
Abstract
Neuropathic pain (NP) and its treatment are considered to constitute an unmet need, with a high-multidimensional impact on society and the sufferer. The broad spectrum of opioid analgesics is considered beneficial for acute pain, yet these drugs pose serious controversial issues due to the potential for adverse behavior and a higher chance of tolerance and addiction in long-term use. Opioids like other first-line medications for NP, will not be useful for every patient suffering from chronic NP. However, due to their possible adverse effects, opioids are considered as second- or third-line medications by various guidelines. Therefore, this literature review was conducted to evaluate the status of opioids in NP and to asses if any recent research has shed further evidence on their efficacy or the contrary. The literature reviewed showed that the mechanisms underlying NP, may themselves contribute to the reduced effect of opioids in this condition. Also, various genetic polymorphisms affecting pharmacokinetic and pharmacodynamic factors are discussed, providing further evidence for the variability in opioid response. Although opioids may reduce NP, nociceptive pain tends to be more responsive to opioids compared to NP. Also, opioids seem to be more effective in intermediate term studies of up to 12 weeks and being mostly effective in peripheral NP compared to supraspinal NP and being least effective in central NP. However, there is still no robust evidence that any specific opioid agent is better than any other one for NP, but it is possible that opioids targeting multiple mechanisms may provide benefit. A limitation of many trials is the lack of consideration for the comorbid psychological aspects of NP, which tend to lower opioid analgesia.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Neuropathic pain (NP) is defined as “pain caused by a lesion or disease affecting the somatosensory system” [1] and it is composed of the emotional, cognitive, and the somatosensory alterations which develop post neuropathy, outlasting the initial cause becoming a disease in its own right [2]. Lesions or diseases of the somatosensory nervous system will negatively effect the ascending sensory impulses reaching the spinal cord, the thalamus, and the cerebral cortex. Consequently, there will be alterations in the perception of touch, temperature, position, movement, vibration, pressure, and pain [3].
Burden of Neuropathic Pain
Using the well-validated EQ-5D, 17% of people with NP from a UK general population sample of 4451, classified its impact on quality of life as “worse than death,” [4]. There is evidence showing that the impact of NP is more dependent on its severity than its underlying cause [5]. NP symptoms tend to be refractory to pharmacological treatment, leading to increased health care utilization and drug prescriptions, as shown in a recent nationwide study in the Japanese population [6]. Chronic NP causes more sleep disturbances, anxiety, depression, and an inferior quality of life when compared to chronic nociceptive pain [7].
Epidemiology of Neuropathic Pain
Chronic NP is prevalent in 7–10% in the general population [8] as assessed by the Douleur Neuropathique 4 questions (DN4) [9] and the Leeds Assessment of Neuropathic Symptoms and Signs (LANSS) [10]. Chronic NP commonly affects the neck and upper limbs, lower back, and lower limbs; it affects more women (8%) than men (5.7%), and it affects patients older than 50 years more [11]. Forty percent of all patients attending German pain clinics have NP characteristics [12].
Pathophysiology of Neuropathic Pain
The mechanisms underlying NP are different from nociceptive pain, even though both types of pain can coexist in the same patient. Due to such mechanistic differences, the diagnosis and treatment of NP differ from nociceptive pain [13]. NP can be divided into two main categories: central NP and peripheral NP. Central NP is characterized by lesions to the brain, e.g., cerebrovascular accident, Parkinson disease, or to the spinal cord, e.g., spinal cord injury, syringomyelia, and demyelinating diseases [14].
Peripheral NP is characterized by pathology to the C, Aβ, and Aδ afferent fibers [15] and it is divided into focal and generalized distribution. Generalized peripheral neuropathies include those caused by diabetes mellitus, metabolic dysfunctions, infectious diseases, chemotherapy, immune and inherited neuropathies, and channelopathies. Such patients often present with a “glove and stocking” distribution due to a die-back, length-dependent neuropathies, featuring distal to proximal sensory loss and pain. Pathology involving one or more peripheral nerves or nerve roots leads to focal NP, for example in postherpetic neuralgia, post-traumatic neuropathy, cervical and lumbar radiculopathies, and trigeminal neuralgia [3]. Rare, painful channelopathies include inherited erythromelalgia [16] and paroxysmal extreme pain disorder [17]. Both are caused by mutations of the SCN9A gene which encodes the voltage-gated sodium channel Nav1.7. Mutations in this channel are found in 30% of patients with idiopathic small fiber neuropathy [18].
Not all patients with peripheral neuropathy will develop NP. Only 21% of patients with diabetic neuropathy suffer from NP, but 60% of the patients with severe diabetic neuropathy suffer from NP [19]. The reason for this inconsistent link in NP is altered neuronal electrogenesis [3]. NP can be caused by a multitude of mechanisms which increase the neuronal hyperexcitability, shifting the plastic changes from the periphery to the brain, leading to pain chronification. Altered ion channel function and expression leads to ectopic impulse generation having a pivotal role in the pathophysiology of peripheral NP. The altered electrogenesis will affect second-order nociceptive neuronal function leading to altered inhibitory interneuronal function [3]. Increased sodium channel expression and concomitant loss of potassium channels enhance neuronal excitability and neurotransmitter release leading to a state of peripheral sensitization. The ongoing peripheral afferent barrage causes changes in second-order nociceptive neurons, e.g., in the N-methyl-D-aspartate (NMDA) and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor pathways. Loss of γ-aminobutyric acid (GABA)-releasing inhibitory interneurons can further enhance the function of excitatory neurons within the spinal cord [20]. This can cause low threshold Aβ and Aδ fibers to communicate with second-order nociceptive pathways, therefore expanding the receptive fields in turn leading to central sensitization [21].
The interactions between the neuron and the immune system can further contribute to the sensitization process [22]. Therefore, NP is considered a neuro-immune disorder [23]. Recent evidence showed that nerve injury leads to supraspinal neuroinflammation especially in the emotional regions of the forebrain [24]. The sensory projections from the thalamus to the cortex and the limbic system heighten the severity of pain and increase the psychological comorbidities, e.g., anxiety and depression, of NP [25]. The presence of NP correlated with fear of pain, perceived danger associated with different activities, higher levels of depression and anxiety, with the latter correlating with pain intensity in chronic pain conditions [26].
Projections from the cingulate cortex and amygdala act on the periaqueductal gray to the brainstem and spinal cord, affecting the descending pain modulatory system [25]. In NP, there is a reduction in noradrenergic inhibitions, while serotonin and its receptors are enhanced. The noradrenergic system is responsible for the diffuse noxious inhibitory controls (DNIC), being the human equivalent of conditioned pain modulation (CPM). Therefore, the reduction in noradrenergic inhibition explains the loss or reduction of DNIC post neuropathy [27]. In NP patients, a reduction in DNIC leads to a pro-nociceptive pain profile coupled with a more enhanced temporal summation of painful stimulations compared to non-NP conditions [28]. Less hypersensitivity was observed in animal models of neuropathy with an intact noradrenergic inhibition [27], therefore justifying the use of medication aimed at manipulating the noradrenergic inhibition to enhance the DNIC. Gabapentinoids can have a role in treating a facilitatory pro-nociceptive profile, while serotonin-noradrenaline reuptake inhibitors (SNRI) can be useful in patients with an inhibitory pro-nociceptive profile. Therefore, CPM can be restored with both duloxetine [29] and tapentadol [30].
Neuropathic Pain Assessment
The neuropathic pain special interest group (NeuPSIG) [15] within the International Association for the Study of Pain (IASP) has devised a revised grading system with an adjusted order that is considered the gold standard for diagnosing NP. A history of related neurological disease or lesion and pain distributed in a plausible neuroanatomical way leads to a “possible” NP diagnosis. The presence of sensory alterations including negative and positive sensory signs in the same neuroanatomical plausible distribution increases the grade to “probable” NP, while a diagnostic test, e.g., neurophysiological techniques and quantitative sensory testing (QST) [31], confirming the lesion in the somatosensory nervous system provides a “definite” NP diagnosis. However, contrary to neurophysiological techniques and QST, laser-evoked potentials (LEPs) are considered the most reliable neurophysiological tool to assess nociceptive function as it selectively activates dermal Aδ and C nociceptors [32].
In the NP grading system, pain descriptors, and therefore symptoms, are suggestive but not pathognomonic of NP; however, their combination has a highly discriminative value for NP [15]. The three most common NP symptoms are an ongoing burning pain (65.4%), paroxysmal electric shock-like pain (57.0%), and brush-evoked pain (54.9%) [33], with most patients reporting a coexistence of heterogeneous sensory signs and symptoms [34]. The typical pain descriptors used for NP has led to the development of several screening tools, e.g., DN4 [9] and painDETECT [12], some of which can differentiate between neuropathic and nociceptive pain with high specificity and sensitivity. Nevertheless, pain descriptors reported in NP and nociceptive pain conditions have exhibited considerable overlap [35], e.g., in fibromyalgia [36]. A systematic review found that the DN4 and Neuropathic Pain Questionnaire were the most suitable for clinical use, but as tools, NP screening questionnaires have limited measurement properties [37].
Management of Neuropathic Pain
Pharmacological recommendations have been proposed either for the condition of NP in general [38, 39] or specific NP conditions, such as painful diabetic neuropathies (PDN) [40] and postherpetic neuralgia (PHN) [41]. Despite the similarities, some of the recommendations [40, 42] encountered discrepancies due to the methodology adopted in assessing the evidence. The latest NeuPSIG recommendations on the pharmacological treatment of NP are based on a systematic review and meta-analysis of drug treatments [38].
Management focuses on symptomatic treatment since etiological treatment, e.g., targeting diabetes mellitus, is typically insufficient to relieve NP [3]. Usually, the efficacy of systemic drug treatment for NP is not dependent on the etiology of the underlying disorder. This has led Finnerup et al. [38] to conclude that the recommendations on the pharmacological treatment of NP applies to the general adult population, except in trigeminal neuralgia where specific guidelines exist [43]. Listed in Table 1 are the drug classes and drugs listed as first-, second-, and third- line medications by the NeuPSIG guideline [38]. However, the NeuPSIG guideline [38] noted that specific cancer populations require different opioid use recommendations.
Contrasting to the guideline provided by the NeuPSIG [38], the European Federation of Neurological Societies (EFNS) provided etiology-specific recommendations [43]. The role of opioids in the different NP etiologies in the EFNS guideline are listed below:
-
For PDN, a level A rating was given to oxycodone and tramadol alone or in combination with acetaminophen as second- or third-line treatment.
-
For PHN, a level A rating was given to morphine, oxycodone, and methadone as second- or third-line treatment.
-
For central NP, a level B rating for efficacy was given to opioids, including tramadol [43].
According to the US Food and Drug Administration (FDA), pregabalin is the only analgesic drug patented explicitly for NP due to PHN and PDN. Nonetheless, in off-label NP syndromes, pregabalin failed in one third of the patients [45]. The NICE guidelines [46] address adult NP in general, except for trigeminal neuralgia for which the first line of treatment is carbamazepine. The latter guidelines propose tramadol as an acute rescue medication only, while morphine and cannabis sativa extract should only be started under supervision of a specialist.
Challenges in the Pharmacological Management of Neuropathic Pain
Epidemiological studies state that many patients with NP do not receive appropriate treatment for their pain [47] possibly due to the lack of diagnosis and relatively ineffective drugs. The clinician’s lack of knowledge about NP medications and their appropriate use in clinical practice augments this problem [48]. The use of diagnostic algorithms for NP [38] and screening tools [32] should contribute to reducing diagnostic heterogeneity.
Due to the multitude of pathophysiological changes in NP, there is currently no drug for NP targeting such a broad spectrum of action [45]. Trials of NP medications have reported an increased placebo response which can lead to an underestimation of drug effects [49]. The NNT for 50% pain relief for most NP medications ranges from around 4 to 10, constituting a modest overall outcome [38]. In addition, NP medications including anticonvulsants, antidepressants, and opioids [50], are hampered by adverse central nervous system (CNS) effects [2]. Those patients who fail such treatment are termed to have refractory pain [51]. The inadequate response of NP to various drug therapies constitutes a substantial unmet need and may have substantial consequences regarding psychological or social adjustment [38]. Most of the abovementioned guidelines seem to underestimate the interaction between cognitive, emotional, sociocultural, and physical factors [52], especially stress, anxiety, depression, and catastrophization. These psychological aspects provide a better correlation with perceived pain intensity as compared to the extent of tissue damage [53].
