
Abstract
Perfluorooctanoic acid (PFOA) is a highly persistent organic pollutant of global concern. A novel nanocomposite composed of ZnO nanoparticles and citric acid-modified g-C3N4 was synthesized by ball milling process. The synthesized nanocomposite was more efficient than pure ball-milled ZnO nanoparticles for PFOA elimination under visible light irradiation. The optimal hybrid photocatalyst, produced by the addition of 5 wt% of citric acid-modified g-C3N4, demonstrated significantly better performance for PFOA removal than pure ZnO nanoparticles under UV irradiation, with the apparent rate constants of 0.468 h−1 and 0.097 h−1, respectively. The addition of peroxymonosulfate (0.53 g L−1) significantly increased PFOA removal, clarifying the crucial effect of sulfate radicals on PFOA photodegradation. In comparison, citric acid-modified g-C3N4 was not effective for PFOA elimination under visible light irradiation, even with the addition of peroxymonosulfate. Further experiments under dark conditions identified surface adsorption on hybrid photocatalyst as a key process in total PFOA removal. In summary, PFOA removal by ZnO@citric acid-modified graphitic carbon nitride nanocomposites is due to the combined action from adsorption and photodegradation, with adsorption as the dominating mechanism.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
The carbon-fluorine bond is considered the most stable and strongest bond in organic chemistry; therefore, the effective dissociation of the C-F bond could be the main challenge in the total remediation of per- and polyfluoroalkyl substances (PFAS) from the polluted water [1]. PFAS residues have been reported in human, plants, and wild animals [2]. Perfluorooctanoic acid (PFOA) is among well-known PFAS widely detected in aquatic environments with concentrations of nanograms per liter to milligrams per liter [3]. Although adsorption technologies are considered a promising remediation process for PFAS removal, their extensive application could suffer from the economic burden of costly adsorbents, regeneration issues after treatment, and mere transfer from water to solid phase [2]. Among advanced oxidation processes, photocatalysis technology has received great attention for the removal of organic pollutants [4]. Various metal oxides including TiO2 [4], In2O3 [5], Ga2O3 [6], CeO2 [3], and CdS [3] have been recently used for the photocatalytic decomposition of PFOA. Fu et al. [3] used five commercial powders (TiO2, In2O3, Ga2O3, CeO2, and CdS) for PFOA photocatalytic degradation under UV irradiation (32 W, 17.1 Wm−2). They found that the photodegradation efficiency increased as CdS < In2O3 < CeO2 < TiO2 < Ga2O3, while the price of these photocatalysts increased as TiO2 < CdS < CeO2 < Ga2O3 < In2O3. The efficiency of catalysts relies on numerous factors such as catalyst properties (particle size, specific surface area (SBET)), catalyst dosage, solution properties (e.g., pH), and the operating conditions (type of reactor, source of light). For instance, Lopes da Silva et al. [7] used commercial TiO2, In2O3, and Ga2O3 powders for the photodegradation of PFOA under UVB irradiation (36 W, 12 Wm−2). Considering their findings, catalysts dosage could highly affect the photocatalytic activity of semiconductors, where the orders of the efficiency at catalyst dosages of 0.1, 0.2, 0.5, and 1 g L−1 were Ga2O3 < In2O3 < TiO2, TiO2 < In2O3 < Ga2O3, In2O3 < Ga2O3 < TiO2, and In2O3 ≈ Ga2O3 < TiO2, respectively. In addition, self-made In2O3 showed higher photocatalytic activity than commercial TiO2, In2O3, and Ga2O3 catalysts. Overall, In2O3 or Ga2O3 has been suggested to be more promising than TiO2 for the photocatalytic decomposition of PFOA in several cases [6, 8,9,10], but In2O3 and Ga2O3 could suffer from the lack of cost-effectiveness. In addition, ZnO has been effectively used for such a purpose [11]. Notably, ZnO is not toxic and could reap the benefits of its chemical inertness and availability [12]. Besides, its electron mobility is superior than TiO2, and its lower refractive index than TiO2 offers ZnO as a highly transparent material with good light harvest and low light scattering [13]. Furthermore, compared with TiO2, wurtzite ZnO could take advantage of its great thermal stability even at high temperatures caused by plasma spraying [12]. Overall, ZnO, which is considered an n-type semiconductor [14], has been used for various applications such as photocatalysis [15], solar cells [16], photoelectrochemical properties [17], and resistive sensors [18].
Despite its advantages, ZnO has a wide band gap energy, restricting its visible light harvest. Designing and preparation of visible light-activated photocatalysts are of high interest, to maximize the use of solar power [19]. Hence, graphitic carbon nitride (g-C3N4 or GCN) has been used for environmental remediation [20]. Notably, g-C3N4 is considered a cost-effective and light-responsive support semiconductor owing to the presence of various anchoring sites, unique optical properties, layered structure, and the feasibility of chemical modification of surface defects [21]. Besides, GCN has the advantages of facile synthetic process and high stability [20] and is of current interest for photocatalytic applications [22]. It has seven phases including pseudocubic C3N4, g-o-triazine, α-C3N4, g-h-heptazine, cubic C3N4, g-h-triazine, and β-C3N4 where triazine and tri-s-triazine/heptazine rings are the basic tectonic units to construct GCN allotropes [21]. Among the various methods used for the synthesis of GCN, thermal condensation is considered a cost-effective method to scale it up for commercialization in which different materials are used as GCN precursors (i.e., dicyanamide, urea, melamine, thiourea, and cyanamide) [23]. Despite its numerous advantages, bulk GCN suffers from major shortcomings including the inefficient solar spectrum harvesting and fast recombination of charge carriers, necessitating the modification of GCN to achieve high photocatalytic activity [20]. Several strategies have been used to modify the chemical and physical properties of pristine g-C3N4. Element doping (metal or non-metal) is among the methods widely used to improve the photocatalytic activity of GCN [22]. The addition of citric acid (CA) to urea has been effectively used to modify GCN [24, 25]. As reported by Zhou et al. [24] and Zhu and Zhou [25], the addition of CA to urea could play an important role in the significantly improved photocatalytic activity, and CA to urea mass ratio of 0.001 yielded the best efficiency in both studies.
Overall, the application of ZnO and g-C3N4 for PFOA photodecomposition is important, yet GCN has not been widely used for such a purpose. Hence, this study aimed to synthesize a novel hybrid photocatalyst, containing ZnO nanoparticles and CA-modified GCN, using the mechanical milling process (as a cost-effective method used to produce different heterostructures) for the photocatalytic removal of PFOA under both UV and visible light irradiation. Importantly, to the best of our knowledge, the application of ZnO in PFOA photodegradation under visible light irradiation has not been investigated. Various strategies are used to generate SO4•− radicals, of which peroxymonosulfate (PMS)-assisted photocatalysis has been effectively employed to increase PFOA removal over metal oxides [4, 6, 26]. Hence, PMS was used to improve PFOA removal over the optimal hybrid photocatalyst.
2 Materials and methods
A detailed description of the materials and methods used in this study is provided in the supplementary information (Text S1).
3 Results and discussion
3.1 Characterization of GCN
XRD patterns of bulk g-C3N4 synthesized by urea are shown in Fig. S1(a), and GCN could be evidenced by the appearance of two sharp XRD peaks at around 13° and 27° [27, 28]. The g-C3N4 was synthesized at annealing temperatures from 450 to 530 °C, while the annealing time was 4 h in all cases. Notably, increasing the annealing temperature to 530 °C enhanced the intensity of the peak at ~ 27° which could be attributed to the improved crystallinity at high calcination temperatures. The peaks at ~ 13° and ~ 27° correspond to (100) and (002) diffraction planes, respectively, attributing to the in-planar structural packing and inter-planar stacking peaks of the aromatic system [29]. Paul et al. [30] synthesized GCN using urea at different calcination temperatures of 350, 400, 450, 500, 550, 600, and 650 °C. Considering their XRD results, raising the annealing temperature increased the intensity of the peak related to the inter-planar stacking of GCN which has been attributed to the improved crystallinity. The optimized range of annealing temperature has been reported to be 450–550 °C.
The FTIR spectra of bulk g-C3N4 synthesized by urea (Fig. S1(b)) showed three characteristic bands in all samples. The adsorption peak at ~ 808 cm−1 is associated with the breathing mode of triazine units. The strong band in the range of ~ 1200–1600 cm−1 corresponds to the stretching vibration of C-N and C=N heterocycles. The wide absorption peak at ~ 3000–3500 cm−1 is related to the stretching vibration of the terminal either N-H2 or N-H and is the result of uncondensed amino groups [31, 32]. Notably, the wide absorption peak at 3000–3500 cm−1 could also be attributed to the stretching vibration of O-H originating from the absorbed water molecule [31]. The photo-absorption ability of bulk g-C3N4 synthesized by urea at different annealing temperatures is compared in Fig. S1(c), which shows that increasing the annealing temperature increased the photo-absorption ability. The redshift caused by increasing the annealing temperature could be attributed to the improvement of polymerization degree [33]. The band gap energies of bulk g-C3N4 synthesized by urea were calculated using the Tauc method (Fig. S1(d)), which were reduced by increasing the calcination temperature which could be attributed to the higher degree of polymerization. In addition, the formation of defects and improved polymerization could widen and narrow the band gap energy, respectively [33]. SBET and pore volume of bulk g-C3N4 synthesized by urea at different annealing temperatures are presented in Table 1.
