
Abstract
Background
QSAR modelling is a powerful technique widely used in the discovery and development of new drugs and chemicals.
The approach involves the use of mathematical and statistical techniques to develop models that relate biological activity to the chemical structure of compounds. An effective strategy for QSAR modelling requires an understanding of the descriptors that are used to represent the chemical structure of compounds and the selection of appropriate prediction algorithms. In this review, we were discussed the importance of developing a well-thought-out strategy for QSAR modelling, including the strategy of appropriate descriptors and physiochemical parameters were used for QSAR. We were also provided the review on the current strategies for conducting QSAR studies and discussing their parameters, descriptors and applications in drug discovery, toxicity testing, and more.
Conclusion
This finding suggests how to develop an effective QSAR modelling approach that can be used to accurately predict the activity of new compounds.


Similar content being viewed by others


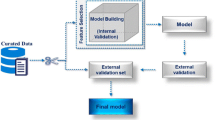
Abbreviations
- QSAR:
-
Quantitative structure activity relationship
- SAR:
-
Structure Activity Relationship
- logP:
-
Log of the partition co-efficient
- pKa:
-
Equilibrium constant
References
Yu Wenbo et al (2017) Computer-aided drug design methods. Methods Mol Biol 15(20):85–106
Da-Yong Lu, Ting-Ren Lu (2020) Anticancer drug development, challenge and dilemma. Nurse Care Open Acces J 7(3):72–75
Cui W, Aouidate A, Wang S, Yu Q, Li Y, Yuan S (2020) Discovering anti-cancer drugs via computational methods. Front Pharmacol 11:733
Surabhi Singh, B. K. (2018) Computer aided drug design: an overview. J Drug Deliv Ther 8(5):504–509
Ece A (2023) Computer-aided drug design. BMC. Chemistry 17:26. https://doi.org/10.1186/s13065-023-00939-w
Tamer N, Jarada Jon, Rokne G, Alhajj Reda (2020) A review of computational drug repositioning: strategies, approaches, opportunities, challenges, and directions. J Cheminform 12:46
Oprea Tudor I, Overington John P (2015) Computational and practical aspects of drug repositioning. Assay Drug Dev Technol 13(6):299–306
Shaker Bilal, Ahmad Sajjad et al (2021) In silico methods and tools for drug discovery. Comput Biol Med 137:104851
Makrynitsa GI, Lykouras M, Spyroulias GA, Matsoukas M.-T (2018) In silico Drug Design. In: eLS. John Wiley & Sons, Ltd (Ed.). https://doi.org/10.1002/9780470015902.a0028112
Lu W, Zhang R, Jiang H, Zhang H, Luo C (2018) Computer-aided drug design in epigenetics. Front Chem 6:57
Achary PGR (2020) Mini Rev Med Chem 20(14):1375–1388
Neves BJ, Braga RC, Melo-Filho CC, Moreira-Filho JT, Muratov EN, Andrade CH (2018) QSAR-based virtual screening: advances and applications in drug discovery. Front Pharmacol 9:1275
Achary P (2014) QSPR modelling of dielectric constants of π-conjugated organic compounds by means of the CORAL software. SAR QSAR Environ Res 25:507–526
Agresti A (2007) An introduction to categorical data analysis, John Wiley. Second Edition, Department of Statistics University of Florida Gainesville, Florida. ISBN 978-0-471-22618-5
Andrews CW, Bennett L, Lawrence XY (2000) Predicting human oral bioavailability of a compound: development of a novel quantitative structure bioavailability relationship. Pharm Res 17:639–644
Balaban AT (1982) Highly discriminating distance-based topological index. Chem Phys Lett 89:399–404
Barber CE, Marshall DA, Mosher DP, Akhavan P, Tucker L, Houghton K, Batthish M, Levy DM, Schmeling H, Ellsworth J, Tibollo H, Grant S, Khodyakov D, Lacaille D (2016) Arthritis alliance of Canada performance measurement development panel. Development of system-level performance measures for evaluation of models of care for inflammatory arthritis in Canada. J Rheumatol 43(3):530–40. https://doi.org/10.3899/jrheum.150839
Barycki M, Sosnowska A, Puzyn T (2017) Which structural features stand behind micelization of ionic liquids? Quantitative structure-property relationship studies. J Colloid Interface Sci 487:475–483
Berinde Z (2013) A QSPR study of hydrophobicity of phenols and 2-(aryloxy-α-acetyl)- phenoxathiin derivatives using the topological index ZEP. Creative Math Inform 22:33–40
Basith Shaherin, Cui Minghua et al (2017) Expediting the design, discovery and development of anticancer drugs using computational approaches. Curr Med Chem 24:4753–4778
Gagic Z, Ruzic D, Djokovic N, Djikic T, Nikolic K (2020) In silico methods for design of kinase inhibitors as anticancer drugs. Front Chem 7:873