How Opioids Modulate Pain
Opioids can be classified depending on their effect on opioid receptors as agonists, partial agonists, or antagonists, where agonists, e.g., morphine, elicit a maximal response from the receptor, while an antagonist, e.g., naxolone, leads to no response from the receptor. Partial agonist, e.g., buprenorphine, elicits only a partial functional response despite increasing dose of the drug [54].
Opioid Receptors
The classical three opioid receptors are the μ- receptor, δ- receptor, and the κ- receptor, all of which modulate the pain experience. These receptors are all G protein–coupled receptors and are found within the CNS and peripheral tissues [54]. There are three prohormone precursors of the endogenous opioid compounds which bind to these receptors. Proenkephalin is cleaved to met-enkephalin and leu-enkephalin which bind to the δ-opioid receptor. Prodynorphin is metabolized to dynorphin A and B, which bind to the κ-opioid receptor. The μ-opioid receptor is agonized by β-endorphin which is derived from pro-opioimelanocortin (POMC). In addition, POMC can be cleaved into adrenocorticotropic hormone (ACTH), lipotropins, and melanotropins, thus the opioid system is connected to the neuroimmunoendocrine system [55].
The μ-opioid receptor apart from its analgesic effects is responsible for sedation, respiratory depression, bradycardia, physical dependence, nausea, vomiting, and a reduction in gastric motility. Consequently, there is no single μ-opioid receptor–binding ligand having an analgesic action that is not associated with side effects; this occurs since the same μ-opioid receptor mediates both analgesic and side effects [56]. Activation of the δ-opioid receptor causes spinal and supraspinal analgesia, reduction in gastric motility, and psychotomimetic and dysphoric effects [57]. κ-opioid receptor activation may produce spinal analgesia, dysphoria [54], sedation, dyspnea, dependence, and respiratory depression [57]. Despite providing analgesia, compounds derived from selective δ- or κ-opioid receptor compounds have failed due to limited analgesic potency and dysphoric effects as shown in rodents [58].
Nociceptin-opioid receptor (NOP), which was previously known as opioid receptor-like-1 (ORL1), is the fourth G protein–coupled endogenous opioid-like receptor which binds to the endogenous ligand nociceptin (NOC). NOC was previously known as orphanin FQ (OFQ) [59] and it behaves similarly to traditional opioids, causing membrane hyperpolarization via the opening of potassium channels [60], despite not acting on the classical opioid receptors, since NOC lacks the N-terminal tyrosine [61].
Previous NP animal studies found an increase in NOP receptor mRNA in the dorsal root ganglia and spinal cord [62, 63]. Such an upregulation increases the analgesic properties of NOC and NOP receptor ligands in NP conditions [64] compared to classical opioids [65]. Similarly, systemic or spinal administration of NOP agonists in rodent and nonhuman primate models of inflammatory and NP show analgesic effects similar to opioids without the opioid-induced side effects [66]. However, there are fundamental differences in the distribution and localization of the NOC-NOP system between species [67,68,69]. These differences can have a significant role in the translation from preclinical animal models to clinical trials.
Opioid Receptor Heteromerization
Although being highly debated, opioid receptors, especially the μ- and δ-opioid receptors, can functionally interact in vivo leading to the formation of heteromers. Therefore, heteromers are macromolecular complexes composed of at least two functional receptors units that have different biochemical properties compared to the individual components. Opioid heteromers are relatively abundant in nociceptive pathways and many are associated with antinociceptive responses [70]. Heteromers can be potentially targeted with bivalent compounds, one of which is the recently identified CYM51010, being a μ-δ receptor-heteromer-biased agonist. This compound exhibits simultaneous δ-opioid receptor antagonism and μ-opioid receptor agonism simultaneously, thereby separating analgesia from side effects. Such a separation will potentially reduce antinociceptive tolerance compared to chronic morphine administration [71].
Intracellular Events
Despite different functional effects, all the opioid receptors lead to similar intracellular responses. Binding of an opioid agonist to the G protein–coupled opioid receptor on the transmembrane portion of the receptor, causes the α subunit of the G protein to the exchange its bound guanosine diphosphate (GDP) molecule with intracellular guanosine triphosphate (GTP). This allows the dissociation of the α-GTP away from the βγ complex, both of which are free to interact with target proteins. Binding of a classical opioid agonist to its G protein receptor causes an inhibition of adenylyl cyclase which decreases the intracellular cyclic adenosine monophosphate (cAMP) levels. These complexes lead to an increase in potassium conductance through its respective ion channel and inhibit calcium conductance. Therefore, the resultant effect is a reduction in intracellular cAMP, hyperpolarization of the cell involved and specifically in neurons, a reduction in neurotransmitter release, and action potential propagation [54].
Opioid-Mediated Analgesia
Supraspinal sites of opioid action are the periaqueductal gray (PAG) and the rostral ventromedial medulla (RVM). Endorphins and μ-opioids block the release of GABA within the PAG, which normally acts tonically via projection fibers to inhibit serotonergic activity in the nucleus raphe magnus and in the RVM. This system provides analgesia via descending modulation upon the dorsal horn of the spinal cord, which can be reversed by PAG GABA activity. More recent research found that μ-ligands can act directly on RVM cells labeled as “ON” and “OFF” cells. OFF cells are antinociceptive neurons within the RVM being inhibited by GABA . Upon activation by opioid agonists, the OFF cells work at the level of the dorsal horn of the spinal cord, via serotonin, to downregulate the ascending pain signals. Conversely, opioid agonists have an inhibitory effect on the ON cells, thereby reducing ascending nociceptive signals within the spinothalamic pathway [72]. In addition, the placebo effect is a manifestation of endorphin activity, being a μ-receptor agonist within the PAG [73].
At the level of the spinal cord, μ-opioid receptor ligands:
-
1.
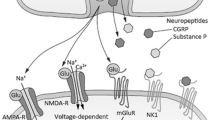
Act directly on the presynaptic neuron within the dorsal horn, specifically Rexed laminae I, II, and V to reduce substance P via suppression of N-type voltage–gated calcium channel activity. Substance P is the primary pain neurotransmitter at this level [74].
-
2.
Inhibit the presynaptic release of glutamate, thereby reducing excitatory neurotransmission [75].
-
3.
In mouse models, μ-ligands reduced the release of calcitonin gene-related peptide (CGRP) from primary afferents [76].
-
4.
At supraphysiologic doses, opioids directly inhibit the voltage-gated sodium channel, and action potential propagation like local anesthetics [77].
-
5.
At the pre-synaptic level, opioids, activate potassium channels on second-order neurons, causing hyperpolarization, thereby reducing action potential transmission [78].
-
6.
Act on nicotinic and muscarinic acetylcholine receptors within the spinal cord, contributing significantly to the analgesic effects [79].
-
7.
Act on the spinal cord through the abovementioned PAG/RVM descending modulation [72].
Genetic Influences on Opioid Analgesia
Opioid analgesia is hindered by a considerable inter-individual difference to obtain the minimal effective analgesic concentration [80]. For example, by using opioid rotation, a better clinical outcome can be obtained in up to 30% of patients who do not respond well to morphine [80]. Differences in pain perception, sociocultural factors, environmental influences, sex, age, and genetics contribute to this interindividual difference [81]. Allelic gene variants involving the opioid and non-opioid systems affect opioid analgesia in humans [82]. However, the number of candidate genes affecting opioid analgesia can increase further since genes coding for pain signaling and modulatory pathways, which affect the individual’s susceptibility to pain can influence overall opioid response [83].
Apart from the analgesic effects of opioids, the adverse effects caused by these analgesics seems to be dependent on genetic alleles which code for the μ- and δ-opioid, dopamine and serotonin receptors [83]. The variability in opioid dosage could have significant clinical consequences due to the narrow therapeutic index on respiratory depression, which can have life-threatening effects. Therefore, the dose needs to be personalized [84].
Pharmacokinetic Factors Affected by Polymorphisms
Drug Transporters
Drug transporters are present in the blood-brain barrier, gastrointestinal tract, liver, and the kidneys. Therefore, drug transporters influence the absorption, distribution, and elimination of opioids [81]. The most well-characterized efflux transporter of the ATP-binding cassette family is the P-glycoprotein (P-gp) transporter. The P-gp is coded by the highly polymorphic ABCB1 (MDR1) gene [85] with over 100 single nucleotide polymorphisms [86]. Therefore, substantial variation in the P-gp expression exists, e.g., hepatic expression of ABCB1 varies 200-fold, and P-gp protein expression varies 20-fold [87, 88]. The pharmacokinetic and pharmacodynamic profile of morphine, methadone, and fentanyl is dependent on P-gp polymorphism which causes varied opioid responses, as shown by in vitro and animal studies [89,90,91].
Drug-Metabolizing Enzymes
Following absorption, most opioids undergo the first-pass effect in the liver through two forms of metabolism, thereby reducing systemic bioavailability [92]:
-
1.
Phase 1 metabolism (modification reactions) involving cytochrome P450 (CYP) enzymes
-
2.
Phase 2 metabolism (conjugation reactions) involving glucuronidation reaction by the enzyme uridine diphosphate glucuronosyltransferase (UGT).
The CYP2D6 pathway is responsible for the metabolism through O-dealkylation of codeine to morphine, tramadol to O-desmethyltramadol, and oxycodone to oxymorphone, being active metabolites too. The CYP2D6 enzyme has more than 63 alleles causing a considerable variation in enzyme function [93] and opioid metabolism [94], leading to a 10,000-fold difference in codeine and tramadol’s analgesia [93]. Approximately, 8–10% of Caucasians and up to 50% of people of Asian descent have an inactive form of this enzyme [95].
CYP2D6 is poorly inducible. Therefore, co-administration of drugs metabolized by CYP2D6 or CYP2D6 inhibitors will alter the enzyme’s metabolic process. Potent CYP2D6 inhibitors can alter a genomic CYP2D6 extensive metabolizer phenotype into a CYP2D6 poor metabolizer phenotype. Strong CYP2D6 inhibitors are bupropion, fluoxetine, paroxetine, cinacalcet, and quinidine, while less potent inhibitors are duloxetine, sertraline, terbinafine, amiodarone, cimetidine, and ritonavir [96]. Consequently, it is important in NP patients with comorbid psychiatric pathologies on pharmacological treatment to avoid drug interactions as these will reduce opioid analgesia.
Despite the expected benefit in extensive or ultra-rapid metabolizers or the expected absence of analgesia in poor metabolizers, the literature provides conflicting evidence. A review [84] found that there is no reliable clinical evidence that codeine provides negligible pain relief in poor metabolizers, while there is enough evidence of an absence of an effect for oxycodone, tramadol, and dihydrocodeine in poor metabolizers. Contrastingly, in health volunteers, oral oxycodone provided 1.5- to 6-fold more analgesia in ultra-rapid metabolizers as compared with extensive metabolizers, while poor metabolizers experienced a 2- to 20-fold less analgesia when compared to extensive metabolizers in experimental pain [97]. Similarly, a randomized, placebo-controlled, double-blinded, crossover experiment in healthy volunteers showed extensive metabolizers using oxycodone obtained more analgesia to electrical stimulation and the cold pressor test when compared to poor metabolizers [98]. Contrarily, in patients who underwent knee arthroscopy, tramadol provided better analgesia in poor metabolizers compared to ultra-rapid and extensive metabolizers [99].
Pharmacodynamic Factors Affected by Polymorphisms
μ-Opioid Receptor Gene (OPRM1)
Different opioids have different relative affinities for each opioid receptor, therefore increasing the analgesic and adverse effect heterogeneity in different persons. Such variation leads to a fivefold difference in morphine dose postoperatively despite similar surgeries [100]. Environmental and genetic factors cause considerable variation in opioid responsiveness. The μ-opioid receptor gene (OPRM1) has over 3000 known polymorphisms [101], causing altered pain sensitivities in 20–30% of the population and varied opioid responses [102].