As evident, increasing the calcination temperature significantly enhanced the SBET of GCN synthesized by urea which can be attributed to the improved polymerization and emission of larger amounts of gases during the condensation and polymerization processes. The presence of oxygen in urea could facilitate the formation of CO2 during the polymerization process, taking into account the enlarged SBET of GCN synthesized by urea [34]. Table 1 signifies that using the high calcination temperature of 530 °C remarkably increased the pore volume of GCN, consistent with the results of SBET reported in the literature. For example, Paul et al. reported the SBET of GCN synthesized by urea at calcination temperatures of 450, 550, and 650 °C (calcination time = 3 h and heating rate = 2 °C min−1) to be 37.8, 73.7, and 65.6 m2 g−1, respectively [30]. Li et al. [35] synthesized GCN using urea, where the SBET of 63.46 m2 g−1 was obtained at 550 °C (calcination time = 4 h and heating rate = 2 °C min−1). In another study, Xia et al. [36] synthesized GCN using urea with the SBET of 108.83 m2 g−1 being obtained at 550 °C (2-h calcination time, 5 °C min−1 heating rate). Noteworthy, a significantly high SBET (122.82 m2 g−1) was achieved at 530 °C (4-h calcination time) where the heating rate was not fixed but was greater than 10 °C min−1. Overall, it is concluded that the SBET value strongly depends on the synthesis procedure of GCN, in terms of calcination time, calcination temperature, and heating rate.
3.2 Characterization of CA-modified GCN
The optimum annealing temperature (530 °C) was used to synthesize CA-modified GCN, while the annealing time was 4 h. As shown in Fig. S2(a), the XRD pattern of CA-modified GCN is identical to that of bare GCN, clarifying that the addition of CA did not significantly affect the molecular framework of bare GCN, which is consistent with other studies [24, 25]. The FTIR spectra of pristine and CA-modified GCN are compared in Fig. S2(b), where the peaks observed for CA-modified GCN are similar to those for pristine GCN which could be assigned to triazine units (1), C-N and C=N heterocycles (2), and N-H, N-H2, and O-H groups (3) as discussed previously. In addition to the XRD patterns, FTIR spectra also confirm that the molecular structure of GCN is retained after CA modification. The marginal shift in the location of peaks could be attributed to element doping by CA, which will be clarified by XPS analysis in the following sections. The photo-absorption ability and band gap energy of bare GCN and CA-modified GCN are compared in Fig. S2(c), (d), respectively. Interestingly, CA-modified GCN showed a remarkably higher photo-absorption ability than bare GCN in wavelengths from 250 to 800 nm. Moreover, modification by CA addition significantly reduced the band gap energy of GCN from 2.69 to 2.33 eV, which could be of high importance when using GCN-based catalysts for the photocatalytic decomposition of pollutants (especially under visible light irradiation). It should be noted that CA-modified GCN showed a higher photo-absorption ability than pristine GCN in the wavelength range (300–800 nm) in a study by Zhou et al. [24]. The significant redshift of absorption edge and reduced band gap energy could be specifically related to the modified structure by CA addition.
The XPS analysis was used to record the surface chemical composition of bare GCN and CA-modified GCN. XPS survey and high-resolution C 1s and N 1s spectra of bare GCN and CA-modified GCN are compared in Fig. S3. The similar spectra of bare GCN and CA-modified GCN signify that both samples were mainly composed of nitrogen and carbon elements [24, 25]. Notably, the minor peak located at ~ 532 eV is assigned to O element originated from thermal annealing in air at high temperature [37]. As evident in Fig. S3(c), (d), both pristine and CA-modified GCN showed similar C 1s spectra, which could be divided into three peaks with different binding energies. The peak at 284.8 eV of both samples can be assigned to C-C bonds of graphitic C impurities or adventitious C and sp2-hybridized C-N, and the peaks at 288.43 eV (GCN) and 288.35 eV (CA-modified GCN) could be attributed to sp2-hybridized C bonded to N in a triazine ring (N-C = N) [24, 25]. The minor peaks at 286.37 eV (GCN) and 286.50 eV (CA-modified GCN) could be assigned to either the bonds of C and amino groups (C-NH2) [37] or C-OH bonding [38]. The peaks at 284.8 eV (3.7%) and 286.50 eV (1.3%) of the CA-modified GCN sample were stronger than those of GCN sample (2.19% at 284.8 eV, 0.92% at 286.37 eV), while the peak at 288.35 eV (37.32%) of CA-modified GCN sample was weaker than that of GCN sample (38.38%). As shown in Fig. S3(e), (f), N 1s spectra of bare GCN and CA-modified GCN samples were also similar, which could be divided into three major peaks with different binding energies. The main peaks at 398.91 eV (GCN) and 398.84 eV (CA-modified GCN) are usually assigned to the sp2-hybridized N in a triazine ring (C-N=C); the peaks at 400.18 eV (GCN) and 400.14 eV (CA-modified GCN) correspond to N-C3 groups; and the peaks at 401.3 eV (GCN) and 401.26 eV (CA-modified GCN) correspond to the uncondensed amino groups (C-N-H) [25, 37]. Noteworthy, all three peaks at 398.91 eV (41.53%), 400.18 eV (8.9%), and 401.3 eV (6.39%) of GCN sample are stronger than those of CA-modified GCN sample at 398.84 eV (41.15%), 400.14 eV (8.81%), and 401.26 eV (6.06%). Considering the XPS results, the C/N atomic ratios of GCN and CA-modified GCN samples were 0.73 and 0.76, respectively. Overall, no remarkable binding-energy shift was observed on C 1s and N 1s between bare GCN and CA-modified GCN samples, clarifying that CA addition did not significantly affect the dominant structure of GCN except with the incorporation of C. Although CA has been used by other researchers to modify the structure of GCN, a unique modification mechanism has not accounted for the improvement. For instance, in preparing a mixture of 20 mg each of CA and urea which was heated at 550 °C in the air for 4 h at 2 °C min−1, Zhou et al. [24] showed that CA addition resulted in the formation of N-doped graphitic carbon-incorporated GCN from XPS results. In another research, Zhu and Zhou [25] mixed 50 mg each of CA monohydrate and urea, with the mixture being heated at 550 °C in the air for 4 h at 2 °C min−1. The XPS results showed that CA addition resulted in the formation of N-doped GCN. As evident in Fig. S4 (despite similar CA to urea ratio, annealing time and temperature, and heating rate), the XPS spectra reported by Zhou et al. [24] and Zhu and Zhou [25] are remarkably different, signifying the occurrence of different modification mechanisms although condensation is considered the dominant mechanism of the formation of GCN in the thermal annealing process. Furthermore, a semi-closed system was used to synthesize GCN in such a process. Hence, the formation of GCN using thermal annealing could be considered a non-equilibrium process in which several parameters, even the type of the crucible used for condensation, could affect the final product. Considering the XPS results reported by Zhu and Zhou [25], the C/N atomic ratios of GCN and CA-modified GCN were 1.99 and 0.92, respectively, compared with values of 0.73 and 0.76 in this research (using 10 mg each of CA and urea). Notably, the ratio of CA to urea in this research is similar to that used by Zhou et al. [24] and Zhu and Zhou [25], but the amount of urea (10 g) is considerably lower than that used by Zhou et al. (20 g) [24] and Zhu and Zhou (50 g) [25]. In addition, the heating rate (> 10 °C min−1) and annealing temperature (530 °C) were different from those used by Zhou et al. [24] and Zhu and Zhou [25] (annealing temperature = 550 °C, heating rate = 2 °C min−1). Hence, it can be concluded that the modification mechanism by CA addition to urea strongly relies on the synthetic process of GCN.
As shown in Fig. S5, CA-modified GCN showed extremely lower photoluminescence (PL) intensity than pristine GCN which could be assigned to the introduction of the C element to the GNC framework [39]. Meanwhile, the emission peak maximum moved from 459 (pristine GCN) to 467 nm (CA-modified GCN), exhibiting a redshift which is consistent with the redshift of the absorption edge of DRS spectra (Fig. S2(c)), and could be attributed to C doping. It has been suggested that attaching aromatic heterocycles and replacement of N with C in triazine units may promote the delocalization of e− in the π-conjugation system, improving light harvest and boosting transfer and separation of charge carriers [40]. Overall, the lower the PL intensity, the higher the efficiency of charge carrier transfer and a higher photocatalytic activity could be expected [19]. Hence, CA-modified GCN with its higher photo-absorption ability, lower band gap energy, and lower recombination rate of charge carriers than those obtained by bare GCN was used to synthesize ZnO-CA-modified GCN nanocomposites for the effective PFOA elimination.
The SEM images of pristine and CA-modified GCN at different magnifications of 10,000, 40,000 and 70,000 (Fig. S6) showed that both samples were fluffy and contained several thin sheets. By using urea as the precursor of GCN, a large amount of gas is released during the annealing process, acting as a pore forming agent and favoring the formation of g-C3N4 with a large specific surface area and fluffy nanostructure [36]. Hence, the production of ultrathin sheets in bare GCN using the facile one-step synthetic process could be responsible for the outstanding SBET of 123 m2 g−1. To clarify the distribution of elements in pristine and CA-modified GCN, EDX analysis (Fig. S7) showed that C and N elements were uniformly distributed in both samples. Notably, the Al element was also detected since the samples were mounted on a piece of Al foil. The C/N atomic ratio of bare and CA-modified GCN was also measured by EDX analysis at 30 different points (Fig. S8), with a higher C/N atomic ratio of CA-modified GCN (0.65) than that of pristine GCN (0.61). The same trend was observed by the results of XPS analysis though with different corresponding values (0.76 and 0.73, respectively), which could be related to the higher accuracy of XPS than EDX analysis.
