
Abstract
Background
Cebranopadol (a.k.a. GRT-6005) is a dually acting nociceptin/orphanin FQ and opioid receptor agonist that has been recently developed in Phase 2 clinical trials for painful diabetic neuropathy or cancer pain. It also showed analgesic properties in various rat models of pain and had a better safety profile as compared to equi-analgesic doses of morphine. Since antinociceptive properties of cebranopadol have been studied mainly in rat models, in the present study, we assessed analgesic activity of subcutaneous cebranopadol (10 mg/kg) in various mouse pain models.
Methods
We used models of acute, tonic, and chronic pain induced by thermal and chemical stimuli, with a particular emphasis on pharmacoresistant chronic neuropathic pain evoked by oxaliplatin in which cebranopadol was used alone or in combination with simvastatin.
Key results
As shown in the hot plate test, the analgesic activity of cebranopadol developed more slowly as compared to morphine (90–120 min vs. 60 min). Cebranopadol displayed a significant antinociceptive activity in acute pain models, i.e., the hot plate, writhing, and capsaicin tests. It attenuated nocifensive responses in both phases of the formalin test and reduced cold allodynia in oxaliplatin-induced neuropathic pain model. Its efficacy was similar to that of morphine. Used in combination and administered simultaneously, 4 or 6 h after simvastatin, cebranopadol did not potentiate antiallodynic activity of this cholesterol-lowering drug. Cebranopadol did not induce any motor deficits in the rotarod test.
Conclusion
Cebranopadol may have significant potential for the treatment of various pain types, including inflammatory and chemotherapy-induced neuropathic pain.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The International Association for the Study of Pain (IASP) defines pain as an unpleasant sensory and emotional experience associated with actual or potential tissue damage, or described in terms of such damage. The sensation of pain involves multiple signaling pathways, numerous neurotransmitters, and other mediators that are involved in the inhibitory or facilitatory control of pain intensity. These mechanisms affect the perception of stimuli as non-painful or painful, respectively, but their positive or negative modulation of pain signaling is strongly dependent on the receptor type involved and its location in the target tissue (Argoff 2011).
In living organisms, endogenous opioids (endorphins, enkephalins, and dynorphins) are key molecules in the descending pain suppression pathways. Recently, it has been discovered that opioid receptors are widely distributed not only in the central but also peripheral nervous system and in the non-neuronal tissues. There is also evidence from animal and human studies for the involvement of peripheral opioid receptors in analgesia, especially in the presence of inflammation (Sehgal et al. 2011), or neuropathy (Plein and Rittner 2017).
The nociceptin/orphanin FQ opioid peptide receptor (NOP receptor) is the most recently discovered member of the opioid receptor family. Together with its endogenous ligand–nociceptin, also known as orphanin FQ (N/OFQ), it forms the fourth member of the opioid receptor family which is abundantly expressed in various body tissues. A large body of evidence shows that the activation of N/OFQ-NOP system regulates functions of the central nervous system being implicated in feeding, body weight homeostasis, stress, stress-related psychiatric disorders—depression, anxiety, drug, and alcohol dependence (Witkin et al. 2014). Data from preclinical studies are also in line with these findings showing that N/OFQ plays an important role in comorbid neuropathic pain and post-traumatic stress disorder (Zhang et al. 2015), acute and chronic restraint stress responses (Delaney et al. 2012), depression (Vitale et al. 2017), and other stress-related conditions (Leggett et al. 2006; Witkin et al. 2014).
Apart from this, the N/OFQ-NOP pathway is also involved in the modulation of inflammatory and immune responses of the body by influencing migration of leucocytes, cytokine secretion, and lymphocyte proliferation. Recent findings showing the involvement of N/OFQ in inflammatory responses (Gavioli and Romão 2011) and the evidence for a role of NOP receptors and N/OFQ in the modulation of neurogenic inflammation, migraine (Tajti et al. 2015), and airway tone (Singh et al. 2016) led to the hypothesis that N/OFQ-NOP system might be an important drug target for analgesic drugs. This is in part supported by the previous findings showing that the blockade of NOP receptors can attenuate inflammation (Gavioli et al. 2015). On the other hand, this issue is not completely clear and unequivocally explored as there is also evidence for analgesic efficacy of NOP agonists in neuropathic and inflammatory pain, both in animal models (Sukhtankar et al. 2013) and clinical trials (Sałat et al. 2015a).
Cebranopadol (a.k.a. GRT-6005; trans-6′-fluoro-4′,9′-dihydro-N,N-dimethyl-4-phenyl-spiro[cyclohexane-1,1′(3′H)-pyrano[3,4-b]indol]-4-amine) is a dually acting nociceptin/orphanin FQ and opioid receptor agonist (Ki (nM)/EC50 (nM)/relative efficacy (%): human NOP receptor 0.9/13.0/89; human mu-opioid peptide (MOP) receptor 0.7/1.2/104; human kappa-opioid peptide (KOP) receptor 2.6/17/67; human delta-opioid peptide (DOP) receptor 18/110/105) (Linz et al. 2014) that has been recently developed in Phase 2 clinical trials for painful diabetic neuropathy or cancer pain (reviewed in Sałat et al. 2015a). It showed analgesic properties in various rat models of acute thermal pain, i.e., tail-flick model, chronic inflammatory pain (CFA-induced arthritis model), bone cancer pain model (Raffa et al. 2017), and neuropathic pain: chronic constriction injury (CCI) and diabetic neuropathic pain models (Raffa et al. 2017) after intraplantar, intracerebroventricular, intrathecal, intravenous (Tzschentke et al. 2017), subcutaneous, or oral route (Linz et al. 2014). Compared to selective MOP receptor agonists, cebranopadol was more potent in models of chronic neuropathic than acute nociceptive pain and its duration of action was long (Linz et al. 2014). Noteworthy, safety pharmacology studies with cebranopadol demonstrated that the development of analgesic tolerance in cebranopadol-treated animals subjected to CCI procedure was delayed as compared to equi-analgesic doses of morphine (Sałat et al. 2015a), and at analgesic doses, cebranopadol did not cause respiratory depression in a rat whole-body plethysmography model, or motor coordination deficits in the rat rotarod test (Linz et al. 2014, 2017; Lambert et al. 2015; Günther et al. 2017).
The data presented above come from rat studies and there is limited knowledge about antinociceptive properties of cebranopadol in mice. Moreover, these previous studies investigated the influence of cebranopadol on tactile allodynia but not thermal (i.e., heat or cold) allodynia and hyperalgesia. Hence, in the present study, we utilized mouse models of acute, tonic, and chronic pain induced by thermal or chemical (inflammatory) stimuli, with a particular emphasis on pharmacoresistant chronic neuropathic pain evoked by oxaliplatin. Oxaliplatin is a third-generation platinum-based anti-tumor drug used to treat advanced colorectal cancer. Compared to other platinum-based drugs, it has lower incidence of hematological adverse effects and gastrointestinal toxicity, but in approximately 95% of patients, oxaliplatin causes severe neuropathic pain episodes and increased sensitivity to cold (Manji 2013) which often lead to dose reduction or even treatment discontinuation. These neuropathic pain episodes can be effectively attenuated by μ opioid peptide (MOP) and NOP receptor agonists (Micheli et al. 2015).