Raval K, Ganatra T (2022) Basics, types and applications of molecular docking: a review. Int J Compr Adv Pharmacol 7(1):12–16
Wang X, Song K, Li L, Chen L (2018) Structure-based drug design strategies and challenges. Curr Top Med Chem 18:998–1006
Fyaz MDI, Nahar L, Sarker SD (2018) High-Throughput screening of phytochemicals: application of computational methods. In: Sarker SD, Nahar L (eds) Computational phytochemistry. Elsevier, pp 165–192
Wenbo Yu et al (2017) Computer-aided drug design methods. Methods Mol Biol 1520:85–106
Surabhi Singh, B. K. (2018) Computer aided drug design: an overview. J Drug Deliv Ther 8(5):504–509
Ece Abdulilah (2023) Computer-aided drug design. Ece BMC Chem 17:26
Shaker Bilal, Ahmad Sajjad et al (2021) In silico methods and tools for drug discovery. Comput Biol Med. 137:104851
Makrynitsa GI, Lykouras M, Spyroulias GA, Matsoukas M-T (2018) In silico drug design. In: eLS. John Wiley & Sons, Ltd, Chichester
Lu W, Zhang R, Jiang H, Zhang H, Luo C (2018) Computer-aided drug design in epigenetics. Front Chem 6:57
Yousefinejad S, Hemmateenejad B (2015) Chemometrics tools in QSAR/QSPR studies: a historical perspective. Chemom Intell Lab Syst 149:177–204
Raies AB, Bajic VB (2016) In silico toxicology: computational methods for the prediction of chemical toxicity. WIREs Comput Mol Sci 6(2):147–172
Carracedo-Reboredo P, Liñares-Blanco J, Rodríguez-Fernández N et al (2021) A review on machine learning approaches and trends in drug discovery. Comput Struct Biotechnol J 19:4538–4558
Vamathevan J, Clark D, Czodrowski P et al (2019) Applications of machine learning in drug discovery and development. Nat Rev Drug Discovery 18(6):463–477
Gupta R, Srivastava D, Sahu M, Tiwari S, Ambasta RK, Kumar P (2021) Artificial intelligence to deep learning: machine intelligence approach for drug discovery. Mol Divers 25(3):1315–1360
Carracedo-Reboredo P, Liñares-Blanco J, Rodríguez-Fernández N et al (2021) A review on machine learning approaches and trends in drug discovery. Comput Struct Biotechnol J 19:4538–4558
Dara S, Dhamercherla S, Jadav SS, Babu ChM, Ahsan MJ (2021) Machine learning in drug discovery: a review. Artif Intell Rev 55(3):1947–1999
Réda C, Kaufmann E, Delahaye-Duriez A (2020) Machine learning applications in drug development. Comput Struct Biotechnol J 18:241–252
Golbraikh A, Wang XS, Zhu H, Tropsha A (2012) Predictive QSAR modeling: Methods and applications in drug discovery and chemical risk assessment. In: Handbook of Computational Chemistry, Springer, Netherlands, pp 1309–1342. https://doi.org/10.1007/978-94-007-0711-5_37
Tropsha A, Gelbukh A (2007) Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr Pharm Des 13(34):3494–3504
Kim E, Yang C (2020) Application of quantitative structure activity relationship models for virtual screening drug libraries: an overview of state-of-the art methodologies and strategies. Drug Des Rev 6(2):75–88
Dablander M et al (2023) Exploring QSAR models for activity-clif prediction. J Cheminformatics 15:47
Sharan S, Swaminathan PN (2020) Exploring new avenues using pharmacophore mapping. Int J Pharm Sci Res 11(11):5270–5279
Manisha YJ, Satya E (2022) Modern paradigm towards potential target identification for antiviral (SARS-nCoV-2) and anticancer lipopeptides: a pharmacophore-based approach. Avicenna J Med Biotechnol 14(1):70–78
Khedkar SA et al (2007) Pharmacophore modeling in drug discovery and development: an overview. Med Chem 3(2):187–97
Verma J, Khedkar VM, Coutinho EC (2010) 3D-QSAR in drug design - a review. Curr Top Med Chem (Print) 10(1):95–115
Toshio F (1995) QSAR and drug design: new developments and applications, vol 23. pp 3–493
Tropsha A, Cho SJ, Zheng W (1999) “New tricks for an old dog”: development and application of novel QSAR methods for rational design of combinatorial chemical libraries and database mining. ACS Symposium Series 719:198–211. https://doi.org/10.1021/bk-1999-0719.ch013
Kleandrova VV et al (2021) QSAR modeling for multi-target drug discovery: designing simultaneous inhibitors of proteins in diverse pathogenic parasites. Front Chem 9:634663
Speck-Planche Alejandro, Valeria V et al (2012) Rational drug design for anti-cancer chemotherapy: multi-target QSAR models for the in silico discovery of anti-colorectal cancer agents. Bioorg Med Chem 20:4848–4855
Zhao L, Teng Q, Liu Y, Chen H et al (2022) Machine learning-based identification of a novel prognosis-related long noncoding RNA signature for gastric cancer. Front Cell Dev Biol 10(1017767):1–20
