
Abstract
The Zig-zag eel (Mastacembelus armatus) is an economically important species in southern China. Its natural resources have declined year by year due to overfishing. Understanding its genetic diversity and population structure is very important for resource conservations. Here, we first successfully developed 28 polymorphic microsatellite markers for zig-zag eels and ten of them were used to examine the genetic diversity and differentiation of 7 populations collected from the major river systems of south China. In total, 224 alleles were found with the 10 microsatellite loci in 7 populations, ranging from 4.6 (Nandujiang: NDJ) to 11.1 (Xijiang, XJ), with an average of 8.871 alleles. The average observed and expected heterozygosity ranged from 0.550 (NDJ) to 0.964 (Yuangjiang, YJ) and from 0.537 (NDJ) to 0.775 (Tanjiang, TJ), respectively. The average polymorphism-information content ranged from 0.472 (NDJ) to 0.757 (TJ). Hardy–Weinberg equilibrium test results revealed the loci showed differing deviation in different populations. In total, low level of genetic diversity was only found in HJ (Hanjiang) and NDJ populations. Besides, evidence of recent bottleneck was found in the HJ populations. Analysis of molecular variation showed that the percent variation within individuals (75.00%) was higher than that among populations (25%). In addition, population structure and the pairwise FST revealed that there was low differentiation among XJ, TJ and YJ populations. These data provide important genetic resources for understanding the population differentiation and facilitating genetic conservation and utilization of this species.
Article highlights
-
(I)
A total of 28 polymorphic microsatellite markers were successfully developed for zig-zag eels.
-
(II)
Low level of genetic diversity was only found in HJ and NDJ populations.
-
(III)
Low differentiation was found among XJ, TJ and YJ populations.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
In recent years, as fossil fuel decreases rapidly and the current crisis of global warming due to the use of fossil fuels, people have to find new renewable “green” energy including wind energy, solar energy, hydroenergy and so on. In order to develop the abundant water resource, control flood and relieve drought, more and more key water-control projects have been constructed especially in south China. However, these projects will affect river ecosystems seriously by disrupting thermal regimes, disconnecting river corridors and modifying aquatic and terrestrial habitats [1, 2]. Meanwhile, with the improvement of people’s living standards, the proportion of fish on the dinner table is also getting higher and higher, which further leads to overfishing. Thus, the wild fishes in river and lakes have been under severe threat.
The Zig-zag eel (Mastacembelus armatus) is an important fish for human consumption that is widely distributed in fresh water rivers of south China. M. armatus belongs to the Mastacembelidae family within the Synbranchiformes order. M. armatus has contributed to the Asian aquaculture economy, with wide distribution in India, Vietnam, Nepal, Malaysia, Southern China, and other parts of South East Asia [3]. As an important economic fish, its populations face some challenges including overfishing, water pollution and habitat destruction. Thus, the wild resource of Zig-zag eel has gradually declined in recent years [4]. Due to these challenges, understanding the present situation of germplasm resources is extremely important for conservation management and sustainable utilization of this species.
Genetic diversity reflects the living and adaptive state of the species, which is critically important for the management and sustainable exploitation of species [5]. High genetic diversity of the species usually means strong adaptability to environmental change. Microsatellites, also named simple sequence repeats, are neutral co-dominant genetic markers [6, 7]. Because of the locus specificity, highly polymorphism and high reproducibility, microsatellites have been widely used in assessment of genetic diversity and population structure in many species [5, 8].
Previous studies have mainly focused on the reproductive biology, breeding, immunity and nutrition of M. armatus [9,10,11,12,13,14,15]. However, few genetic studies have been published, and information regarding genetic variation and population structure for this species is scarce. Yang et al. [16] examined genetic diversity in 11 populations of M. armatus using ISSR (inter-simple sequence repeat), and the results indicated a relatively high diversity of M. armatus. Cytochrome c oxidase sub-unit, cytochrome b and D-loop region of mtDNA and ribosomal S7 introns 1 and 2 of nuclear genome have been used to evaluate its phylogeny and population structure [17,18,19]. Although, its complete mitochondrial and nuclear genomes have been published [20, 21], genetic markers were not well mined. Thus, it’s necessary to get more information on the genetic diversity and population structure of this zig-zag eel.