The single nucleotide polymorphism, A118G nucleotide substitution is the most widely studied of all the OPRM1 polymorphisms. This causes an exchange of asparagine to aspartate at the site of amino acid 40, leading to reduced signaling efficacy and possibly reduced OPRM1 expression [103]. This allele occurs in 10–15% of the Caucasian population [98] and in 40–50% of Asian persons [84]. Therefore, this allele is of considerable importance due to ethnic differences in opioid effectiveness. In the acute postoperative period, morphine dose needs a 30% escalation in heterozygotes variants and a 50% escalation in homozygous variants [104]. A118G alleles pose a more significant contribution in opioid dosage variability, while CYP2D6 ultra metabolizers have a more significant threat from potentially life-threatening adverse effects since both of these polymorphisms are commonest in Asian populations compared to Caucasians [84].
Different G protein–coupled opioid receptor subtypes may be partly responsible for the different inter-individual opioid analgesia. Therefore, if one opioid is unsuccessful, e.g., morphine, opioid rotation using one with different G protein interactions, e.g., buprenorphine and methadone, is warranted [105]. It is hypothesized that compounds promoting G signaling will produce analgesia while avoiding the β-arrestin complex–dependent effects leads to less respiratory depression [106]. The presence of several OPRM1 splice variants leads to a different efficacy of each opioid receptor [107], where each splice variant may exhibit a distinct pharmacological profile explaining the different opioid consumptions between individuals [108]. μ- opioid receptor heterodimerization further diversifies the possible opioid response since dimerization will affect intracellular pathways [109, 110].
Catechol-O-Methyltransferase (COMT) Gene
COMT metabolizes dopamine, epinephrine, and norepinephrine. The most studied polymorph causes an amino acid substitution of valine to methionine causing a threefold to fourfold reduction in enzyme activity [111]. Importantly, allelic variants of the COMT gene can influence opioid effectiveness [82].
Phenotyping
The challenging nature of NP treatment stems from the unpredictable response to drugs to alleviate this condition [43]. This could have occurred due to the heterogeneity of patient phenotypes in clinical trials, reflecting various NP mechanisms [112]. It is increasingly recognized that phenotyping, stratification, sensory profiling [113, 114], and endogenous pain modulation measures [115] might allow for a personalized pain medicine approach, thus enhancing the positive outcomes of NP drug trials.
Phenotyping of patients with PHN predicted a better opioid response in patients having relatively higher heat pain threshold at baseline, loss of peripheral terminals, and younger age [113]. A systematic review [116] showed that intermediate-term studies demonstrate the efficacy of opioids for evoked NP, while in the short-term, opioids can reduce the intensity of dynamic mechanical allodynia and possibly cold allodynia in peripheral NP. Similarly, a review [117] found that opioids can reduce mechanical and cold allodynia as well as pinprick hyperalgesia in NP.
A limitation of many randomized controlled trials (RCTs) is that comorbid psychological conditions are not sufficiently taken into consideration, since poor coping skills and catastrophizing tend to be associated with persistent NP in PHN and with an inadequate response to drugs [118]. Besides, psychological factors including anxiety, depression, fear, stress, fatigue, and sleepiness reduce opioid analgesia [119]. Therefore, future research could provide evidence for the efficacy of psychological interventions as it does for medications [3].
The Problems of Opioid Treatment in Neuropathic Pain
Despite opioids being second- or third-line medications for NP [38], IASP, supports the use of opioids for the short-term treatment of severe acute pain and at the end of life, e. g., in cancer pain since no oral medication provides such immediate and effective analgesia. For non-cancer pain, several guidelines [120, 121] state that opioids should be considered when alternative treatments did not provide enough analgesia, when the patient’s well-being is drastically reduced due to pain and the potential benefit outweighs the possible adverse effects. Furthermore, opioids should only be continued if the patient benefits from the treatment.
There is evidence portraying the effectiveness of morphine [122], methadone [122], hydromorphone [123], levorphanol [124], and transdermal fentanyl [125] in treating NP. Yet, various Cochrane reviews [126,127,128,129,130,131,132] have found insufficient evidence to support or decline the use of the individual opioids in treating NP.
Smith [105] states that “all opioids are not created equally” therefore distinctive opioids which additionally bind to non-opioid receptors may be useful for NP. A systematic review [133] confirmed that opioids could provide clinically meaningful analgesia for a significant proportion of patients with NP. This does not imply that opioids will abolish all NP, just as opioids will not ameliorate all nociceptive pain. Therefore, the aim is not complete analgesia but reducing pain to a tolerable range. They found that short-term studies provide only equivocal evidence regarding the analgesic efficacy of opioids in NP while intermediate-term studies demonstrate significant efficacy of opioids over placebo for NP, which is likely to be clinically significant. This systematic review [133] found that morphine , oxycodone, methadone, and levorphanol provided 20–30% pain intensity reduction in NP. Regardless of providing analgesia, opioids fail to improve physical functioning and disability in NP patients [134].
Despite opioids, e.g., oxycodone and morphine being mildly efficacious [38], the most recent NeuPSIG guideline rated strong opioids as third-line treatment medications [38]. This contrasts with previous recommendations that considered opioids as first- or second-line treatments [135, 136]. The NeuPSIG guideline justified this rating due to the increased risk of abuse especially at high doses [137] and due to the increase in prescription opioid-associated overdose mortality, diversion, misuse, and other opioid-related morbidity [138,139,140,141]. The risk of misuse or addiction in chronic pain, although low (2.6%) in systematic studies [142], may represent a concern about long-term use [143]. However, for short-term analgesia, addiction is a rare occurrence, except among a few highly susceptible individuals [45]. In addition, the long-term use of opioids is limited by various side effects such as constipation, nausea, tolerance, and physical dependence [144]. Opioid dependence is associated with structural and functional changes in brain regions implicated in the regulation of emotions, reward, and motivational functions [145]. Long-term morphine administration may be associated with the development of opioid tolerance [146], hypogonadism, and immunosuppressive changes [143]. The latter occurs since immune cells possess μ-opioid receptors which are activated by morphine, being the most immunosuppressive opioid [147].
Furthermore, opioids can lower the CPM effect, causing a paradoxical increase in pain since opioids can switch descending inhibitory controls to facilitatory ones in the brainstem [148]. A similar increase in pain sensitivity can occur in long-term opioid use due to opioid-induced hyperalgesia in the absence of disease progression [149], where pain escalates as opioid doses are increased [150]. The use of adjunctive analgesia or opioid rotation having antagonism to NMDA or κ-opioid receptor can provide a favorable outcome [151]. Therefore, the NeuPSIG guideline recommends that opioid prescription should be strictly monitored in patients necessitating high doses with risk assessment tools, treatment agreements, and dose tracking [38].
Why Are Opioids Ineffective/Effective in Neuropathic Pain?
Thirty percent of opioid receptors are located in the postsynaptic terminals of interneurons and the dendrites of projection neurons, while the remaining 70% are present in the presynaptic location of Aδ and C-fiber, excluding the Aβ-fibers [152, 153]. This can provide a rationale for the reduced efficacy of opioids in dynamic allodynia, being mediated by Aβ fibers [117].
In an animal model, the μ-opioid receptor agonist, [D-Ala2, N-MePhe4, Gly-ol]-enkephalin (DAMGO), exhibited a higher potency and efficacy at C-fiber synapses rather than at Aδ fiber synapses, explaining the observation that opioids control well C-fiber mediated pain, while higher doses are required to control Aδ driven nociception [154]. This happens since in the resting state only a limited number of Aδ receptors are present on the cell membrane’s surface in nociceptive afferent neurons, while μ-opioid receptors are abundantly present. Therefore, a high dose of δ-opioid receptor agonist is required to activate the μ-opioid receptors, while a low dose of Aδ agonist is required in stimulated neurons, e.g., in NP, in which the δ-opioid receptor is transferred to the cell membrane in a stimulus-dependent manner [155].
There are conflicting studies about whether a neuropathy leads to the upregulation [156] or downregulation of spinal opioid receptors [105]. Functional downregulation or desensitization of dorsal horn μ-opioid receptors, particularly in laminae I and II, was evidenced in nerve-injury neuropathy [157] and PDN [79]. Further evidence for the lack of available functional spinal μ-opioid receptors, either due to downregulation or desensitization, stems from the reduced analgesia of intrathecal opioids compared to peritoneal opioid injection [158] and from the failure of intrathecal opioids in reducing allodynia following neuropathy [157].
In the brain, the μ-opioid receptors are functionally downregulated or desensitized in NP states, which may contribute to the reduced efficacy of opioids in NP [159] evidenced in the thalamus of animal models of NP [160]. In a rat model of NP, there was an inhibition of endogenous analgesia in the PAG due to an increase in presynaptic GABA release. Thus, exogenous opioids will provide less analgesia in NP [161].
Contemporarily, pain is facilitated due to:
-
1.
Increased expression of the α2-δ1 subunit of voltage-dependent calcium channels [162].
-
2.
Activation of the N-methyl-D-aspartate receptor (NMDAR) due to the prolonged ectopic impulses from the injured C-fibers causes the release of glutamate [163]. NMDAR boosts synaptic efficacy, possibly leading to the development of central sensitization in NP models. Therefore, NMDAR blockade could reduce NP and pain due to central sensitization [164].
-
3.
A possible impairment of G-transducer proteins function [165].
-
4.
Opioid receptor heterodimerization leads to a reduction in opioid analgesia in NP [110].
-
5.
An increase in dynorphin A reduces opioid analgesia [166].
-
6.
Enhanced synthesis of cholecystokinin reduces morphine analgesia [167].
Opioids tend to be more effective in nociceptive pain [168], followed by peripheral NP, spinal central NP, and least useful in supraspinal central NP [169]. A higher opioid dose beyond those effective against nociceptive pain can overcome the above problems [168, 170,171,172,173]. The necessity for a higher dose was confirmed by Moulin, [172], in post-thoracotomy pain, where double the dose of buprenorphine was needed to provide 50% pain relief for NP compared to nociceptive pain. However, high-opioid doses increase the risk of tolerance [146].
Various Cochrane reviews focusing on specific opioids, buprenorphine [126], tramadol [127], morphine [128], fentanyl [129], hydromorphone [130], oxycodone [131], and methadone [132], have found insufficient evidence to support or decline the use of the individual opioids in treating NP. However multiple investigators have reported that opioids have similar efficacy for NP [39]. A Cochrane review [175] and a systematic review [176] both concluded that opioid therapy for NP was most effective within the first 12 weeks.
Distinct Pharmacological Aspects of Opioids in Neuropathic Pain
Single-agent opioids most often fail to treat NP effectively. Distinctive opioids that bind to non-opioid receptors that are relevant to the pathophysiology of NP may conceivably be particularly effective against NP, e.g., levorphanol, methadone [174], oxycodone, buprenorphine, tapentadol, and tramadol [105].
Morphine
Morphine’s metabolism through glucuronidation produces morphine-6-glucuronide (M6G) and morphine-3-glucuronide (M3G) in a ratio of 6:1. M6G provides some additional analgesic effects [177], while M3G can cause hyperalgesia [178] and modulate the Toll-like receptor 4 [179], causing a pro-inflammatory effect by the microglia, therefore opposing morphine analgesia [180].
Hydromorphone
Hydromorphone undergoes glucuronidation to hydromorphone-3-glucuronide (H3G), being similar to M3G. H3G is devoid of any analgesic effects, but it can evoke allodynia, myoclonus, and seizures in animal models [181]. A Cochrane review, [130] evaluating hydromorphone for chronic NP only found one small study with a high risk of bias and was deemed “very low quality of evidence.” The authors, therefore, concluded that there was an insufficient evidence to support or deny its use in chronic NP.
