CC2D2A as a Meckel syndrome gene adds an important piece to the ciliopathy puzzle. Am J Hum Genet. 2008;82(6):1361–7. https://doi.org/10.1016/j.ajhg.2008.05.004.
Chih B, Liu P, Chinn Y, Chalouni C, Komuves LG, Hass PE, Sandoval W, Peterson AS. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat Cell Biol. 2011;14(1):61–72. https://doi.org/10.1038/ncb2410.
Nagy E, Maquat LE. A rule for termination-codon position within intron-containing genes: when nonsense affects RNA abundance. Trends Biochem Sci. 1998;23(6):198–9.
Yamada Y, Kato K, Oguri M, Horibe H, Fujimaki T, Yasukochi Y, Takeuchi I, Sakuma J. Identification of 13 novel susceptibility loci for early-onset myocardial infarction, hypertension, or chronic kidney disease. Int J Mol Med. 2018;42(5):2415–36. https://doi.org/10.3892/ijmm.2018.3852.
Echelard Y, Epstein DJ, St-Jacques B, Shen L, Mohler J, McMahon JA, McMahon AP. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75(7):1417–30.
Yam PT, Charron F. Signaling mechanisms of non-conventional axon guidance cues: the Shh, BMP and Wnt morphogens. Curr Opin Neurobiol. 2013;23(6):965–73. https://doi.org/10.1016/j.conb.2013.09.002.
Patel SS, Tomar S, Sharma D, Mahindroo N, Udayabanu M. Targeting sonic hedgehog signaling in neurological disorders. Neurosci Biobehav Rev. 2017;74(Pt A):76–97. https://doi.org/10.1016/j.neubiorev.2017.01.008.
Porter JA, Young KE, Beachy PA. Cholesterol modification of hedgehog signaling proteins in animal development. Science. 1996;274(5285):255–9.
Briscoe J, Small S. Morphogen rules: design principles of gradient-mediated embryo patterning. Development. 2015;142(23):3996–4009. https://doi.org/10.1242/dev.129452.
Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins DN, Chen JK, Cooper MK, Taipale J, Olson JM, Beachy PA. Medulloblastoma growth inhibition by hedgehog pathway blockade. Science. 2002;297(5586):1559–61.
Dahmane N, Lee J, Robins P, Heller P, Ruiz i Altaba A. Activation of the transcription factor Gli1 and the sonic hedgehog signalling pathway in skin tumours. Nature. 1997;389(6653):876–81.
Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol. 2000;20(23):9055–67.
Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Science. 2007;317(5836):372–6.
McCarthy RA, Barth JL, Chintalapudi MR, Knaak C, Argraves WS. Megalin functions as an endocytic sonic hedgehog receptor. J Biol Chem. 2002;277(28):25660–7.
Lee J, Platt KA, Censullo P, Ruiz i Altaba A. Gli1 is a target of Sonic hedgehog that induces ventral neural tube development. Dev. 1997;124(13):2537–52.
Acknowledgements
We thank to the probands and members from the two families we investigated. We are thankful to the Reproductive Medicine Center of Zhongnan Hospital of Wuhan University, the Department of Pathology, and the Department of Ultrasound. We are thankful to Dr. Yan Xiong from the Hubei Transplantation Medicine Key Laboratory of Zhongnan Hospital of Wuhan University, and Pf. Songmei Liu, from the Department of Clinical Laboratory of Zhongnan Hospital of Wuhan University. We are thankful to NCBI ClinVar and SMART database. We are also thankful to Anhui Microanaly Genetech Co., Ltd.
Funding
This study was supported by the Health Commission of Hubei Province Scientific Research Project (WJ2019C002).
Health Commission of Hubei Province Scientific Research Project,WJ2019C002,Yuanzhen Zhang
Author information
Authors and Affiliations
Contributions
Zhidan Hong, Yuanzhen Zhang, and Xuanyi He designed the research. Zhidan Hong and Yuanzhen Zhang made genetic consulting. Zhidan Hong and Xuanyi He performed the experiments. Zhidan Hong made WES and sanger sequencing and analyzed results. Zhidan Hong and Xuanyi He wrote and edited manuscript. Xuanyi He searched the data about reported variants. Fang Yu did autopsy and HE pathologic examination. Huanyu Liu collected and conserved bodies and extracted DNA. Zhidan Hong and Huanyu Liu collected clinical data. Xiaoli Zhang performed ultrasound examination.
Corresponding author
Ethics declarations
Ethical Approval and Consent
The research was approved by the Ethics Committee of Zhongnan Hospital of Wuhan University [2020053 K]. All the procedures performed in the present study were in accordance with the ethical standards of the 1964 Helsinki Declaration. We obtained written informed consent from all the participant of this study.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Conflict of Interest
The authors declare no competing interests.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hong, Z., He, X., Yu, F. et al. Three Novel Variants of CEP290 and CC2D2DA and a Link Between ZNF77 and SHH Signaling Pathway Are Found in Two Meckel-Gruber Syndrome Fetuses. Reprod. Sci. 29, 2322–2332 (2022). https://doi.org/10.1007/s43032-021-00835-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s43032-021-00835-5