In this study, we successfully discovered multiple novel microsatellites based on RADseq data. These microsatellites were first tested in 35 individuals from seven wild populations and then ten microsatellites were further used to study the genetic diversity and population differentiation of seven populations of M. armatus. The genetic information obtained would provide valuable information for developing conservation and management strategies of Zig-zag eel.
2 Materials and methods
2.1 Compliance with ethical standards
Guidelines for the care and use of animals at Guangzhou University were followed. All experimental protocols were approved by Special Committee on Science Ethics of Guangzhou University, Guangzhou, China. An Institutional Review Board (IRB) approval (#115/2020, dated 25/10/2020) was obtained.
2.2 Samples and DNA extraction
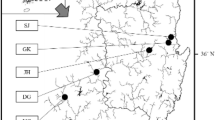
A total of 188 individuals of M. armatus were collected from 7 locations in different river systems in south China (Table S1, Fig. 1), including Beijiang (BJ; n = 30), Xijiang (XJ; n = 30), Dongjiang (DJ; n = 30), Hanjiang (HJ; n = 30), Tanjiang (TJ; n = 21), Nandujiang (NDJ; n = 22) and Yuanjiang (YJ; n = 25). Sampling information including the date acquired, location, and river was shown in Table 1. The XJ, DJ, BJ, and TJ rivers belong to the Pearl River System. Muscle tissues were preserved in 95% ethanol and stored at − 80 °C. Genomic DNA was extracted using the Ezup Column Animal Genomic DNA Purification Kit (Sangon Biotech, Shanghai, China).

Map of the sampling sites where the M. armatus populations were obtained. BJ: Beijiang; XJ: Xijiang; DJ: Dongjiang, HJ: Hanjiang; TJ: Tanjiang; NDJ: Nandujiang; and YJ: Yuanjiang
2.3 Development and characterization of microsatellite markers
According to RADseq data of M. armatus (unpublished data), the potential SSR motifs were identified using MicroSAtellite identification tool (MISA, http://pgrc.ipk-gatersleben.de/misa/) [22]. Twenty-eight primers were randomly selected and synthesized by Sangon Biotech (Shanghai, China) (Table S2). The primers were labelled with a fluorochrome (FAM) and verified on a subset of random 35 individuals from seven wild populations (five in each population). Polymerase chain reaction (PCR) was performed in 25-μl reactions containing 20 ng genomic DNA, 0.5 μM of each primer, 7.5 μl of ddH2O, and 12.5 μl 2 × Taq Master Mix (GDSbio, China). The amplification conditions were as follows: an initial denaturation at 94ºC for 5 min; followed by 30 cycles of 94 °C for 45 s, 58 °C for 25 s, and 72 °C for 30 s; and a final extension at 72 °C for 10 min. The PCR products were first separated by 1.0% agarose gel electrophoresis and then analyzed on ABI3730XL DNA Analyzer (Applied Biosystems). The PCR products were sized using GeneMapper software (version 4.0) with size standards GS500 or GS500Liz. Polymorphic primers were selected for subsequent experiments.
Ten primer sets with high PIC were selected for genotyping all samples in the 7 studied populations (Table 1). One primer in each set was labelled with a fluorophore (FAM, HEX or TAMRA) and used for PCR. PCR products were separated using an ABI3130 DNA Sequencer (Applied Biosystems). PCR was performed by first mixing 20 ng of template DNA, 0.5 μM of forward primer and reverse primer, 5 μl 2 × Taq Master Mix, and 3 μl ddH2O in a total volume of 10 µl. Amplification was performed using the following thermocycling conditions: 94 °C for 5 min; 30 cycles of 94 °C for 30 s, the appropriate annealing temperature (Table 1) for 30 s, and 72 °C for 30 s; with a final extension at 72 °C for 10 min. PCR products were first separated by 1.0% agarose gel electrophoresis and then analyzed on ABI3730XL DNA Analyzer (Applied Biosystems). The PCR products were sized using GeneMapper software (version 4.0) and the size standard was GS500 or GS500Liz.
