
Abstract
Rare-earth disilicates are promising candidates for thermal and environmental barrier coatings (TEBC) in gas turbines that safeguard SiCf/SiC ceramic matrix composites (CMCs) from thermal degradation and environmental attacks. Here, we report a systematic investigation on novel TEBC material, γ-Y1.5Yb0.5Si2O7. The γ-phase quarter molar ytterbium–doped yttrium disilicate exhibited low thermal conductivity (1.72 W·m−1·K−1 at 1200 °C) and reduced intrinsic thermal expansion (3.17 ± 0.22 × 10−6 K−1 up to 1000 °C), ensuring promisingly effective thermal insulation and minimized thermal stress with CMC substrates. Using density functional theory (DFT), the heat capacity of γ-Y1.5Yb0.5Si2O7 was predicted higher than that of undoped γ-Y2Si2O7. Comparing these predictions to results calculated using the Neumann–Kopp (NK) rule revealed only minor variations. A metastable CMAS interaction byproduct, cyclosilicate phase Ca3RE2(Si3O9)2, was identified based on energy dispersive X-ray spectrometer (EDS) and electron backscatter diffraction (EBSD) techniques, appearing at 1300 °C but disappearing at 1400 °C. The γ-Y1.5Yb0.5Si2O7 exhibited good CMAS resistance on both dense pellets and sprayed coatings, forming a protective apatite (Ca2RE8(SiO4)6O2) interlayer that effectively hindered CMAS infiltration at evaluated temperatures. The relatively higher Y:Yb atomic ratio (> 3) in the apatite grains indicate differential reactivity with molten CMAS and provides crucial insights into the CMAS corrosion mechanism. These findings highlight the potential of γ-Y1.5Yb0.5Si2O7 as a CMC coating material, emphasizing the need for tailored microstructural optimization as a thermal sprayed coating to enhance long-term performance in extreme gas turbine environments.
Similar content being viewed by others