3.3 Synthesis of ZnO@CA-modified GCN nanocomposites
The XRD patterns of ZnO@CA-modified GCN nanocomposites and ball-milled ZnO nanoparticles were compared (Fig. 1a) which presented no significant difference and followed the pattern of wurtzite ZnO as the main phase in all samples. Notably, the main peak of CA-modified GCN (around 27°) was not observed in any of the nanocomposites. Wang et al. synthesized ZnO hybridized with GCN using different mass ratios of GCN to ZnO (1%, 2%, 3%, 4%, 6%, and 8%), where the main peak of GCN was observed only in samples with high mass ratio of GCN/ZnO (≥ 4%) [41]. More recently, in synthesizing hybrid ZnO-GCN using ball milling with different GCN/ZnO mass ratios of 5%, 10%, 15%, and 20%, Chen et al. [42] did not detect the main peak of GCN. They suggested that vigorous mechanical force initiated by the milling process could result in the uniform distribution of GCN on the surface of ZnO particles, which could not be easily achieved by other synthetic techniques. Hence, the disappearance of the main peak of GCN, even at the high mass ratio of GCN/ZnO (i.e., 20%), was attributed to the mechanical milling process to produce hybrid structures [42]. Meanwhile, the main peak of GCN has not been detected in other studies (GCN/ZnO nanocomposites), due to the high dispersion of GCN [43].

Comparing a XRD patterns, b FTIR spectra, c photo-absorption ability, and d band gap energy of pure ball-milled ZnO nanoparticles and ZnO@CA-modified GCN nanocomposites
The FTIR spectra of ZnO@CA-modified GCN nanocomposites and pure ball-milled ZnO nanoparticles are compared (Fig. 1b). Similar to the XRD patterns, there is no significant difference between nanocomposites which all follow the pattern of pure ball-milled ZnO as the main phase in all samples. The vibration band from 400 to 500 cm−1 is generally attributed to the stretching mode of the Zn-O bond, and the broad bending peaks from 1330 to 1670 cm−1 are usually assigned to the presence of surface hydroxyl groups [44]. The broad peaks at 3607 and 3726 cm−1 could be assigned to OH groups the non-H bonded hydroxyl groups of H2O molecules, consecutively [45]. Photo-absorption ability and band gap energy of ZnO@CA-modified GCN nanocomposites and pure ball-milled ZnO nanoparticles are compared in Fig. 1c and d. Pure ball-milled ZnO nanoparticles showed a considerable absorption below the absorption edge (420 nm) which could be related to the electron transfer from the valence band (VB) to the CB of ZnO, whereas the absorbance was insignificant in the visible range. All nanocomposites provided broad absorption in the visible range in contrast to pure ball-milled ZnO nanoparticles, which is related to the formation of hybrid nanostructures by the addition of CA-modified GCN to ZnO nanoparticles, favoring the application of ZnO@CA-modified GCN nanocomposite for photocatalytic applications under visible light irradiation. Among those, the hybrid catalyst produced by 5 wt% of CA-modified GCN exhibited the highest photo-absorption ability. Considering Fig. S2(c), (d), the potential visible light harvest of CA-modified GCN and its extremely narrower band gap energy than ZnO could be easily verified. Meanwhile, the band gap energy remarkably reduced from 3.07 (for pure ball-milled ZnO nanoparticles) to 2.89 eV (for ZnO@CA-modified GCN nanocomposite produced by the addition of 50 mg of CA-modified GCN to 1 g of ZnO nanoparticles), but the nanocomposite still suffers from a relatively high band gap energy to provide excellent performance under visible light irradiation. The significant redshift of absorption edge and reduced band gap energy of nanocomposites could be specifically related to the formation of a hybrid nanostructured material composed of ZnO nanoparticles and CA-modified GCN.
The SEM morphology of ZnO@CA-modified GCN nanocomposite (5 wt%) at different magnifications (Fig. 2) demonstrates that ZnO nanoparticles were extensively embedded in the sheet structures. The formation of a hybrid nanostructure could be evidenced by SEM images at high magnifications (Fig. 2c).

SEM images of ZnO@CA-modified GCN nanocomposite (5 wt%) at different magnifications
The EDX analysis was performed to study the distribution of elements in the hybrid nanostructured material produced by mixing 50 mg of CA-modified GCN and 1 g of ZnO nanoparticles. Notably, EDX mapping analysis was carried out in two different areas (Fig. S9), which shows that the graphitic phase is mainly composed of C (rather than N). Unexpectedly, the C/N atomic ratio throughout the analyzed areas was 12.6 and 5, offering the possibility of nitrogen removal from the GCN framework during the 5-h ball milling process followed by the annealing treatment at 400 °C for 2 h. Nitrogen removal from the GCN framework has also been reported by Paul et al. [46] in the synthesis of ZnO-modified GCN composite for the photocatalytic degradation of methylene blue, whereas zinc carbonate basic dehydrate ([ZnCO3]2.[Zn(OH)2]3) and urea were used as ZnO and GCN precursors, respectively. The C/N atomic ratio of pure GCN and GCN-ZnO composite (0.4 mmol [ZnCO3]2.[Zn(OH)2]3 in 10 g of urea) was 0.85 and 1.5, consecutively. Deamination during thermal polymerization, caused by the addition of [ZnCO3]2.[Zn(OH)2]3, has been accounted for the probable nitrogen removal from the GCN framework. The creation of nitrogen vacancy has also been reported by Lee et al. [47], where the post-solvothermal treatment of Mg-doped GCN resulted in the production of nitrogen vacancy-rich GCN which has been related to the formation of numerous amine sites in GCN during urea polycondensation with the addition of Mg precursor. Our findings confirm that even using chemically inert ZnO nanoparticles, unlike [ZnCO3]2.[Zn(OH)2]3 which was used as the ZnO precursor [46], could result in nitrogen removal from the GCN framework during the milling process. Hence, the milling process in the presence of ZnO nanoparticles followed by annealing could facilitate nitrogen removal due to the probably damaged GCN framework, although further research is required to clarify the mechanism. Although nitrogen removal was not expected, point-defect engineering by the creation of nitrogen vacancy is considered a promising approach to improve the photocatalytic activity of GCN by mitigating the recombination rate of electron-hole pairs [47].
Some CA-modified GCN powder was milled for 5 h and annealed at 400 °C for 2 h and then analyzed by XPS to clarify the effect of ball milling on the stability of CA-modified GCN. XPS survey and high-resolution C 1s and N 1s spectra of CA-modified and ball-milled CA-modified GCN are compared (Fig. S10), with no significant difference between samples. The C/N atomic ratio of CA-modified GCN (0.76) and ball-milled CA-modified GCN (0.84) was similar, suggesting that the milling process (for 5 h) followed by annealing treatment (400 °C for 2 h) did not remarkably affect the chemical composition of CA-modified GCN (without the addition of ZnO nanoparticles). Hence, nitrogen removal from the GCN framework could be mainly related to the presence of ZnO nanoparticles in the milling process followed by the annealing treatment. Although ball milling was performed at room temperature, the temperature inside the grinding jar could increase considerably during milling due to the frequent and severe collision of zirconia balls.
XPS survey and high-resolution Zn 2p and O 1s spectra of initial ZnO nanoparticles, ball-milled ZnO nanoparticles (after annealing at 400 °C for 2 h), and ZnO@CA-modified GCN nanocomposite produced by the addition of 50 mg of CA-modified GCN to 1 g of ZnO nanoparticles (after annealing at 400 °C for 2 h) are compared in Fig. 3. As shown, XPS spectra of ZnO and ball-milled ZnO nanoparticles are thoroughly similar with slight differences in the intensity of peaks. The main peaks at 1021.21 eV and 1021.09 eV in the high-resolution Zn 2p spectra of initial ZnO and ball-milled ZnO nanoparticles, respectively, signify +2 oxidation state of Zn ions; and the main peaks at 530.07 eV and 530.02 eV in the high-resolution O 1s spectra of initial ZnO and ball-milled ZnO nanoparticles clarify the presence of O element in Zn-O bonding of ZnO (wurtzite phase) with −2 oxidation state. The peaks at 531.42 eV and 531.34 eV were due to the presence of O vacancy in ZnO and ball-milled ZnO nanoparticles, respectively [48], whereas the peaks at 532.26 eV and 532.17 eV could be related to the chemisorbed oxygen species, and those at 530.07 eV and 530.02 eV could be assigned to the O22− species in the lattice [43]. Considering the Zn2p3 A, O1s A, O1s B, and O1s C peaks (Fig. 3b, c, e, f), the Zn/O atomic ratios of initial ZnO and ball-milled ZnO nanoparticles were 1.07 and 1.02, respectively, clarifying that the ball milling process followed by annealing (400 °C for 2 h) did not considerably affect the surface chemical composition of ZnO nanoparticles. Meanwhile, the XPS spectra of initial and ball-milled ZnO nanoparticles are comparable to those of ZnO@CA-modified GCN nanocomposite (5 wt%). Noteworthy, the synthesized nanocomposite was mainly composed of ZnO nanoparticles. In addition, as confirmed by the XPS results of GCN and CA-modified GCN samples, the intensity of N 1s peak is remarkably higher than that of C 1s peak (Fig. S10(a, b)), while N 1s was not detected in the XPS analysis of ZnO@CA-modified GCN nanocomposite (5 wt%). The findings signified the probable nitrogen removal from the GCN framework which was also confirmed by EDX analysis (Fig. S9).