In the present study, we investigated the effect of cebranopadol on cold nociceptive threshold of oxaliplatin-treated mice. We used two protocols of its administration: the first one which utilized this drug alone, and the second one in which cebranopadol was used in combination with simvastatin. Available data show potential effectiveness of simvastatin in several animal models of pain (Shi et al. 2011; Miranda et al. 2011; Chen et al. 2013; Mansouri et al. 2017), including inflammatory (Chen et al. 2013) and neuropathic pain models (Shi et al. 2011). First, in the previous studies (Bhalla et al. 2015), simvastatin effectively reversed vincristine-induced neuropathic pain by anti-inflammatory effects and it was able to attenuate vincristine-induced increase in myeloperoxidase activity. Second, anti-inflammatory and anti-oxidant effects of this drug also resulted in reduction of cisplatin-induced nephrotoxicity and hepatotoxicity in rats (Işeri et al. 2007) and simvastatin protected Sertoli cells against cisplatin cytotoxicity (Wang et al. 2015). Third, it has been also shown that simvastatin was able to attenuate neuropathic pain induced by CCI in rats and it significantly decreased the ratio of membrane/cytosolic RhoA by reducing RhoA/LIMK/cofilin pathway activity (Qiu et al. 2016). Furthermore, it exerted antihyperalgesic and antiallodynic effects through the inhibition of spinal RhoA activation and its daily intrathecal administration before nerve injury prevented the development of neuropathy in nerve-ligated mice (Ohsawa et al. 2016). Interestingly, the RhoA-dependent pathway is implicated in the regulation of Transient receptor potential melastatin subtype 8 (TRPM8), a cold-sensing cation channel (Sun et al. 2014) which is also required for cold-related symptoms of oxaliplatin-induced peripheral neurotoxicity (Knowlton et al. 2011; Kono et al. 2012). Taken together, these data clearly suggest that statins are effective in neuropathic pain conditions and they might modulate pain sensitivity of cold-exposed subjects. This justifies the rationale to undertake this part of research which aimed to assess if combined use of simvastatin and cebranopadol could attenuate cold hypersensitivity of oxaliplatin-treated mice.
Materials and methods
Animals and housing conditions
Experiments were carried out at the Department of Pharmacodynamics, Faculty of Pharmacy, Jagiellonian University Medical College in Krakow. The investigators involved in behavioral assays were blinded to the experimental groups to avoid potential bias in data recording. Adult male Albino Swiss (CD-1) mice weighing 18–22 g were purchased from the Animal Breeding Farm of the Jagiellonian University Faculty of Pharmacy. Before behavioral tests, the animals were kept in groups of 10 mice in standard plastic cages and housed under controlled conditions (room temperature of 22 ± 2 °C, light/dark (12:12) cycle, lights on at 8.00 AM, humidity 50–60% and free access to food and water). Experimental groups consisted of 8–10 animals/dose. For the tests, the animals were selected randomly. After the assay, the mice were immediately euthanized by cervical dislocation. All experiments were performed between 9 AM and 3 PM. The procedures for in vivo tests were approved by the Local Ethics Committee of the Jagiellonian University in Krakow (Approval No. 4/2016; 22.03.2016) and the treatment of animals was in full accordance with ethical standards laid down in respective Polish and EU regulations (Directive No. 86/609/EEC).
Chemicals
Cebranopadol and morphine hydrochloride were purchased from MedChem Express (NJ, USA) and Polfa Kutno (Poland), respectively. These drugs at a fixed dose of 10 mg/kg were administered subcutaneously before behavioral tests. This dose was selected based on our previous pilot study which revealed that full antinociceptive efficacy of morphine used as a reference drug was observed at doses 6–10 mg/kg. For in vivo tests, cebranopadol was prepared in a mixture of 100% DMSO (Polskie Odczynniki Chemiczne, Poland) and 0.9% saline (1:1), then being diluted in saline to achieve a proper concentration and was injected 120 min before testing (for details, see Sect. 3.1.1). Morphine hydrochloride was dissolved in 0.9% saline solution and it was administered 60 min before the tests. Control animals received vehicle. Acetic acid, ethanol, 0.9% natrium chloride solution, 5% glucose solution, and 37% formaldehyde solution were provided by Polskie Odczynniki Chemiczne (Poland). Capsaicin, simvastatin, and oxaliplatin were purchased from Sigma-Aldrich (Germany). For the in vivo experiments, capsaicin was dissolved in ethanol (100%) at 5% of the final desired volume, and then, 0.9% saline was added. This mixture was vortexed for 10 min (Sałat et al. 2014). Simvastatin was suspended in 0.9% saline solution. The dose of simvastatin used in the present research (100 mg/kg, p.o.) was chosen on the basis of the previous studies published by other authors (Mansouri et al. 2015).
Behavioral tests
Acute pain models (thermal pain, inflammatory, and chemogenic pain models)
Thermal pain—hot plate test
Antinociceptive properties of cebranopadol and morphine in the hot plate test were assessed as described previously (Eddy and Leimbach 1953) with some minor modification (Sałat et al. 2015b). Briefly, 1 day before the proper pharmacological experiment, the animals were tested to establish baseline pain sensitivity threshold (baseline latency) for each animal. For further pain tests, only mice with baseline latencies ≤ 20 s were used. On the test day, the animals were subcutaneously treated either with the test drugs, or vehicle 60 min before placing the animal on a hot/cold plate apparatus (Bioseb, France). This apparatus can generate heat or cold and is supplied with a temperature controller that maintains surface temperature to a set point. Herein, the temperature was set at 55–56 °C. Latency time to pain reaction, i.e., the time until the animal licked its hind paws or jumped was recorded by means of a stop-watch (Q&Q HS-46, Japan, precision: 1/100 s). In this assay, a cut-off time (60 s) was enforced to avoid paw tissue damage, and mice not responding within 60 s were removed from the apparatus and assigned a score of 60 s.
Inflammatory acute pain—writhing test
In this test, mice were placed individually into glass beakers and were allowed to habituate for the next 30 min. Then, each mouse was weighed, injected with the test compound or vehicle, and then placed back into the glass beaker for 60 min. To induce inflammatory pain, 0.9% acetic acid solution prepared in saline was injected by the intraperitoneal route. Mice were placed in the beakers once again and were observed continuously for the next 30 min. Stereotypical writhes (lengthwise constrictions of the torso with a concomitant concave arching of the back) were counted over this period in drug-treated and control mice (Sałat et al. 2013a).
Neurogenic pain—capsaicin test
After the adaptation period (15 min), 1.6 μg of capsaicin dissolved in 20 μl of a mixture containing 0.9% saline and ethanol (5% of the final volume) was injected intraplantarly in the ventral surface of the right-hind paw of each mouse. The animals were observed individually for 5 min following capsaicin injection. In all experimental groups, the amount of time spent on licking, biting, or lifting the injected paw was recorded with a chronometer and was considered as an indicator of nociception (Sałat et al. 2014).
Inflammatory tonic pain model—formalin test
In rodents, the injection of diluted formalin solution evokes a biphasic nocifensive behavioral response (licking or biting the injected paw) of experimental animals. The first (acute) nociceptive phase of the test lasts for 5 min, whilst the second (late) one occurs between 15 and 30 min after formalin injection. In this assay, 20 μl of 5% formalin solution was injected into the dorsal surface of the right-hind paw of each mouse. Then, the mice were placed separately in glass beakers and were observed for the next 30 min. Total time spent on licking or biting the formalin-treated paw was measured during the first 5 min of the test, and then between 15 and 30 min of the test in drug-treated and vehicle-treated mice (Sałat et al. 2015b).
Chemotherapy-induced neuropathic pain model
Induction phase
To develop chemotherapy-induced peripheral neuropathy (CIPN) and neuropathic pain mice were injected intraperitoneally with a single dose of oxaliplatin (10 mg/kg prepared in 5% glucose solution). Control mice received 5% glucose solution as vehicle. Pain threshold of experimental animals was assessed using the cold plate test 3 h and 7 days after oxaliplatin administration to establish its effect on acute-phase and late-phase cold allodynia, respectively.
Influence on cold allodynia (cold plate test)—single-drug administration protocol
The cold plate test was performed using the hot/cold plate apparatus set at 2 °C. In this assay (Fig. 1a), the animals were tested first to obtain baseline latencies to pain reaction (i.e., lifting, biting, shaking of hind paws, jumping, and movement deficits) before oxaliplatin injection (referred to as latencies ‘before oxaliplatin’). Then, oxaliplatin was injected and latencies to pain reaction were measured again (referred to as ‘pre-drug latencies’). Subsequently, test drugs were administered and post-drug latencies were measured again. In this assay, a cut-off time of 60 s was established to avoid potential paw tissue damage and animals not responding within 60 s were removed from the apparatus and assigned a score of 60 s.