Hydrocodone
Hydrocodone is structurally similar to codeine, being a weak μ-opioid receptor agonist. Hydrocodone is a prodrug being dependent on the highly polymorphic CYP2D6 enzyme which demethylates it into hydromorphone. The latter opioid has a much stronger binding to μ-opioid receptors [182].
Oxycodone
Oxycodone is a semisynthetic opioid analgesic with high oral bioavailability. It has similar analgesic properties to morphine when administered orally, despite having less intrinsic activity and less than 20 times the affinity for the μ-opioid receptor compared to morphine [183]. Also, oxycodone needs 3–8 times more concentration compared to morphine, to activate the G protein [184]. Despite all this, oxycodone is equianalgesic compared to morphine when the differences in bioavailability are considered [185].
Oxycodone has predictable pharmacokinetics, fewer side effects in the elderly and in mild-moderate renal impairment [186]. Oxycodone has a low affinity for the δ- and κ-opioid receptors too, thus giving it a unique anti-sedative effect [187]. The major difference between oxycodone and morphine is the passage through the blood-brain barrier. Both opioids are equally hydrophilic, yet oxycodone is actively transported through the endothelial cells of the blood-brain barrier in animal models by a cation-proton antiporter [188] six times more than morphine [189]. This unique mechanism produces a concentration of oxycodone three times higher in the brain compared to the blood [190].
Oxycodone is probably the best-studied opioid in NP. Oxycodone has level 1 evidence of efficacy in PHN and PDN [191] using 10–120 mg doses [43]. A non-systematic, yet comprehensive review of the literature, found three RCTs and one open-label study on oxycodone and NP which supported a significant role for oxycodone in neuropathic cancer pain [192]. Similarly, in NP, oxycodone in combination with anticonvulsants improves health-related quality of life and diminishes the impact of pain on physical activity and improves sleep [191]. However as discussed in the above sections, the doses necessary to reach clinical efficacy may be higher in NP than in nociceptive pain [193].
In an animal model of NP, either spinal or oral administration, of the combined oxycodone with an ultra-low dose of naltrexone markedly decreased mechanical and thermal hypersensitivities over a period of 7 days compared to oxycodone alone while minimizing tolerance to these effects [194]. Controlled-release oxycodone is effective for the management of constant pain, paroxysmal spontaneous pain, and allodynia, which frequently characterize PHN [195]. However, a Cochrane review on modified release oxycodone concluded that there was no good evidence that oxycodone MR works in PDN or PHN or for other NP conditions [131].
Tramadol
Tramadol is an atypical synthetic analog of codeine. It is a racemic mixture of two enantiomers. (+)-tramadol is a μ-opioid agonist and a serotonin reuptake inhibitor, while (−)-tramadol inhibits norepinephrine reuptake. Therefore, the racemic mixture acts as a dual μ-opioid agonist and SNRI. The (+)-tramadol is metabolized by the highly polymorphic CYP2D6 pathway to produce (+)-O-dimethyl tramadol, which has six times higher affinity for the μ-opioid receptor compared to the parent drug [196]. The potency ratio of tramadol compared to morphine is better in NP rather than in nociceptive pain models [197]. A placebo-controlled double-blind trial showed the effectiveness of tramadol in PDN [198]. Also, the combination of tramadol and acetaminophen was more effective than placebo in the management of PDN [199]. Tramadol is useful especially in peripheral NP, while its efficacy is less established in central NP [38]. Therefore, tramadol 200–400 mg/day is categorized as second-line treatment [38, 43]. However, a recent Cochrane review found that 53% of the participants in the reviewed studies experienced at least 50% pain relief with tramadol compared to 30% with placebo, while the calculated NNT was 4.4. Yet, this review concluded that there was low-quality evidence that oral tramadol has a significant beneficial effect in moderate or severe NP [127].
Tapentadol
It provides analgesia by being a dual μ-opioid receptor agonist and norepinephrine reuptake inhibitor [200]. Its action on the μ-opioid receptor is responsible for tapentadol’s antinociceptive effects, while the noradrenaline reuptake inhibition function mediates its antihypersensitive effects [65]. Animal models of NP have portrayed the spinal cord as the critical site of tapentadol’s actions due to the shift from mostly opioid inhibitory mechanisms to noradrenergic inhibition in NP animal models [201]. Compared to tramadol, tapentadol is not dependent on enzymes to provide active metabolites since it is the parent drug that provides the analgesic effect [202].
Methadone
Methadone consists of a racemic mixture of two enantiomers; the R-methadone form has a tenfold higher affinity for μ-opioid receptors with low affinity for δ- and κ-opioid receptor [203]. The S-methadone is an SNRI and a modest NMDA antagonist, therefore potentially useful in severe NP and in reversing opioid tolerance [204]. The SNRI function of S-methadone should be anticipated especially when using methadone in combination with selective serotonin reuptake inhibitors (SSRIs) and tricyclic antidepresants (TCAs). Methadone is dissimilar to standard opioids thereby being potentially useful in patients with “true” morphine allergies. Methadone lacks profound euphoria; however, it has a much shorter analgesic action (4–8 h) compared to its elimination half-life (up to 150 h), potentially increasing the chance of respiratory depression and death. Its metabolism is always variable being primarily metabolized by CYP3A4 and secondarily by CYP2D6 [57]. The metabolites of methadone are not active thereby leading to less hyperalgesia, myoclonus, and neurotoxicity compared to morphine. In the case of NP, R-methadone has been shown to yield greater anti-allodynia action when compared to morphine and oxycodone in animal models [205]. By targeting multiple mechanisms, methadone may be useful to combat NP [206], yet a Cochrane review [132] found no good evidence to support or reject the suggestion that methadone works in any NP condition since this review only found three small studies with different methodolgies and with few participants.
Levorphanol
It agonizes μ-, δ-, and κ-opioid receptors, antagonizes NMDAR (more than ketamine and methadone) and it is an SNRI, thus being potentially useful in NP [207, 208]. Compared to methadone it has a shorter plasma half-life yet longer duration of action and no QTc prolongation risk. It is a viable option in the elderly and in palliative care and it causes less visitis to the emergency departments compared to other opioids [209].
Buprenorphine
Buprenorphine is a partial μ-opioid receptor agonist, κ- and δ-receptor antagonist, an NMDAR antagonist [105], a partial agonist of NOP, it has a different receptor - G protein interaction and selectively activates the neuronal ATP sensitive potassium channels. The partial agonism at NOP mediates analgesic and antinociceptive functions at the spinal cord being useful in NP states where NOP receptors may be upregulated [105]. The concomitant deficient opening of potassium channels in NP conditions is counteracted by buprenorphine, thus contributing further to its analgesic efficacy where other opioids fail [210]. The high lipophilicity permits buprenorphine to act as a potent local anesthetic by blocking voltage-gated sodium channels more than lidocaine, or even bupivacaine, when evaluated under identical experimental conditions [211]. Buprenorphine blocked the generation of action potentials in isolated C-fibers with higher potency and with a slower onset and offset kinetics versus lidocaine [212]. The above multimechanistic features of buprenorphine make it particularly attractive in NP conditions. Through these mechanisms, buprenorphine may provide analgesia in NP [213]. A consensus panel [214] stated that there is significant evidence that buprenorphine effectively relieves NP and offers a distinct benefit. However, a Cochrane review [126] on buprenorphine in NP found insufficient evidence to support or decline it in any NP condition.
Cebranopadol
A novel drug is cebranopadol, which agonizes all opioid and NOP receptors [54]. In animal models, cebranopadol administered through peripheral, spinal, and supraspinal routes exerted potent and efficacious antihyperalgesic, antiallodynic, and antinociceptive effects, therefore being useful in NP [215, 216]. Additionally, animal studies have portrayed a better safety profile of cebranopadol compared to equianalgesic doses of morphine [217]. In a phase II randomized, double-blind, placebo- and active-controlled trial of cebranopadol in LBP patients, with and without NP component, cebranopadol was safe, and it displayed a good analgesic efficacy similar to tapentadol [218].
Drug Combinations and the Cannabinoid Agonists
Owing to the multitude of mechanisms in NP, there is no single drug able to provide complete analgesia. Therefore, the combination of agents acting at different sites and by different mechanisms especially in patients who are unable to receive the maximal dose of a drug forms the basis for rational multimodal polypharmacy [51, 219] increasing the importance of opioids [186]. In NP, the combination of opioids with either an anticonvulsant [220, 221] or a tricyclic antidepressant, e.g., nortriptyline [222] yielded better analgesia over the respective monotherapy while using lower doses of both drugs, thus resulting in less side effects [221].
A recent Cochrane review [223] on cannabis-derived products for chronic NP found that despite all cannabis-based products reduce pain intensity, improve sleep, and reduce psychological distress, yet more people had to stop the medications due to cannabis side effects when compared to placebo. However, the review concluded that there is lack of good evidence that cannabis-derived products reduce chronic NP. Similarly, a systematic review showed that cannabinoids are unlikely to be highly effective for chronic non-cancer pain [224]. In addition, the NeuPSIG found a weak recommendation against the use of cannabinoids in NP due to negative results, potential misuse, abuse, diversion, and long-term mental health risks particularly in susceptible individuals [38].
Despite such negative reviews on cannabinoids, endocannabinoid activity may be critical for morphine’s action [225]. Moreover, there are several similarities between the opioid and the cannabinoid receptor systems:
-
1.
Activation of either receptor leads to similar behavior effects including antinociception, sedation, motor depression, and decreased intestinal motility [226].
-
2.
Colocalization of the μ-opioid receptor and cannabinoid type I receptor (CB1R) on the same neurons in the superficial dorsal horn of the spinal cord increasing the chance of interactions between these receptors [227].
-
3.
Both receptors have similar transduction properties, being G protein–coupled receptors, both activate mitogen-activated protein kinases and inhibit neurotransmitter release through inhibition of calcium channels and activating potassium channels [228].
-
4.
Both receptors are generally found on presynaptic terminals, leading to the inhibition of neurotransmitter release [228].
The acute administration of cannabinoid receptor agonists leads to opioid peptide release, while chronic tetrahydrocannabinol (THC) administration increases endogenous opioid peptide gene expression [229]. Therefore, the combination of low doses of cannabinoid agonists with opioid agonists is attractive. Animal and human studies have shown that there are additive effects between the agonists of these two classes [230,231,232]. The combined use of chronic low doses of THC and morphine leads to an upregulation of opioid receptor protein which may underlie the analgesic synergism of these two agonists [233].
Conclusion
Despite much research conducted on opioids in NP conditions portraying the effectiveness of opioids in NP, previous multiple high-quality reviews provide limited evidence on the effectiveness of opioids in NP [126,127,128,129,130,131,132]. There are instances where opioids can provide analgesia especially in the first 12 weeks of administration [175, 176] and in PHN patients exhibiting a higher heat pain threshold at baseline, loss of peripheral terminals, and younger age [113]. Also, rational polypharmacy, combining opioids with anticonvulsants and tricyclic antidepressants, is advocated in patients who are unable to receive the maximal dose of first-line medications [220,221,222].
The reduced opioid effectiveness can be explained by the vast array of physiological changes that occur in NP causing the concomitant reduction of opioid receptors, while facilitating pro-nociceptive mechanisms which are not targeted by most opioids [3]. The higher prevalence of psychological comorbidities in NP lowers opioid effectiveness [119]. Future studies need to take the emotional and psychological aspects of pain in consideration especially when devising RCTs to honestly asses the biopsychosocial experience of pain and how opioids affect it, and not just the sensory aspects.
Some of the main issues of opioids in NP are the difficulty in translating preclinical to clinical studies, the rise of the opioid epidemic, the associated long-term use of opioids potentially leading to tolerance and dependence [143], coupled with the lack of education on NP in general, let alone the use of opioids in NP [48]. The dual function of the μ-opioid receptor in mediating the analgesic and the adverse side effects [54] presents an obstacle that needs to be targeted by future research, possibly with the use of bivalent compounds [71]. The presence of several genetic polymorphisms acting on opioid function makes a mechanism-based treatment approach harder than predicted [170]. In the future genotyping of NP can provide further information [234], since phenotyping is still far from providing mechanisms, and thus the role of opioids in NP will probably remain in the shadows [235].