a XPS survey spectra, b Zn 2p spectra, and c O 1s spectra of initial ZnO nanoparticles; d XPS survey spectra, e Zn 2p spectra, and f O 1s spectra of ball-milled ZnO nanoparticles; and g XPS survey spectra, h Zn 2p spectra, and i O 1s spectra of ZnO@CA-modified GCN nanocomposite (5 wt%)
The PL intensity of the catalysts (Fig. 4) showed that ZnO@CA-modified GCN nanocomposite with 5 wt% of CA-modified GCN presented the minimal PL intensity among the nanocomposites. Hence, compared with pure ball-milled ZnO nanoparticles, the minimal recombination rate of charge carriers was obtained by the hybrid catalyst (5 wt% of CA-modified GCN) which could be related to the formation of the heterostructure with a strong conjugation between the graphitic sheets decorated by ZnO nanoparticles (Fig. 2) as suggested by Javed et al. [49]. Meanwhile, the emission peak for pure ball-milled ZnO nanoparticles (466 nm) marginally moved to a lower value for the nanocomposites (e.g., 464 nm for ZnO@CA-modified GCN (5 wt%)), exhibiting a blueshift which is consistent with the results obtained by Kalisamy et al. where the emission peak of ZnO@S-doped GCN composite (with 39 wt% of S-doped GCN) blueshifted compared with S-doped GCN and has been allocated to the defect trap state originated from the quantum confinement effect [50]. Overall, due to the low recombination rate of charge carriers, the synthesized nanocomposite produced by mixing 50 mg of CA-modified GCN and 1 g of ZnO nanoparticles is considered an efficient hybrid catalyst for PFOA elimination.

Comparing the PL intensity of ZnO@CA-modified GCN nanocomposites and pure ball-milled ZnO nanoparticles (excitation wavelength = 325 nm, room temperature)
3.4 Photocatalytic activity
For screening, the performance of the synthesized nanocomposites for PFOA elimination under visible light irradiation was compared to that of pure ball-milled ZnO nanoparticles (for control) and bare CA-modified GCN. As shown in Fig. S11, the synthesized nanocomposites remarkably improved PFOA removal, whereas CA-modified GCN was approximately inert to PFOA degradation. As mentioned previously, bulk GCN could suffer from inefficient solar spectrum harvest and fast recombination of charge carriers [20]. Although GCN was significantly modified by CA addition, it was not efficient for PFOA removal due to the weak oxidation potential of g-C3N4 which is a more suitable photocatalyst for reduction applications (Fig. S12). Notably, compared with pure ball-milled ZnO nanoparticles, ZnO@CA-modified GCN nanocomposite produced by the addition of 50 mg of CA-modified GCN to 1 g of ZnO nanoparticles showed the highest efficiency which was attributed to its superior photo-absorption ability and lower recombination rate of charge carriers. To confirm the promising performance of ZnO for PFOA removal under visible light irradiation, the photocatalytic activities of initial ZnO nanoparticles, TiO2 P25, and In2O3 nanoparticles were compared (Fig. S13). ZnO nanoparticles showed significantly higher performance than TiO2 P25 and In2O3, inducing PFOA removal of 64%, 10%, and 5%, respectively, within 6 h under visible light irradiation.
To elucidate the efficiency of ZnO@CA-modified GCN (5 wt%) nanocomposite, the photocatalytic activity of initial ZnO nanoparticles was compared to that of ZnO@CA-modified GCN (5 wt%) nanocomposite under visible light irradiation within 2 h. As shown in Fig. 5a, the overall PFOA elimination over ZnO@CA-modified GCN (5 wt%) nanocomposite was insignificantly higher than that over initial ZnO nanoparticles at 2 h. Even by increasing the irradiation time from 2 to 6 h, ZnO@CA-modified GCN (5 wt%) nanocomposite was only marginally more efficient than initial ZnO nanoparticles (Figs. S11 and S13). The synthesized nanocomposite was mainly composed of ZnO phase, which possesses a wide band gap energy, restricting its high photocatalytic performance under visible light irradiation. However, it is obvious that PFOA removal using the nanocomposite was remarkably higher than that using the initial ZnO nanoparticles (especially in the early stages), resulting in the remarkably higher apparent rate of PFOA removal over the nanocomposite (Fig. 5b). The activation of ZnO by using a powerful source of visible light (300-W Xenon lamp) could be considered as shown in Eq. (1) [12]:

Comparing the photocatalytic activity of initial ZnO nanoparticles and ZnO@CA-modified GCN (5 wt%) nanocomposite under a, b visible and c, d UV irradiation (initial PFOA concentration, 53 mg L−1; dosage of catalysts, 0.53 g L−1). The graph regarding initial ZnO nanoparticles in c has been adapted from [26] https://pubs.rsc.org/en/content/articlelanding/2023/en/d3en00241a/unauth#!divAbstract
The energy of photons was calculated by Eq. (2) [4]:
where E0, h, c, and λ are the energy of one photon, Plank’s constant (i.e., 6.63 × 10−34 Js), light speed (i.e., 3 × 108 m s−1), and wavelength, respectively. By considering the minimum wavelength of visible light (i.e., 400 nm), E0 is 5.0 × 10−19 J. The band gap energy of initial ZnO nanoparticles is ~ 3.23 eV (~ 5.2 × 10−19 J) which is very close to E0. Therefore, using a powerful visible light source as in this study could provide photons with sufficient energy to activate ZnO even under visible light illumination despite its wide band gap energy. The same finding has been reported by Xu et al. [4] using TiO2 under visible light irradiation.
The photocatalytic activity of initial ZnO nanoparticles was also compared to that of ZnO@CA-modified GCN (5 wt%) nanocomposite under UV irradiation (Fig. 5c, d). As evident, the synthesized nanocomposite showed superior efficiency than initial ZnO nanoparticles for PFOA elimination which could be related to the formation of a potential heterojunction in the synthesized nanocomposite. The large band gap energy of the synthesized nanocomposite necessitates using UV irradiation to produce sufficient photons. Overall, ZnO@CA-modified GCN (5 wt%) nanocomposite is considered highly promising for PFOA removal especially under UV irradiation.
3.4.1 Effect of PMS on PFOA elimination
ZnO@CA-modified GCN nanocomposite (5 wt%) with its superior efficiency than other nanocomposites was used to evaluate the effect of PMS addition on PFOA removal. As shown in Fig. S14, the addition of PMS (0.27 g L−1 and 0.53 g L−1) yielded similar PFOA removal, whereas using the high concentration of 1.06 g L−1 remarkably reduced PFOA removal. The cleavage of PMS could result in the production of SO4•− and •OH radicals (Eq. (3)) [51]:
However, on the downside, high concentrations of PMS could quench the photocatalytic activity due to the self-combination reaction of SO4•− radicals, leading to the production of S2O82−, or some other side reactions [52]. Furthermore, high concentrations of PMS could facilitate ZnO corrosion [26]. The apparent rate constants of PFOA elimination by the addition of PMS (0.27 and 0.53 g L−1) were also similar. However, as evident in Fig. S14(a), PFOA removal at the initial stages (first 40 min) by using 0.53 g L−1 PMS was remarkably higher than that obtained by 0.27 g L−1 PMS. Hence, the PMS concentration of 0.53 g L−1 was chosen for further experiments.
The plots of C/C0 and ln(C0/C) versus t (h) for PFOA elimination over ZnO@CA-modified GCN nanocomposite (5 wt%) with the addition of PMS under visible light irradiation are shown in Fig. 6a, b. The ZnO-GCN nanocomposite (5 wt%) is much more efficient than pure ball-milled ZnO nanoparticles for PFOA removal under visible light irradiation which is consistent with the results obtained from the screening experiments (Fig. S11). Furthermore, it is clarified that the addition of PMS can remarkably improve PFOA removal over ZnO@CA-modified GCN nanocomposite, where the apparent rate constant of PFOA removal considerably increased from 0.447 (ZnO@CA-modified GCN nanocomposite) to 0.796 h−1 (the same catalyst with PMS), which is attributed to the production of sulfate radicals [4, 6]. Furthermore, when compared with the results shown in Fig. 5, it could be concluded that pure ball-milled ZnO nanoparticles showed significantly lower efficiency than initial ZnO nanoparticles under visible light irradiation. Notably, the ball milling process could result in the agglomeration of ZnO nanoparticles, and some structural changes as the color of ZnO nanoparticles changed from white (initial ZnO) to yellowish (ball-milled ZnO). Notably, it did not turn white even after annealing at 400 °C for 2 h. Hence, in addition to the reduced SBET, some unexpected structural changes occurred during the milling process, causing inferior efficiency of ball-milled ZnO nanoparticles than the initial ZnO nanoparticles under visible light.