2.4 Genetic diversity and population structure analysis
Genotyping errors and the possible existence of null alleles were estimated by Microchecker [23]. Genepop (version 4.2) was used to calculate the observed heterozygosity (HO) and expected heterozygosity (HE), the number of alleles at each locus (NA), the effective number of alleles (NE), Nei’s genetic diversity, and the Hardy–Weinberg equilibrium (HWE) of each locus within each population [24]. The polymorphism-information content (PIC) was estimated using PIC-CALC (version 0.6). Population pairwise FST values and analysis of molecular variation (AMOVA) were calculated using Arlequin (version 3.0) [25]. All results for multiple tests were adjusted using Bonferroni's corrections [26]. The gene flow (Nm) between localities was estimated according to the FST using the formula Nm = (1 − FST)/(4 × FST) [25]. Significant correlations between population genetic distances and linear straight geographical distances were assessed using the Mantel test, implemented in TFPGA [27]. The geographic distance was estimated as the shortest water surface distance using Google Maps.
Population differentiation was also examined using the Structure program, with K ranging from 1 to 7 [28]. Ten independent runs for each K value were performed under the admixture-ancestry model with 1,000,000 Markov chain Monte Carlo replications after a burn-in of 100,000 repetitions. The most likely K value was determined using the Structure Harvester program. The number of groups was determined using the delta K method [29]. Recent population-size changes were detected as bottlenecks using the Bottleneck program (version 1.2) [30]. Analysis was performed using a two-phased model of mutation (TPM), a stepwise mutation model (SMM), and an infinite allele model (IAM). Significance was tested using the 2-tailed Wilcoxon signed-rank test and the sign test (1000 iterations, variance for TPM = 50).
3 Results
3.1 Identification of microsatellites and polymorphic analysis
Based on RADseq data, a lot of microsatellites were identified. Then, we randomly selected 28 primer pairs for further polymorphic analysis and the results showed that these microsatellites were all polymorphic. The primer sequences and repeat motifs are listed in Table S2. The average NA per locus was 7.857, ranging from 3 alleles (Mat03, Mat07, Mat15, Mat20, Mat23, Maf07, Maf09, Maf10) to 21 alleles (Mad15). The mean HO and HE values for each locus were 0.513 (0.133–0.867) and 0.604 (0.245–0.944), respectively. Four loci (Mat03, Mat08, Mat20, Mat23) showed significant departure from HWE because of heterozygote deficiency.
The PIC of each locus ranged from 0.236 to 0.928 (mean PIC = 0.596). Seventeen loci were highly informative (PIC > 0.5), 10 loci were reasonably informative (0.5 > PIC > 0.25), and only the Mat20 locus showed a low degree of polymorphism (PIC < 0.25). No evidence of null alleles was detected in the 28 loci.
3.2 Genetic diversity in 7 populations of M. armatus
All 188 eels were successfully genotyped at each of the 10 microsatellite loci. No evidence of genotyping errors was detected across the whole data set. In total, 224 alleles were obtained at the 10 microsatellite loci in several populations of M. armatus. The NA at each locus among the populations ranged from 1 (locus Mad18 in population BJ) to 22 (locus Mad10 in population BJ), with an average number of 8.871 alleles (Table S3). Among all 7 populations, XJ showed the highest NA, with an average number of 11.100 alleles, whereas NDJ had the smallest mean number of alleles (4.600). The HO and HE ranged from 0.550 (NDJ) to 0.964 (YJ) and 0.537 (NDJ) to 0.799 (TJ), respectively. The average HE for all loci was > 0.5 for all populations.
Population genetic analysis of departure from HWE was undertaken to assess the suitability of the 10 selected markers for assessing genetic variability in the 7 M. armatus populations. Loci Mad05 and Mad15 showed a significant departure from HWE in more than 3 populations with heterozygotic deficiency or excess. The numbers of loci that deviated from HWE in the BJ, XJ, TJ, DJ, NDJ, and YJ samples were 3 (Mad02, Mad09, Mad27), 5 (Mad05, Mad10, Mad15, Mad02, Mad27, Mat03), 3 (Mad15, Mad18, Mad29), 3 (Mad05, Mad15, Mad18), 5 (Mad02, Mad05, Mad10, Mad22, Mad29), and 4 (Mad02, Mad05, Mad15, Mat03), respectively. HJ population showed only one locus that deviated from HWE (Mad02). PIC values ranged from 0.033 to 0.937 (mean PIC = 0.664), indicating that most loci have high information content. The highest mean PIC (0.757) and HE values (0.799) were observed in TJ. YJ showed the maximum HO value (0.964).