Cox PB et al. (2022) Contemporary computational applications and tools in drug discovery. ACS Med Chem Lett 13, 7, 1016–1029
Ojha Probir Kumar et al (2021) Recent advances in quantitative structure-activity relationship models of antimalarial drugs. Expert Opin Drug Discov 16(6):659–695
Chen B, Zhang T, Bond T, Gan Y (2015) Development of quantitative structure activity relationship (QSAR) model for disinfection byproduct (DBP) research: a review of methods and resources. J Hazard Mater (Print) 299:260–279
Yousefinejad S, Hemmateenejad B (2015) Chemometrics tools in QSAR/QSPR studies: a historical perspective. Chemom Intell Lab Syst 149:177–204
He L, Jurs PC (2005) Assessing the reliability of a QSAR model’s predictions. J Mol Graph Model 23(6):503–523
Maruca A et al (2019) Computer-based techniques for lead identification and optimization I: basics. Phys Sci Rev 11
Carels N, Tilli T, Tuszynski JA (2015) A computational strategy to select optimized protein targets for drug development toward the control of cancer diseases. PLoS One 10(1):e0115054
Ferreira AK, Kawamura B, Jorge SD, de Azevedo RA, Zaim MH et al (2017) Developing novel anticancer drug candidates regarding the integration of three main knowledge fields: computeraided drug design, chemical synthesis, and pharmacological evaluation. J Drug Des Res 4(2):1035
Sugrue MF (2000) Pharmacological and ocular hypotensive properties of topical carbonic anhydrase inhibitors. Prog Retin Eye Res 19(1):87–112
Monroe D (2016) Looking for chinks in the armor of bacterial biofilms. PLoS Biol 5(11):e307. 5
Jagiełło K, Grzonkowska M, Swirog M et al (2016) Advantages and limitations of classic and 3D QSAR approaches in nano-QSAR studies based on biological activity of fullerene derivatives. J Nanopart Res 18(9)
Cherkasov A, Muratov E, Fourches D et al (2014) QSAR modeling: Where have you been? Where are you going to? J Med Chem 57(12):4977–5010
Roy K, Kar S, Ambure P (2015) On a simple approach for determining applicability domain of QSAR models. Chemom Intell Lab Syst (Print) 145:22–29
Ghasemi F, Mehridehnavi A, Pérez-Garrido A, Pérez-Sánchez H (2018) Neural network and deep-learning algorithms used in QSAR studies: merits and drawbacks. Drug Discov Today 23(10):1784–1790
Nicotia (2021) Introduction to (Quantitative) Structure Activity Relationships. J Anat Physiol 2:224–42
Selassie C, Verma RP (2016) History of quantitative structure– activity relationships. In: Abraham DJ (editor) Burger's medicinal chemistry and drug discovery. 1–96
Mishra V, Siva-Prasad CV (2011) Ligand based virtual screening to find novel inhibitors against plant toxin Ricin by using the ZINC database. Bioinformation 7:46–51
Song CM, Lim SJ, Tong JC (2009) Recent advances in computeraided drug design. Brief Bioinform 10:579–591
Jorgensen WL (2004) The many roles of computation in drug discovery. Science 303:1813
Martin YC, Kofron JL, Traphagen LM (2002) Do structurally similar molecules have similar biological activity? J Med Chem 45:4350
Tropsha A (2010) Best Practices for QSAR model development, validation, and exploitation. Mol Inf 29(6–7):476–488
Neves BJ, Braga RC, Melo-Filho CC, Moreira-Filho JT, Muratov E, Andrade CH (2018) QSAR-based virtual screening: advances and applications in drug discovery. Front Pharmacol 9
Kitchen DB, Decornez H, Furr JR, Bajorath J (2004) Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 3(11):935–949
Jorgensen WL (2004) The many roles of computation in drug discovery. Science 303:1813
Martin YC, Kofron JL, Traphagen LM (2002) Do structurally similar molecules have similar biological activity? J Med Chem 45:4350
Gramatica P (2012) On the development and validation of QSAR models. In: Methods in molecular biology. 499–526
Tropsha A, Gelbukh A (2007) Predictive QSAR modeling workflow, model applicability domains, and virtual screening. Curr Pharml Des (Print) 13(34):3494–3504
Devillers J, Balaban AT (2000) Topological indices and related descriptors in QSAR and QSPR
Eriksson L, Johansson E (1996) Multivariate design and modeling in QSAR. Chemom Intell Lab Syst 34(1):1–19
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflicts of Interest
There is no conflict of interest in this study.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vijayalakshmi, M.K., Srinivasan, R. Review of Contemporary QSAR Study Approach. Chemistry Africa (2024). https://doi.org/10.1007/s42250-024-00983-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42250-024-00983-6