1 Introduction
The development of aerospace gas turbine engine materials has undergone significant advancements over the years [1, 2]. The operational temperature of gas turbines with current nickel-based alloys can only be increased incrementally by utilizing improved cooling methods and advanced thermal barrier coatings whereas the incorporation of SiC-based CMCs can bring a step change in performance [3]. SiC-based CMCs possess superior thermal and mechanical properties, enabling engines to withstand higher temperatures (> 1600 °C) while reducing weight-to-flight ratio (about 1/3 that of Ni-based super-alloys) enhance fuel efficiency, reduce emissions, and improve overall performance [4, 5]. However, turbine engines operate under environments that consist of corrosive gases and elevated oxygen levels. In combustion environments, material temperatures can reach between 1100 and 1400 °C, with gas velocities ranging from 100 to 668 m/s, total pressures of 6 to 31 atm, and a 10% water vapor atmosphere [6]. When subjected to a fast-flowing water vapor atmosphere at high temperatures, the SiC-based components experience a rapid recession due to the transformation of the protective SiO2 into gaseous Si(OH)4 [7]. Consequently, the reaction product becomes volatile, leading to significant material degradation. In addition, the occurrence of corrosive species arises from ingesting debris during take-off and landing, flying over arid regions, or encountering airborne volcanic ash [8]. Given the diverse characteristics and compositions of these corrosive elements, which typically encompass CaO, MgO, Al2O3, and SiO2 in varying proportions, the term CMAS was coined as a comprehensive descriptor to encompass this group of corrosive species.
The development of environment barrier coatings (EBCs) aims to safeguard SiC-based CMCs from degradation caused by oxidizing and water vapor environments [9], which includes a bond coat as an interlayer to ensure strong adhesion between the substrate and the topcoat. Successful EBCs should possess a high melting point, phase and thermal stability at temperatures above 1200 °C, together with a dense microstructure to inhibit infiltration of water vapor and oxygen. Moreover, it is crucial to minimize the difference in the coefficient of thermal expansion (CTE) between the EBCs and the substrate (bond coat and SiC CMCs) since the high thermal stresses can potentially lead to the failure of the protective system [10,11,12]. Current research on EBCs has primarily focused on rare earth (RE) disilicates owing to their remarkable high-temperature stability and exceptional chemical compatibility with thermally grown SiO2 (TGO). Among them, yttrium and ytterbium disilicates possess closely matching CTE values (γ-Y2Si2O7: ~ 4 \(\times\) 10−6 K−1; β-Yb2Si2O7: 3.7–4.5 \(\times\) 10−6 K−1) with both the Si bond coat and SiC/SiC CMC [4, 13,14,15]. This advantageous similarity aids in reducing inter-layer thermal stress and preventing coating detachment during thermal cycling. Moreover, by employing Clark’s model [16] and simulated elastic properties, Sun et al. [13] respectively predicted minimum theoretical thermal conductivities of 1.35 W·(m·K)−1 for γ-Y2Si2O7. Their experimental results provided supporting evidence for the predicted low thermal conductivity of 1.90 W·(m·K)−1 for γ-Y2Si2O7 at 1400 K. Luo et al. [17] elucidated the mechanism by utilizing the DFT calculations to explain the structural origin of low-lying optic phonons and the subsequent phonon anharmonicity in γ-Y2Si2O7: the intrinsic bonding heterogeneity within the lattice structure. Tian et al. [18] revealed that the γ-structured yttrium disilicate, 1664/T W·(m·K)−1, has lower intrinsic lattice thermal conductivities than both its β-phase and β-Yb2Si2O7, 2304/T W·(m·K)−1 and 1740/T W·(m·K)−1 that is attributed to the enhanced complexity of the γ-phase crystal structure, suggesting a promising application as TEBC candidates.
Numerous studies have documented the interaction between yttrium and ytterbium disilicates and CMAS. Markus et al. [19] conducted a thorough investigation into the interaction of four pure rare earth silicate EBC materials (Yb2SiO5, β-Yb2Si2O7, Y2SiO5, γ-Y2Si2O7) with CMAS (50%SiO2-38%CaO-5%MgO-4%Al2O3-1%Ka2O-1%Na2O-1%Fe2O3) at 1400 °C. Their findings revealed that the recession of ytterbium silicates is significantly lower compared to the recession observed in yttrium silicates when subjected to CMAS attack. Similar results were observed by Turcer et al. [20,21,22]. They pointed out that the CMAS (51.5%SiO2-39.2%CaO-4.1%Al2O3-5.2%MgO) resistance of β-Yb2Si2O7 was higher when compared to γ-Y2Si2O7. This was attributed to the smaller ionic radius of Yb, which resulted in a lower tendency to form secondary phases. These studies highlight the intricate nature of the interactions between various RE disilicates and CMAS, with differing conclusions regarding their respective resistance properties. Contrasting results from Liu et al. [23] demonstrated that among a series of RE2Si2O7 (RE = Yb, Lu, La, Eu, Gd, and Y), Y2Si2O7 exhibited superior resistance to a standard CMAS composition (33%CaO-9%MgO-12%Al2O3-45%SiO2) at 1400 °C for 10 h. In the context of the CMAS corrosion mechanism of yttrium and ytterbium disilicates, the widely accepted process is dissolution–precipitation [24], wherein coatings are dissolved by molten CMAS, and various stable crystalline products precipitate from the melt. The formation of apatite phases is believed to serve as a thermodynamically stable barrier, effectively preventing further CMAS melt penetration at high temperatures. Stokes et al. [25] and Summers et al. [26] emphasized the significant influence of the Ca to Si ratio in CMAS compositions on the resultant reaction products in yttrium and ytterbium disilicates. At Ca to Si ratios above 0.72, primarily apatite phases are formed, whereas, for lower ratios, γ-Y2Si2O7 shows a propensity to generate more apatite phase compared with β-Yb2Si2O7. However, the emergence of a metastable cyclosilicate phase, Ca3Y2(Si3O9)2, was revealed by Sleeper et al. [27] in their comprehensive study on the thermochemical interactions between Y-apatite phase, Ca2Y8Si6O26, and CMAS (23.3%CaO-6.4%MgO-3.1%Al2O3-62.5%SiO2-4.1%Na2O-0.5%K2O-0.04%Fe2O3). Intriguingly, this cyclosilicate phase was not detected at higher temperatures of 1400 °C and 1500 °C. The evidence presented above indicates that the Yb-doped yttrium disilicate may offer a potential solution to compensate for their respective shortcomings in CMAS corrosion resistance.
Recent studies have been conducted by employing a solid solution design between yttrium and ytterbium disilicates. Fernández-Carrión et al. [28] reported the solid solubility of Yb in Y2Si2O7, showing that a complete YxYb(2−x)Si2O7 solid solution can form at 1300 °C. Notably, the YxYb(2−x)Si2O7 solid solution with x values below 1.2 maintains a stable β-RE2Si2O7 phase at temperatures lower than 1650 °C, making it a promising EBC candidate. Stack et al. [29] conducted thermal cycling investigations on APS Si/β-Y1.2Yb0.8Si2O7 EBCs coated with SiC at 1350 °C in air. The topcoat β-Y1.2Yb0.8Si2O7 layer exhibited the β structured disilicate phase with limited Y2SiO5 monosilicate after 500 h of cyclic oxidation in air, while a thinner TGO layer was observed at the β-Y1.2Yb0.8Si2O7/SiC interface. This indicated that the coating offered effective protection to SiC at 1350 °C in dry air. Zhang et al. [30] reported a novel two-layer bond coat and topcoat structured EBCs (50%HfO2-50%SiO2)/β-YxYb(2−x)Si2O7. After undergoing 200 h of exposure in dry air and 100 h in steam conditions, the EBCs demonstrated strong bonding to the SiC substrate, showcasing its potential as a promising candidate for EBCs. Remarkably, it effectively protected the SiC substrate in both air and steam conditions at 1475 °C. Turcer et al. [31] discovered that the β-phase was restored with more than half Yb doping in YxYb(2−x)Si2O7 solid solutions. However, there is limited research on the thermal properties and CMAS resistance of Yb-doped γ-structured Y2Si2O7. Further studies in this area are needed.
Suspension plasma spray (SPS) is a promising coating deposition technique that can produce bespoke architecture and microstructure [32]. By employing liquid suspensions rather than powders (in atmospheric plasma spray), SPS offers enhanced material flexibility and homogeneity, leading to enhanced adhesion and finer microstructures. These advancements position SPS as a viable option for the development of Y1.5Yb0.5Si2O7 in TEBC applications. It excels in providing diverse microstructures, from columnar and porous to dense formations depending on the application requirements. Crucial processing parameters in SPS, such as plasma power, plasma gas mixture, plasma gas flow, stand-off distance, robotic program, substrate temperature, accelerating nozzle geometry along with suspension medium, particle size distribution, additives, and solid loading play pivotal roles in controlling the SPS microstructures. Vassen et al. [33] fabricated Yb2Si2O7 coatings by using the SPS technique with different stand-off distances of 50, 70, and 90 mm. The shorter stand-off distance yielded a high degree of crystallinity and denser microstructure; however, the challenge lies in mitigating segmentation cracks. The reason for segmentation crack formation in the SPS coating is associated with the high torch power used to evaporate the suspension liquid and short spray distances in this process [34].
In this study, a promising TEBC material with a unique composition design, γ-Y1.5Yb0.5Si2O7, was fabricated for the first-time using SPS and conventional sintering techniques. The quarter molar Yb-doped γ-Y2Si2O7 is chosen because < 0.5 molar ratio replacement in the Y site not only helps improve CMAS resistance but maintains the intrinsic low thermal conductivity γ-phase structure as well. The lattice thermal expansion behavior of the γ-Y1.5Yb0.5Si2O7 was investigated through hot X-ray diffraction (hot-XRD). The volumetric thermal expansion coefficient was calculated and compared with that of yttrium and ytterbium disilicates. Furthermore, the thermal diffusivity of the γ-Y1.5Yb0.5Si2O7 dense pellet was measured. The thermal conductivity of γ-Y1.5Yb0.5Si2O7 was calculated using two different approaches based on specific heat (Cp) data: the laser flash comparative method and the Neumann–Kopp rule, providing valuable insights into its thermal insulation performance. Subsequently, the CMAS interaction of both the coatings and pellets was examined at temperatures of 1300–1400 °C for durations up to 60 min. While focusing on the influence of composition design on CMAS attack, especially utilizing dense γ-Y1.5Yb0.5Si2O7 pellets in the context of CMAS chemistry, considerations related to microstructural factors such as pores and microcracks were deferred. Microstructure and reaction products after CMAS interactions were systematically studied via energy dispersive X-ray spectrometer (EDS) and electron backscatter diffraction (EBSD) techniques. Possible CMAS reactions and resistance mechanism were proposed. The primary objective of this research was to gain valuable insights into the underlying thermo-chemo-mechanical mechanisms of novel EBC material: γ-Y1.5Yb0.5Si2O7. This knowledge will then be utilized to guide the design and development of future CMAS-resistant TEBCs.