Plots of C/C0 versus t (h) and ln (C0/C) versus t (h) for the PFOA removal over ZnO@CA-modified GCN nanocomposite with the addition of PMS under a, b visible light and c, d UV irradiation (initial PFOA concentration, 53 mg L−1; dosage of ZnO and PMS, 0.53 g L−1)
The plots of C/C0 and ln(C0/C) versus t (h) for PFOA removal over ZnO@CA-modified GCN nanocomposite (5 wt%) with PMS addition under UV irradiation are shown (Fig. 6c, d). Like the experiments performed under visible light irradiation, ZnO@CA-modified GCN nanocomposite (5 wt%) showed much higher efficiency than pure ball-milled ZnO nanoparticles, clarifying the vital role of the formation of a heterostructure in improving the performance of PFOA elimination. Besides, despite the considerably higher power of the Xenon lamp than UV lamps, all catalysts showed higher performances under UV irradiation. Both pure ball-milled ZnO nanoparticles and ZnO@CA-modified GCN nanocomposite (5 wt%) are not intrinsically visible light-activated catalysts, since ZnO has a wide band gap energy. Hence, a higher performance is expected under UV irradiation. The apparent rate of PFOA elimination was significantly enhanced from 0.468 (ZnO@CA-modified GCN nanocomposite) to 0.868 h−1 (the same catalyst with PMS) which is consistent with the results obtained under visible light irradiation, confirming the vital role of sulfate radials in PFOA elimination. Compared to the results in Fig. 5, pure ball-milled and initial ZnO nanoparticles showed similar performances under UV irradiation (unlike under visible light). Compared with UV irradiation, the stronger effect of ZnO structural changes/defects from milling on PFOA removal under visible light irradiation could be related to the wide band gap of ZnO which is not a visible light-activated semiconductor.
Overall, the synthesis of an efficient hybrid photocatalyst for PFOA removal is of high importance. Firstly, this hybrid nanomaterial was produced by ZnO and g-C3N4 which are both among the most economical semiconductors used for photocatalytic applications. Secondly, ZnO@CA-modified GCN nanocomposite was highly promising for PFOA elimination under visible light irradiation (even without PMS), compared to TiO2 [4, 8, 9]. More importantly, In2O3 with its certain solar light absorption is not efficient for PFOA removal under visible light irradiation (without PMS) which has been attributed to its fast recombination rate of charge carriers [5]. Although Ga2O3 has been introduced to be very promising for PFOA degradation, its application was restricted to UV irradiation due to its very large band gap energy. Hence, it was not efficient for PFOA removal under visible light irradiation without the addition of PMS [6]. The performance of ZnO@CA-modified GCN nanocomposite for PFOA elimination is compared to that of main oxide semiconductors (Table 2). By far, ZnO@CA-modified GCN nanocomposite showed a higher efficiency than other oxide semiconductors under visible light irradiation (with and without PMS). The superior efficiency of the synthesized nanocomposite over Ga2O3 under visible light irradiation was predictable due to the large band gap energy of Ga2O3 [3], restricting its visible light harvest, but the findings were more exciting where the performance of the hybrid photocatalyst was considerably higher than that of TiO2. Notably, the band gap energy of ZnO and TiO2 is similar, but ZnO could take advantage of its higher visible light harvest than TiO2 [12], making ZnO more attractive than TiO2 for photocatalytic applications under visible light irradiation. Under UV irradiation, the synthesized nanocomposite again showed good performance compared with other main oxide semiconductors, but PMS addition can make several oxide semiconductors more proficient than the ZnO@CA-modified GCN nanocomposite for PFOA elimination. Meanwhile, catalyst morphology can highly affect the photocatalytic activity, e.g., different In2O3 morphologies (nanostructures) showed significantly different performances (Table 2). Overall, considering cost-effectiveness and efficiency, ZnO@CA-modified GCN nanocomposite is promising for PFOA removal under UV and especially visible light irradiation. Further improvement of the performance of these nanocomposites should be studied in future by using different ZnO morphologies and crystal structures.
3.4.2 Probable mechanism of photocatalytic degradation of PFOA
The main active species responsible for the photocatalytic degradation of PFOA over the ZnO@CA-modified GCN nanocomposite were studied by the addition of BQ, BuOH, and EDTA-Na2 to the initial PFOA solution to scavenge O2•−, •OH, and the photogenerated holes (h+), respectively, and the results are shown in Fig. 7a.

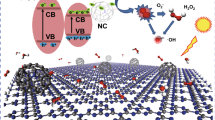
a Effects of the addition of scavengers on PFOA removal (C0: 53 mg L−1) under UV irradiation. b Comparing the VB of CA-modified GCN and initial ZnO nanoparticles (before mechanical milling). c Schematic of the probable separation pathways of charge carriers in the ZnO@CA-modified nanocomposite
The performance of ZnO@CA-modified GCN nanocomposite was suppressed by adding BQ and EDTA-Na2, signifying the vital role of O2•− and h+ in PFOA removal. In addition to O2•− and h+, sulfate radicals played an important role in PFOA elimination over the hybrid photocatalyst as confirmed previously (Fig. 6). The probable production of sulfate radicals has accounted for the considerably improved PFOA degradation in PMS-assisted photocatalysis in several studies [4, 6, 26]. Unlike BQ and EDTA-Na2, t-BuOH did not quench PFOA removal over the hybrid catalyst. Noteworthy, t-BuOH could boost PFOA decomposition due to its less polarity than water, resulting in the higher reactivity of superoxide radicals [53].
It is suggested that PFOA photodegradation over the ZnO@CA-modified GCN nanocomposite follows a stepwise manner which has been reported in other studies [4, 6, 11]. Considering our findings, the primary PFOA decomposition mechanism could be explained by the following stages. Firstly, PFOA could dissociate to become an anionic compound (C7F15COO−) and adsorbed on the surface of the hybrid photocatalyst as shown in Eq. (4) [54]:
C7F15COO− could react with h+ and/or SO4•− to generate C7F15COO• (perfluoroperoxy radicals) according to Eqs. (5) [11] and (6) [55], respectively:
Notably, the photogenerated holes could drive the oxidation of water, leading to the production of •OH radicals, and the photogenerated electrons could react with HSO5−, enabling the production of both SO4•− and •OH radicals (Eq. (3)).
After the production of C7F15COO•, the Kolbe decarboxylation (by the collapse of C-C bonds between C7F15− and −COO•) could result in the formation of C7F15• (perfluoroalkyl radicals), as shown in Eq. (7) [4, 56]:
Perfluoroalkyl radicals could also be produced in the presence of superoxide radicals [6]:
Then, C7F15• could react with H2O, generating C7F15OH according to Eq. (9) [4]:
The conversion of C7F15OH to C6F13COF, due to the instability of C7F15OH, followed by hydrolysis could finally yield C6F13COOH (PFHpA) as shown in Eqs. (10) and (11), respectively [11, 57]:
Other shorter-chain PFAS including PFHxA, PFPeA, PFBA, PFPrA, and TFA could be produced during the process. It has been suggested that PFOA could be thoroughly mineralized in such a stepwise manner to generate fluoride ions and CO2 as the final products [4].
The interfacial contact of at least two semiconductors could result in the construction of a heterostructure, facilitating the effective separation of charge carriers [58]. The VB of CA-modified GCN (before mechanical milling) and the VB of initial ZnO nanoparticles (before mechanical milling) are compared in Fig. 7b. As evident, the VB of initial ZnO nanoparticles seems to be lower than that of CA-modified GCN. It should be noted that the band structure of ZnO nanoparticles and CA-modified GCN could vary by the ball milling process. Noteworthy, the color of ZnO changed from white to yellowish after the milling process, leading to a decrease of the band gap energy from 3.23 (initial ZnO nanoparticles) to 3.07 eV (ball-milled ZnO nanoparticles). For CA-modified GCN, its nitrogen content was significantly reduced after the milling process, leading to the probable change of VB and CB positions. Overall, it is not accurate to consider the VB and CB positions of CA-modified GCN and initial ZnO nanoparticles (before milling) to infer the mechanism of the separation of charge carriers in the hybrid photocatalyst produced after milling. However, by considering the probable positions of VB and CB of CA-modified GCN and ZnO (in the synthesized nanocomposite), the schematic of the possible mechanisms of the separation of charge carriers (type I and type II) is provided in Fig. 7c. In both mechanisms, the transfer of the photogenerated holes from VB of ZnO to VB of CA-modified GCN is required, which could not be expected due to some reasons. Importantly, the photogenerated holes were considered among the main active species responsible for PFOA removal over the synthesized nanocomposite under UV irradiation (Fig. 7a). Meanwhile, CA-modified GCN with/without PMS addition was efficient for PFOA elimination under visible light irradiation (Figs. S11 and S16), whereas ball-milled ZnO nanoparticles were efficient for PFOA removal under UV and visible light irradiation (Fig. 6). The hybrid photocatalyst produced by the addition of 50 mg of CA-modified GCN to 1 g of ZnO nanoparticles showed promising performance where the apparent rate constant of PFOA removal significantly increased from 0.097 (ball-milled ZnO nanoparticles) to 0.468 h−1 (ZnO@CA-modified GCN nanocomposite) and from 0.046 (ball-milled ZnO nanoparticles) to 0.447 h−1 (ZnO@CA-modified GCN nanocomposite) under UV and visible light irradiation, respectively. Determination of VB and CB positions of ZnO and nitrogen vacancy-rich CA-modified GCN, which are essential to infer the mechanism of the separation of photogenerated e−/h+ pairs, needs further deep investigations. However, considering our findings, the probable separation mechanism of charge carriers is estimated to follow an S-scheme heterojunction rather than type I and type II heterojunctions (Fig. 7c). The formation of S-scheme heterojunction has been recently reported when using numerous hybrid photocatalysts composed of ZnO and g-C3N4 [59,60,61,62]. Noteworthy, S-scheme heterojunctions can take advantage of the simultaneous effects of reduced recombination rate of charge carriers and retained redox potential of the photogenerated electrons and holes [59, 60]; and they are generally composed of two n-type semiconductors, representing oxidation and reduction photocatalysts [63]. Both ZnO [14] and g-C3N4 [22] are among n-type semiconductors. In the S-scheme heterojunction, an internal electric field is generated between semiconductors after their contact [61, 62]. Then, the photogenerated electrons of the oxidation photocatalyst are combined with the photogenerated holes of the reduction photocatalyst, retarding the recombination of charge carriers itself [62]. To show the contact between ZnO nanoparticles and the graphitic phase, the TEM image and EDX mapping analysis of the hybrid photocatalyst are provided in Fig. 8.