The results of the bottleneck analysis of M. armatus using 3 tests (IAM, TPM, and SMM) were shown in Table 2. Both Sign and Wilcoxon rank tests revealed that HJ had experienced a recent bottleneck event (P < 0.05). No obvious evidence of a bottleneck was observed in any other samples, including BJ, XJ, YJ, and NDJ.
3.3 Population structure
Pairwise FST values were used to estimate the degree of genetic differentiation among the 7 M. armatus populations, using the 10 microsatellite markers (Table 3). The average FST value was 0.190, and all pairwise FST values among the populations were highly significant (P < 0.01). The largest differentiation was observed between populations BJ and NDJ (0.405), while the FST value between XJ and YJ was smallest haing a value of 0.009. AMOVA showed that 75% of the genetic variation occurred within individuals, whereas 25% of the variation occurred among populations (Table 4). The pairwise Nm value (Table S4) between populations varied between 0.367 (BJ and NDJ) and 27.528 (YJ and XJ). The Nm values between NDJ population and other populations were all substantially less than 1.0.
The population structure was further studied using the Bayesian program Structure Harvester, and the most likely number of genetic clusters was revealed to be 5 (Fig. 2). Structure results supported the division of M. armatus populations into 5 groups: group 1 (XJ, TJ and YJ), group 2 (BJ), group 3 (DJ), group 4 (HJ) and group 5 (NDJ) (Fig. 3). Interestingly, we observed that each of these 5 groupss was significantly differentiated from each other.

Variation trend of lnP (D) values and ΔK values

Structure bar plots (K = 5) for the 10 microsatellite loci of the 7 populations
4 Discussion
Random amplified polymorphic DNA, ISSR, and amplified fragment length polymorphism are valuable markers for analysing the genetic diversity at the species level, but they also have many limitations, such as a low mutation rate. Previous research has indicated that the most appropriate genetic markers for determining the genetic diversity of fish species may be microsatellite markers because they are co-dominantly inherited and are often highly polymorphic [31]. Various studies have used microsatellite markers to conduct genetic diversity and population structure analysis of teleosts, such as Tanakia somjinensis and Trachidermus fasciatus [32, 33]. However, only one such study has been reported for M. armatus [16]. The current study aimed at developing more polymorphic microsatellites and assessing the genetic diversity and population structure of M. armatus populations in 7 geographic sites using polymorphic microsatellites.
4.1 Developing of polymorphic microsatellites
In this study, 28 polymorphic SSR markers of M. armatus were successfully developed. The average Na per locus (7.857), ranging from 3 to 21 alleles was higher than the previous report using ISSR (Na = 1.321, I = 0.168) [16]. Besides, the mean HO and HE values for each locus were 0.513 (0.133–0.867) and 0.604 (0.245–0.944), which were a little lower than the average values (Na = 9.1, Ho = 0.54) reported by DeWoody based on 13 kinds of freshwater fish [34], suggesting relatively low genetic diversity of M. armatus. According to previous reports, lower genetic diversity was also found in other fishes in the Pearl River like Cranoglanis bouderius (Na = 2.04, Ho = 0.448 and He = 0.462) [35], Paramisgurnus dabryanus (Na = 4.833, Ho = 0.265 and He = 0.504) [36] and Misgurnus anguillicaudatus [37], indicating M. armatus might have relatively stronger adaptability than other fishes in the rivers of south China. The PIC of each locus ranged from 0.236 to 0.928 (mean PIC = 0.596), indicating they were highly informative (PIC > 0.5) [38] and could be further used to evaluate the genetic diversity of population.