Protocol of the administration of cebranopadol alone (a), simvastatin alone (b), or both drugs in combination (c–e) in oxaliplatin-induced neuropathic pain model. The scheme shows specific time-points at which the data measured as latencies to pain reaction in the cold plate test were collected. CPT cold plate test, OXA oxaliplatin, CEB cebranopadol, SIM simvastatin
Influence on cold allodynia (cold plate test)—combined drug administration protocol (simvastatin and cebranopadol)
In the cold plate test, we additionally investigated the antiallodynic activity of cebranopadol used in combination with simvastatin. The protocol of the administration of these both drugs (alone or in combination) is shown in Fig. 1. It served to establish whether cebranopadol could enhance antiallodynic properties of simvastatin in the oxaliplatin model of neuropathic pain.
Influence on motor coordination—rotarod test
Before the test, the animals were trained for 3 consecutive days on the rotarod apparatus (Rotarod apparatus, May Commat RR0711, Turkey; rod diameter: 2 cm) that was rotating at a fixed speed of 18 rotations per minute (rpm). In each session, the mice were placed on the rotating rod for 3 min with an unlimited number of trials. The proper experiment was performed 24 h after the last training session. After the administration of the test drugs or vehicle, the mice were tested on the rod that revolved at 6, 18, or 24 rpm. Motor impairments in mice were defined as the inability to remain on the rotarod apparatus for 1 min. The results are expressed as the mean time spent on the rotarod (Sałat et al. 2015b).
Data analysis
Data analysis was carried out using GraphPad Prism software (v.5.0, CA, USA). Numerical results are expressed as the mean ± SEM. Statistical analysis was carried out using one-way analysis of variance (ANOVA), followed by Tukey’s, or Dunnett’s post hoc comparisons, or repeated-measures analysis of variance, followed by Bonferroni’s multiple comparison. P < 0.05 was considered significant.
Results
Acute pain models
Thermal acute pain—hot plate test
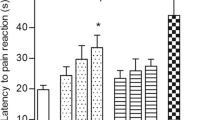
To establish at which time-points subcutaneous cebranopadol exerts antinociceptive activity, the mouse hot plate test was used. In this assay, an overall effect of treatment was observed (F[4,28] = 6.676, p < 0.001). The post hoc analysis revealed that the antinociceptive activity of cebranopadol was significant 90 min and 120 min after its administration (p < 0.01 and p < 0.001, respectively; Fig. 2). Taking this into consideration, further pain tests were performed 90 and 120 min after cebranopadol injection.

Antinociceptive activity of subcutaneous cebranopadol in the mouse hot plate test measured at various time-points. Results are shown as latency time to pain reaction (±SEM) in response to thermal stimulation (temperature of 55–56 °C). Statistical analysis: repeated-measures analysis of variance, followed by Bonferroni’s multiple comparison. Significance vs. pre-drug latency: **p < 0.01, ***p < 0.001
Previously obtained results for subcutaneous morphine in the hot plate test performed under the same experimental conditions (Sałat et al. 2009) demonstrated its significant antinociceptive properties. The dose of 6 mg/kg was highly effective and this activity was noted earlier than that of cebranopadol (between 30 and 60 min after administration).
Inflammatory acute pain—writhing test
As shown in Fig. 3a, cebranopadol and morphine were equally effective in reducing acetic acid-induced writhing behavior (F[2,20] = 52.86, p < 0.0001).

Antinociceptive activity of cebranopadol (CEB) and morphine (MOR) in the acetic acid-induced writhing test (a) and in capsaicin-induced neurogenic pain model (b). Results are shown as the mean number of writhes (±SEM) (a), or duration of the licking/biting response (±SEM) (b). Statistical analysis: one-way analysis of variance, followed by Dunnett’s post hoc test. Significance vs. vehicle-treated mice: ***p < 0.001
Neurogenic pain—capsaicin test
In the intraplantar capsaicin test (Fig. 3b), the pretreatment with cebranopadol and morphine completely abolished the licking response of capsaicin-treated mice (F[2,21] = 50.18, p < 0.0001). This confirmed strong antinociceptive properties of both agents in this pain model.
Inflammatory tonic pain model—formalin test
The results obtained in the first phase of the intraplantar formalin test confirmed that both drugs were able to attenuate neurogenic pain (F[2,21] = 14.92, p < 0.0001) with morphine being slightly more effective (Fig. 4a). The antinociceptive activity of both tested agents was also shown in the second (inflammatory) phase which demonstrated that cebranopadol and morphine significantly (at p < 0.01 and p < 0.001, respectively) reduced pain responses in formalin-treated animals (F[2,21] = 11.81, p < 0.001; Fig. 4b).

Antinociceptive activity of cebranopadol (CEB) and morphine (MOR) in the tonic pain model (i.e., the intraplantar formalin test). Influence on the first, neurogenic phase responses (a) and on the second, inflammatory phase responses (b). Statistical analysis: one-way analysis of variance, followed by Dunnett’s post hoc test. Significance vs. vehicle-treated mice: **p < 0.01, ***p < 0.001
Chemotherapy-induced neuropathic pain model
Influence on cold allodynia (single-drug administration protocol)
In this experiment, an overall effect of treatment was observed (F[16,127] = 15.58, p < 0.0001). As shown in Fig. 5, during the early phase cold allodynia, both cebranopadol and morphine demonstrated strong and statistically significant (p < 0.01) antiallodynic properties. Their antiallodynic activity was also confirmed during the late phase of cold allodynia—7 days after oxaliplatin injection (significant at p < 0.05 and p < 0.01 for cebranopadol and morphine, respectively).

Antiallodynic activity of cebranopadol and morphine in oxaliplatin-induced neuropathic pain model. Cold allodynia was measured using the cold plate test in the early phase (3 h after oxaliplatin administration) and in the late phase (7 days after oxaliplatin administration). Results are shown as latency time to pain reaction (±SEM) in response to thermal stimulation (temperature of 2 °C). Statistical analysis: one-way analysis of variance, followed by Tukey’s post hoc test. Significance vs. latencies of vehicle-treated mice (i.e., mice not treated with oxaliplatin): ### p < 0.001, and vs. pre-drug latencies at the respective time-point: *p < 0.05, **p < 0.01, ***p < 0.001
Influence on cold allodynia (combined treatment protocol: cebranopadol and simvastatin)
Preliminary experiments (data not shown) performed using the cold plate test in mice not treated with oxaliplatin (i.e., non-neuropathic mice) revealed overall effects of single-dose and repeated-dose simvastatin treatments (single-dose simvastatin: F[5,32] = 8.366, p < 0.0001; repeated-dose simvastatin: F[7,46] = 4.209, p < 0.01). Post hoc analyses revealed that there were significant (p < 0.001) differences between the latencies of naïve mice and those of mice treated with a single dose of simvastatin. The latencies of the latter group were significantly reduced as compared to naïve mice. Similar effects were observed in mice repeatedly treated with simvastatin (p < 0.01 vs. naïve mice). No significant differences were noted when the latencies of mice treated with single-dose and repeated-dose simvastatin were compared (F[5,46] = 0.3205, p > 0.05).
The administration of oxaliplatin significantly reduced cold sensitivity threshold in mice (F[8,31] = 11.48, p < 0.0001). Significantly decreased latency time to pain reaction (p < 0.001 vs. values before oxaliplatin administration) was observed 3 h after oxaliplatin injection. This indicated for the development of cold allodynia in oxaliplatin-treated, neuropathic mice (Fig. 6a). Cold allodynia was maintained even for 7 days (significant at p < 0.05 vs. values before oxaliplatin administration).