a TEM and b EDX mapping analysis of ZnO@CA-modified GCN (5 wt%) nanocomposite
For control, ZnO@GCN nanocomposite (5 wt%) was also produced by mixing 50 mg of GCN and 1 g of ZnO nanoparticles, which was milled for 5 h and annealed at 400 °C for 2 h. The efficiencies of bare GCN, ZnO@GCN nanocomposite (5 wt%), and ZnO@CA-modified GCN nanocomposite (5 wt%) for PFOA removal under visible light irradiation are compared (Fig. S17). The bare GCN was ineffective for PFOA removal, clarifying that neither degradation nor adsorption occurred on GCN surface. The same result was also observed for CA-modified GCN (Figs. S11 and S16). In addition, ZnO@CA-modified GCN nanocomposite (5 wt%) was more effective than ZnO@GCN nanocomposite (5 wt%) for PFOA elimination under visible light irradiation that was related to the superior properties of CA-modified GCN than GCN. The SBET of the initial ZnO nanoparticles and ZnO@CA-modified GCN nanocomposite (5 wt%) is compared in Table 3.
The significantly reduced SBET of ZnO@CA-modified GCN nanocomposite (5 wt%) than initial ZnO nanoparticles was related to the milling process used to synthesize the nanocomposite, resulting in the compression of particles. PFOA removal over initial ZnO nanoparticles and ZnO@CA-modified GCN nanocomposite (5 wt%) was investigated under dark condition. As shown in Fig. S18, the ratio of C/C0 reduced considerably over both samples, signifying the high adsorption of PFOA on both ZnO and ZnO@CA-modified GCN nanocomposite (5 wt%). Besides, the addition of CA-modified GCN to ZnO did not remarkably affect the amount of PFOA adsorption. Wu et al. [11] also showed that the addition of reduced graphene oxide (0.2–2 wt%) to ZnO did not considerably affect PFOA adsorption; however, the amount of PFOA adsorption on the surface of the photocatalysts (8.1–10.2%) is remarkably lower than that observed in this research. It could be related to the different experimental conditions (catalyst dosage, initial PFOA concentration) and using catalysts with different morphology, particle size, specific surface area, and surface properties.
By comparing the experiments carried out in the dark and under light irradiation, it is concluded that PFOA adsorption on the surface of ZnO nanoparticles and nanocomposites played the dominant role in total PFOA removal under light irradiation. Noteworthy, adsorption has been considered an unavoidable topic in photocatalytic experiments [64]. Although there was not a significant difference between the amount of PFOA removal over initial ZnO nanoparticles and ZnO@CA-modified GCN nanocomposite (5 wt%) under dark condition, the amount of PFOA elimination over these photocatalysts was considerably different under visible light irradiation especially at the initial stages (Fig. 5a) and under UV irradiation (Fig. 5c). Besides, the efficiency of ball-milled ZnO and ZnO@CA-modified GCN nanocomposite (5 wt%) for PFOA elimination was significantly different under UV and visible irradiation (Fig. 6). The reason of such a significant difference should be further studied, but likely related to the synergy of high adsorption and photocatalysis of PFOA over the synthesized hybrid nanocomposite. Notably, it has been suggested that such a synergy could improve the efficiency of pollutant elimination [65].
Interestingly, PFOA removal over initial ZnO nanoparticles in the dark (Fig. S18) was higher than that under UV irradiation (Fig. 5). The water temperature could remarkably affect PFOA removal. For instance, Wu et al. [64] evaluated the effect of temperature on PFOA degradation by ZnO assisted-photocatalytic ozonation, where the minor increase of temperature from 298 to 308 K markedly reduced PFOA removal from 70.9 to 52.9% in 4 h (under UV irradiation). Wu et al. [11] studied the effect of temperature on PFOA decomposition using persulfate-assisted photocatalytic ozonation over ZnO-reduced graphene oxide, where increasing the temperature from 298 to 318 K reduced PFOA elimination from 99.2 to 87.1% in 4 h under UV. It has been stated that the adsorption capacity of ZnO could be reduced at high temperatures [64]. Therefore, the heat input from light irradiation to the solution could decrease PFOA adsorption on ZnO surface, subsequently reducing its efficiency for PFOA removal under light irradiation. In addition to the effect of temperature, the mechanism of adsorption in the dark could be different from that under light irradiation. The determination of the mechanism of adsorption and the proportion of adsorption in total PFOA removal over ZnO nanocomposite under light irradiation should be further studied by experiments at constant temperatures over long reaction time to reach the adsorption-desorption equilibrium.
4 Conclusions
The removal of PFOA as one of the most persistent organic pollutants has been studied using ZnO, GCN, and ZnO@CA-modified GCN nanocomposites, under both UV and visible irradiation. ZnO nanoparticles showed promising PFOA removal of up to 64% within 6 h of visible light irradiation, which is significantly better than TiO2 P25 and In2O3 nanoparticles with PFOA elimination up to 10% and 5%, respectively. CA was effectively used to modify g-C3N4 synthesized by thermal condensation at 530 °C for 4 h. Importantly, PFOA removal was not effective by CA-modified GCN but was substantially improved over the ZnO@CA-modified GCN nanocomposites under visible light irradiation, where a remarkable amount of nitrogen vacancy was introduced in the GCN framework. In addition, the optimal hybrid photocatalyst produced by mixing 50 mg of CA-modified GCN with 1 g of ZnO nanoparticles showed a significantly higher efficiency (k = 0.468 h−1) than pure ball-milled ZnO nanoparticles (k = 0.097 h−1) within 2 h of UV irradiation, which could be explained by the formation of a probable S-scheme heterojunction. Furthermore, the addition of 0.53 g L−1 PMS considerably improved PFOA removal under both UV and visible irradiation, confirming the vital role played by sulfate radicals in PFOA removal. In comparing the results between the dark and under light irradiation, it can be concluded that the adsorption of PFOA over ZnO surface played a dominant role, surpassing photodegradation, in PFOA elimination under light irradiation. Therefore, ZnO@CA-modified GCN nanocomposites could effectively facilitate the photocatalytic degradation of PFOA by PMS addition or could function as effective adsorbents for PFOA removal in the absence of light.
Availability of data and materials
The data and materials are available on request.