2 Experimental methods
2.1 Materials
A Y1.5Yb0.5Si2O7 suspension (Treibacher Industrie AG), with a D90 value of 1.8 μm, D50 value of 0.9 μm, and D10 value of 0.5 μm, was used in this study. The composition of the suspension is listed in Table 1 using inductively coupled plasma (ICP) spectrometry that was provided by the supplier.
2.2 Coating manufacture
The coating was produced by a high power, high efficiency, three cathode axial injection plasma spray torch (Axial III, M600, Mettech Corp., Surrey, Canada) with a continuous suspension feeder system (Nanofeed 350, Mettech Corp., Surrey, Canada). A plasma exit nozzle of 3/8 inch in diameter was used, and the feedstock was injected axially into the plasma through an injector of 0.02 inch in diameter. The detailed spray parameters are listed in Table 2. A hydrogen-free plasma parameter was used to deposit the EBC in line with other works in this area. The samples were mounted on a fixed jig with front and back cooling, and the SPS was mounted on a six-axis robot IRB2400 (ABB, Zurich, Switzerland), which followed a raster pattern on the sample surface. The speed of the robot was 1000 mm/s, and the stand-off distance was fixed at 100 mm. In total, 60 passes were conducted to build the coating.
Graphite substrates were sized at approximately 60 × 25 × 2 mm3. All substrates were grit blasted (Guyson, Dudley, UK) with fine F100 brown alumina (0.125–0.149 mm) particles at 2 bars. After spraying, heat treatment was conducted at 650 °C for 6 h to help the detachment and to obtain the free-standing coatings. Annealing of detached free-standing coatings was conducted in a box furnace at 1250 °C and 1450 °C for 2 h using a heating rate of 10 °C/min. The annealing temperatures were chosen based on the crystallization and phase transformation behaviors of Y2Si2O7 [35].
The dense pellet was fabricated from the dried feedstock powder, drying from a small amount of suspension in a box furnace at 100 °C overnight. Then, the powder was pressed into pellets at 20 MPa for 1 min to obtain Y1.5Yb0.5Si2O7 green bodies with a thickness of 3 mm. The green bodies were placed in an alumina crucible and sintered at 1450 °C in the air for 4 h using a heating rate of 2.5 °C/min.
2.3 Material characterization
2.3.1 Room temp and hot XRD diffraction
XRD analysis of the dried feedstock powder and the as-sprayed and annealed coatings was conducted using a D8 Advance DaVinci system (Bruker, Coventry, UK), equipped with a lynx eye detector. The diffractograms were obtained with Cu-Kα radiation with a wavelength of 1.54 Å in Bragg–Brentano scanning mode. The scanning range was set from 10 to 55° 2θ, and a slow scan rate was used (0.02° step size and 0.2 s of counting time per step). DIFFRACT.SUITE EVA software (Bruker, Coventry, UK) was used for the phase identification with the international center for diffraction data (ICDD) PDF4 database. Hot-XRD of Y1.5Yb0.5Si2O7 was conducted in a vacuum from room temperature to 1000 °C using a D8 Advance Series 2 (Bruker, Coventry, UK), equipped with an MTC-HIGHTEMP heating stage (Bruker AXS GmbH, Karlsruhe, Germany). The scanning was done at 200 °C, 400 °C, 600 °C, 800 °C and 1000 °C in the 2 \(\theta\) range of 10 to 90° with the same scan rate above. Rietveld refinement (GSAS software [36]) was applied for the obtained XRD patterns. The coefficients of thermal expansion (CTE) were calculated from change of lattice parameters (a, b, c, and β) with increased temperatures. The variation of the lattice parameters was fitted to the polynomial equation with the least squares method:
where χ is the cell dimensions at elevated temperatures, χrt the cell dimensions at room temperature (30 °C), and ΔT = (T-30). The volumetric coefficient of thermal expansion at measured temperatures, αT, at constant pressure is defined using the expression reported by Ziqi Sun et al. [13], under the assumption of thermal expansion measurements being a constant when the temperature higher than the material’s Debye temperature (θD).
The θD of γ-Y2Si2O7 and β-Yb2Si2O7 were reported to be 577 K and 450 K respectively [18]. Hence, the θD of γ-Y1.5Yb0.5Si2O7 was assumed as 545.25 K (272.1 °C) based on the rule of mixture.
2.3.2 Thermal conductivity
Thermal diffusivity (α) was measured from room temperature to 1200 °C on an as-sintered Y1.5Yb0.5Si2O7 pellet (porosity of 8.8%, 9.8 mm in diameter and 2.1 mm in thickness) under an N2 atmosphere using a laser-flash technique (LFA 467 Nanoflash, Netzsch, Selb, Germany) by Parker et al. [37] in 1961. The square of the sample thickness d in mm and the half-life period of the temperature rise t1/2 in s (time value at half signal height) are used to calculate the thermal diffusivity α in mm2s−1:
The specific heat (Cp) of Y1.5Yb0.5Si2O7 was determined and compared by means of LFA measurement and the Neumann–Kopp rule, respectively.
The technique for determining specific heat (\({c}_{p}^{test}\)) of as-sintered Y1.5Yb0.5Si2O7 pellet using LFA measurements is comprehensively outlined in ASTM E1461-07, Annex X2 with an accuracy of ± 5–7% [38]. This standard mandates the use of a reference material with a known specific heat value (\({c}_{p}^{ref}\)) as one of its essential prerequisites. The POCO graphite (AXM-5QA, Netzsch) was the reference material in this study.
where Tmax was the maximum temperature recorded on a sample, ρ is the density, d is the sample thickness, and the superscripts ref and test refer to the reference sample and the test sample respectively.
The Neumann–Kopp rule [39, 40] calculated Cp values of Y1.5Yb0.5Si2O7 were determined based on literature data considering the constituent oxides (Y2O3, Yb2O3, and SiO2) [41]:
The molar heat capacities of the binary oxides (Y2O3, Yb2O3, and SiO2) at elevated temperatures were taken from the database. It has been established that the Neumann–Kopp rule is a reliable method for calculating the heat capacity of complex ternary oxides with errors typically within ± 3% compared to experimental measurements of heat capacity [42,43,44].
The bulk density of the as-sintered Y1.5Yb0.5Si2O7 pellet was measured by the Archimedes method.
The thermal conductivity (κ) was calculated from the equation below using the LFA and theoretical Cp respectively:
2.3.3 Metallographic sample preparation
The cold mounting of samples was achieved by mixing Epo-set resin, which comprises 70–90 wt% bisphenol-A-co-epichlorohydrin and 10–30 wt% 2-ethylhexyl glycidyl ether, with Epo-set hardener. The hardener is a combination of triethylenetetramine (propoxylated) and trientine, with a 4:1 volume ratio. This blend is provided by MetPrep Ltd. in Coventry, UK. The cross-sectioned samples were sequentially grounded with SiC grinding papers (MetPrep, Coventry, UK), and polished to 1 μm finish by diamond and non-crystalizing colloidal silica polishing. For the scanning electron microscope (SEM) analysis of the coatings and pellets, all samples were carbon-coated to obtain a conducting surface. All images were taken using JEOL 7100F field emission gun scanning electron microscope (JEOL, Japan) under a high vacuum, spot size of 4, and a working distance of 10 mm with energy dispersive X-ray spectroscopy (EDS, X-max 150, Oxford Instruments, UK) and electron backscatter diffraction (EBSD, Oxford Instruments, UK) capabilities. To understand the CMAS attack on dense pellets, EBSD was used with an accelerating voltage of 15 kV on the ~ 70° tiled specimen. EBSD data acquisition was performed at a step size of 0.15–0.20 µm. The scan area chosen was about 80 μm × 80 μm. AZtec Crystal was used to analyze the EBSD project data. The grains were identified based on 10° mis-orientation criteria; i.e., the continuous presence of a mis-orientation of 10° or greater was defined as a grain boundary.
2.4 DFT modelling
Quantum mechanical first-principle plane wave calculations were carried out within the DFT formalism as employed in the CASTEP code to minimize the unit-cell total energy [45]. The exchange–correlation potential is estimated by Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA) approach using on-the-fly generated (OTFG) norm-conserving pseudopotential [46]. In the geometric optimization step, the atomic position and lattice parameters were iteratively refined in a mixed space of element internal parameters and element degrees of freedom. Electronic wave functions were expanded by using a plane-wave basis set with an optimized cutoff energy (Ecut) of 500 eV. Brillouin zone integration was performed using a 3 × 1 × 2 k-mesh. All the calculations have converged on the energy convergence standard 10−5 eV and the force convergence standard 0.01 eV Å−1 per atom. The maximum stress and maximum displacement are 0.05 GPa and 0.001 Å, respectively. Then, the temperature dependencies of enthalpy, entropy, Gibbs energy, and heat capacity of γ-Y2Si2O7 and γ-Y1.5Yb0.5Si2O7 were calculated using the optimized structure.
2.5 CMAS test
The CMAS slurry was prepared by mixing the CMAS powder (from Oerlikon Metco, Cheshire, UK) with ethanol at a 1:9 ratio. The CMAS powder adhered to a nominal composition of 35%CaO-10%MgO-7%Al2O3-48%SiO2 in mol%, consistent with relevant literature [47,48,49]. A uniform distribution of CMAS was then deposited on both the free-standing coating and pellet with an airbrush kit where an aerosolized jet of liquid containing CMAS powder was sprayed directly on the sample located on the hot plate. Due to flash evaporation, the medium evaporates leaving a thin layer of CMAS. The slurry was constantly agitated with a magnetic stirrer on an Isotemp hot plate (Fisher Scientific, Loughborough, UK). A CMAS concentration of 15 mg/cm2 was chosen according to the previously established protocol, and standard practice from the high-temperature corrosion community [50]. After depositing the CMAS slurry, the sample was placed on a hot plate and heated to approximately 100 °C to evaporate the ethanol. The sample was weighted before and after the CMAS deposition using an analytical balance (Readability of 0.0001 g, Fisher Scientific, UK). The process of depositing CMAS and sample weighting were repeated until the desired concentration was obtained.
3 Results and discussion
3.1 Coating characterization
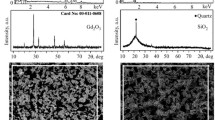
Figure 1a–b illustrates a secondary electron image of the Y1.5Yb0.5Si2O7 powder dried from suspension. The observed powder showed an agglomerate morphology with the size slightly higher than the D50 value of 0.9 μm. The occurrence is likely attributed to the suspension drying process at 100 °C overnight. XRD patterns of the suspension dried powders, the as-sprayed coatings, and the annealed coatings are demonstrated in Fig. 1c. The characteristic peaks of as-dried Y1.5Yb0.5Si2O7 suspension powder could be indexed to γ-Y2Si2O7 (PDF#00–040-0034). Alongside the identified crystalline peaks, the as-sprayed coatings exhibit a broad amorphous hump within the 25–35° range which is in line with thermal sprayed coatings due to rapid cooling of the splats upon impact [51]. Upon annealing at 1250 °C, the amorphous content undergoes crystallization, resulting in fully crystalline samples. After annealing at 1450 °C, the polymorphic formation of Y1.5Yb0.5Si2O7 was maintained as the γ phase which is consistent with the as-reported phase transformation diagram of Y2Si2O7 [28].