Influence of oxaliplatin (10 mg/kg, i.p.; OXA), simvastatin (100 mg/kg, oral route; SIM) alone, or in combination with subcutaneous cebranopadol (10 mg/kg, s.c.; CEB) on cold sensitivity threshold and cold allodynia measured in the cold plate test in oxaliplatin-induced neuropathic pain model. Effect of oxaliplatin on the development of cold allodynia (a). Effect of 7-day administration of oral simvastatin alone (OXA + SIM), or in combination with single-dose cebranopadol added on day 7: simultaneously (b OXA + SIM + CEB), 4 h (c OXA + SIM + CEB (4 h)), or 6 h (d OXA + SIM + CEB (6 h)) after simvastatin. Results are shown as latency time to pain reaction (±SEM) in response to thermal (cold) stimulation (2 °C). In this test, the data were collected at two time-points: on day 1 to measure the latencies to pain reaction during the acute-phase cold allodynia that developed after oxaliplatin injection, and on day 7 to measure the latencies to pain reaction during the late-phase cold allodynia. Day 1: grey bars indicate baseline latencies of oxaliplatin-treated mice measured before drug treatment (i.e., before vehicle or simvastatin administration; defined as ‘pre-drug’) and black bars depict latencies after drug administration (i.e., after vehicle or simvastatin administration; referred to as ‘post-drug’ values). Day 7: grey bars indicate baseline latencies of oxaliplatin-treated mice measured before drug treatment (i.e., before vehicle, simvastatin or cebranopadol administration; defined as ‘pre-drug’) and black bars depict latencies after drug administration (i.e., after vehicle, simvastatin only, or simvastatin and cebranopadol administration; referred to as ‘post-drug’ values). A detailed protocol used is also shown in Fig. 1. Statistical analysis: one-way analysis of variance, followed by Tukey’s post hoc test. Significance vs. latency to pain reaction before oxaliplatin injection: ## p < 0.01, ### p < 0.001.
On day 1 in neuropathic (oxaliplatin-treated) mice, simvastatin administration (black bars—Figs. 6b–d: sim + oxa) did not influence cold allodynia significantly, although we observed a slight prolongation of latency time to pain reaction after a single dose of this drug. Noteworthy, comparing pre-drug latencies in the cold plate test (grey bars—Figs. 6b–d: sim + oxa) during the early (on day 1) and the late (on day 7) phases of cold allodynia to the latency of untreated controls (a white bar—Figs. 6b–d: sim + oxa), it can be concluded that the repeated (7-day) administration of simvastatin partially reversed the decrease of pain threshold caused by oxaliplatin and partially restored it to values of mice not treated with oxaliplatin.
In general, the use of cebranopadol as ‘add-on’ therapy with simvastatin in neuropathic, oxaliplatin-treated mice showed no benefits. However, we noted some differences among groups treated with ‘add-on’ cebranopadol used simultaneously (F[9,70] = 6.657, p < 0.0001; Fig. 6B: sim + ceb), 4 h (F[9,65] = 11.37, p < 0.0001; Fig. 6C: sim + ceb (4 h)) or 6 h (F[9,65] = 9.875, p < 0.0001; Fig. 6d: sim + ceb (6 h)) after simvastatin. Post hoc analyses showed no significance of these results but interestingly, we observed a trend towards the prolongation of latency time to pain reaction in mice treated with combined simvastatin and cebranopadol, when cebranopadol was administered 4 h (Fig. 6c), or 6 h after simvastatin (Fig. 6d), but not if these two drugs were given simultaneously (Fig. 6b).
Influence on motor coordination (rotarod test)
In the rotarod test, neither cebranopadol nor morphine influenced animals’ motor coordination at 6, 18, or 24 rpm which means that the drugs tested did not cause motor deficits in mice.
Discussion
In this study, we implemented mouse models of acute, tonic, and chronic pain to assess antinociceptive properties of cebranopadol, a dually acting nociceptin/orphanin FQ and opioid receptor agonist. Antinociceptive efficacy of cebranopadol was compared to that of morphine used at an equal dose. A summary of results obtained for both drugs in various mouse pain tests is presented in Table 1.
In the hot plate test, both cebranopadol and morphine demonstrated strong and statistically significant antinociceptive properties. Of note, the activity of cebranopadol was delayed as compared to that previously shown for morphine (90–120 min vs. 30–60 min for cebranopadol and morphine, respectively) (Gades et al. 2000; Sałat et al. 2012). The hot plate assay is a rodent model of acute pain. The paws of mice are very sensitive to heat at temperatures that are not harmful to the skin. The characteristic responses such as jumping, licking of the paws are of central origin and it is thought that drugs with antinociceptive properties in the hot plate test act primarily in the spinal medulla and/or higher central nervous system levels (Vogel and Vogel 1997). In this assay, peripherally acting analgesics are generally not active (Vogel and Vogel 1997). Thus, the results obtained in the present study confirmed the role of central opioidergic system in mediating analgesia caused by cebranopadol (and morphine).
Available literature data indicate that functional NOP and MOP receptors are expressed not only at spinal and supraspinal sites of the ascending and descending pain pathways but also in the periphery. NOP receptors and their ligand—N/OFQ have been found in many peripheral organs (e.g., airways and cardiovascular system) and in the immune system in rodents and humans (Schröder et al. 2014). In a rat model of inflammatory pain (i.e., trinitrobenzene sulfonic acid (TNBS)-induced colonic hyperalgesia), N/OFQ demonstrated antihypersensitive effects after peripheral administration and it was antinociceptive in the capsaicin test in mice (Sakurada et al. 2005). N/OFQ exerted analgesic properties in the tail-flick test in rats (Xu et al. 1996; Tian et al. 1997) and mice (King et al. 1997). Moreover, spinal N/OFQ potentiated analgesia caused by systemic morphine (Tian et al. 1997), while a selective non-peptide NOP receptor agonist, SCH-221510, showed anti-inflammatory and analgesic properties in a mouse model of TNBS-induced inflammatory bowel disease after systemic administration (Sobczak et al. 2013, 2014).
In line with these findings, in our present study, both cebranopadol and morphine attenuated chemogenic, inflammatory acute pain responses induced by acetic acid. Since the writhing test is regarded a rodent model of pain mediated by peripheral mechanisms related to inflammation (Vogel and Vogel 1997), it seems plausible that peripheral NOP and MOP receptors might play a role in the observed activity of both drugs. The involvement of NOP receptors in the pathophysiology of inflammation, arterial hypertension, and cardiac or brain circulatory ischemia has been reported previously (reviewed in Schröder et al. 2014). In the rat model of carrageenan-induced inflammation, i.t. N/OFQ inhibited thermal hyperalgesia (Yamamoto et al. 1997b; Hao et al. 1998), and NOP receptors and their endogenous ligand—N/OFQ modulated neurogenic inflammation and other functions, such as airway tone and caliber (Singh et al. 2016).
The influence of cebranopadol and morphine on peripherally expressed opioid receptors might also explain their activity observed in the capsaicin test. This pain test reflects acute inflammatory pain responses related to neurogenic inflammation. Capsaicin is an exogenous activator of the TRPV1 channels present in sensory neurons, mainly in C-fibers and, to a lesser extent, Aδ. It shows a biphasic effect, i.e., it stimulates TRPV1 located in sensory neurons, resulting in a rapid phase of neurogenic pain with a burning sensation, local vascular and extravascular responses, after which persistent desensitization with concomitant long lasting analgesia appears (reviewed in Sałat et al. 2013b; Marwaha et al. 2016). Previously, it was shown that NOP receptor activation abolished capsaicin-induced contraction of guinea pig airways (Shah et al. 1998; Corboz et al. 2000), reduced capsaicin-induced bronchoconstriction, and increased airway hyper-responsiveness in ovalbumin-sensitized mice (D’Agostino et al. 2010). Of note, in the peripheral nervous system, N/OFQ inhibited neurotransmitter release (Giuliani et al. 2000) and inhibited substance P-mediated nociception in mice (Inoue et al. 1999). Considering an important role of substance P in neurogenic inflammation caused by capsaicin, the antinociceptive activity of cebranopadol in the capsaicin test can be explained in terms of its influence on neurogenic inflammation.