References
Yamijala SSRKC, Shinde R, Wong BM (2020) Real-time degradation dynamics of hydrated per- and polyfluoroalkyl substances (PFASs) in the presence of excess electrons. Phys Chem Yami Jala Phys 22:6804–6808. https://doi.org/10.1039/C9CP06797C
Zhang Z, Sarkar D, Datta R, Deng Y (2021) Adsorption of perfluorooctanoic acid (PFOA) and perfluorooctanesulfonic acid (PFOS) by aluminum-based drinking water treatment residuals. J Hazard Mater Lett 2:100034. https://doi.org/10.1016/j.hazl.2021.100034
Fu C, Xu X, Zheng C, Liu X, Zhao D, Qiu W (2022) Photocatalysis of aqueous PFOA by common catalysts of In2O3, Ga2O3, TiO2, CeO2 and CdS: influence factors and mechanistic insights. Environ Geochem Health 44:2943–2953. https://doi.org/10.1007/s10653-021-01127-2
Xu B, Ahmed MB, Zhou JL, Altaee A (2020) Visible and UV photocatalysis of aqueous perfluorooctanoic acid by TiO2 and peroxymonosulfate: process kinetics and mechanistic insights. Chemosphere 243:125366. https://doi.org/10.1016/j.chemosphere.2019.125366
Yuan Y, Feng L, He X, Liu X, Xie N, Ai Z, Zhang L, Gong J (2022) Efficient removal of PFOA with an In2O3/persulfate system under solar light via the combined process of surface radicals and photogenerated holes. J Hazard Mater 423:127176. https://doi.org/10.1016/j.jhazmat.2021.127176
Xu B, Zhou JL, Altaee A, Ahmed MB, Johir MAH, Ren J, Li X (2020) Improved photocatalysis of perfluorooctanoic acid in water and wastewater by Ga2O3/UV system assisted by peroxymonosulfate. Chemosphere 239:124722. https://doi.org/10.1016/j.chemosphere.2019.124722
Lopes da Silva F, Laitinen T, Pirilä M, Keiski RL, Ojala S (2017) Photocatalytic degradation of perfluorooctanoic acid (PFOA) from wastewaters by TiO2, In2O3 and Ga2O3 catalysts. Top Catal 60:1345–1358. https://doi.org/10.1007/s11244-017-0819-8
Li Z, Zhang P, Shao T, Wang J, Jin L, Li X (2013) Different nanostructured In2O3 for photocatalytic decomposition of perfluorooctanoic acid (PFOA). J Hazard Mater 260:40–46. https://doi.org/10.1016/j.jhazmat.2013.04.042
Li X, Zhang P, Jin L, Shao T, Li Z, Cao J (2012) Efficient photocatalytic decomposition of perfluorooctanoic acid by indium oxide and its mechanism. Environ Sci Technol 46:5528–5534. https://doi.org/10.1021/es204279u
Li Z, Zhang P, Li J, Shao T, Wang J, Jin L (2014) Synthesis of In2O3 porous nanoplates for photocatalytic decomposition of perfluorooctanoic acid (PFOA). Catal Commun 43:42–46. https://doi.org/10.1016/j.catcom.2013.09.004
Wu D, Li X, Zhang J, Chen W, Lu P, Tang Y, Li L (2018) Efficient PFOA degradation by persulfate-assisted photocatalytic ozonation. Sep Purif Technol 207:255–261. https://doi.org/10.1016/j.seppur.2018.06.059
Navidpour AH, Kalantari Y, Salehi M, Salimijazi HR, Amirnasr M, Rismanchian M, Azarpour Siahkali M (2017) Plasma-sprayed photocatalytic zinc oxide coatings. J Therm Spray Technol 26:717–727. https://doi.org/10.1007/s11666-017-0541-x
Kumar S, Kumar A, Kumar A, Krishnan V (2020) Nanoscale zinc oxide based heterojunctions as visible light active photocatalysts for hydrogen energy and environmental remediation. Catal Rev 62:346–405. https://doi.org/10.1080/01614940.2019.1684649
Saravanan S, Dubey RS, Sarma GVS (2023) Electrochemically deposited ZnO and sol-gel spin-coated Zn-doped TiO2 thin films on FTO: an optical investigation. ES Energy Environ 22:1014. https://doi.org/10.30919/esee1014
Kadam PA, Gadve KM, Gave S, Jadkar eR, Kadam VS, Jagtap CV, (2023) C:ZnO composites for improving catalytic activity of ZnO. ES Energy Environ 21:94. https://doi.org/10.30919/esee946
Shaikh KS, Mujawar AM, Supekar AT, Lokhande PE, Gunjakar JL, Pathan HM (2023) Deposition of nickel doped zinc oxide/titanium oxide films and its applications towards light-harvesting device. ES Energy Environ 22:992. https://doi.org/10.30919/esee992
More PB, Bansode SB, Aleksandrova M, Jadkar S, Pathan HM (2023) Synthesis of ZnO thin films using chemical bath and investigation of physicochemical properties. ES Energy Environ 22:983. https://doi.org/10.30919/esee983
Prabhu NN, Chandra RBJ, Rajendra BV, George G, Mourad A-HI, Shivamurthy B (2022) Electrospun ZnO nanofiber based resistive gas/vapor sensors -a review. Eng Sci 19:59–82. https://doi.org/10.30919/es8d612
Akhundi A, Habibi-Yangjeh A, Abitorabi M, Rahim Pouran S (2019) Review on photocatalytic conversion of carbon dioxide to value-added compounds and renewable fuels by graphitic carbon nitride-based photocatalysts. Catal Rev 61:595–628. https://doi.org/10.1080/01614940.2019.1654224
Xiao Y, Wang Z, Yao B, Chen Y, Chen T, Wang Y (2023) Hollow g-C3N4@Cu0.5In0.5S core-shell S-scheme heterojunction photothermal nanoreactors with broad-spectrum response and enhanced photocatalytic performance. Catalysts 13:723. https://doi.org/10.3390/catal13040723
Das MR, Hussain N, Duarah R, Sharma N, Sarmah P, Thakur A, Bhattacharjee P, Bora U, Boukherroub R (2022) Metal nanoparticles decorated two-dimensional nanosheets as heterogeneous catalysts for coupling reactions. Catal Rev. https://doi.org/10.1080/01614940.2022.2100633
Navidpour AH, Hao D, Li X, Li D, Huang Z, Zhou JL (2023) Key factors in improving the synthesis and properties of visible-light activated g-C3N4 for photocatalytic hydrogen production and organic pollutant decomposition. Catal Rev. https://doi.org/10.1080/01614940.2023.2228130
Alaghmandfard A, Ghandi K (2022) A comprehensive review of graphitic carbon nitride (g-C3N4)–metal oxide-based nanocomposites: potential for photocatalysis and sensing. Nanomaterials 12:294. https://doi.org/10.3390/nano12020294
Zhou Y, Zhang L, Huang W, Kong Q, Fan X, Wang M, Shi J (2016) N-doped graphitic carbon-incorporated g-C3N4 for remarkably enhanced photocatalytic H2 evolution under visible light. Carbon 99:111–117. https://doi.org/10.1016/j.carbon.2015.12.008
Zhu D, Zhou Q (2021) Nitrogen doped g-C3N4 with the extremely narrow band gap for excellent photocatalytic activities under visible light. Appl Catal B: Environ 281:119474. https://doi.org/10.1016/j.apcatb.2020.119474
Navidpour AH, Safaei J, Zhang G, Mojiri A, Ni B-J, Huang Z, Zhou J (2023) Photocatalytic and photoelectrocatalytic degradation of perfluorooctanoic acid by immobilised ZnO nanoparticles using electrophoretic deposition. Environ Sci Nano 10:1955–1965. https://doi.org/10.1039/D3EN00241A
Lahootifar Z, Habibi-Yangjeh A, Rahim Pouran S, Khataee A (2023) G-C3N4 dots decorated with hetaerolite: visible-light photocatalyst for degradation of organic contaminants. Catalysts 13:346. https://doi.org/10.3390/catal13020346
Fao GD, Catherine HN, Huang C-H, Lee Y-L, Jiang J-C, Hu C (2023) Unraveling the effects of P and S doping over g-C3N4 in strengthening Lewis basicity for CO2/glycerol conversion: a theoretical and experimental study. Carbon 201:129–140. https://doi.org/10.1016/j.carbon.2022.09.011
Shen L, Xing Z, Zou J, Li Z, Wu X, Zhang Y, Zhu Q, Yang S, Zhou W (2017) Black TiO2 nanobelts/g-C3N4 nanosheets laminated heterojunctions with efficient visible-light-driven photocatalytic performance. Sci Rep 7:41978. https://doi.org/10.1038/srep41978
Paul DR, Sharma R, Nehra SP, Sharma A (2019) Effect of calcination temperature, pH and catalyst loading on photodegradation efficiency of urea derived graphitic carbon nitride towards methylene blue dye solution. RSC Adv 9:15381–15391. https://doi.org/10.1039/C9RA02201E
Chen L-Y, Zhang W-D (2014) In2O3/g-C3N4 composite photocatalysts with enhanced visible light driven activity. Appl Surf Sci 301:428–435. https://doi.org/10.1016/j.apsusc.2014.02.093
Zhang C, Liu J, Huang X, Chen D, Xu S (2019) Multistage polymerization design for g-C3N4 nanosheets with enhanced photocatalytic activity by modifying the polymerization process of melamine. ACS Omega 4:17148–17159. https://doi.org/10.1021/acsomega.9b01510
Mo Z, She X, Li Y, Liu L, Huang L, Chen Z, Zhang Q, Xu H, Li H (2015) Synthesis of g-C3N4 at different temperatures for superior visible/UV photocatalytic performance and photoelectrochemical sensing of MB solution. RSC Adv 5:101552–101562. https://doi.org/10.1039/c5ra19586a
Martha S, Nashim A, Parida KM (2013) Facile synthesis of highly active g-C3N4 for efficient hydrogen production under visible light. J Mater Chem A 1:7816–7824. https://doi.org/10.1039/C3TA10851A