a, b. Secondary electron (SE) image of suspension-dried Y1.5Yb0.5Si2O7 powder; c XRD patterns of the Y1.5Yb0.5Si2O7 powder, the as-sprayed coatings, and the annealed coatings
The as-sprayed coating underwent annealing to crystallize the amorphous content, and the SEM images of the free-standing coatings cross-section are depicted in Fig. 2. It can be seen that the cross-section average thickness of the as-sprayed sample (Fig. 2a) decreased after annealing at 1250 °C and 1450 °C. The shrinkage of the coatings implied likely the presence of sintering phenomena during annealing. Although macro cracks remained among all the free-standing coatings, relatively denser microstructures with less micro-cracks were acquired according to the magnified SE images in Fig. 2d, e, and f. The sintering behavior of the as-sprayed coatings was observed with the gradual reduction in pores (as can be seen in the higher magnification images in Fig. 2d–f). A second phase with a lighter contrast was distributed among all the free-standing coatings. EDS point analysis, listed in Table 3, suggests the RE to Si ratio being 1.6 for the secondary phase with a lighter contrast. In contrast, the matrix phase with grey contrast possesses a RE to Si ratio close to 1. Also, the existing fraction of the observed secondary phase can be below the detection limit of XRD since no impurity peaks are observed in the XRD patterns.