In the formalin test—a tonic pain model, cebranopadol was slightly less active than morphine. This was observed both in the acute (neurogenic) phase, and in the late (inflammatory) phase of this test. Intraplantar formalin induces chemogenic pain that results from neurogenic inflammation, sensory C-fibers activation, as well as sensitization within the spinal cord dorsal horn and the brain (Hunskaar and Hole 1987; Tjølsen et al. 1992; Yashpal and Coderre 1998), but there is also evidence that formalin is a potent stimulator of TRPA1 channels (McNamara et al. 2007; Nassini et al. 2015; Sałat and Filipek 2015). Our present study shows that NOP/MOP receptor activation might also mediate analgesia in the formalin test. It was previously demonstrated that in the formalin test, N/OFQ was antinociceptive after intrathecal administration, whereas it exerted pronociceptive effects and antagonized opioid analgesia when administered intracerebroventricularly (Erb et al. 1997; Yamamoto et al. 1997a; Zhu et al. 1997; Hao and Ogawa 1998; Wang et al. 1999). This bidirectional and site-specific modulation of nociception was also confirmed in the mouse formalin test in which UFP-101, a peptide antagonist selective for NOP receptors, exerted antinociceptive and pronociceptive effects after intracerebroventricular and intrathecal administration, respectively (Rizzi et al. 2006). Antihyperalgesic activity in the formalin test was also observed after systemic administration of selective, non-peptide NOP receptor agonist GRT-TA2210 (Linz et al. 2013).
In animals treated with oxaliplatin, the pain threshold for cold nociception is significantly lower as compared to non-treated mice. This was confirmed in our study in the cold plate test and indicated for the development of cold allodynia in oxaliplatin-treated mice. Recently, it has been shown that tactile allodynia and cold allodynia in rodents treated with oxaliplatin are mediated by TRPA1 stimulation (Nassini et al. 2011; Zhao et al. 2012; Sałat et al. 2013b; Marwaha et al. 2016) and a single dose of oxaliplatin induces acute cold hypersensitivity associated with an enhanced responsiveness of TRPA1 channels (Zhao et al. 2012). The antiallodynic activity of cebranopadol (and morphine) in the oxaliplatin model of neuropathic pain, in particular in the acute phase, together with the results obtained in the formalin test indicate that both drugs tested are able to attenuate TRPA1-mediated pain responses in mice. To the best of our knowledge, this drug has not been evaluated in these pathological conditions previously and our present study confirmed its potential utility for patients suffering from oxaliplatin-induced neuropathic pain. This is a potentially interesting finding as classical opioid drugs have limited efficacy in neuropathic pain conditions and they are regarded the third-line (morphine) or the second-line (tramadol) treatment for neuropathic pain (Finnerup et al. 2015), which is in part due to a diversity of mechanisms underlying the development of neuropathic pain (Torrance et al. 2013).
The expression of NOP receptors is up-regulated in chronic (neuropathic and inflammatory) pain conditions (Briscini et al. 2002; Chen and Sommer 2006; Schröder et al. 2014) and N/OFQ showed antihypersensitive effects in various rodent models of neuropathic pain, including rat CCI model (Yamamoto et al. 1997b; Corradini et al. 2001; Courteix et al. 2004), spinal nerve ligation (SNL) model (Ju et al. 2013), as well as the mouse diabetic neuropathic pain model (Kamei et al. 1999). In addition, some non-peptide NOP receptor agonists (SR14150 and SR16835) displayed NOP receptor-dependent antiallodynic activity in SNL model in mice (Khroyan et al. 2011), whereas in the mouse CCI model, GRT-TA2210 and Ro65-6570 demonstrated strong antiallodynic effects after spinal, supraspinal, and systemic administration (Linz et al. 2013).
Strong anti-inflammatory activity of cebranopadol, as well as the previously reported role of MOP, NOP receptors and N/OFQ in the attenuation of inflammation led us to investigate, if cebranopadol might potentiate/modulate the antiallodynic activity of other drugs with anti-inflammatory properties in chronic pain conditions. For this purpose, we chose the oxaliplatin neuropathic pain model and we used simvastatin as a potential novel treatment option for this pharmacoresistant pain type. Among numerous pathomechanism inflammation has been discovered as one of the key factors underlying neurotoxicity of oxaliplatin (Massicot et al. 2013; Waseem and Parvez 2016). Anti-inflammatory and analgesic properties of simvastatin in neuropathic pain conditions have been widely described in the literature (Miranda et al. 2011; Jaiswal and Sontakke 2012). Moreover, it has been shown that statins have protective effect on ultrastructural alterations induced by cold stress in rats (Bombig et al. 2003). They are also able to attenuate mitochondrial injury induced by cold exposure, inhibit the elevation of blood pressure in cold-treated mice via the downregulation of Bcl-2 pathway (Liang et al. 2017), and they are effective in the attenuation of Raynaud’s phenomenon (Baumhäkel and Böhm 2010). This activity of simvastatin is complex and it involves various pleiotropic effects, such as modulation of endothelial functions and Rho-kinase inhibition, which, in turn, affects TRPM8 activity (Sun et al. 2014). Taken together, these data clearly suggest that statins might modulate body functioning of cold-exposed subjects.
In the cold plate test in oxaliplatin-treated mice compared to non-treated controls, a significant reduction of latency time to pain reaction was observed both 3 h and 7 days after its administration. This indicated the development of cold allodynia in oxaliplatin-treated mice. Cold allodynia in neuropathic mice was not influenced by a single dose of simvastatin administered alone (see results for the early phase), but in contrast to this, a 7-day treatment with this drug partially reversed allodynia induced by cold and the difference between latencies of non-neuropathic mice and oxaliplatin + repeated simvastatin-treated mice was no longer significant. Thus, it might indicate that the repeated administration of simvastatin elevated pain threshold of animals with CIPN. The addition of cebranopadol to simvastatin did not demonstrate additional benefits. Of note, a simultaneous treatment with both drugs reduced latency time to pain reaction. Numerous studies have shown that the hot/cold plate test latencies might decrease with a repeated testing and this decrease may involve learning abilities, differences in animals’ weight, habituation time, and other unknown factors (Czopek et al. 2016). Of note, cebranopadol added to oxaliplatin + repeated simvastatin-treated mice 4 h or 6 h after the last dose of simvastatin (day 7) did not reduce the latency time to pain reaction and a tendency towards the prolongation of this parameter was noted. This is an interesting finding which requires further studies, but it may potentially suggest some pharmacodynamic interaction at the common site of action.
The rotarod test was involved for a proper interpretation of data obtained in pain tests to avoid the possibility of false positive results (Vogel and Vogel 1997). The results of our study showed no motor deficits in mice treated with cebranopadol and this finding is in line with the previous literature data (Bird and Lambert 2015).
To conclude, in this study using mouse models of acute, tonic, and chronic pain, we demonstrated antinociceptive and antiallodynic properties of cebranopadol—a novel, first-in-class agonist at nociceptin/orphanin FQ and opioid receptors. Of note, the antiallodynic activity of cebranopadol in neuropathic pain related to CIPN was shown for the first time, indicating a potential novel treatment option for this type of chronic pain. Apart from this, the results from our present study suggest that NOP receptors might be an important drug target for analgesics used not only in neuropathic pain conditions but also in inflammatory pain. This is of particular relevance in terms of pharmacoresistance of these types of chronic pain to currently available analgesic drugs.