4.2 Genetic diversity
The genetic diversity of population has an important influence on the environmental adaptation of species. Endangered, endemic and narrowly distributed species usually have low genetic diversities [39]. Of course, not all endangered species have low genetic diversity [40]. Despite the wild resource of zig-zag eel have declined a lot in recent years, our results showed a relatively high-level genetic diversity. Similarly, based on ISSR and part of mtDNA sequence, previous studies also suggested a high level of genetic diversity of zig-zag eel in south China [16, 41]. Besides, the average numbers of Na (4.600–11.100) and Ne (3.470–6.531) in this study were obviously higher than that reported based on ISSR (1.232–1.402, 1.146–1.277) [16], but similar to that reported based on SSR in BJ (4.000–20.000, 1.544–12.766) [42]. Based on our results, the genetic diversity of YJ and TJ populations was relatively higher among seven populations. High genetic diversity of M. armatus in EnPing and Red River, with similar geographic location to TJ and YJ population, was also revealed [16]. However, the level of genetic diversity of NDJ population in Hainan province was the lowest, which was different from previous findings [16]. Zig-zag eels have been cultured for many years in Hainan and wild eels are captured by farmers and used as breeding parents. Thus, low genetic diversity may be due to overfishing. Besides, the genetic diversity of HJ population was also relatively lower and the bottleneck effect was detected. HJ basin is small, and there are problems of overfishing and damming [43]. Previous study also revealed low genetic diversity of Squaliobarbus curriculus and Macropodus opercularis in HJ [44]. Thus, lower genetic diversity of many fish species in this river may be due to the overfishing and damming.
4.3 Population structure
According to AMOVA analysis, there was 25% of variance among populations. Wright (1978) defined the genetic differentiation coefficient, namely FST < 0.05 indicates low genetic differentiation, 0.05 ≤ FST < 0.15 indicates medium genetic differentiation, and FST ≥ 0.15 indicates high genetic differentiation [45]. There was a high degree of genetic differentiation among most populations of zig-zag eel, namely FST ≥ 0.15. NDJ population significantly differentiated from other populations with low gene flow. From a geographical point of view, NDJ is far away from other water systems and only distributes on Hainan Island. Hainan Island has separated from mainland for a long time (about one million years ago). Thus, long periods of geographic isolation led to higher levels of species differentiation from species distributed in mainland [46]. HJ population also showed high genetic differentiation from other populations, which might be related to overfishing and damming [43]. Also, acting as a dependent river may also cause large differentiation of species in HJ from other populations. Strangely, very low genetic differentiation was found between YJ and XJ population, which suggested the two populations of zig-zag eel existed a close correlation. As we can see, YJ locates very closely to the upper reaches of the XJ, it was reasonable to infer that there might be frequent human activity or some unkown connections between YJ and XJ [47]. Genetic structure analysis also suggested that the M. armatus populations could be split into 5 genetic clusters. XJ, TJ, and YJ population grouped together, while other 3 populations formed a separate group, indicating the genetic relationships among YJ, TJ, and XJ populations were very close.
4.4 Conservation implications
Population genetic diversity is very important for the long-term survival and sustainable utilization of species [48]. After a population experiences the bottleneck effect, the genetic variation of its offspring will be included in the few individuals, which further leads to the loss of some alleles, the reduction of genetic diversity, and the increasing frequency of inbreeding, resulting in germplasm degradation. Thus, for HJ and NDJ populations, it’s necessary to perform in situ conservation, and establish nature reserve. Even, artificial breeding should be performed to transplant a lot of juveniles to HJ and NDJ to increase the size of local population. Besides, increasing minimum flows, installation or improvement of fish ladders could also decrease the effect of dam on fish surviving. For other populations with higher level of genetic diversity, only by controlling the fishing intensity, the population size can be effectively restored. Moreover, these populations with high genetic diversity were also used as good resource for aquaculture and breeding of M. armatus. Besides, solving the problem of artificial breeding and aquaculture is also an important means to solve the fishing pressure of wild zig-zag eel.
5 Conclusion
M. armatus is an economically and ecologically important freshwater fish, which is mainly distributed in South China. However, in recent years, the wild population decreased due to a lot of human activities especially the construction of dams and overfishing. In this study, for the first time, we successfully developed 28 polymorphic microsatellite markers for zig-zag eels. Further, ten of them were used to evaluate the genetic diversity and population structure of 7 populations distributed in south China. The results showed low level of genetic diversity was only found in HJ and NDJ populations and evidence of recent bottleneck was found in the HJ populations. Besides, there was low differentiation among XJ, TJ and YJ populations. These results provided important data of genetic diversity of zig-zag eel, which would further facilitate the germplasm conservation and utilization.