Overall and central zoom-in SE images of as-sprayed (a, d) and 2-h annealed (b, e, at 1250 °C; c, f, at 1450 °C) γ-Y1.5Yb0.5Si2O7 free-standing coating
3.2 Lattice thermal expansion and thermal conductivity
Subsequently, the 1250 °C annealed sample was utilized to perform hot-XRD measurement in situ from 200 to 1000 °C (Fig. 3a). The characteristic peaks of the annealed Y1.5Yb0.5Si2O7 sample were well indexed to γ-Y2Si2O7 (PDF#00–040-0034), and no phase transformation appeared in the heating process. The lattice parameters of Y1.5Yb0.5Si2O7 at evaluated temperatures, including a, b, c, beta angle, and V, were calculated according to hot-XRD patterns using the GSAS software as shown in Table 4. Figure 3b illustrates the change of lattice parameters with temperatures. During heating, the a-axis slightly decreased to 400 °C, followed by an increase during further heating. The b- and c-axes steadily increased while the beta angle hardly changed with temperatures up to 1000 °C. The curves for variation of normalized cell parameters were fitted using Eq. 1 and plotted in Fig. 3b. Least-squares fitting results of the normalized cell dimensions for γ-Y1.5Yb0.5Si2O7, including constants A and B, are given in Table 5.

a In situ hot-XRD patterns illustrating the annealing process of γ-Y1.5Yb0.5Si2O7 from 200 to 1000 °C; b variation of normalized lattice parameters (a, b, c, beta angle, and V) with increasing temperatures
By linear fitting the data in the plot of ln(V/Vrt) vs. (T-Trt) above the temperature, the volumetric thermal expansion coefficient of γ-Y1.5Yb0.5Si2O7 as 3.17 ± 0.22 × 10−6 K−1 up to 1000 °C, lower than both reported intrinsic CTE values of γ-Y2Si2O7 (6.68 ± 0.35 × 10−6 K−1) and β-Yb2Si2O7 (4.82 × 10−6 K−1) [13, 52]. It is inferred that the quarter molar Yb doping in the Y site induced localized lattice distortion around [O3Si-OB-SiO3] units may create a negative contribution to the lattice thermal expansion of γ structured pyrosilicate [53]. As a result, the γ-Y1.5Yb0.5Si2O7 material synthesized in this study exhibited a thermal expansion coefficient value (3.17 ± 0.22 × 10−6 K−1) that closely matches that of the Si bond coat (3.5–4.5 × 10−6 K−1) [54]. This characteristic ensures reduced stress levels during subsequent thermal cycling at both high and low temperatures, aligning with the fundamental criteria for thermal expansion coefficient compatibility. Hence, γ-Y1.5Yb0.5Si2O7 holds significant promise as a material with practical coating applications.
Temperature dependences of the thermal diffusivities, heat capacity, and thermal conductivity of γ-Y1.5Yb0.5Si2O7 and β-Yb2Si2O7 pellets are shown in Fig. 4. They generally decreased with the increase of temperature. The as-sintered γ-Y1.5Yb0.5Si2O7 pellet possessed a low thermal diffusivity in the range of 1.34, 0.50 m2/s from room temperature to 1200 °C. The T−1-dependent thermal diffusivity of γ-Y1.5Yb0.5Si2O7 ceramic was determined based on the phonon heat transfer mechanism, which is prevalent in polycrystalline materials. This phenomenon can be elucidated as follows: as the temperature rises, the intensity of phonon–phonon collisions increases, leading to enhanced phonon scattering and a significant reduction in the mean free path of phonons. Consequently, the thermal diffusivity decreases with the rise in temperature from room temperature to 1200 °C.

a Thermal diffusivity plotted against temperature; b specific heat Cp plotted versus temperature in two sets; c microstructure of as-sintered γ-Y1.5Yb0.5Si2O7 pellet, and d thermal conductivity with/without density normalization obtained using Cp in b at evaluated temperatures
As shown in Fig. 4b, two sets of Cp were acquired using the LFA measurement with a reference material and the Neumann–Kopp rule, respectively. The large error bar for the Cp measurement at 1200 °C may be related to the N2 shield gas used. In the Neumann–Kopp (NK) rule primary form, it describes the theoretical molar heat capacity (Cp, m) of a compound as, by a mass fraction, a weighted sum of the heat capacities of its components. Hence, the NK Cp results of γ-Y1.5Yb0.5Si2O7 were obtained for multi-element compounds in a complex form using Eq. 6. The molar heat capacity is converted to the specific heat capacity by division with the molar mass of γ-Y1.5Yb0.5Si2O7. The NK Cp increased gradually from 0.50 J·g−1·K−1 at room temperature to 0.72 J·g−1·K−1 at 1200 °C. The deduced LFA Cp with standard reference material had a similar value of 0.49 J·g−1·K−1 at ambient temperature but grew higher to 0.90 J·g−1·K−1 at 1200 °C. The deviation between the two sets of Cp data increases at higher temperatures, resulting in significant discrepancies.
The thermal conductivity (TC) before and after normalizing to theoretically dense is plotted in Fig. 4d. The TC values of γ-Y1.5Yb0.5Si2O7 with LFA Cp varied in the range of 3.14, 2.15 W·m−1·K−1 from room temperature to 1200 °C. While TC values were achieved using NK Cp of γ-Y1.5Yb0.5Si2O7, decreasing from 3.20 W·m−1·K−1 at room temperature to even lower 1.72 W·m−1·K−1 at 1200 °C, which was lower than the reported TC values of γ-Y2Si2O7 (1.90 W·m−1·K−1 at 1127 °C/1400 K) [13] and β-Yb2Si2O7 (2.10 W·m−1·K−1 at 1000 °C) [55].
Xiang et al. [56] reported that the rigid [SiO4] units in ytterbium disilicate were beneficial to the phonon conduction, while the weakly bonded [REO6] units could effectively enhance the phonon scattering and reduce the phonon transport. While in this case as shown in Fig. 5a, the doping of Yb into YO6 units reduced the order of the original octahedral structure and intensified the phonon scattering, resulting in even lower thermal conductivity of γ-Y1.5Yb0.5Si2O7. The thermal stability of the studied materials can be predicted by investigating the thermodynamic properties like enthalpy, free energy, and entropy. For the plots of γ-Y1.5Yb0.5Si2O7 (Fig. 5b), blue colored solid line represents enthalpy while free energy and temperature time's entropy are indicated by red-and green-colored solid lines, respectively. A similar trend was observed regardless of the undoped γ-Y2Si2O7 lattice. This slight difference in the Yb-doped γ-Y2Si2O7 indicated a faster decay in the Gibbs energy according to the γ-Y1.5Yb0.5Si2O7. Hence, the γ-Y1.5Yb0.5Si2O7 structure is energetically stable with increasing temperature. Figure 5c demonstrates the behavior of the DFT-simulated heat capacity as a function of the temperature in °C, considering the Yb-doping as γ-Y1.5Yb0.5Si2O7 (solid line) and γ-Y2Si2O7 (dash line). It was estimated that the increases rapidly as the temperature increases in a range from − 273 to 100 °C, reaching the Dulong–Petit limit at around 270 °C. By comparing the heat capacity values calculated using DFT simulated and the NK rules (Fig. 5d), it was inferred that the synthesis process is non-spontaneous. Therefore, it clearly shows that γ-Y1.5Yb0.5Si2O7 with a more energetically stable structure exhibits a low value of thermal conductivity of only 1.72 W·m−1·K−1 at 1200 °C, suggesting this material serves as a promising TEBC material.

a Lattice schematic presenting the crystal structure of γ-Y1.5Yb0.5Si2O7; b DFT-calculated enthalpy, Gibbs free energy, and entropy with temperature variation of γ-Y1.5Yb0.5Si2O7; c heat capacity curves with temperature variation for γ-Y1.5Yb0.5Si2O7 (solid line) and γ-Y2Si2O7 (dash line); d comparison between DFT and NK method calculated heat capacity Cp with temperatures
3.3 CMAS exposure on γ-Y1.5Yb0.5Si2O7 pellets
The CMAS corrosion mechanism of RE disilicates has been well established over recent years, primarily relying on the reaction-crystallization process, which is contingent on the ionic size of RE elements. With the smaller RE ion doping (Yb3+ 0.0868 nm) in γ-Y2Si2O7 (Y3+ 0.1011 nm), the CMAS resistance capability of γ-Y1.5Yb0.5Si2O7 from further damage is expected to be improved. In addition to erosion, CMAS attack in the mechanical aspect is primarily associated with thermal stress. A thorough examination and the implementation of mitigation measures are essential for effectively utilizing EBCs in light of this multifaceted challenge. The CMAS powder used in our work has a melting point of 1220 °C [57]. Hence, during the exposure at 1300 °C, the CMAS is expected to be fully molten.
The XRD patterns of CMAS interacted γ-Y1.5Yb0.5Si2O7 pellet at 1300 and 1400 °C for 30 and 60 min are plotted in Fig. 6a. Hexagonal apatite silicate phase (PDF#04–005-7025) was detected among all the samples after the CMAS interaction. It was worth noting that the relative intensity of a set of parallel crystal planes, (010), (020), (030), and (040), of the apatite phase was apparently higher than other characteristic peaks after the CMAS interaction at 1400 °C for 60 min. In the view of the x-ray incidence on the top surface of the CMAS reaction region, it was assumed the preferential (010) crystal plane orientated towards the x-ray incident direction (Fig. 6b).