References
Argoff C (2011) Mechanisms of pain transmission and pharmacologic management. Curr Med Res Opin 27(10):2019–2031. doi:10.1185/03007995.2011.614934
Baumhäkel M, Böhm M (2010) Recent achievements in the management of Raynaud’s phenomenon. Vasc Health Risk Manag 6:207–214
Bhalla S, Singh N, Jaggi AS (2015) Dose-related neuropathic and anti-neuropathic effects of simvastatin in vincristine-induced neuropathic pain in rats. Food Chem Toxicol 80:32–40. doi:10.1016/j.fct.2015.02.016
Bird MF, Lambert DG (2015) Simultaneous targeting of multiple opioid receptor types. Curr Opin Support Palliat Care 9:98–102. doi:10.1097/SPC.0000000000000129
Bombig MT, Ferreira C, Mora O et al (2003) Pravastatin protection from cold stress in myocardium of rats. Jpn Heart J 44(2):243–255
Briscini L, Corradini L, Ongini E, Bertorelli R (2002) Up-regulation of ORL-1 receptors in spinal tissue of allodynic rats after sciatic nerve injury. Eur J Pharmacol 447:59–65. doi:10.1016/S0014-2999(02)01833-2
Chen Y, Sommer C (2006) Nociceptin and its receptor in rat dorsal root ganglion neurons in neuropathic and inflammatory pain models: implications on pain processing. J Peripher Nerv Syst 11:232–240. doi:10.1111/j.1529-8027.2006.0093.x
Chen XY, Li K, Light AR, Fu KY (2013) Simvastatin attenuates formalin-induced nociceptive behaviors by inhibiting microglial RhoA and p38 MAPK activation. J Pain 14:1310–1319. doi:10.1016/j.jpain.2013.05.011
Corboz MR, Rivelli MA, Egan RW et al (2000) Nociceptin inhibits capsaicin-induced bronchoconstriction in isolated guinea pig lung. Eur J Pharmacol 402:171–179. doi:10.1016/S0014-2999(00)00505-7
Corradini L, Briscini L, Ongini E, Bertorelli R (2001) The putative OP4 antagonist, [Nphe1]nociceptin(1-13)NH2, prevents the effects of nociceptin in neuropathic rats. Brain Res 905:127–133. doi:10.1016/S0006-8993(01)02520-3
Courteix C, Coudoré-Civiale MA, Privat AM et al (2004) Evidence for an exclusive antinociceptive effect of nociceptin/orphanin FQ, an endogenous ligand for the ORL1 receptor, in two animal models of neuropathic pain. Pain 110:236–245. doi:10.1016/j.pain.2004.03.037
Czopek A, Sałat K, Byrtus H et al (2016) Antinociceptive activity of novel amide derivatives of imidazolidine-2,4-dione in a mouse model of acute pain. Pharmacol Rep 68:529–535. doi:10.1016/j.pharep.2015.12.007
D’Agostino B, Orlotti D, Calò G et al (2010) Nociceptin modulates bronchoconstriction induced by sensory nerve activation in mouse lung. Am J Respir Cell Mol Biol 42:250–254. doi:10.1165/rcmb.2008-0488OC
Delaney G, Dawe KL, Hogan R et al (2012) Role of nociceptin/orphanin FQ and NOP receptors in the response to acute and repeated restraint stress in rats. J Neuroendocrinol 24:1527–1541. doi:10.1111/j.1365-2826.2012.02361.x
Eddy NB, Leimbach D (1953) Synthetic analgesics. II. Dithienylbutenyl- and dithienylbutylamines. J Pharmacol Exp Ther 107:385–393
Erb K, Liebel JT, Tegeder I et al (1997) Spinally delivered nociceptin/orphanin FQ reduces flinching behaviour in the rat formalin test. NeuroReport 8:1967–1970
Finnerup NB, Attal N, Haroutounian S et al (2015) Pharmacotherapy for neuropathic pain in adults: a systematic review and meta-analysis. Lancet Neurol 14:162–173. doi:10.1016/S1474-4422(14)70251-0
Gades NM, Danneman PJ, Wixson SK, Tolley EA (2000) The magnitude and duration of the analgesic effect of morphine, butorphanol, and buprenorphine in rats and mice. Contemp Top Lab Anim Sci 39(2):8–13
Gavioli EC, Romão PRT (2011) NOP receptor ligands as potential agents for inflammatory and autoimmune diseases. J Amino Acids 2011:836569. doi:10.4061/2011/836569
Gavioli EC, de Medeiros IU, Monteiro MC et al (2015) Nociceptin/orphanin FQ-NOP receptor system in inflammatory and immune-mediated diseases. Vitam Horm 97:241–266. doi:10.1016/bs.vh.2014.11.003
Giuliani S, Lecci A, Maggi CA (2000) Nociceptin and neurotransmitter release in the periphery. Peptides 21:977–984. doi:10.1016/S0196-9781(00)00237-0
Günther T, Dasgupta P, Mann A et al (2017) Targeting multiple opioid receptors—improved analgesics with reduced side effects? Br J Pharmacol. doi:10.1111/bph.13809
Hao S, Ogawa H (1998) Naltrexone, but not atropine or yohimbine, antagonizes suppression of formalin-induced spinal sensitization by intrathecal nociceptin. Life Sci 63:PL 167–73
Hao J-X, Xu IS, Wiesenfeld-Hallin Z, Xu X-J (1998) Anti-hyperalgesic and anti-allodynic effects of intrathecal nociceptin/orphanin FQ in rats after spinal cord injury, peripheral nerve injury and inflammation. Pain 76:385–393. doi:10.1016/S0304-3959(98)00071-2
Hunskaar S, Hole K (1987) The formalin test in mice: dissociation between inflammatory and non-inflammatory pain. Pain 30:103–114. doi:10.1016/0304-3959(87)90088-1
Inoue M, Shimohira I, Yoshida A et al (1999) Dose-related opposite modulation by nociceptin/orphanin FQ of substance P nociception in the nociceptors and spinal cord. J Pharmacol Exp Ther 291:308–313
Işeri S, Ercan F, Gedik N, Yüksel M, Alican I (2007) Simvastatin attenuates cisplatin-induced kidney and liver damage in rats. Toxicology 230(2–3):256–264
Jaiswal SR, Sontakke SD (2012) Experimental evaluation of analgesic and anti-inflammatory activity of simvastatin and atorvastatin. Indian J Pharmacol 44:475–479. doi:10.4103/0253-7613.99311
Ju J, Shin DJ, Na YC, Yoon MH (2013) Role of spinal opioid receptor on the antiallodynic effect of intrathecal nociceptin in neuropathic rat. Neurosci Lett 542:118–122. doi:10.1016/j.neulet.2013.03.026
Kamei J, Ohsawa M, Kashiwazaki T, Nagase H (1999) Antinociceptive effects of the ORL1 receptor agonist nociceptin/orphanin FQ in diabetic mice. Eur J Pharmacol 370:109–116. doi:10.1016/S0014-2999(99)00112-0
Khroyan TV, Polgar WE, Orduna J et al (2011) Differential effects of NOP receptor agonists in acute versus chronic pain: studies with bifunctional NOP/mu receptor agonists in the sciatic nerve ligation chronic pain model in mice. J Pharmacol Exp Ther 339:687–693. doi:10.1124/jpet.111.184663
King MA, Rossi GC, Chang AH et al (1997) Spinal analgesic activity of orphanin FQ/nociceptin and its fragments. Neurosci Lett 223:113–116. doi:10.1016/S0304-3940(97)13414-0
Knowlton WM, Daniels RL, Palkar R, McCoy DD, McKemy DD (2011) Pharmacological blockade of TRPM8 ion channels alters cold and cold pain responses in mice. PLoS One 6(9):e25894. doi:10.1371/journal.pone.0025894
Kono T, Satomi M, Suno M et al (2012) Oxaliplatin induced neurotoxicity involves TRPM8 in the mechanism of acute hypersensitivity to cold sensation. Brain Behav 2(1):68–73. doi:10.1002/brb3.34