References
Poff NL, Allan JD, Bain MB, Karr JR, Prestegaard KL, Richter BD, Sparks RE, Stromberg JC (1997) The natural flow regime. Bioscience 47(11):769–784. https://doi.org/10.2307/1313099
Ward JV, Stanford JA (1983) The serial discontinuity concept of lotic ecosystems. Dyn Lotic Ecosyst 10:29–42
Froese R, Pauly D (2017) Editors. FishBase. World Wide Web electronic publication. www.fishbase.org
Hossain MY, Hossen MA, Yahya K, Ahmed ZF, Sarder MRI, Islam MA, Ahmed KKU (2016) Threatened fishes of the world: Mastacembelus armatus (lacépède, 1800) (synbranchiformes: mastacembelidae). Croat J Fish 73:137–139. https://doi.org/10.14798/73.3.820
Gu S, Yi MR, He XB, Lin PS, Liu WH, Luo ZS, Lin HD, Yan YR (2021) Genetic diversity and population structure of cutlassfish (Lepturacanthus savala) along the coast of mainland China, as inferred by mitochondrial and microsatellite DNA markers. Reg Stud Mar Sci 43:101702. https://doi.org/10.1016/j.rsma.2021.101702
Wambulwa MC, Meegahakumbura MK, Chalo R, Kamunya S, Muchugi A, Xu JC, Liu J, Li DZ, Gao LM (2016) Nuclear microsatellites reveal the genetic architecture and breeding history of tea germplasm of east Africa. Tree Genet Genom 12:1–10. https://doi.org/10.1007/s11295-015-0963-x
Wu FQ, Shen SK, Zhang XJ, Wang YH, Sun WB (2015) Genetic diversity and population structure of an extremely endangered species: the world’s largest Rhododendron. AoB Plants 7:10696–10700. https://doi.org/10.1093/aobpla/plu082
Zhu W, Fu J, Luo M, Wang L, Wang P, Liu Q, Dong Z (2022) Genetic diversity and population structure of bighead carp (Hypophthalmichthys nobilis) from the middle and lower reaches of the Yangtze River revealed using microsatellite markers. Aqua Rep 27:101377. https://doi.org/10.1016/j.aqrep.2022.101377
Bhargava HN (1959) The morphology of the chondrocranium of Mastacembelus armatus (Cuv. & Val.). J Morphol 104:237–267
Zaccone G (1980) Studies on the structure and histochemistry of the epidermis in the air-breathing teleost Mastacembelus armatus (Mastacembelidae, Pisces). Z fur mikroskopisch-anatomische Forsch 95:809–826
Panikkar P, Khan MF, Sharma AP, Jha BC, Vijaykumar ME (2013) Index of relative importance of diet components in Mastacembelus armatus (Lacepède, 1800) from Karapuzha Reservoir, Wayanad, Kerala, India. Indian J Fish 60:37–40
Vankara AP, Vijayalakshmi C (2015) Population dynamics of cestode, Circumonchobothrium shindei (Cestoda: Pseudophyllidea Carus, 1863) in the freshwater eel, Mastacembelus armatus Lacépède, 1800 from River Godavari, Rajahmundry. J Parasit Dis 39:287–291. https://doi.org/10.1007/s12639-013-0345-y
Han C, Li Q, Chen Q, Zhou GF, Huang JR, Zhang Y (2019) Transcriptome analysis of the spleen provides insight into the immunoregulation of Mastacembelus armatus under Aeromonas veronii infection. Fish Shellfish Immunol 88:272–283. https://doi.org/10.1016/j.fsi.2019.02.020
Han C, Li Q, Liu J, Hao ZQ, Huang JR, Zhang Y (2019) Characterization, evolution, and expression analysis of TLR7 gene subfamily members in Mastacembelus armatus (Synbranchiformes: Mastacembelidae). Dev Comp Immunol 95:77–88. https://doi.org/10.1016/j.dci.2019.02.002
Verma SK, Alim A (2015) Ultimobranchial gland respond in a different way in male and female fresh water teleost Mastacembelus armatus (Lacepede) during reproductive cycle. Anim Reprod Sci 156:111–117. https://doi.org/10.1016/j.anireprosci.2015.03.007
Yang HQ, Li Q, Shu H, Yue L, Lin TT, Liu YB (2016) Genetic diversity of Mastacembelus armatus in southern China and surrounding areas based on ISSR analysis. Acta Hydrobiol Sin 40:63–70. https://doi.org/10.7541/2016.9
Khedkar GD, Jamdade R, Naik S, David L, Haymer D (2014) DNA barcodes for the fishes of the Narmada, one of India’s longest rivers. PLoS ONE 9:e101460. https://doi.org/10.1371/journal.pone.0101460