a XRD patterns of CMAS interacted γ-Y1.5Yb0.5Si2O7 pellet at 1300–1400 °C for 30–60 min; b lattice schematic illustrating the Ca2(Y/Yb)8(SiO4)6O2 apatite phase
There remained a dark grey “bump” on the surface of the as-sintered γ-Y1.5Yb0.5Si2O7 pellet after exposure at 1300 °C for 30 min (Fig. 7a). A more detailed view of the CMAS/pellet interface is depicted in Fig. 7b, where the reaction layer between the CMAS and the matrix can be observed. Referring to the EDS mapping in Fig. 6c, it was clear that the CMAS presented above a well-defined interlayer with the “V”-shaped-like phase scattering within the CMAS layer. The Y and Yb elements were found distributed within the “V”-shaped-like phase as well. While the interlayer was composed of elongated grains between the matrix and CMAS layers with an average thickness of about 13 µm. EDS point analysis of the CMAS interacted γ-Y1.5Yb0.5Si2O7 pellet at 1300 °C for 30 min is listed in Table 6. The Ca to Si ratio of the “V”-shaped-like morphology phase was all close to 0.5. While the elongated grains had a Ca to Si ratio close to 0.3. It was speculated that the cyclosilicate phase, Ca3RE2(Si3O9)2 Ca to Si = 0.5, is the “V”-shaped-like morphology phase within the CMAS layer (Fig. 7b). In contrast, the highlighted interlayer possessed a similar Ca to Si ratio with that of apatite phase, Ca2RE8(SiO4)6O2.

a, b Microstructure images and c EDS mapping of CMAS interacted γ-Y1.5Yb0.5Si2O7 pellet at 1300 °C for 30 min
In Fig. 8, EBSD-EDS analysis was conducted to rigorously identify the phase after CMAS interaction at 1300 °C for 30 min. Not only was elemental stoichiometry confirmed by EDS point analysis (Table 6, 1–3#), but the crystal structure of the cyclosilicate Ca3RE2(Si3O9)2 was verified in the EBSD phase map as well (Fig. 8e). Speculation might be due to the low content of cyclosilicate phase, which is beyond the detection limit of XRD. In the EDS mapping (Fig. 8a–d), minimal traces of calcium were observed in the matrix, suggesting that the presumed apatite interlayer effectively functions as a barrier, inhibiting CMAS corrosion. It is noteworthy that, not only in the apatite interlayer but also within the cyclosilicate phase, the elemental distribution trend of Y elements closely mirrors that of Yb elements. This suggests that during the high-temperature corrosion process, the reactions of Y3+ and Yb3+ with CMAS ran simultaneously. It was hypothesized that the formation of the cyclosilicate phase needed to consume more calcium oxide and silica, which was consistent with its existing environment within the residual CMAS layer. Hence, possible reactions at 1300 °C that would happen during the CMAS corrosion are shown below:

a–d EDS mapping and e EBSD phase map of CMAS interacted γ-Y1.5Yb0.5Si2O7 pellet at 1300 °C for 30 min. The scale bar is 25 µm
Based on the EDS quantitative analysis (Table 6) and EBSD phase identification (Fig. 8) mentioned earlier, the formation of different CMAS interaction crystallization products appears to depend on the concentrations of CaO and SiO2. Specifically, the cyclosilicate phase, Ca3(Y,Yb)2(Si3O9)2, was observed embedded within the molten CMAS, while the Ca2(Y,Yb)8(SiO4)6O2 apatite phase remained localized with lower CaO and SiO2 concentrations, closer to the matrix (Fig. 8). At temperatures exceeding the CMAS melting point, γ-Y1.5Yb0.5Si2O7 predominantly reacted with CMAS, leading to the precipitation of the CMAS barrier apatite interlayer (Eq. 8). The crystallization of the cyclosilicate phase, Ca3(Y,Yb)2(Si3O9)2, likely occurred on the initially formed apatite grains, serving as one of the reaction materials (Eq. 9). It tended to propagate and grow predominantly towards the direction of the residual CMAS, where higher concentrations of CaO and SiO2 were present.
As depicted in Fig. 9, the apatite interlayer thickness expanded to approximately 17 µm following a prolonged exposure time (60 min) at 1300 °C. Notably, the “V”-shaped cyclosilicate phase remained within the CMAS layer, however exhibiting a significantly higher aspect ratio, implying its sustained stability at evaluated temperatures. This raises interest in whether the cyclosilicate phase exhibits beneficial high-temperature thermal stability, potentially enhancing the γ-Y1.5Yb0.5Si2O7’s resistance under continuous CMAS chemical corrosion at high temperatures, improving its durability.

a Microstructure image and b Ca element mapping of CMAS interacted γ-Y1.5Yb0.5Si2O7 pellet at 1300 °C for 60 min
Subsequently, an experiment involving higher CMAS exposure temperature was conducted. Figure 10 shows the deposited CMAS remained on the pellet surface after 60 min exposure at 1400 °C. The magnified microstructure of the CMAS/pellet reaction area was selected at the center location (Fig. 10a). EBSD grain orientation analysis of the apatite phase (Fig. 10c) in the interlayer (Fig. 10b) supported the (010) preferenced orientation distribution hypothesis based on the XRD patterns in Fig. 6. The thickness of the apatite barrier layer grew to about 90 µm. Several particles are scattered within the outer CMAS layer, having a comparable morphology with the apatite phase. EDS point analysis (Table 7, 1–3#) confirmed the presence of grains embedded within the CMAS layer as the apatite phase rather than the cyclosilicate phase, which was in line with our conjecture on the worse thermal stability of the cyclosilicate phase. It is inferred that the cyclosilicate phase, 4Ca3(Y/Yb)2(Si3O9)2, decomposed into the more stable apatite phase at 1400 °C. Herein, this result confirmed that the reaction (Eq. 9) is reversible under the molten CMAS layer at 1400 °C:

a Microstructure image and b magnified band contrast image of CMAS interacted γ-Y1.5Yb0.5Si2O7 pellet at 1400 °C for 60 min; c apatite grain orientation of selected grains related to b (4#, 5#, 6#)
The decomposition of the cyclosilicate phase was therefore believed to contribute to the expansion of the apatite barrier layer at 1400 °C. The newly crystalized apatite grains with finer size spread across the pre-existing apatite barrier layer and resulted in a more compact microstructure, which could be advantageous for CMAS resistance. In the case of CMAS interaction of γ-Y2Si2O7 [20], a reaction layer approximately 400 μm deep formed from the ceramic surface after 1 h dwell at 1500 °C, consisting of needle-shaped Y-Ca–Si apatite and CMAS (51.5%SiO2-39.2%CaO-4.1%Al2O3-5.2%MgO) glass. Despite its favorable CTE match with SiC and lightweight properties, concerns arise due to the formation of a significantly thicker reaction layer, potentially compromising its effectiveness against high-temperature CMAS attacks. In contrast, β-Yb2Si2O7 exhibited minimal reaction-recrystallization of apatite under the same experimental condition [21]. The limited formation of apatite in β-Yb2Si2O7 is attributed to the smaller ionic radius of Yb3+ compared to Y3+, resulting in a reduced affinity for Ca and a diminished driving force for reaction-crystallization. Instead of reaction-crystallization, CMAS glass seems to have penetrated the grain boundaries of dense β-Yb2Si2O7 EBC ceramic pellets. This slow penetration process creates a dilatation gradient, leading to the expansion of the top layer, ultimately causing “blister” cracking when compressive strain accumulates during prolonged heat treatment. Understanding these mechanisms is crucial for optimizing EBC ceramics to enhance resistance against CMAS-induced corrosion.
3.4 CMAS exposure on γ-Y1.5Yb0.5Si2O7 coating
CMAS exposure evaluation of γ-Y1.5Yb0.5Si2O7 coating was also performed to understand the effect of CMAS in coating microstructure. The CMAS exposure reaction product was identified through XRD analysis (Fig. 11) of the coatings exposed to temperatures of 1300 °C for 5, 30, and 60 min. Following the beginning stage of CMAS infiltration (1300 °C for 5 min), the γ-Y1.5Yb0.5Si2O7 free-standing coating already underwent a reaction with the CMAS, resulting in the formation of apatite (PDF#04–005-7025) within the coatings. While prolonging the dwelling time at 1300 °C, the relative intensity of γ phase disilicate characteristic peaks decreased gradually.