Lambert DG, Bird MF, Rowbotham DJ (2015) Cebranopadol: a first in-class example of a nociceptin/orphanin FQ receptor and opioid receptor agonist. Br J Anaesth 114:364–366. doi:10.1093/bja/aeu332
Leggett JD, Harbuz MS, Jessop DS, Fulford AJ (2006) The nociceptin receptor antagonist [Nphe1, Arg14, Lys15]nociceptin/orphanin FQ-NH2 blocks the stimulatory effects of nociceptin/orphanin FQ on the HPA axis in rats. Neuroscience 141:2051–2057. doi:10.1016/j.neuroscience.2006.05.036
Liang J, Yin K, Cao X et al (2017) Attenuation of low ambient temperature-induced myocardial hypertrophy by atorvastatin via promoting Bcl-2 expression. Cell Physiol Biochem 41(1):286–295. doi:10.1159/000456111
Linz K, Christoph T, Schiene K, Koch T, Englberger W (2013) GRT-TA2210, a selective NOP receptor agonist, is active in mouse models of inflammatory and neuropathic pain. In: EFIC–8th “Pain in Europe” Congress, 2013 October 9–12, Florence, Italy. Abstract 599
Linz K, Christoph T, Tzschentke TM et al (2014) Cebranopadol: a novel potent analgesic nociceptin/orphanin FQ peptide and opioid receptor agonist. J Pharmacol Exp Ther 349:535–548. doi:10.1124/jpet.114.213694
Linz K, Schröder W, Frosch S, Christoph T (2017) Opioid-type respiratory depressant side effects of cebranopadol in rats are limited by its nociceptin/orphanin FQ Peptide receptor agonist activity. Anesthesiology 126:708–715. doi:10.1097/ALN.0000000000001530
Manji H (2013) Drug-induced neuropathies. Handb Clin Neurol 115:729–742. doi:10.1016/B978-0-444-52902-2.00042-4
Mansouri MT, Khodayar MJ, Tabatabaee A, Ghorbanzadeh B, Naghizadeh B (2015) Modulation of morphine antinociceptive tolerance and physical dependence by co-administration of simvastatin. Pharmacol Biochem Behav 137:38–43. doi:10.1016/j.pbb.2015.08.002
Mansouri MT, Naghizadeh B, Ghorbanzadeh B, Alboghobeish S (2017) Systemic and local anti-nociceptive effects of simvastatin in the rat formalin assay: role of peroxisome proliferator-activated receptor γ and nitric oxide. J Neurosci Res. doi:10.1002/jnr.24008
Marwaha L, Bansal Y, Singh R et al (2016) TRP channels: potential drug target for neuropathic pain. Inflammopharmacology 24:305–317
Massicot F, Hache G, David L et al (2013) P2X7 cell death receptor activation and mitochondrial impairment in oxaliplatin-induced apoptosis and neuronal injury: cellular mechanisms and approach. PLoS One 8:e66830. doi:10.1371/journal.pone.0066830
McNamara CR, Mandel-Brehm J, Bautista DM et al (2007) TRPA1 mediates formalin-induced pain. Proc Natl Acad Sci 104:13525–13530. doi:10.1073/pnas.0705924104
Micheli L, Di Cesare Mannelli L, Rizzi A et al (2015) Intrathecal administration of nociceptin/orphanin FQ receptor agonists in rats: a strategy to relieve chemotherapy-induced neuropathic hypersensitivity. Eur J Pharmacol 766:155–162. doi:10.1016/j.ejphar.2015.10.005
Miranda HF, Noriega V, Olavarria L et al (2011) Antinociception and Anti-Inflammation Induced by Simvastatin in Algesiometric Assays in Mice. Basic Clin Pharmacol Toxicol 109:438–442. doi:10.1111/j.1742-7843.2011.00746.x
Nassini R, Gees M, Harrison S et al (2011) Oxaliplatin elicits mechanical and cold allodynia in rodents via TRPA1 receptor stimulation. Pain 152:1621–1631. doi:10.1016/j.pain.2011.02.051
Nassini R, Fusi C, Materazzi S et al (2015) The TRPA1 channel mediates the analgesic action of dipyrone and pyrazolone derivatives. Br J Pharmacol 172:3397–3411. doi:10.1111/bph.13129
Ohsawa M, Ishikura K, Mutoh J, Hisa H (2016) Involvement of inhibition of RhoA/Rho kinase signaling in simvastatin-induced amelioration of neuropathic pain. Neuroscience 333:204–213. doi:10.1016/j.neuroscience.2016.07.029
Plein LM, Rittner HL (2017) Opioids and the immune system—friend or foe. Br JPharmacol. doi:10.1111/bph.13750
Qiu Y, Chen WY, Wang ZY et al (2016) Simvastatin attenuates neuropathic pain by inhibiting the RhoA/LIMK/Cofilin pathway. Neurochem Res 41(9):2457–2469. doi:10.1007/s11064-0161958-1
Raffa RB, Burdge G, Gambrah J et al (2017) Cebranopadol: novel dual opioid/NOP receptor agonist analgesic. J Clin Pharm Ther 42:8–17. doi:10.1111/jcpt.12461
Rizzi A, Nazzaro C, Marzola GG et al (2006) Endogenous nociceptin/orphanin FQ signalling produces opposite spinal antinociceptive and supraspinal pronociceptive effects in the mouse formalin test: pharmacological and genetic evidences. Pain 124:100–108. doi:10.1016/j.pain.2006.03.021
Sakurada T, Komatsu T, Moriyama T et al (2005) Effects of intraplantar injections of nociceptin and its N-terminal fragments on nociceptive and desensitized responses induced by capsaicin in mice. Peptides 26:2505–2512. doi:10.1016/j.peptides.2005.05.022
Sałat K, Filipek B (2015) Antinociceptive activity of transient receptor potential channel TRPV1, TRPA1, and TRPM8 antagonists in neurogenic and neuropathic pain models in mice. J Zhejiang Univ B 16:167–178. doi:10.1631/jzus.B1400189
Sałat K, Filipek B, Wiȩckowski K, Malawska B (2009) Analgesic activity of 3-mono-substituted derivatives of dihydrofuran-2-one in experimental rodent models of pain. Pharmacol Rep 61:807–818. doi:10.1016/S1734-1140(09)70136-7
Sałat K, Librowski T, Moniczewski A et al (2012) Analgesic, antioedematous and antioxidant activity of γ-butyrolactone derivatives in rodents. Behav Pharmacol 23:407–416. doi:10.1097/FBP.0b013e3283566042
Sałat K, Kulig K, Gajda J et al (2013a) Evaluation of anxiolytic-like, anticonvulsant, antidepressant-like and antinociceptive properties of new 2-substituted 4-hydroxybutanamides with affinity for GABA transporters in mice. Pharmacol Biochem Behav 110:145–153. doi:10.1016/j.pbb.2013.06.013
Sałat K, Moniczewski A, Librowski T (2013b) Transient receptor potential channels—emerging novel drug targets for the treatment of pain. Curr Med Chem 20:1409–1436. doi:10.2174/09298673113209990107
Sałat K, Cios A, Wyska E et al (2014) Antiallodynic and antihyperalgesic activity of 3-[4-(3-trifluoromethyl- phenyl)-piperazin-1-yl]-dihydrofuran-2-one compared to pregabalin in chemotherapy-induced neuropathic pain in mice. Pharmacol Biochem Behav 122:173–181. doi:10.1016/j.pbb.2014.03.025
Sałat K, Jakubowska A, Kulig K (2015a) Cebranopadol: a first-in-class potent analgesic agent with agonistic activity at nociceptin/orphanin FQ and opioid receptors. Expert Opin Investig Drugs 24:837–844. doi:10.1517/13543784.2015.1036985
Sałat K, Podkowa A, Kowalczyk P et al (2015b) Anticonvulsant active inhibitor of GABA transporter subtype 1, tiagabine, with activity in mouse models of anxiety, pain and depression. Pharmacol Rep 67:465–472. doi:10.1016/j.pharep.2014.11.003