Khedkar GD, Jamdade R, Kalyankar A, Tiknaik A, Ron TB, Haymer D (2014) Genetic fragmentation in India’s third longest river system, the Narmada. Springerplus 3:385. https://doi.org/10.1186/2193-1801-3-385
Brown KJ, Rüber L, Bills R, Day JJ (2010) Mastacembelid eels support Lake Tanganyika as an evolutionary hotspot of diversification. BMC Evol Biol 10:188. https://doi.org/10.1186/1471-2148-10-188
Li Q, Xu R, Shu H, Chen Q, Huang J (2016) The complete mitochondrial genome of the zig-zag eel Mastacembelus armatus (teleostei, mastacembelidae). Mitochondr DNA Part A 27(1):330–331. https://doi.org/10.3109/19401736.2014.892102
Xue L, Gao Y, Wu M, Tian T, Fan H, Huang Y, Huang Z, Li D, Xu LH (2021) Telomere-to-telomere assembly of a fish Y chromosome reveals the origin of a young sex chromosome pair. Genom Biol 22(1):1–20. https://doi.org/10.1186/s13059-021-02430-y
Thiel T, Michalek W, Varshney R, Graner A (2004) Exploiting EST databasesfor the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor Appl Genet 106:411–422. https://doi.org/10.1007/s00122-002-1031-0
Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
Rousset F (2008) Genepop: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106. https://doi.org/10.1111/j.1471-8286.2007.01931.x
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 1:47. https://doi.org/10.1177/117693430500100003
Rice WR (1989) Analyzing tables of statistical tests. Evolution 43:223–225
Miller MP (1997) Tools for population genetic analyses (TFPGA) 1.3: A Windows program for the analysis of allozyme and molecular population genetic data. Comput Softw Distrib Author 4:157
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Earl DA (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Luikart G, Allendorf FW, Sherwin CJM, WB (1998) Distortion of allele frequency distributions provides a test for recent population bottlenecks. J Heredity 89:238–247. https://doi.org/10.1093/jhered/89.3.238
Powell W, Morgante M, Andre C, Hanafey M, Vogel J, Tingey S, Rafalski A (1996) The comparison of RFLP, RAPD, AFLP and SSR (microsatellite) markers for germplasm analysis. Mol Breed 2:225–238. https://doi.org/10.1007/BF00564200
Jeon HB, An J, Kweon SM, Kim S, Yu JN, Kim BJ, Kawase S, Suk HY (2016) Development of novel microsatellite loci and analyses of genetic diversity in the endangered Tanakia somjinensis. Biochem System Ecol 66:344–350. https://doi.org/10.1016/j.bse.2016.05.006
Li YL, Xue DX, Gao TX, Liu JX (2016) Genetic diversity and population structure of the roughskin sculpin (Trachidermus fasciatus Heckel) inferred from microsatellite analyses: implications for its conservation and management. Conserv Genet 17:921–930
DeWoody JA, Avise JC (2000) Microsatellite variation in marine, freshwater and anadromous fishes compared with other animals. J Fish Biol 56(3):461–473. https://doi.org/10.1007/s10592-016-0832-7
Cheng F, Ye W, Ye FL, Xie SG, Long CH (2007) Genetic diversity of Cranglanis bouderius with microsatellite marker. Freshw Fish 37(6):61–64. https://doi.org/10.1111/j.1095-8649.2000.tb00748.x
You CH, Tong JG, Yu XM (2012) Microsatellite DNA analysis on genetic diversity of seven populations of Paramisgurnus dabryanus. J Hydroecol 33(1):84–91. https://doi.org/10.15928/j.1674-3075.2012.01.024
Ke X, Liu J, Gao F, Cao J, Liu Z, Lu M (2022) Analysis of genetic diversity among six dojo loach (Misgurnus anguillicaudatus) populations in the Pearl River Basin based on microsatellite and mitochondrial DNA markers. Aquac Rep 27:101346. https://doi.org/10.1016/j.aqrep.2022.101346
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Zhang ZY, Chen YY, Li DZ (2005) Detection of low genetic variation in a critically endangered Chinese pine, Pinus squamata, using RAPD and ISSR markers. Biochem Genet 43:239–249. https://doi.org/10.1007/s10528-005-5215-6