XRD patterns of CMAS interacted γ-Y1.5Yb0.5Si2O7 free-standing coating at 1300 °C for 5–60 min
To ascertain the depth of CMAS penetration within the coating and differentiate it from pore structure, EDS point analysis and elemental mapping were conducted. Ca mapping was specifically selected as it serves as a reliable indicator of the presence of CMAS within the coating due to its consistent association with CMAS reaction products. Based on the Ca distribution depicted in Fig. 12a and b, the estimated thickness of the CMAS infiltration layer was approximately ~ 80 μm at 1300 °C for 5 min. Notably, the microstructure of the free-standing coating infiltrated by CMAS exhibited greater density with reduced pores and cracks. Several grains with a relatively higher aspect ratio were observed, indicating the formation of the apatite phase (Fig. 12c); however, the EDS point analysis in Table 8 did not show evidence of the formation of the apatite phase based on the consideration of Ca to Si ratio far less than 0.3. Besides, all the EDS point analyses had a Y to Yb atomic ratio close to 3 which is consistent with the stoichiometry of γ-Y1.5Yb0.5Si2O7. While extending the exposure time to 30 min at 1300 °C, the overall microstructure of the γ-Y1.5Yb0.5Si2O7 free-standing coating became more compact (Fig. 12d). The nearly homogenous distribution of Ca element in Fig. 12e confirmed that the infiltration of CMAS went throughout the entire coating. Light grey grains with obviously higher aspect ratio and black amorphous phase were observed, as shown in Fig. 12f. With the assistance of EDS analysis (Table 8), γ-Y1.5Yb0.5Si2O7 matrix with the Y to Yb ratio close to 3, Ca2RE8(SiO4)6O2 apatite with the Ca to Si ratio close to 0.3, and amorphous CMAS phase were confirmed. Interestingly, the Y to Yb ratio in Ca2RE8(SiO4)6O2 apatite grains was relatively higher than 3 indicating the possible difference in CMAS reaction activities between Y and Yb.

Microstructure image, Ca element mapping, and high-resolution images of CMAS interacted γ-Y1.5Yb0.5Si2O7 free-standing coating at 1300 °C for 5 min (a, b, c) and 30 min (d, e, f)
Significantly different microstructure features of the CMAS-interacted free-standing coating were observed from 60-min dwelling at 1300 °C. The shape of the coating did not retain and consisted of a porous shell covering a relatively dense core (Fig. 13a). Figure 13b suggests a relatively higher concentration of Ca in the “shell” part. It was inferred that the inhomogeneous distribution of the Ca element mainly resulted from the amorphous CMAS phase after comparing the magnified SEM images between the “shell” and “core” part in Fig. 13c, d. The EDS point analysis of the selected area from the “shell” and “core” parts in Table 9 further confirmed that similar phase composition appeared compared with those results after CMAS interaction for 30 min at 1300 °C. As to the analysis above, the amorphous CMAS phase can infiltrate along cracks and pores and remain within the microstructure. The coating was, therefore, nearly completely consumed by the aggressive CMAS.

Microstructure image and Ca element mapping of CMAS interacted γ-Y1.5Yb0.5Si2O7 free-standing coating at 1300 °C for 60 min
Figure 14 schematically illustrates the distinct interactions of γ-Y1.5Yb0.5Si2O7 in its pellet and coating forms. Starting at 1300 °C, the CMAS melts. Molten CMAS reacts with the different forms of γ-Y1.5Yb0.5Si2O7, producing two phenomena. Due to the dense microstructure of the as-sintered pellet, it is easier to constrain the molten CMAS on its surface. The metastable Ca-RE cyclosilicate phase appears as an intermediate product during CMAS interaction. Its decomposition into newly crystalized Ca2RE8(SiO4)6O2 results in a more compact apatite barrier layer to hinder the further corrosion of molten CMAS. For its coating form, a profound comprehension related to a chemo-thermo-mechanical CMAS corrosion mechanism was proposed. At an early stage, molten CMAS infiltration into the coating is intensified by its porous structure. Apatite forms particularly along the micro-cracks. The crystallized apatite grains with elongated morphology facilitate the construction of diffusion microchannels, resulting in the intensified capillary-driven absorption of molten CMAS into the coating’s interior. With the prolonging of the reaction time, coatings are consumed from the outside in and finally turn into a porous “shell” covering the small “core.” Although the composition design of γ-Y1.5Yb0.5Si2O7 is reliable for impeding CMAS continuous corrosion, the dense microstructure is also an indispensable parameter to achieve thermal and environmental functionality. Although pellets are fully dense and provide a much better performance in CMAS, applying sintering to highly engineered CMC parts is not feasible. EBCs can only be applied on CMCs using sophisticated manufacturing routes like SPS, and further work is ongoing to optimize the SPS coating microstructure for CMAS.