References
Treede R, Jensen T, Campbell J, Cruccu G, Dostrovsky J, Griffin J, et al. Neuropathic pain: redefinition and a grading system for clinical and research purposes. Neurology. 2007;70(18):1630–5.
Machelska H. Control of neuropathic pain by immune cells and opioids. CNS Neurol Disord Drug Targets. 2011;10(5):559–70.
Colloca L, Ludman T, Bouhassira D, Baron R, Dickenson A, Yarnitsky D, et al. Neuropathic pain. Nat Rev Dis Prim. 2017;3:17002.
Torrance N, Lawson K, Afolabi E, Bennett M, Serpell M, Dunn K, et al. Estimating the burden of disease in chronic pain with and without neuropathic characteristics: does the choice between the EQ-5D and SF-6D matter? Pain. 2014;155(10):1996–2004.
Doth A, Hansson P, Jensen M, Taylor R. The burden of neuropathic pain: a systematic review and meta-analysis of health utilities. Pain. 2010;149(2):338–44.
Inoue S, Taguchi T, Yamashita T, Nakamura M, Ushida T. The prevalence and impact of chronic neuropathic pain on daily and social life: a nationwide study in a Japanese population. Eur J Pain. 2017;21(4):727–37.
Smith B, Torrance N, Bennett M, Lee A. Health and quality of life associated with chronic pain of predominantly neuropathic origin in the community. Clin J Pain. 2007;23(2):143–9.
van Hecke, O., Austin, S., Khan, R., Smith, B. and Torrance, N. (2014). Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain, 155(4), pp.654–662.
Bouhassira D, Attal N, Alchaar H, Boureau F, Brochet B, Bruxelle J, et al. Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). Pain. 2005;114(1):29–36.
Bennett M. The LANSS Pain Scale: the Leeds assessment of neuropathic symptoms and signs. Pain. 2001;92(1):147–57.
Bouhassira D, Lantéri-Minet M, Attal N, Laurent B, Touboul C. Prevalence of chronic pain with neuropathic characteristics in the general population. Pain. 2008;136(3):380–7.
Freynhagen R, Baron R, Gockel U, Tölle T. painDETECT: a new screening questionnaire to identify neuropathic components in patients with back pain. Curr Med Res Opin. 2006;22(10):1911–20.
Costigan M, Scholz J, Woolf C. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32(1):1–32.
Watson J, Sandroni P. Central neuropathic pain syndromes. Mayo Clin Proc. 2016;91(3):372–85.
Finnerup N, Haroutounian S, Kamerman P, Baron R, Bennett D, Bouhassira D, et al. Neuropathic pain: an updated grading system for research and clinical practice. PAIN. 2016;157(8):1599–606.
Yang Y. Mutations in SCN9A, encoding a sodium channel alpha subunit, in patients with primary erythermalgia. J Med Genet. 2004;41(3):171–4.
Fertleman C, Baker M, Parker K, Moffatt S, Elmslie F, Abrahamsen B, et al. SCN9A mutations in paroxysmal extreme pain disorder: allelic variants underlie distinct channel defects and phenotypes. Neuron. 2006;52(5):767–74.
Faber C, Hoeijmakers J, Ahn H, Cheng X, Han C, Choi J, et al. Gain of function NaV1.7 mutations in idiopathic small fiber neuropathy. Ann Neurol. 2011;71(1):26–39.
Abbott, C., Malik, R., van Ross, E., Kulkarni, J. and Boulton, A. (2011). Prevalence and characteristics of painful diabetic neuropathy in a large community-based diabetic population in the U.K. Diabetes Care, 34(10), pp.2220–2224.
Gagnon, M., Bergeron, M., Lavertu, G., Castonguay, A., Tripathy, S., Bonin, R., Perez-Sanchez, J., Boudreau, D., Wang, B., Dumas, L., Valade, I., Bachand, K., Jacob-Wagner, M., Tardif, C., Kianicka, I., Isenring, P., Attardo, G., Coull, J. and De Koninck, Y. (2013). Chloride extrusion enhancers as novel therapeutics for neurological diseases. Nat Med, 19(11), pp.1524–1528.
Woolf C. Central sensitization: implications for the diagnosis and treatment of pain. Pain. 2011;152(Supplement):S2–S15.
Tsuda M, Beggs S, Salter M, Inoue K. Microglia and intractable chronic pain. Glia. 2012;61(1):55–61.
Martini R, Willison H. Neuroinflammation in the peripheral nerve: cause, modulator, or bystander in peripheral neuropathies? Glia. 2015;64(4):475–86.
Fiore N, Austin P. Are the emergence of affective disturbances in neuropathic pain states contingent on supraspinal neuroinflammation? Brain Behav Immun. 2016;56:397–411.
Navratilova E, Atcherley C, Porreca F. Brain circuits encoding reward from pain relief. Trends Neurosci. 2015;38(11):741–50.
Radat F, Margot-Duclot A, Attal N. Psychiatric co-morbidities in patients with chronic peripheral neuropathic pain: a multicentre cohort study. Eur J Pain. 2013;17(10):1547–57.
Bannister K, Dickenson A. What the brain tells the spinal cord. PAIN. 2016;157(10):2148–51.
Yarnitsky D. Role of endogenous pain modulation in chronic pain mechanisms and treatment. PAIN. 2015;156:S24–31.
Yarnitsky D, Granot M, Nahman-Averbuch H, Khamaisi M, Granovsky Y. Conditioned pain modulation predicts duloxetine efficacy in painful diabetic neuropathy. Pain. 2012;153(6):1193–8.
Niesters M, Proto P, Aarts L, Sarton E, Drewes A, Dahan A. Tapentadol potentiates descending pain inhibition in chronic pain patients with diabetic polyneuropathy. Br J Anaesth. 2014;113(1):148–56.
Backonja M, Attal N, Baron R, Bouhassira D, Drangholt M, Dyck P, et al. Value of quantitative sensory testing in neurological and pain disorders: NeuPSIG consensus. Pain. 2013;154(9):1807–19.
Haanpää M, Attal N, Backonja M, Baron R, Bennett M, Bouhassira D, et al. NeuPSIG guidelines on neuropathic pain assessment. Pain. 2011;152(1):14–27.
Truini A, Garcia-Larrea L, Cruccu G. Reappraising neuropathic pain in humans—how symptoms help disclose mechanisms. Nat Rev Neurol. 2013;9(10):572–82.
Attal N, Fermanian C, Fermanian J, Lanteri-Minet M, Alchaar H, Bouhassira D. Neuropathic pain: are there distinct subtypes depending on the aetiology or anatomical lesion? Pain. 2008;138(2):343–53.
Turk D. and Melzack R (2011) Handbook of pain assessment. New York: London, pp.326–353.
Koroschetz J, Rehm S, Gockel U, Brosz M, Freynhagen R, Tölle T, et al. Fibromyalgia and neuropathic pain - differences and similarities. A comparison of 3057 patients with diabetic painful neuropathy and fibromyalgia. BMC Neurol. 2011;11(1).
Mathieson S, Maher C, Terwee C, Folly de Campos T, Lin C. Neuropathic pain screening questionnaires have limited measurement properties. A systematic review. J Clin Epidemiol. 2015;68(8):957–66.
Finnerup N, Attal N, Haroutounian S, McNicol E, Baron R, Dworkin R, et al. Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. The Lancet Neurology. 2015;14(2):162–73.
Moulin D, Boulanger A, Clark A, Clarke H, Dao T, Finley G, et al. Pharmacological management of chronic neuropathic pain: revised consensus statement from the Canadian Pain Society. Pain Research and Management. 2014;19(6):328–35.
Bril, V., England, J., Franklin, G., Backonja, M., Cohen, J., Del Toro, D., Feldman, E., Iverson, D., Perkins, B., Russell, J. and Zochodne, D. (2011). Evidence-based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology, 76(20), pp.1758–1765.
Argoff C. Review of current guidelines on the care of postherpetic neuralgia. Postgrad Med. 2011;123(5):134–42.
Tan T, Barry P, Reken S, Baker M. Pharmacological management of neuropathic pain in non-specialist settings: summary of NICE guidance. BMJ. 2010;340(mar24 1):c1079–9.
Attal N, Cruccu G, Baron R, Haanpää M, Hansson P, Jensen T, et al. EFNS guidelines on the pharmacological treatment of neuropathic pain: 2010 revision. Eur J Neurol. 2010;17(9):1113–e88.
Balshem H, Helfand M, Schünemann H, Oxman A, Kunz R, Brozek J, et al. GRADE guidelines: 3. Rating the quality of evidence. J Clin Epidemiol. 2011;64(4):401–6.
Yan Y, Li C, Zhou L, Ao L, Fang W, Li Y. Research progress of mechanisms and drug therapy for neuropathic pain. Life Sci. 2017;190:68–77.
Nice.org.uk. (2018). Neuropathic pain in adults: pharmacological management in non-specialist settings | guidance and guidelines | NICE. [online] Available at: https://www.nice.org.uk/guidance/cg173/chapter/Update-information [Accessed 3 Oct. 2018].
Torrance N, Ferguson J, Afolabi E, Bennett M, Serpell M, Dunn K, et al. Neuropathic pain in the community: more under-treated than refractory? Pain. 2013;154(5):690–9.
Martinez V, Attal N, Vanzo B, Vicaut E, Gautier J, Bouhassira D, et al. Adherence of French GPs to chronic neuropathic pain clinical guidelines: results of a cross-sectional, randomized, “e” case-vignette survey. PLoS One. 2014;9(4):e93855.
Lund K, Vase L, Petersen G, Jensen T, Finnerup N. Randomised controlled trials may underestimate drug effects: balanced placebo trial design. PLoS One. 2014;9(1):e84104.
Cruccu G, Truini A. A review of neuropathic pain: from guidelines to clinical practice. Pain and Therapy. 2017;6(S1):35–42.
Gilron I, Baron R, Jensen T. Neuropathic pain: principles of diagnosis and treatment. Mayo Clin Proc. 2015;90(4):532–45.
Torta R, Munari J. Symptom cluster: depression and pain. Surg Oncol. 2010;19(3):155–9.
Castelnuovo G, Giusti E, Manzoni G, Saviola D, Gatti A, Gabrielli S, et al. Psychological considerations in the assessment and treatment of pain in neurorehabilitation and psychological factors predictive of therapeutic response: evidence and recommendations from the Italian consensus conference on pain in neurorehabilitation. Front Psychol. 2016;7.
Pathan H, Williams J. Basic opioid pharmacology: an update. Br J Pain. 2012;6(1):11–6.
Cawley N, Li Z, Loh Y. 60 YEARS OF POMC: biosynthesis, trafficking, and secretion of pro-opiomelanocortin-derived peptides. J Mol Endocrinol. 2016;56(4):T77–97.
Kieffer B, Gavériaux-Ruff C. Exploring the opioid system by gene knockout. Prog Neurobiol. 2002;66(5):285–306.
Deer T, Leong M. Treatment of chronic pain by medical approaches. 1st ed. Springer. In: Pp.116–117; 2015.
Bodnar R. Endogenous opiates and behavior: 2009. Peptides. 2010;31(12):2325–59.
Reinscheid R, Higelin J, Henningsen R, Monsma F, Civelli O. Structures that delineate orphanin fq and dynorphin a pharmacological selectivities. J Biol Chem. 1998;273(3):1490–5.
Connor M, Vaughan C, Chieng B, Christie M. Nociceptin receptor coupling to a potassium conductance in rat locus coeruleus neurones in vitro. Br J Pharmacol. 1996;119(8):1614–8.
Meunier J, Mouledous L, Topham C. The nociceptin (ORL1) receptor: molecular cloning and functional architecture. Peptides. 2000;21(7):893–900.
Briscini L, Corradini L, Ongini E, Bertorelli R. Up-regulation of ORL-1 receptors in spinal tissue of allodynic rats after sciatic nerve injury. Eur J Pharmacol. 2002;447(1):59–65.
