
Abstract
In the FFC-Cambridge process, the cathodic dissociation of oxide and CO/CO2 production on carbon anode is the basis for metal production in a CaCl2 bath. Using an inert intermetallic anode, the CO2 evolution can be eliminated altogether with acceleration in the electro-reduction kinetics. In the presence of a carbon anode, the cathodic dissociation of TiO2 suffers from slow reduction kinetics of TiO2 to Ti metal, which can be enhanced significantly by the incorporation of alkali species in the TiO2 pellet at the cathode and in the CaCl2 bath in the presence of an intermetallic inert anode. With inert anode and incorporation of K+-ion in the TiO2 matrix and in the salt bath, nearly full metallization with greater than 99% of Ti metal containing 1500 ppm of oxygen was possible to achieve in less than 16 h of electro-reduction. The microstructural and chemical analysis of the metallic phase and its morphology revealed the presence of a layer of titanium metal that forms in the fast reduction reaction step in less than 5 h, after which the reaction rate slows down significantly before terminating in 16 h. The investigation showed that two different types of microstructures of metallic titanium were evident—a thin sheet-like material on the outer periphery of the reduced pellet and the dendritic core which was found to be under the peripheral sheet of the metallic layer. The mechanism of morphological and microstructural changes in the reduced form of titanium metal is explained.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
Production of metals and alloys without greenhouse gas (GHG) emission has become essential to for meeting the United Nations Climate Change for achieving net zero target by 20501. Amongst metal producers, the iron and steel and aluminium industries are some of the largest consumers of fossil-fuel based energy, which contributes to GHG emission producers of CO2.According to the 2019 statistics, total production of crude steel was 1869 million tonnes(t), and the net energy consumption was 21–23 GJ/t reflecting 1.2 t and 1.0t of CO2 for direct and indirect emissions, respectively, for each tonne of crude steel produced2. By comparison, the carbon footprint for aluminium production varies between 3.5 and 4.2 t of CO2 for each tonne of cast aluminium. For metallurgical grade silicon, the figure for tonnage carbon emission is 6.3t3. The FFC-Cambridge process, developed in 2000, was a breakthrough in reactive metal processing, which raised awareness whether the Kroll process via chloride: \({\text{MCl}}_{{4}} \;{ + }\;{\text{2Mg}}\;{ = }\;{\text{2MgCl}}_{{2}} \;{ + }\;{\text{M}}\), might be the only course for future of reactive metal processing. Here M defines a reactive metal such as titanium. The FFC process uses high purity metal oxide for cathodic dissociation—a basis for sustainable route to reactive metal production4. In FFC, however, carbon anode is used for removing oxygen which produces CO2 at the molten salt temperature.
In the FFC process, the cathodic dissociation of TiO2 in a CaCl2 bath with carbon anode leads to the evolution of CO2, which is expected, as explained in the FFC-Cambridge process4. However, the cyclic voltammetric5,6 and calcium thermic5 have enabled the characterization of slow reaction kinetics due to the formation of interfacial CaTiO3 phase as a chemical barrier for the metallization at cathode4,5,6,7,8. The complementary interfacial reactions are shown below arising from the dissociation of TiO2 and due to the residual solubility of O2− ions in the CaCl2 liquid.
Note that the reactions (1a)–(1f) are interconnected for ionic transport and electronic charge balance in the overall electro-reduction of TiO2. The O2− ions, when discharge at the carbon cathode, produce CO2 via reaction (1f), and the electrons liberated reduce the Ti4+ to Ti metal, as shown in reaction (1d). In this investigation the use of inert metallic anode is discussed below which eliminates the CO2 evolution at anode and yields pure oxygen via reaction (1g). The production of commercial grade titanium metal powder and pure oxygen is, therefore, far more attractive than what has been demonstrated in the literature4,5,6,7,8,9 using carbon anode.
The main motivation for this article is to demonstrate a laboratory-scale proof-of-principle of the production of metallic titanium and commercial purity oxygen generation for a range of applications, including further purification of evolved oxygen for hospital use. In this article, we have also analysed the microscopic reason for the formation of calcium titanate (CaTiO3) perovskite structure, in which the O2− ion lattice diffusion is known to be slow (1.323 × 10–6—4.033 × 10–5 cm2s−1) at 900 °C 9. By comparison, the value of the atomic diffusion of oxygen in β-titanium alloy is of the order of (~ 10−6cm2s−1)10 which forms as an intermediate phase before the metallization with ultra-low oxygen in Ti metal is achieved at the cathode. The report on the formation of alkaline earth perovskite (CaTiO3) is significant because the presence of alkaline earth perovskites on the surface of partially reduced pellets acts as an insulating barrier for the O2− charge transport into the CaCl2 bath, which is known to have a limited solubility of O2−ions6,7,8 at the partially reduced oxide and perovskite interface. Also, the perovskite phase is an insulator which means during the cathodic dissociation (reaction 1d), the electronic transport is impeded and leads to slowing down of the overall metal reduction process. In previous investigations7, the porosity of the pellets of TiO2 was increased by incorporating fugitive agent polyethylene, which were burnt before commencing the reaction, so that a large surface area were possible to maintain for a uniform rapid electroreduction, when compared with the pellets of TiO2 without polyethylene. The investigation7 concluded that when the potential at 3.0 V (below the decomposition potential of CaCl2) was applied the minimum level of oxygen achieved was approximately 3000 ppm after 48 h using a carbon anode. Furthermore, when the potential was raised from 3 to 3.15 V the time to achieve the residual oxygen concentrations at 3000 ppm in the reduced pellet was halved from 48 to 24 h. The experimental evidence, shown in the literature, confirm although the speed of reaction increases, a complete or near complete metallization is not possible by enhancing the in-situ porosity alone, as the insulating perovskite was thermodynamically more stable under the cathodic dissociation condition, due to the difference in the Gibbs energies for the formation of (− 821 kJ per mole of CaTiO3 versus − 733 kJ per mole of TiO2). Table 1 below summarizes the Gibbs energy ((ΔGo, kJ mol−1) change11 for the molecular form of the chemical reactions which may be relevant in explaining the perovskite stability in the absence and presence of K+-ions in the salt bath present at the interface between the pressed pellets of TiO2 and CaCl2-KCl. In this article, the cathodic dissociation of TiO2 in the presence of K+-ion in the salt mixture and in the pellet has been characterized. At the end of this article, we also compare and give a critical summary of other electro-reduction techniques used in the lab for reactive metal production.