CMAS corrosion resistance mechanism of γ-Y1.5Yb0.5Si2O7 coating and pellet, illustrating stages from the initial to the final stage
The γ-Y1.5Yb0.5Si2O7 coatings fabricated using the SPS method with some defects are inevitable. When comparing CMAS infiltration between the dense pellet and coating, the detrimental impact of microstructural defects, particularly cracks and a porous network, on CMAS infiltration becomes evident. These microstructural defects significantly affect the CMAS resistance behavior of coatings; however, achieving a defect-free coating is impractical, and a certain level of porosity is necessary to manage the thermal stresses of the coating system. This comprehensive examination provided valuable insights into the current effectiveness of γ-Y1.5Yb0.5Si2O7 coatings in real-world conditions and guided further advancements in coating technology. To further advance this research, we will optimize γ-Y1.5Yb0.5Si2O7 coatings to reduce porosity levels and eliminate cracks in a follow-on study.
4 Conclusion
The novel γ-structured disilicate, γ-Y1.5Yb0.5Si2O7, exhibited remarkable dual functionality as a thermal and environment barrier ceramic coating. The outstanding thermal and CMAS barrier properties, coupled with the low thermal expansion coefficient and thermal conductivity at high temperatures, position γ-Y1.5Yb0.5Si2O7 as an ideal material for advanced TEBC applications, effectively addressing current challenges in gas turbine technology.
γ-Y1.5Yb0.5Si2O7 displays a low CTE value of 3.17 ± 0.22 × 10−6 K−1 up to 1000 °C, demonstrating its potential for reducing thermal load on the Si bond coat. The γ-Y1.5Yb0.5Si2O7 with a more energetically stable structure exhibits a low value of thermal conductivity of only 1.72 W·m−1·K−1 at 1200 °C. Temperature dependences of enthalpy, entropy, and Gibbs energy of γ-Y1.5Yb0.5Si2O7 and γ-Y2Si2O7 are predicted based on the first-principle calculations of crystal structure. Subtle disparities have emerged between the experimentally observed and DFT-predicted heat capacity values for γ-Y1.5Yb0.5Si2O7, thereby underscoring the reliability of theoretical predictions in elucidating the intrinsic thermodynamic characteristics of this compound.
The systematic investigation of its CMAS resistance confirms the formation of a protective apatite, Ca2RE8(SiO4)6O2, on both sprayed coatings and dense pellets during CMAS infiltration at evaluated temperatures. The “core–shell” structure of the coating is acquired after CMAS exposure at 1300 °C for 60 min. EDS point analysis reveals that the Y to Yb ratio in apatite grains is relatively higher than 3, indicating the reactivity difference between Y and Yb with molten CMAS. For the CMAS exposure on dense pellet, it is crucial to acknowledge that the cyclosilicate phase’s stability, Ca3RE2(Si3O9)2, is temperature-dependent, forming at 1300 °C but disappearing at 1400 °C. Through a comprehensive analysis of microstructural characteristics in γ-Y1.5Yb0.5Si2O7, ranging from as-sprayed coatings to nearly dense pellets, we have unveiled promising avenues for optimizing the fabrication process. These enhancements have the potential to improve both microstructural features and resistance to CMAS infiltration. This discovery underscores the critical importance of precise temperature management in practical applications, ensuring the long-term performance and reliability of γ-Y1.5Yb0.5Si2O7 coatings and, consequently, extending the lifespan of gas turbine engines.
Data availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Padture NP (2016) Advanced structural ceramics in aerospace propulsion. Nat Mater 15(8):804–809. https://doi.org/10.1038/nmat4687
Eswarappa Prameela S, Pollock TM, Raabe D, Meyers MA, Aitkaliyeva A, Chintersingh K-L, Cordero ZC, Graham-Brady L (2023) Materials for extreme environments. Nat Rev Mater 8(2):81–88. https://doi.org/10.1038/s41578-022-00496-z
Bansal NP, Lamon J (2014) Ceramic matrix composites: materials, modeling and technology. John Wiley & Sons, pp 217–233
Zhu D (2018) Aerospace ceramic materials: thermal, environmental barrier coatings and SiC/SiC ceramic matrix composites for turbine engine applications. https://ntrs.nasa.gov/api/citations/20180002984/downloads/20180002984.pdf
Lee KN, Zhu D, Lima RS (2021) Perspectives on environmental barrier coatings (EBCs) manufactured via air plasma spray (APS) on ceramic matrix composites (CMCs): a tutorial paper. J Therm Spray Technol 30:40–58. https://doi.org/10.1007/s11666-021-01168-0
Ridley MJ, Tomko KQ, Tomko JA, Hoglund ER, Howe JM, Hopkins PE, Opila EJ (2022) Tailoring thermal and chemical properties of a multi-component environmental barrier coating candidate (Sc0.2Nd0.2Er0.2Yb0.2Lu0.2)2Si2O7. Materialia. https://doi.org/10.1016/j.mtla.2023.101729
Richards BT, Young KA, de Francqueville F, Sehr S, Begley MR, Wadley HN (2016) Response of ytterbium disilicate–silicon environmental barrier coatings to thermal cycling in water vapor. Acta Mater 106:1–14. https://doi.org/10.1016/j.actamat.2015.12.053
Drexler JM, Gledhill AD, Shinoda K, Vasiliev AL, Reddy KM, Sampath S, Padture NP (2011) Jet engine coatings for resisting volcanic ash damage. Adv Mater 23(21):2419–2424. https://doi.org/10.1002/adma.201004783
Tejero-Martin D, Bennett C, Hussain T (2021) A review on environmental barrier coatings: history, current state of the art and future developments. J Euro Ceram Soc 41(3):1747–1768. https://doi.org/10.1016/j.jeurceramsoc.2020.10.057
Lu Y, Luo L, Liu J, Zhu C, Wang Y (2016) Failure mechanism associated with the thermally grown silica scale in environmental barrier coated C/SiC composites. J Am Ceram Soc 99(8):2713–2719. https://doi.org/10.1111/jace.14273
Lv X, Yue M, Feng X, Li X, Wang Y, Wang J, Zhang J, Wang J (2022) Rare earth monosilicates as oxidation resistant interphase for SiCf/SiC CMC: investigation of SiCf/Yb2SiO5 model composites. J Adv Ceram 11(5):702–711. https://doi.org/10.1007/s40145-021-0560-4
Chen S, Chen P, Duan J, Chen M, Xiao P, Li Y (2022) Thermal cycling behavior of bi-layer Yb2Si2O7/SiC EBC-coated Cf/SiC composites in burner rig tests. Adv Compos Hybrid Mater 5(3):2184–2192. https://doi.org/10.1007/s42114-021-00270-9
Sun Z, Zhou Y, Wang J, Li M (2008) Thermal properties and thermal shock resistance of γ-Y2Si2O7. J Am Ceram Soc 91(8):2623–2629. https://doi.org/10.1111/j.1551-2916.2008.02470.x
Zhong X, Niu Y, Li H, Zhou H, Dong S, Zheng X, Ding C, Sun J (2018) Thermal shock resistance of tri-layer Yb2SiO5/Yb2Si2O7/Si coating for SiC and SiC-matrix composites. J Am Ceram Soc 101(10):4743–4752. https://doi.org/10.1111/jace.15713
Zhou YC, Zhao C, Wang F, Sun YJ, Zheng LY, Wang XH (2013) Theoretical prediction and experimental investigation on the thermal and mechanical properties of bulk β-Yb2Si2O7. J Am Ceram Soc 96(12):3891–3900. https://doi.org/10.1111/jace.12618
Clarke DR (2003) Materials selection guidelines for low thermal conductivity thermal barrier coatings. Sur Coat Tech 163:67–74. https://doi.org/10.1016/S0257-8972(02)00593-5
Luo Y, Wang J, Li Y, Wang J (2016) Giant phonon anharmonicity and anomalous pressure dependence of lattice thermal conductivity in Y2Si2O7 silicate. Sci Rep 6(1):29801. https://doi.org/10.1038/srep29801
Tian Z, Zheng L, Li Z, Li J, Wang J (2016) Exploration of the low thermal conductivities of γ-Y2Si2O7, β-Y2Si2O7, β-Yb2Si2O7, and β-Lu2Si2O7 as novel environmental barrier coating candidates. J Euro Ceram Soc 36(11):2813–2823. https://doi.org/10.1016/j.jeurceramsoc.2016.04.022
Wolf M, Mack DE, Guillon O, Vaßen R (2020) Resistance of pure and mixed rare earth silicates against calcium-magnesium-aluminosilicate (CMAS): a comparative study. J Am Ceram Soc 103(12):7056–7071. https://doi.org/10.1111/jace.17328
Turcer LR, Krause AR, Garces HF, Zhang L, Padture NP (2018) Environmental-barrier coating ceramics for resistance against attack by molten calcia-magnesia-aluminosilicate (CMAS) glass: part I, YAlO3 and γ-Y2Si2O7. J Euro Ceram Soc 38(11):3905–3913. https://doi.org/10.1016/j.jeurceramsoc.2018.03.021
Turcer LR, Krause AR, Garces HF, Zhang L, Padture NP (2018) Environmental-barrier coating ceramics for resistance against attack by molten calcia-magnesia-aluminosilicate (CMAS) glass: part II, β-Yb2Si2O7 and β-Sc2Si2O7. J Euro Ceram Soc 38(11):3914–3924. https://doi.org/10.1016/j.jeurceramsoc.2018.03.010
Turcer LR, Padture NP (2020) Rare-earth pyrosilicate solid-solution environmental-barrier coating ceramics for resistance against attack by molten calcia–magnesia–aluminosilicate (CMAS) glass. J Mater Res 35(17):2373–2384. https://doi.org/10.1557/jmr.2020.132
Liu J, Zhang L, Liu Q, Cheng L, Wang Y (2013) Calcium–magnesium–aluminosilicate corrosion behaviors of rare-earth disilicates at 1400°C. J Euro Ceram Soc 33(15–16):3419–3428. https://doi.org/10.1016/j.jeurceramsoc.2013.05.030
Poerschke DL, Shaw JH, Verma N, Zok FW, Levi CG (2018) Interaction of yttrium disilicate environmental barrier coatings with calcium-magnesium-iron alumino-silicate melts. Acta Mater 145:451–461. https://doi.org/10.1016/j.actamat.2017.12.004
Stokes JL, Harder BJ, Wiesner VL, Wolfe DE (2019) High-temperature thermochemical interactions of molten silicates with Yb2Si2O7 and Y2Si2O7 environmental barrier coating materials. J Euro Ceram Soc 39(15):5059–5067. https://doi.org/10.1016/j.jeurceramsoc.2019.06.051
Summers WD, Poerschke DL, Park D, Shaw JH, Zok FW, Levi CG (2018) Roles of composition and temperature in silicate deposit-induced recession of yttrium disilicate. Acta Mater 160:34–46. https://doi.org/10.1016/j.actamat.2018.08.043
Sleeper J, Garg A, Wiesner VL, Bansal NP (2019) Thermochemical interactions between CMAS and Ca2Y8 (SiO4)6O2 apatite environmental barrier coating material. J Euro Ceram Soc 39(16):5380–5390. https://doi.org/10.1016/j.jeurceramsoc.2019.08.040
Fernández-Carrión A, Alba M, Escudero A, Becerro A (2011) Solid solubility of Yb2Si2O7 in β-, γ-and δ-Y2Si2O7. J Solid State Chem 184(7):1882–1889. https://doi.org/10.1016/j.jssc.2011.05.034
Stack P, Kane K, Sweet M, Parker C, Lance M, Ridley M, Pint B (2022) Dry air cyclic oxidation of mixed Y/Yb disilicate environmental barrier coatings and bare silica formers. J Euro Ceram Soc 42(7):3345–3350. https://doi.org/10.1016/j.jeurceramsoc.2022.02.009
Zhang Z, Park Y, Xue Z, Ji X, Park H, Zhang S, Byon E, Koo BH (2023) High-temperature oxidation performance of novel environmental barrier coating 50HfO2-50SiO2/YxYb(2–x)Si2O7 at 1475°C. J Euro Ceram Soc 43(3):1127–1140. https://doi.org/10.1016/j.jeurceramsoc.2022.10.082
Turcer LR, Sengupta A, Padture NP (2021) Low thermal conductivity in high-entropy rare-earth pyrosilicate solid-solutions for thermal environmental barrier coatings. Scr Mater 191:40–45. https://doi.org/10.1016/j.scriptamat.2020.09.008
Kassner H, Siegert R, Hathiramani D, Vassen R, Stoever D (2008) Application of suspension plasma spraying (SPS) for manufacture of ceramic coatings. J Therm Spray Tech 17:115–123. https://doi.org/10.1007/s11666-007-9144-2
Vaßen R, Bakan E, Gatzen C, Kim S, Mack DE, Guillon O (2019) Environmental barrier coatings made by different thermal spray technologies. Coatings 9(12):784. https://doi.org/10.3390/coatings9120784
Bakan E, Marcano D, Zhou D, Sohn YJ, Mauer G, Vaßen R (2017) Yb2Si2O7 environmental barrier coatings deposited by various thermal spray techniques: a preliminary comparative study. J Therm Spray Tech 26:1011–1024. https://doi.org/10.1007/s11666-017-0574-1
Luo Y, Sun L, Wang J, Du T, Zhou C, Zhang J, Wang J (2023) Phase formation capability and compositional design of β-phase multiple rare-earth principal component disilicates. Nat Comm 14(1):1275. https://doi.org/10.1038/s41467-023-36947-6
Toby BH (2001) EXPGUI, a graphical user interface for GSAS. J Appl Cryst 34(2):210–213. https://doi.org/10.1107/S0021889801002242
Parker W, Jenkins R, Butler C, Abbott G (1961) Flash method of determining thermal diffusivity, heat capacity, and thermal conductivity. J Appl Phys 32(9):1679–1684. https://doi.org/10.1063/1.1728417
Zajas J, Heiselberg P (2013) Measurements of thermal diffusivity, specific heat capacity and thermal conductivity with LFA 447 apparatus. Aalb Univ 144:5–11
Leitner J, Voňka P, Sedmidubský D, Svoboda P (2010) Application of Neumann-Kopp rule for the estimation of heat capacity of mixed oxides. Thermo Acta 497(1–2):7–13. https://doi.org/10.1016/j.tca.2009.08.002
Kopp H (1865) III. Investigations of the specific heat of solid bodies. Philos Trans R Soc Lond 155:71–202. https://doi.org/10.1098/rstl.1865.0003
Barin I, Platzki G (1989) Thermochemical data of pure substances, vol 304. vol 334. Wiley Online Library.
Wu R, Pan W, Ren X, Wan C, Qu Z, Du A (2012) An extremely low thermal conduction ceramic: RE9.33 (SiO4)6O2 silicate oxyapatite. Acta Mater 60(15):5536–5544. https://doi.org/10.1016/j.actamat.2012.06.051
Limarga AM, Shian S, Baram M, Clarke DR (2012) Effect of high-temperature aging on the thermal conductivity of nanocrystalline tetragonal yttria-stabilized zirconia. Acta Mater 60(15):5417–5424. https://doi.org/10.1016/j.actamat.2012.06.054
Wu J, Wei X, Padture NP, Klemens PG, Gell M, García E, Miranzo P, Osendi MI (2002) Low-thermal-conductivity rare-earth zirconates for potential thermal-barrier-coating applications. J Am Ceram Soc 85(12):3031–3035. https://doi.org/10.1111/j.1151-2916.2002.tb00574.x
Clark SJ, Segall MD, Pickard CJ, Hasnip PJ, Probert MI, Refson K, Payne MC (2005) First principles methods using CASTEP. Z Kristallogr Cryst Mater 220(5–6):567–570. https://doi.org/10.1524/zkri.220.5.567.65075
Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77(18):3865. https://doi.org/10.1103/PhysRevLett.77.3865
Gao Z, Zhang X, Chen Y, Chalk C, Nicholls J, Brewster G, Xiao P (2024) Strain tolerance evolution of EB-PVD TBCs after thermal exposure or CMAS attack. J Euro Ceram Soc 44(1):426–434. https://doi.org/10.1016/j.jeurceramsoc.2023.08.047
Fang H, Zhou P, Wang Y, Di C, Pu J (2023) Research on aggressiveness of CMAS+ NaVO3 mixtures towards thermal barrier coatings from the perspective of physical and chemical characteristics. Corro Sci 223:111463. https://doi.org/10.1016/j.corsci.2023.111463
Ahlborg NL, Zhu D (2013) Calcium–magnesium aluminosilicate (CMAS) reactions and degradation mechanisms of advanced environmental barrier coatings. Surf Coat Technol 237:79–87. https://doi.org/10.1016/j.surfcoat.2013.08.036
Leng K, Romero AR, Hussain T (2023) Multilayer GZ/YSZ thermal barrier coating from suspension and solution precursor plasma spray. J Euro Ceram Soc 43(11):44991–45003. https://doi.org/10.1016/j.jeurceramsoc.2023.04.014
McPherson R (1980) On the formation of thermally sprayed alumina coatings. J Mater Sci 15:3141–3149. https://doi.org/10.1007/BF00550387
Stokes JL (2021) Thermal expansion coefficients of Ca2Y8(SiO4)6O2 and Ca2Yb8(SiO4)6O2 apatite-type silicates. https://ntrs.nasa.gov/api/citations/20210009690/downloads/TM-20210009690.pdf
Luo Y, Wang J, Li J, Hu Z, Wang J (2015) Theoretical study on crystal structures, elastic stiffness, and intrinsic thermal conductivities of β-, γ-, and δ-Y2Si2O7. J Mater Res 30(4):493–502. https://doi.org/10.1557/jmr.2015.1
Zhang Z, Park Y, Xue Z, Zhang S, Byon E, Koo BH (2022) Research status of bond coats in environmental barrier coatings. Inter J Appl Ceram Tech 19(4):1841–1859. https://doi.org/10.1111/ijac.14042
Liu B, Liu Y, Zhu C, Xiang H, Chen H, Sun L, Gao Y, Zhou Y (2019) Advances on strategies for searching for next genersation thermal barrier coating materials. J Mater Sci Tech 35(5):833–851. https://doi.org/10.1016/j.jmst.2018.11.016
Xiang H, Feng Z, Zhou Y (2014) Mechanical and thermal properties of Yb2SiO5: first-principles calculations and chemical bond theory investigations. J Mater Res 29(15):1609–1619. https://doi.org/10.1557/jmr.2014.201
Tejero-Martin D, Romero AR, Wellman RG, Hussain T (2022) Interaction of CMAS on thermal sprayed ytterbium disilicate environmental barrier coatings: a story of porosity. Ceram Inter 48(6):8286–8296. https://doi.org/10.1016/j.ceramint.2021.12.033
Acknowledgements
The authors would like to thank John Kirk for his assistance during the suspension plasma spray. The authors acknowledge the Nanoscale and Microscale Research Centre (nmRC) at the University of Nottingham for providing access to SEM and FEG-SEM facilities.
Funding
This work was supported by the Engineering and Physical Sciences Research Council (EPSRC) (grant no. EP/V010093/1; grant no. EP/L022494/1).
Author information
Authors and Affiliations
Contributions
Buhao Zhang: methodology, investigation, data curation, writing—original draft, visualization; Hangfeng Zhang, Aochen Du: software, data curation; Ebenezer Owusu, Alex Lynam, Acacio Rincon Romero: methodology, investigation; Mingwen Bai, Nicholas Curry: writing—reviewing and editing; methodology, investigation; Tanvir Hussain: conceptualization, supervision, resource, writing—reviewing and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, B., Zhang, H., Bai, M. et al. Thermal properties and calcium-magnesium-alumino-silicate (CMAS) interaction of novel γ-phase ytterbium-doped yttrium disilicate (γ-Y1.5Yb0.5Si2O7) environmental barrier coating material. Adv Compos Hybrid Mater 7, 64 (2024). https://doi.org/10.1007/s42114-024-00879-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42114-024-00879-6