Chen Y, Sommer C. Nociceptin and its receptor in rat dorsal root ganglion neurons in neuropathic and inflammatory pain models: implications on pain processing. J Peripher Nerv Syst. 2006;11(3):232–40.
Abdulla F, Smith P. Axotomy reduces the effect of analgesic opioids yet increases the effect of nociceptin on dorsal root ganglion neurons. J Neurosci. 1998;18(23):9685–94.
Schröder W, Vry J, Tzschentke T, Jahnel U, Christoph T. Differential contribution of opioid and noradrenergic mechanisms of tapentadol in rat models of nociceptive and neuropathic pain. Eur J Pain. 2010;14(8):814–21.
Zaveri N. Nociceptin opioid receptor (NOP) as a therapeutic target: progress in translation from preclinical research to clinical utility. J Med Chem. 2016;59(15):7011–28.
Florin S, Meunier J, Costentin J. Autoradiographic localization of [3H]nociceptin binding sites in the rat brain. Brain Res. 2000;880(1–2):11–6.
Kimura Y, Fujita M, Hong J, Lohith T, Gladding R, Zoghbi S, et al. Brain and whole-body imaging in rhesus monkeys of 11C-NOP-1A, a promising PET radioligand for nociceptin/orphanin FQ peptide receptors. J Nucl Med. 2011;52(10):1638–45.
Lohith T, Zoghbi S, Morse C, Araneta M, Barth V, Goebl N, et al. Brain and whole-body imaging of nociceptin/orphanin FQ peptide receptor in humans using the PET ligand 11C-NOP-1A. J Nucl Med. 2012;53(3):385–92.
Massotte D. In vivo opioid receptor heteromerization: where do we stand? Br J Pharmacol. 2014;172(2):420–34.
Gomes I, Fujita W, Gupta A, Saldanha S, Negri A, Pinello C, et al. Identification of a μ-δ opioid receptor heteromer-biased agonist with antinociceptive activity. Proc Natl Acad Sci. 2013;110(29):12072–7.
Heinricher M, Tavares I, Leith J, Lumb B. Descending control of nociception: specificity, recruitment and plasticity. Brain Res Rev. 2009;60(1):214–25.
Eippert F, Bingel U, Schoell E, Yacubian J, Klinger R, Lorenz J, et al. Activation of the opioidergic descending pain control system underlies placebo analgesia. Neuron. 2009;63(4):533–43.
Takasusuki T, Yaksh T. Regulation of spinal substance P release by intrathecal calcium channel blockade. Anesthesiology. 2011;115(1):153–64.
Kumamoto E, Mizuta K, Fujita T. Opioid actions in primary-afferent fibers—involvement in analgesia and anesthesia. Pharmaceuticals. 2011;4(2):343–65.
Baillie L, Schmidhammer H, Mulligan S. Peripheral μ-opioid receptor mediated inhibition of calcium signaling and action potential-evoked calcium fluorescent transients in primary afferent CGRP nociceptive terminals. Neuropharmacology. 2015;93:267–73.
Smith H. Opioid therapy in the 21st century. 1st ed. New York: Oxford University Press; 2013. p. 135–44.
Goodman L, Gilman A, Brunton L, Chabner B, Knollmann B. Goodman & Gilman’s the pharmacological basis of therapeutics. New York: McGraw Hill Medical; 2011. p. 481–525.
Chen S, Pan H. Antinociceptive effect of morphine, but not μ opioid receptor number, is attenuated in the spinal cord of diabetic rats. Anesthesiology. 2003;99(6):1409–14.
Aubrun F, Langeron O, Quesnel C, Coriat P, Riou B. Relationships between measurement of pain using visual analog score and morphine requirements during postoperative intravenous morphine titration. Anesthesiology. 2003;98(6):1415–21.
Nielsen L, Olesen A, Branford R, Christrup L, Sato H, Drewes A. Association between human pain-related genotypes and variability in opioid analgesia: an updated review. Pain Practice. 2014;15(6):580–94.
Smith H. Variations in opioid responsiveness. Pain Physician. 2008;11:237–48.
Branford R, Droney J, Ross J. Opioid genetics: the key to personalized pain control? Clin Genet. 2012;82(4):301–10.
Somogyi A, Coller J, Barratt D. Pharmacogenetics of opioid response. Clinical Pharmacology & Therapeutics. 2014;97(2):125–7.
Somogyi A, Barratt D, Coller J. Pharmacogenetics of opioids. Clinical Pharmacology & Therapeutics. 2007;81(3):429–44.
Marzolini C. Polymorphisms in human MDR1 (P-glycoprotein): recent advances and clinical relevance. Clinical Pharmacology & Therapeutics. 2004;75(1):13–33.
Owen A, Goldring C, Morgan P, Chadwick D, Park B, Pirmohamed M. Relationship between the C3435T and G2677T(a) polymorphisms in the ABCB1 gene and P-glycoprotein expression in human liver. Br J Clin Pharmacol. 2005;59(3):365–70.
Kerb R. Implications of genetic polymorphisms in drug transporters for pharmacotherapy. Cancer Lett. 2006;234(1):4–33.
Drewe J, Ball H, Beglinger C, Peng B, Kemmler A, Schächinger H, et al. Effect of P-glycoprotein modulation on the clinical pharmacokinetics and adverse effects of morphine. Br J Clin Pharmacol. 2001;50(3):237–46.
Kharasch E, Hoffer C, Whittington D. The effect of quinidine, used as a probe for the involvement of P-glycoprotein, on the intestinal absorption and pharmacodynamics of methadone. Br J Clin Pharmacol. 2004;57(5):600–10.
Kharasch E, Hoffer C, Altuntas T, Whittington D. Quinidine as a probe for the role of P-glycoprotein in the intestinal absorption and clinical effects of fentanyl. J Clin Pharmacol. 2004;44(3):224–33.
Drewes A, Jensen R, Nielsen L, Droney J, Christrup L, Arendt-Nielsen L, et al. Differences between opioids: pharmacological, experimental, clinical and economical perspectives. Br J Clin Pharmacol. 2012;75(1):60–78.
Zhou S. Polymorphism of human cytochrome P450 2D6 and its clinical significance. Clin Pharmacokinet. 2009;48(12):761–804.
Ingelman-Sundberg M, Sim S, Gomez A, Rodriguez-Antona C. Influence of cytochrome P450 polymorphisms on drug therapies: pharmacogenetic, pharmacoepigenetic and clinical aspects. Pharmacol Ther. 2007;116(3):496–526.
Stamer U, Stüber F. Genetic factors in pain and its treatment. Curr Opin Anaesthesiol. 2007;20(5):478–84.
Haufroid V, Hantson P. CYP2D6 genetic polymorphisms and their relevance for poisoning due to amfetamines, opioid analgesics and antidepressants. Clin Toxicol. 2015;53(6):501–10.
Samer C, Daali Y, Wagner M, Hopfgartner G, Eap C, Rebsamen M, et al. Genetic polymorphisms and drug interactions modulating CYP2D6 and CYP3A activities have a major effect on oxycodone analgesic efficacy and safety. Br J Pharmacol. 2010;160(4):919–30.
Zwisler S, Enggaard T, Noehr-Jensen L, Pedersen R, Mikkelsen S, Nielsen F, et al. The hypoalgesic effect of oxycodone in human experimental pain models in relation to the CYP2D6 oxidation polymorphism. Basic & Clinical Pharmacology & Toxicology. 2009;104(4):335–44.
Slanar O, Dupal P, Matouskova O, Vondrackova H, Pafko P, Perlik F. Tramadol efficacy in patients with postoperative pain in relation to CYP2D6 and MDR1 polymorphisms. Bratislava Medical Journal. 2012;113(03):152–5.
Klepstad P, Kaasa S, Skauge M, Borchgrevink P. Pain intensity and side effects during titration of morphine to cancer patients using a fixed schedule dose escalation. Acta Anaesthesiol Scand. 2000;44(6):656–64.
Hwang I, Park J, Myung S, Ahn H, Fukuda K, Liao Q. OPRM1 A118G gene variant and postoperative opioid requirement. Anesthesiology. 2014;121(4):825–34.
Klepstad P. Polymorphism in the μ-opioid receptor gene OPRM1 A118G —an example of the enigma of genetic variability behind chronic pain syndromes. Scand J Pain. 2017;5(1):8–9.
Kroslak T, LaForge K, Gianotti R, Ho A, Nielsen D, Kreek M. The single nucleotide polymorphism A118G alters functional properties of the human mu opioid receptor. J Neurochem. 2007;103(1):77–87.
Sia A, Lim Y, Lim E, Ocampo C, Lim W, Cheong P, et al. Influence of mu-opioid receptor variant on morphine use and self-rated pain following abdominal hysterectomy. J Pain. 2013;14(10):1045–52.
Smith H. Opioids and neuropathic pain. Pain Physician. 2012;15:ES93–ES110.
DeWire S, Yamashita D, Rominger D, Liu G, Cowan C, Graczyk T, et al. A G protein-biased ligand at the -opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344(3):708–17.
Pan L, Xu J, Yu R, Xu M, Pan Y, Pasternak G. Identification and characterization of six new alternatively spliced variants of the human μ opioid receptor gene, Oprm. Neuroscience. 2005;133(1):209–20.
Gretton S, Droney J. Splice variation of the mu-opioid receptor and its effect on the action of opioids. Br J Pain. 2014;8(4):133–8.
Hojo M, Sudo Y, Ando Y, Minami K, Takada M, Matsubara T, et al. μ-Opioid receptor forms a functional heterodimer with cannabinoid CB1 receptor: electrophysiological and FRET assay analysis. J Pharmacol Sci. 2008;108(3):308–19.
Jordan B, Devi L. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399(6737):697–700.
Jensen K, Lonsdorf T, Schalling M, Kosek E, Ingvar M. Increased sensitivity to thermal pain following a single opiate dose is influenced by the COMT val158met polymorphism. PLoS One. 2009;4(6):e6016.
Baron R, Förster M, Binder A. Subgrouping of patients with neuropathic pain according to pain-related sensory abnormalities: a first step to a stratified treatment approach. The Lancet Neurology. 2012;11(11):999–1005.
Edwards R, Haythornthwaite J, Tella P, Max M, Raja S. Basal heat pain thresholds predict opioid analgesia in patients with postherpetic neuralgia. Anesthesiology. 2006;104(6):1243–8.
Baron R, Maier C, Attal N, Binder A, Bouhassira D, Cruccu G, et al. Peripheral neuropathic pain: a mechanism-related organizing principle based on sensory profiles. PAIN. 2017;158(2):261–72.
Nir R, Yarnitsky D. Conditioned pain modulation. Current Opinion in Supportive and Palliative Care. 2015;9(2):131–7.
Eisenberg E, McNicol E, Carr D. Efficacy of mu-opioid agonists in the treatment of evoked neuropathic pain: systematic review of randomized controlled trials. Eur J Pain. 2006;10(8):667–7.
Jensen T, Finnerup N. Allodynia and hyperalgesia in neuropathic pain: clinical manifestations and mechanisms. The Lancet Neurology. 2014;13(9):924–35.
Haythornthwaite J, Clark M, Pappagallo M, Raja S. Pain coping strategies play a role in the persistence of pain in post-herpetic neuralgia. Pain. 2003;106(3):453–60.
Edwards R, Dworkin R, Turk D, Angst M, Dionne R, Freeman R, et al. Patient phenotyping in clinical trials of chronic pain treatments. PAIN. 2016;157(9):1851–71.
Dowell D, Haegerich T, Chou R. CDC guideline for prescribing opioids for chronic pain—United States, 2016. JAMA. 2016;315(15):1624.
Chou R, Fanciullo G, Fine P, Adler J, Ballantyne J, Davies P, et al. Clinical guidelines for the use of chronic opioid therapy in chronic noncancer pain. The Journal of Pain. 2009;10(2), pp.113-130.e22.
Raja S, Haythornthwaite J, Pappagallo M, Clark M, Travison T, Sabeen S, et al. Opioids versus antidepressants in postherpetic neuralgia: a randomized, placebo-controlled trial. Neurology. 2002;59(7):1015–21.