Li K, Chen M, Chen L, Zhao S, Xue W, Han Z, Han Y (2023) Synthesis of g-C3N4 derived from different precursors for photodegradation of sulfamethazine under visible light. Processes 11:528. https://doi.org/10.3390/pr11020528
Xia P, Li G, Li X, Yuan S, Wang K, Huang D, Ji Y, Dong Y, Wu X, Zhu L, He W, Qiao L (2022) Synthesis of g-C3N4 from various precursors for photocatalytic H2 evolution under the visible light. Crystals 12:1719. https://doi.org/10.3390/cryst12121719
Wu M, Yan J-M, Tang X-n, Zhao M, Jiang Q (2014) Synthesis of potassium-modified graphitic carbon nitride with high photocatalytic activity for hydrogen evolution. Chemsuschem 7:2654–2658. https://doi.org/10.1002/cssc.201402180
Gong S, Jiang Z, Zhu S, Fan J, Xu Q, Min Y (2018) The synthesis of graphene-TiO2/g-C3N4 super-thin heterojunctions with enhanced visible-light photocatalytic activities. J Nanopart Res 20:310. https://doi.org/10.1007/s11051-018-4399-8
Jiang Y, Lin Z, Zhang Y, Lai Y, Liang D, Yang C (2020) Facile synthesis of porous C-doped C3N4: fast charge separation and enhanced photocatalytic hydrogen evolution. New J Chem 44:17891–17898. https://doi.org/10.1039/D0NJ04169F
Chuang P-K, Wu K-H, Yeh T-F, Teng H (2016) Extending the π-conjugation of g-C3N4 by incorporating aromatic carbon for photocatalytic H2 evolution from aqueous solution. ACS Sustain Chem Eng 4:5989–5997. https://doi.org/10.1021/acssuschemeng.6b01266
Wang Y, Shi R, Lin J, Zhu Y (2011) Enhancement of photocurrent and photocatalytic activity of ZnO hybridized with graphite-like C3N4. Energy Environ Sci 4:2922–2929. https://doi.org/10.1039/C0EE00825G
Chen Q, Hou H, Zhang D, Hu S, Min T, Liu B, Yang C, Pu W, Hu J, Yang J (2018) Enhanced visible-light driven photocatalytic activity of hybrid ZnO/g-C3N4 by high performance ball milling. J Photochem Photobiol A: Chem 350:1–9. https://doi.org/10.1016/j.jphotochem.2017.09.015
Zhang S, Su C, Ren H, Li M, Zhu L, Ge S, Wang M, Zhang Z, Li L, Cao X (2019) In-situ fabrication of g-C3N4/ZnO nanocomposites for photocatalytic degradation of methylene blue: synthesis procedure does matter. Nanomaterials 9:215. https://doi.org/10.3390/nano9020215
Nagaraju G, Udayabhanu S, Prashanth SA, Shastri M, Yathish KV, Anupama C, Rangappa D (2017) Electrochemical heavy metal detection, photocatalytic, photoluminescence, biodiesel production and antibacterial activities of Ag–ZnO nanomaterial. Mater Res Bull 94:54–63. https://doi.org/10.1016/j.materresbull.2017.05.043
Zhang H, Sun J, Liu C, Wang Y (2015) Distinct water activation on polar/non-polar facets of ZnO nanoparticles. J Catal 331:57–62. https://doi.org/10.1016/j.jcat.2015.08.016
Paul DR, Gautam S, Panchal P, Nehra SP, Choudhary P, Sharma A (2020) ZnO-modified g-C3N4: a potential photocatalyst for environmental application. ACS Omega 5:3828–3838. https://doi.org/10.1021/acsomega.9b02688
Lee Y-J, Jeong YJ, Cho IS, Park S-J, Lee C-G, Alvarez PJJ (2023) Facile synthesis of N vacancy g-C3N4 using Mg-induced defect on the amine groups for enhanced photocatalytic •OH generation. J Hazard Mater 449:131046. https://doi.org/10.1016/j.jhazmat.2023.131046
Gayathri M, Sakar M, Satheeshkumar E, Sundaravadivel E (2022) Insights into the mechanism of ZnO/g–C3N4 nanocomposites toward photocatalytic degradation of multiple organic dyes. J Mater Sci Mater Electron 33:9347–9357. https://doi.org/10.1007/s10854-021-07302-6
Javed M, Qamar MA, Shahid S, Alsaab HO, Asif S (2021) Highly efficient visible light active Cu–ZnO/S-g-C3N4 nanocomposites for efficient photocatalytic degradation of organic pollutants. RSC Adv 11:37254–37267. https://doi.org/10.1039/D1RA07203J
Kalisamy P, Lallimathi M, Suryamathi M, Palanivel B, Venkatachalam M (2020) ZnO-embedded S-doped g-C3N4 heterojunction: mediator-free Z-scheme mechanism for enhanced charge separation and photocatalytic degradation. RSC Adv 10:28365–28375. https://doi.org/10.1039/D0RA04642F
Olmez-Hanci T, Arslan-Alaton I (2013) Comparison of sulfate and hydroxyl radical based advanced oxidation of phenol. Chem Eng J 224:10–16. https://doi.org/10.1016/j.cej.2012.11.007
Zhu K, Wang J, Wang Y, Jin C, Ganeshraja AS (2016) Visible-light-induced photocatalysis and peroxymonosulfate activation over ZnFe2O4 fine nanoparticles for degradation of Orange II. Catal Sci Technol 6:2296–2304. https://doi.org/10.1039/C5CY01735A
He X, Sun B, He M, Chi H, Wang Z, Zhang W, Ma J (2020) Highly efficient simultaneous catalytic degradation and defluorination of perfluorooctanoic acid by the H2O2-carbon/MnO2 system generating O2- and OH synchronously. Appl Catal B: Environ 277:119219. https://doi.org/10.1016/j.apcatb.2020.119219
Chen M-J, Lo S-L, Lee Y-C, Huang C-C (2015) Photocatalytic decomposition of perfluorooctanoic acid by transition-metal modified titanium dioxide. J Hazard Mater 288:168–175. https://doi.org/10.1016/j.jhazmat.2015.02.004
Li F, Wei Z, He K, Blaney L, Cheng X, Xu T, Liu W, Zhao D (2020) A concentrate-and-destroy technique for degradation of perfluorooctanoic acid in water using a new adsorptive photocatalyst. Water Res 185:116219. https://doi.org/10.1016/j.watres.2020.116219
Wang S, Yang Q, Chen F, Sun J, Luo K, Yao F, Wang X, Wang D, Li X, Zeng G (2017) Photocatalytic degradation of perfluorooctanoic acid and perfluorooctane sulfonate in water: a critical review. Chem Eng J 328:927–942. https://doi.org/10.1016/j.cej.2017.07.076
Barisci S, Suri R (2020) Electrooxidation of short and long chain perfluorocarboxylic acids using boron doped diamond electrodes. Chemosphere 243:125349. https://doi.org/10.1016/j.chemosphere.2019.125349
Guo X, Duan J, Li C, Zhang Z, Wang W (2020) Highly efficient Z-scheme g-C3N4/ZnO photocatalysts constructed by co-melting-recrystallizing mixed precursors for wastewater treatment. J Mater Sci 55:2018–2031. https://doi.org/10.1007/s10853-019-04097-0
Zhang C, Jia M, Xu Z, Xiong W, Yang Z, Cao J, Peng H, Xu H, Xiang Y, Jing Y (2022) Constructing 2D/2D N-ZnO/g-C3N4 S-scheme heterojunction: efficient photocatalytic performance for norfloxacin degradation. Chem Eng J 430:132652. https://doi.org/10.1016/j.cej.2021.132652
Sayed M, Zhu B, Kuang P, Liu X, Cheng B, Ghamdi AAA, Wageh S, Zhang L, Yu J (2022) EPR investigation on electron transfer of 2D/3D g-C3N4/ZnO S-scheme heterojunction for enhanced CO2 photoreduction. Adv Sustain Syst 6:2100264. https://doi.org/10.1002/adsu.202100264
Liu B, Bie C, Zhang Y, Wang L, Li Y, Yu J (2021) Hierarchically porous ZnO/g-C3N4 S-scheme heterojunction photocatalyst for efficient H2O2 production. Langmuir 37:14114–14124. https://doi.org/10.1021/acs.langmuir.1c02360
Lee J-T, Lee S-W, Wey M-Y (2022) S-scheme g-C3N4/ZnO heterojunction photocatalyst with enhanced photodegradation of azo dye. J Taiwan Inst Chem Eng 134:104357. https://doi.org/10.1016/j.jtice.2022.104357
Li X, Kang B, Dong F, Zhang Z, Luo X, Han L, Huang J, Feng Z, Chen Z, Xu J, Peng B, Wang ZL (2021) Enhanced photocatalytic degradation and H2/H2O2 production performance of S-pCN/WO2.72 S-scheme heterojunction with appropriate surface oxygen vacancies. Nano Energy 81:105671. https://doi.org/10.1016/j.nanoen.2020.105671
Wu D, Li X, Tang Y, Lu P, Chen W, Xu X, Li L (2017) Mechanism insight of PFOA degradation by ZnO assisted-photocatalytic ozonation: efficiency and intermediates. Chemosphere 180:247–252. https://doi.org/10.1016/j.chemosphere.2017.03.127
Zhang L, Ma P, Dai L, Bu Z, Li X, Yu W, Cao Y, Guan J (2022) Removal of pollutants via synergy of adsorption and photocatalysis over MXene-based nanocomposites. Chem Eng J Adv 10:100285. https://doi.org/10.1016/j.ceja.2022.100285
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions This work was supported by the University of Technology Sydney (UTS President’s and International Scholarships).
Author information
Authors and Affiliations
Contributions
AHN: investigation, writing-original draft, writing-review and editing. JS: experiments. MAHJ: research support. BJN: writing-review and editing. AD: experiments. XL: writing-review and editing. JLZ: supervision, writing-review and editing.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Navidpour, A.H., Safaei, J., Johir, M.A.H. et al. Zinc oxide@citric acid-modified graphitic carbon nitride nanocomposites for adsorption and photocatalytic degradation of perfluorooctanoic acid. Adv Compos Hybrid Mater 7, 53 (2024). https://doi.org/10.1007/s42114-024-00867-w
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s42114-024-00867-w