Wu Q, Zang F, Ma Y, Zheng Y, Zang D (2020) Analysis of genetic diversity and population structure in endangered Populus wulianensis based on 18 newly developed EST-SSR markers. Glob Ecol Conserv 24:e01329. https://doi.org/10.1016/j.gecco.2020.e01329
Li Fen, Chen QP, He PY, Huang XQ, Chen ZK, Zhou HQ, Zhong DM, Shu H (2019) The karyotype pattern and genetic diversity of mitochondrial DNA cyt b gene and D-loop sequence in Mastacembelus armatus Beijing River, south China. Oceanol Limnol Sin 50(2):449–454. https://doi.org/10.11693/hyhz20181100275
Fang ZY, Chen XD, Wu YS, Tan JR, Zhang WE, Wang ZY, Lin TT, Zha GC, Shu H (2018) Screening and characteristic analysis on di-/tri-/tetra-nuclerotide-repeated microsatellites in Mastacembelus armatus. Oceanol et Limnol Sin 19(1):174–182. https://doi.org/10.11693/hyhz20170600172
Zhao S, Yue X (2009) Genetic variation of mitochondrial cytochome b of 3 Sequaliobarbus curriculus populations in south China. Ecol Sci 28(6):528–531. https://doi.org/10.3969/j.issn.1008-8873.2009.06.008
Wang PX, Bai JJ, Hu YC, Mo XD, Wang XJ, Li XH, Song HM, Yang YX, Luo JR (2011) Population genetic variations and phylogeography of Macropodus opercularis. Acta Ecolog Sin 31(2):441–448
Wright S (1978) Evolution and the genetics of populations, vol 1. University of Chicago Press
Macholán M (2001) Multivariate analysis of morphometric variation in Asian Mus and Sub-Saharan Nannomys (Rodentia: Muridae). Zool Anz-J Comp Zool 240(1):7–14. https://doi.org/10.1078/0044-5231-00002
Kang B, Deng J, Huang X, Chen L, Feng Y (2013) Explaining freshwater fish biogeography: history versus environment versus species personality. Rev Fish Biol Fish 23:523–536. https://doi.org/10.1007/s11160-013-9314-x
Qiu YX, Hong DY, Fu CX, Cameron KM (2004) Genetic variation in the endangered and endemic species Changium smyrnioides (Apiaceae). Biochem Syst Ecol 32(6):583–596. https://doi.org/10.1016/j.bse.2003.08.004
Acknowledgements
This research was supported by grants from Seed industry revitalization project funded by the Provincial Rural Revitalization Strategy in 2022 (2022-SPY-00-017), Science and Technology Program of Guangdong Province (2019B030316022), China-ASEAN Maritime Cooperation Fund (CAMC-2018F).
Funding
This research was supported by grants from Seed industry revitalization project funded by the Provincial Rural Revitalization Strategy in 2022 (2022-SPY-00-017), Science and Technology Program of Guangdong Province (2019B030316022), China-ASEAN Maritime Cooperation Fund (CAMC-2018F).
Author information
Authors and Affiliations
Contributions
HS and CH conceived and coordinated the project. TL and MC performed the morphological identification of M. armatus breeds from their respective sample collection areas and collected the samples. JY and ZY performed DNA extraction. TL and JY designed the computational framework of this study. TL and ZY performed the computational analyses and constructed the figures. YF and WQ constructed the SSR sequences. TL, BL, and CH interpreted the results. TL wrote the manuscript. All authors have contributed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no relevant financial or non-financial interests to disclose.
Ethical approval
Guidelines for the care and use of animals at Guangzhou University were followed. All experimental protocols were approved by Special Committee on Science Ethics of Guangzhou University, Guangzhou, China. An Institutional Review Board (IRB) approval (#115/2020, dated 25/10/2020) was obtained.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lin, T., Yang, J., Yu, Z. et al. Genetic diversity and population structure of Mastacembelus armatus in the river systems of southern China revealed by microsatellites. SN Appl. Sci. 5, 307 (2023). https://doi.org/10.1007/s42452-023-05534-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42452-023-05534-1