Moulin D, Richarz U, Wallace M, Jacobs A, Thipphawong J. Efficacy of the sustained-release hydromorphone in neuropathic pain management: pooled analysis of three open-label studies. Journal of Pain & Palliative Care Pharmacotherapy. 2010;24(3):200–12.
Rowbotham M, Twilling L, Davies P, Reisner L, Taylor K, Mohr D. Oral opioid therapy for chronic peripheral and central neuropathic pain. N Engl J Med. 2003;348(13):1223–32.
Agarwal S, Polydefkis M, Block B, Haythornthwaite J, Raja S. Transdermal fentanyl reduces pain and improves functional activity in neuropathic pain states. Pain Med. 2007;8(7):554–62.
Wiffen P, Derry S, Moore R, Stannard C, Aldington D, Cole P, et al. Buprenorphine for neuropathic pain in adults. Cochrane Database Syst Rev. 2015.
Duehmke R, Derry S, Wiffen P, Bell R, Aldington D, Moore R. Tramadol for neuropathic pain in adults. Cochrane Database Syst Rev. 2017.
Cooper T, Chen J, Wiffen P, Derry S, Carr D, Aldington D, et al. Morphine for chronic neuropathic pain in adults. Cochrane Database Syst Rev. 2017.
Derry S, Stannard C, Cole P, Wiffen P, Knaggs R, Aldington D, et al. Fentanyl for neuropathic pain in adults. Cochrane Database Syst Rev. 2016.
Stannard C, Gaskell H, Derry S, Aldington D, Cole P, Cooper T, et al. Hydromorphone for neuropathic pain in adults. Cochrane Database Syst Rev. 2016.
Gaskell H, Derry S, Stannard C, Moore R. Oxycodone for neuropathic pain in adults. Cochrane Database Syst Rev. 2016.
McNicol E, Ferguson M, Schumann R. Methadone for neuropathic pain in adults. Cochrane Database Syst Rev. 2017.
Eisenberg E, McNicol E, Carr D. Efficacy and safety of opioid agonists in the treatment of neuropathic pain of nonmalignant origin. JAMA. 2005;293(24):3043.
Bostick G, Toth C, Carr E, Stitt L, Morley-Forster P, Clark A, et al. Physical functioning and opioid use in patients with neuropathic pain. Pain Med. 2015;16(7):1361–8.
Dworkin R, Backonja M, Rowbotham M, Allen R, Argoff C, Bennett G, et al. Advances in neuropathic pain. Arch Neurol. 2003;60(11):1524.
Guyatt G, Oxman A, Vist G, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ. 2008;336(7650):924–6.
Edlund M, Martin B, Russo J, Devries A, Braden J and Sullivan M. The role of opioid prescription in incident opioid abuse and dependence among individuals with chronic non-cancer pain. The Clinical Journal of Pain. 2013 p.1.
Fischer B, Jones W, Urbanoski K, Skinner R, Rehm J. Correlations between prescription opioid analgesic dispensing levels and related mortality and morbidity in Ontario, Canada, 2005-2011. Drug and Alcohol Review. 2013;33(1):19–26.
Bohnert A, Ilgen M, Trafton J, Kerns R, Eisenberg A, Ganoczy D and Blow F. Trends and regional variation in opioid overdose mortality among veterans health administration patients, fiscal year 2001 to 2009. The Clinical Journal of Pain, 2013 p.1.
Giraudon I, Lowitz K, Dargan P, Wood D, Dart R. Prescription opioid abuse in the UK. Br J Clin Pharmacol. 2013;76(5):823–4.
Ray W, Chung C, Murray K, Hall K, Stein C. Prescription of long-acting opioids and mortality in patients with chronic noncancer pain. JAMA. 2016;315(22):2415.
Portenoy R, Farrar J, Backonja M, Cleeland C, Yang K, Friedman M, et al. Long-term use of controlled-release oxycodone for noncancer pain: results of a 3-year registry study. Clin J Pain. 2007;23(4):287–99.
Dworkin R, O'Connor A, Audette J, Baron R, Gourlay G, Haanpää M, et al. Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc. 2010;85(3):S3–S14.
Bailey C, Connor M. Opioids: cellular mechanisms of tolerance and physical dependence. Curr Opin Pharmacol. 2005;5(1):60–8.
Upadhyay J, Maleki N, Potter J, Elman I, Rudrauf D, Knudsen J, et al. Alterations in brain structure and functional connectivity in prescription opioid-dependent patients. Brain. 2010;133(7):2098–114.
Mika J, Wawrzczak-Bargiela A, Osikowicz M, Makuch W, Przewlocka B. Attenuation of morphine tolerance by minocycline and pentoxifylline in naive and neuropathic mice. Brain Behav Immun. 2009;23(1):75–84.
Mellon R, Bayer B. Evidence for central opioid receptors in the immunomodulatory effects of morphine: review of potential mechanism(s)of action. J Neuroimmunol. 1998;83(1–2):19–28.
Goubert D, Danneels L, Cagnie B, Van Oosterwijck J, Kolba K, Noyez H, et al. Effect of pain induction or pain reduction on conditioned pain modulation in adults: a systematic review. Pain Practice. 2014;15(8):765–77.
Crofford L. Adverse effects of chronic opioid therapy for chronic musculoskeletal pain. Nat Rev Rheumatol. 2010;6(4):191–7.
Vorobeychik Y, Chen L, Bush M, Mao J. Improved opioid analgesic effect following opioid dose reduction. Pain Med. 2008;9(6):724–7.
Velayudhan A, Bellingham G, Morley-Forster P. Opioid-induced hyperalgesia. Continuing Education in Anaesthesia Critical Care & Pain. 2014;14(3):125–9.
Besse D, Lombard M, Zajac J, Roques B, Besson J. Pre- and postsynaptic distribution of μ, δ and κ opioid receptors in the superficial layers of the cervical dorsal horn of the rat spinal cord. Brain Res. 1990;521(1–2):15–22.
Besse D, Lombard M, Zajac J, Roques B, Besson J. Pre- and postsynaptic location of mu, delta and kappa opioid receptors in the superficial layers of the dorsal horn of the rat spinal cord. Prog Clin Biol Res. 1990;328:183–6.
Heinke B, Gingl E, Sandkuhler J. Multiple targets of -opioid receptor-mediated presynaptic inhibition at primary afferent a - and C-fibers. J Neurosci. 2011;31(4):1313–22.
Zhang X, Bao L, Li S. Opioid receptor trafficking and interaction in nociceptors. Br J Pharmacol. 2014;172(2):364–74.
Walczak J, Pichette V, Leblond F, Desbiens K, Beaulieu P. Behavioral, pharmacological and molecular characterization of the saphenous nerve partial ligation: a new model of neuropathic pain. Neuroscience. 2005;132(4):1093–102.
Porreca F, Tang Q, Bian D, Riedl M, Elde R, Lai J. Spinal opioid mu receptor expression in lumbar spinal cord of rats following nerve injury. Brain Res. 1998;795(1–2):197–203.
Lee Y, Chaplan S, Yaksh T. Systemic and supraspinal, but not spinal, opiates suppress allodynia in a rat neuropathic pain model. Neurosci Lett. 1995;199(2):111–4.
Niikura K, Narita M, Butelman E, Kreek M, Suzuki T. Neuropathic and chronic pain stimuli downregulate central μ-opioid and dopaminergic transmission. Trends Pharmacol Sci. 2010;31(7):299–305.
Hoot M, Sim-Selley L, Selley D, Scoggins K, Dewey W. Chronic neuropathic pain in mice reduces μ-opioid receptor-mediated G-protein activity in the thalamus. Brain Res. 2011;1406:1–7.
Hahm E, Kim Y, Lee J, Cho Y. GABAergic synaptic response and its opioidergic modulation in periaqueductal gray neurons of rats with neuropathic pain. BMC Neurosci. 2011;12(1):41.
Narita M, Nakajima M, Miyoshi K, Narita M, Nagumo Y, Miyatake M, et al. Role of spinal voltage-dependent calcium channel α2δ-1 subunit in the expression of a neuropathic pain-like state in mice. Life Sci. 2007;80(22):2015–24.
Larsen B, Mau K, Padilla M, Enciso R. NMDA receptor antagonists for the treatment of neuropathic pain compared to placebo: a systematic review and meta-analysis. Open Journal of Dentistry and Oral Medicine. 2017;5(4):59–71.
Hashimoto K. The NMDA receptors. 1st ed. Cham: Springer International Publishing; 2017.
Sánchez-Blázquez P, Gómez-Serranillos P, Garzón J. Agonists determine the pattern of G-protein activation in μ-opioid receptor-mediated supraspinal analgesia. Brain Res Bull. 2001;54(2):229–35.
Vanderah T, Ossipov M, Lai J, Malan P, Porreca F. Mechanisms of opioid-induced pain and antinociceptive tolerance: descending facilitation and spinal dynorphin. Pain. 2001;92(1):5–9.
Wiesenfeld-Hallin Z, de Araúja Lucas G, Alster P, Xu X, Hökfelt T. Cholecystokinin/opioid interactions. Brain Res. 1999;848(1–2):78–89.
Mercadante S, Gebbia V, David F, Aielli F, Verna L, Casuccio A, et al. Tools for identifying cancer pain of predominantly neuropathic origin and opioid responsiveness in cancer patients. J Pain. 2009;10(6):594–600.
Smith H, Meek P. Pain responsiveness to opioids: central versus peripheral neuropathic pain. Journal of Opioid Management. 2011;7(5):391.
Gilron I, Jensen T, Dickenson A. Combination pharmacotherapy for management of chronic pain: from bench to bedside. The Lancet Neurology. 2013;12(11):1084–95.
Mehta A, Halder S, Khanna N, Tandon O, Singh U, Sharma K. Role of NMDA and opioid receptors in neuropathic pain induced by chronic constriction injury of sciatic nerve in rats. J Basic Clin Physiol Pharmacol. 2012;23(2).
Moulin D. Opioid analgesics in the management of neuropathic pain. European Journal of Pain Supplements. 2007;1(S1):57–60.
Mizoguchi H, Watanabe C, Yonezawa A, Sakurada S. Chapter 19 new therapy for neuropathic pain. Int Rev Neurobiol. 2009:249–60.
Zorn K, Fudin J. Treatment of neuropathic pain: the role of unique opioid agents. Practical Pain Management. 2011;11(4):26–33.
McNicol E, Midbari A, Eisenberg E. Opioids for neuropathic pain. Cochrane Database Syst Rev. 2013.
Sommer C, Welsch P, Klose P, Schaefert R, Petzke F, Häuser W. Opioide bei chronischem neuropathischem Schmerz. Schmerz. 2014;29(1):35–46.
Sverrisdóttir E, Lund T, Olesen A, Drewes A, Christrup L, Kreilgaard M. A review of morphine and morphine-6-glucuronide’s pharmacokinetic–pharmacodynamic relationships in experimental and clinical pain. Eur J Pharm Sci. 2015;74:45–62.
Smith M. Neuroexcitatory effects of morphine and hydromorphone: evidence implicating the 3-glucuronide metabolites. Clin Exp Pharmacol Physiol. 2000;27(7):524–8.
Hutchinson M, Shavit Y, Grace P, Rice K, Maier S, Watkins L. Exploring the Neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev. 2011;63(3):772–810.
Due M, Piekarz A, Wilson N, Feldman P, Ripsch M, Chavez S, et al. Neuroexcitatory effects of morphine-3-glucuronide are dependent on Toll-like receptor 4 signaling. J Neuroinflammation. 2012;9(1):200.
Wright A, Mather L, Smith M. Hydromorphone-3-glucuronide: a more potent neuro-excitant than its structural analogue, morphine-3-glucuronide. Life Sci. 2001;69(4):409–20.
Otton S, Schadel M, Cheung S, Kaplan H, Busto U, Sellers E. CYP2D6 phenotype determines the metabolic conversion of hydrocodone to hydromorphone. Clin Pharmacol Ther. 1993;54(5):463–72.