Schröder W, Lambert DG, Ko MC, Koch T (2014) Functional plasticity of the N/OFQ-NOP receptor system determines analgesic properties of NOP receptor agonists. Br J Pharmacol 171:3777–3800. doi:10.1111/bph.12744
Sehgal N, Smith HS, Manchikanti L (2011) Peripherally acting opioids and clinical implications for pain control. Pain Physician 14(3):249–258
Shah S, Page CP, Spina D (1998) Nociceptin inhibits non-adrenergic non-cholinergic contraction in guinea-pig airway. Br J Pharmacol 125:510–516. doi:10.1038/sj.bjp.0702068
Shi XQ, Lim TKY, Lee S et al (2011) Statins alleviate experimental nerve injury-induced neuropathic pain. Pain 152:1033–1043. doi:10.1016/j.pain.2011.01.006
Singh SR, Sullo N, Matteis M et al (2016) Nociceptin/orphanin FQ (N/OFQ) modulates immunopathology and airway hyperresponsiveness representing a novel target for the treatment of asthma. Br J Pharmacol 173:1286–1301. doi:10.1111/bph.13416
Sobczak M, Salaga M, Storr M, Fichna J (2013) Nociceptin/orphanin FQ (NOP) receptors as novel potential target in the treatment of gastrointestinal diseases. Curr Drug Targets 14:1203–1209
Sobczak M, Pilarczyk A, Jonakowski M et al (2014) Anti-inflammatory and antinociceptive action of the dimeric enkephalin peptide biphalin in the mouse model of colitis: new potential treatment of abdominal pain associated with inflammatory bowel diseases. Peptides 60:102–106. doi:10.1016/j.peptides.2014.08.005
Sukhtankar DD, Zaveri NT, Husbands SM, Ko M-C (2013) Effects of spinally administered bifunctional nociceptin/orphanin fq peptide receptor/-Opioid receptor ligands in mouse models of neuropathic and inflammatory pain. J Pharmacol Exp Ther 346:11–22. doi:10.1124/jpet.113.203984
Sun J, Yang T, Wang P et al (2014) Activation of cold-sensing transient receptor potential melastatin subtype 8 antagonizes vasoconstriction and hypertension through attenuating RhoA/Rho kinase pathway. Hypertension 63(6):1354–1363. doi:10.1161/HYPERTENSIONAHA.113.02573
Tajti J, Szok D, Majlath Z et al (2015) Migraine and neuropeptides. Neuropeptides 52:19–30. doi:10.1016/j.npep.2015.03.006
Tian J-H, Xu W, Fang Y et al (1997) Bidirectional modulatory effect of orphanin FQ on morphine-induced analgesia: antagonism in brain and potentiation in spinal cord of the rat. Br J Pharmacol 120:676–680. doi:10.1038/sj.bjp.0700942
Tjølsen A, Berge O-G, Hunskaar S et al (1992) The formalin test: an evaluation of the method. Pain 51:5–17. doi:10.1016/0304-3959(92)90003-T
Torrance N, Ferguson JA, Afolabi E et al (2013) Neuropathic pain in the community: more under-treated than refractory? Pain 154:690–699. doi:10.1016/j.pain.2012.12.022
Tzschentke TM, Linz K, Frosch S, Christoph T (2017) Antihyperalgesic, antiallodynic, and antinociceptive effects of cebranopadol, a novel potent nociceptin/orphanin FQ and opioid receptor agonist, after peripheral and central administration in rodent models of neuropathic pain. Pain Pract. doi:10.1111/papr.12558
Vitale G, Filaferro M, Micioni Di Bonaventura MV et al (2017) Effects of [Nphe 1, Arg 14, Lys 15] N/OFQ-NH 2 (UFP-101), a potent NOP receptor antagonist, on molecular, cellular and behavioural alterations associated with chronic mild stress. J Psychopharmacol 31:691–703. doi:10.1177/0269881117691456
Vogel HG, Vogel WH (1997) Analgesic, anti-inflammatory, and antipyretic activity. Drug discovery and evaluation. Springer, Berlin, pp 360–420
Wang JL, Bin ZhuC, Cao XD, Wu GC (1999) Distinct effect of intracerebroventricular and intrathecal injections of nociceptin/orphanin FQ in the rat formalin test. Regul Pept 79:159–163. doi:10.1016/S0167-0115(98)00161-X
Wang L, Peng J, Huang H et al (2015) Simvastatin protects Sertoli cells against cisplatin cytotoxicity through enhanced gap junction intercellular communication. Oncol Rep 34(4):2133–2141. doi:10.3892/or.2015.4192
Waseem M, Parvez S (2016) Neuroprotective activities of curcumin and quercetin with potential relevance to mitochondrial dysfunction induced by oxaliplatin. Protoplasma 253:417–430. doi:10.1007/s00709-015-0821-6
Witkin JM, Statnick MA, Rorick-Kehn LM et al (2014) The biology of Nociceptin/Orphanin FQ (N/OFQ) related to obesity, stress, anxiety, mood, and drug dependence. Pharmacol Ther 141:283–299
Xu XJ, Hao JX, Wiesenfeld-Hallin Z (1996) Nociceptin or antinociceptin: potent spinal antinociceptive effect of orphanin FQ/nociceptin in the rat. NeuroReport 7:2092–2094
Yamamoto T, Nozaki-Taguchi N, Kimura S (1997a) Analgesic effect of intrathecally administered nociceptin, an opioid receptor-like receptor agonist, in the rat formalin test. Neuroscience 81:249–254. doi:10.1016/S0306-4522(97)00166-8
Yamamoto T, Nozaki-Taguchi N, Kimura S (1997b) Effects of intrathecally administered nociceptin, an opioid receptor-like1 (ORL1) receptor agonist, on the thermal hyperalgesia induced by unilateral constriction injury to the sciatic nerve in the rat. Neurosci Lett 224:107–110. doi:10.1016/S0304-3940(97)13475-9
Yashpal K, Coderre TJ (1998) Influence of formalin concentration on the antinociceptive effects of anti-inflammatory drugs in the formalin test in rats: separate mechanisms underlying the nociceptive effects of low-and high-concentration formalin. Eur J Pain 2:63–68. doi:10.1016/S1090-3801(98)90047-7
Zhang Y, Simpson-Durand CD, Standifer KM (2015) Nociceptin/orphanin FQ peptide receptor antagonist JTC-801 reverses pain and anxiety symptoms in a rat model of post-traumatic stress disorder. Br J Pharmacol 172:571–582. doi:10.1111/bph.12701
Zhao M, Isami K, Nakamura S et al (2012) Acute cold hypersensitivity characteristically induced by oxaliplatin is caused by the enhanced responsiveness of TRPA1 in mice. Mol Pain 8:55. doi:10.1186/1744-8069-8-55
Zhu CB, Cao XD, Xu SF, Wu GC (1997) Orphanin FQ potentiates formalin-induced pain behavior and antagonizes morphine analgesia in rats. Neurosci Lett 235:37–40. doi:10.1016/S0304-3940(97)00704-0
Acknowledgements
This study was financially supported by the National Science Centre grant UMO-2015/17/B/NZ7/02937.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None declared.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. All procedures performed in studies involving animals were in accordance with the ethical standards of the institution at which the present study was conducted.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sałat, K., Furgała, A. & Sałat, R. Evaluation of cebranopadol, a dually acting nociceptin/orphanin FQ and opioid receptor agonist in mouse models of acute, tonic, and chemotherapy-induced neuropathic pain. Inflammopharmacol 26, 361–374 (2018). https://doi.org/10.1007/s10787-017-0405-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10787-017-0405-5