Narita M, Nakamura A, Ozaki M, Imai S, Miyoshi K, Suzuki M, et al. Comparative pharmacological profiles of morphine and oxycodone under a neuropathic pain-like state in mice: evidence for less sensitivity to morphine. Neuropsychopharmacology. 2007;33(5):1097–112.
Thompson C. Activation of G-proteins by morphine and codeine congeners: insights to the relevance of O- and N-Demethylated metabolites at - and -opioid receptors. J Pharmacol Exp Ther. 2003;308(2):547–54.
Silvasti M, Rosenberg P, Seppälä T, Svartling N, Pitkänen M. Comparison of analgesic efficacy of oxycodone and morphine in postoperative intravenous patient-controlled analgesia. Acta Anaesthesiol Scand. 1998;42(5):576–80.
Allen S. Neuropathic pain; the case for opioid therapy. Oncology. 2008;74(1):76–82.
Poyhia R, Seppala T, Olkkola K, Kalso E. The pharmacokinetics and metabolism of oxycodone after intramuscular and oral administration to healthy subjects. Br J Clin Pharmacol. 1992;33(6):617–21.
Seleman M, Chapy H, Cisternino S, Courtin C, Smirnova M, Schlatter J, et al. Impact of P-glycoprotein at the blood-brain barrier on the uptake of heroin and its main metabolites: behavioral effects and consequences on the transcriptional responses and reinforcing properties. Psychopharmacology. 2014;231(16):3139–49.
Boström E, Hammarlund-Udenaes M, Simonsson U. Blood–brain barrier transport helps to explain discrepancies in in vivo potency between oxycodone and morphine. Anesthesiology. 2008;108(3):495–505.
Bostrom E. In vivo blood-brain barrier transport of oxycodone in the rat: indications for active influx and implications for pharmacokinetics/pharmacodynamics. Drug Metab Dispos. 2006;34(9):1624–31.
Barrera-Chacon J, Mendez-Suarez J, Jáuregui-Abrisqueta M, Palazon R, Barbara-Bataller E, García-Obrero I. Oxycodone improves pain control and quality of life in anticonvulsant-pretreated spinal cord-injured patients with neuropathic pain. Spinal Cord. 2010;49(1):36–42.
Olarte J. Oxycodone and the challenge of neuropathic cancer pain: a review. Oncology. 2008;74(1):83–90.
Benedetti F, Vighetti S, Amanzio M, Casadio C, Oliaro A, Bergamasco B, et al. Dose-response relationship of opioids in nociceptive and neuropathic postoperative pain. Pain. 1998;74(2):205–11.
Largent-Milnes T, Guo W, Wang H, Burns L, Vanderah T. Oxycodone plus ultra-low-dose naltrexone attenuates neuropathic pain and associated μ-opioid receptor–Gs coupling. J Pain. 2008;9(8):700–13.
Watson N, C, Babul N. Efficacy of oxycodone in neuronathic pain: a randomized trial in postherpetic neuralgia. Neurology. 1998;50(6):1837–41.
Minami K, Ogata J, Uezono Y. What is the main mechanism of tramadol? Naunyn Schmiedeberg's Arch Pharmacol. 2015;388(10):999–1007.
Christoph T, Kogel B, Strassburger W, Schug S. Tramadol has a better potency ratio relative to morphine in neuropathic than in nociceptive pain models. Drugs in R & D. 2007;8(1):51–7.
Harati Y, Gooch C, Swenson M, Edelman S, Greene D, Raskin P, et al. Maintenance of the long-term effectiveness of tramadol in treatment of the pain of diabetic neuropathy. J Diabetes Complicat. 2000;14(2):65–70.
Freeman R, Raskin P, Hewitt D, Vorsanger G, Jordan D, Xiang J, et al. Randomized study of tramadol/acetaminophen versus placebo in painful diabetic peripheral neuropathy. Curr Med Res Opin. 2006;23(1):147–61.
Meske D, Xie J, Oyarzo J, Badghisi H, Ossipov M, Porreca F. Opioid and noradrenergic contributions of tapentadol in experimental neuropathic pain. Neurosci Lett. 2014;562:91–6.
Bee L, Bannister K, Rahman W, Dickenson A. Mu-opioid and noradrenergic α2-adrenoceptor contributions to the effects of tapentadol on spinal electrophysiological measures of nociception in nerve-injured rats. Pain. 2011;152(1):131–9.
Vadivelu N, Timchenko, Hyang and Sinatra. Tapentadol extended-release for treatment of chronic pain: a review. J Pain Res 2011, p.211.
Kristensen K. The mu1, mu2, delta, kappa opioid receptor binding profiles of methadone stereoisomers and morphine. Life Sci. 1995;56(2):PL45–50.
Posa L, Accarie A, Noble F. and Marie N. Methadone reverses analgesic tolerance induced by morphine pretreatment. Int J Neuropsychopharmacol, 2015;19(7), P.pyv108.
Lemberg K, Kontinen V, Viljakka K, Kylnlahti I, Yli-Kauhaluoma J, Kalso E. Morphine, oxycodone, methadone and its enantiomers in different models of nociception in the rat. Anesth Analg. 2006;102(6):1768–74.
Aiyer R, Mehta N, Gungor S. and Gulati A. A systematic review of NMDA receptor antagonists for treatment of neuropathic pain in clinical practice. Clin J Pain, 2017; p.1.
Prommer, E. (2014). Levorphanol: revisiting an underutilized analgesic. Palliative Care: Research and Treatment, 8, p.PCRT.S13489.
Gudin J, Fudin J, Nalamachu S. Levorphanol use: past, present and future. Postgrad Med. 2015;128(1):46–53.
Pham T, Fudin J, Raffa R. Is levorphanol a better option than methadone? Pain Med. 2015;16(9):1673–9.
Weiner M, Sarantopoulos C, Gordon E. Case study. Transdermal buprenorphine controls central neuropathic pain. Journal of Opioid Management. 2012;8(6):414–5.
Leffler A, Frank G, Kistner K, Niedermirtl F, Koppert W, Reeh P, et al. Local anesthetic-like inhibition of voltage-gated Na+ channels by the partial μ-opioid receptor agonist buprenorphine. Anesthesiology. 2012;116(6):1335–46.
Kistner K, Zimmermann K, Ehnert C, Reeh P, Leffler A. The tetrodotoxin-resistant Na+ channel Nav1.8 reduces the potency of local anesthetics in blocking C-fiber nociceptors. Pflugers Arch - Eur J Physiol. 2010;459(5):751–63.
Pillarisetti S, Khanna I. Buprenorphine; an attractive opioid with underutilized potential in treatment of chronic pain. J Pain Res. 2015;8:859–70.
Pergolizzi J, Böger R, Budd K, Dahan A, Erdine S, Hans G, et al. Opioids and the management of chronic severe pain in the elderly: consensus statement of an international expert panel with focus on the six clinically most often used World Health Organization step III opioids (buprenorphine, fentanyl, hydromorphone, methadone, morphine, oxycodone). Pain Practice. 2008;8(4):287–313.
Tzschentke T, Linz K, Frosch S, Christoph T. Antihyperalgesic, antiallodynic, and antinociceptive effects of cebranopadol, a novel potent nociceptin/orphanin fq and opioid receptor agonist, after peripheral and central administration in rodent models of neuropathic pain. Pain Practice. 2017;17(8):1032–41.
Raffa R, Burdge G, Gambrah J, Kinecki H, Lin F, Lu B, et al. Cebranopadol: novel dual opioid/NOP receptor agonist analgesic. J Clin Pharm Ther. 2016;42(1):8–17.
Sałat K, Furgała A, Sałat R. Evaluation of cebranopadol, a dually acting nociceptin/orphanin FQ and opioid receptor agonist in mouse models of acute, tonic, and chemotherapy-induced neuropathic pain. Inflammopharmacology. 2017;26(2):361–74.
Christoph A, Eerdekens M, Kok M, Volkers G, Freynhagen R. Cebranopadol, a novel first-in-class analgesic drug candidate. PAIN. 2017;158(9):1813–24.
Backonja M, Irving G, Argoff C. Rational multidrug therapy in the treatment of neuropathic pain. Curr Pain Headache Rep. 2006;10(1):34–8.
Eisenberg E, Suzan E. Drug combinations in the treatment of neuropathic pain. Curr Pain Headache Rep. 2014;18(12).
Chaparro L, Wiffen P, Moore R, Gilron I. Combination pharmacotherapy for the treatment of neuropathic pain in adults. Cochrane Database Syst Rev. 2012.
Rowbotham M. Tricyclic antidepressants and opioids—better together? PAIN. 2015;156(8):1373–4.
Mücke M, Phillips T, Radbruch L, Petzke F, Häuser W. Cannabis-based medicines for chronic neuropathic pain in adults. Cochrane Database Syst Rev. 2018.
Stockings E, Campbell G, Hall W, Nielsen S, Zagic D, Rahman R, et al. Cannabis and cannabinoids for the treatment of people with chronic non-cancer pain conditions: PAIN; 2018. p. 1.
da Fonseca Pacheco D, Klein A, de Castro Perez A, da Fonseca Pacheco C, de Francischi J. and Duarte I. The μ-opioid receptor agonist morphine, but not agonists at δ- or κ-opioid receptors, induces peripheral antinociception mediated by cannabinoid receptors. Br J Pharmacol. 2008;154(5), pp.1143–1149.
Manzanares J, Corchero J, Romero J, Fernández-Ruiz J, Ramos J, Fuentes J. Pharmacological and biochemical interactions between opioids and cannabinoids. Trends Pharmacol Sci. 1999;20(7):287–94.
Salio C, Fischer J, Franzoni M, Mackie K, Kaneko T, Conrath M. CB1-cannabinoid and μ-opioid receptor co-localization on postsynaptic target in the rat dorsal horn. Neuroreport. 2001;12(17):3689–92.
Vigano D, Rubino T, Parolaro D. Molecular and cellular basis of cannabinoid and opioid interactions. Pharmacol Biochem Behav. 2005;81(2):360–8.
Bushlin I, Rozenfeld R, Devi L. Cannabinoid–opioid interactions during neuropathic pain and analgesia. Curr Opin Pharmacol. 2010;10(1):80–6.
Cichewicz D. Synergistic interactions between cannabinoid and opioid analgesics. Life Sci. 2004;74(11):1317–24.
Tham S, Angus J, Tudor E, Wright C. Synergistic and additive interactions of the cannabinoid agonist CP55,940 with μopioid receptor andα2-adrenoceptor agonists in acute pain models in mice. Br J Pharmacol. 2005;144(6):875–84.
Lynch M, Clark A. Cannabis reduces opioid dose in the treatment of chronic non-cancer pain. J Pain Symptom Manag. 2003;25(6):496–8.
Cichewicz D, Haller V, Welch S. Changes in opioid and cannabinoid receptor protein following short-term combination treatment with delta(9)-tetrahydrocannabinol and morphine. J Pharmacol Exp Ther. 2001;297(1):121–7.
van Hecke O, Kamerman P, Attal N, Baron R, Bjornsdottir G, Bennett D, et al. Neuropathic pain phenotyping by international consensus (NeuroPPIC) for genetic studies. PAIN, 2015;156(11), pp.2337–2353.
Forstenpointner J, Otto J, Baron R. Individualized neuropathic pain therapy based on phenotyping. PAIN. 2018;159(3):569–75.
Acknowledgments
This work is an expanded version of a previous document completed as part of the MSc in the Clinical Management of Pain program at the University of Edinburgh, UK.
The research work disclosed in this publication is partially funded by the Endeavor Scholarship Scheme (Malta). Scholarships are part-financed by the European Union—European Social Fund (ESF)—Operational Programme II—Cohesion Policy 2014-2020 “Investing in human capital to create more opportunities and promote the well-being of society.”
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The author declares that he has no conflict of interest.
Statement of Human Rights and Animal Welfare
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
This article is part of the Topical Collection on Medicine
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Schembri, E. Are Opioids Effective in Relieving Neuropathic Pain?. SN Compr. Clin. Med. 1, 30–46 (2019). https://doi.org/10.1007/s42399-018-0009-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42399-018-0009-4