The role of alkali (Na, K) ions on the chemical breakdown of crystalline structures of ferruginous ilmenite (FeTiO3), tantalite, and chromite minerals above 700 °C in oxidizing and reducing conditions have been extensively studied recently12,13,14,15,16 in the context of the selective separation of metal oxides as water-soluble and insoluble products. In these investigations, the evidence for the formation of alkali-rich titanate liquid and lattice strain-induced fracture in situ of crystalline alkali complexes (e.g. titanate and chromite) of above 750 °C have also been explained and reported15,16,17. Both the evidence for lattice-induced cracking and formation of alkali-titanate based liquid18 during chemical reactions of ferruginous mineral concentrate with alkali are relevant in the context of cathodic dissociation of TiO2 and promoting decomposition of CaTiO3 in situ. The thermodynamic basis for selecting the incorporation of LiCl in the CaCl2 salt bath and K+-ions in the TiO2 pellet at the outset of electroreduction process is demonstrated via the chemical reactions 2c––2d in Table 1, from which it is evident that at high temperatures (e.g. 900 °C), both the lime and calcium perovskite are unstable with respect to the thermodynamic stabilities of CaCl2 and K2TiO3 in the molten salt mixture, respectively. Also, note that in Table in reaction 2e that Li2O + KCl is more stable than LiCl + K2O.The K2O-TiO2 phase diagram also confirms the evidence for the presence of alkali-titanate liquid above 768 °C in the K2TiO3 phase composition range19
Based on the literature on alkali-titanate complexes12,13,14,15,16,17, the molecular representation of the analysis of thermodynamic equilibrium reactions in Table 1, which correspond to the cathodic and anodic reactions (1a–1f), may help in describing the overall electro-reduction of TiO2, from which the resulting microstructure and composition of titanium metal may be explained. Experimental evidences from previous studies on carbon anode show that the current decreases gradually, and finally stabilizes at a low value between 0.2 and 0.4 A, which has been attributed due to the presence of an insulating perovskite barrier. On the other hand the thermodynamic equilibrium conditions for reactions, shown in Table 1, show that the when alkali ions (Li+, K+) are present, these ions enhance the electro-reduction the TiO2 pellets at cathode both with and without the inert anodes. In this investigation the effect of mixed K+/Li+ ions in breaking down the perovskite phase, which may be decomposed under the favourable thermodynamic alkali-cation exchange condition. The data for potassium ion exchange reactions are also presented for comparison with the lithium ions only in reactions 2c–2e in Table 1. We also show for the first time, a comparative study on the use of an intermetallic anode with carbon anode for Ti-metal production using the FFC-Cambridge process. Nearly full metallization was achieved with the use of inert anode and the reaction mechanism is explained with the help of phase stability analysis based on thermodynamic equilibrium and microstructural evidence.
2 Experimental
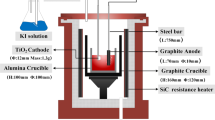
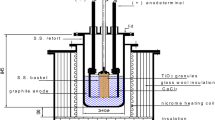
The electrolysis experiments were performed in a molten salt mixture of CaCl2-LiCl (180gms CaCl2 and 20gms LiCl) inside an alumina crucible. The chloride salts were initially dried for 24 h at 320 °C by removing moisture, followed by slowly heating it to 900 °C at a rate of 1 °C min−1 in a flowing atmosphere of argon, maintained at a rate of 500 ml min−1. We used two different types of anodes: a carbon anode for comparison, followed by replacement of carbon anode with an intermetallic Al-Ti-Cu anode20 for enhanced current carrying capacity. The oxide pellet (1 g) at the cathode was prepared by mixing 99.95% pure TiO2 with analytical grade (99.9% pure) KHCO3 in a molar ratio of 1:0.5 so that at the electro-reduction temperature of 900 °C, so that the reaction between TiO2 and the decomposed KHCO3, which yields K2TiO3 and K4TiO4 which then forms a liquid phase, as shown in the isothermal ternary section19. Both the cathode and the anode were fixed on to a steel holder and fastened using a molybdenum wire, ensuring a good electrical contact. Once the mixture of 90 wt%CaCl2–10 wt%LiCl was molten, the electrodes were lowered down into the salt bath. The cell was continuously maintained with a flow of argon gas atmosphere. A constant voltage of 3.1 V was applied through the electrodes and the current was continuously monitored using a computer-controlled software. After 15 h of electrolysis, the electrodes were extracted from the salt bath and the reduced pellet at the cathode was thoroughly washed using water. The reduced pellets were analysed for the presence of phases and the resulting microstructural changes were examined using a Cambridge Camscan 4 FEG SEM, which was operated at 20 kV. The SEM was equipped with an energy dispersive X-ray spectrometer (EDS Link Oxford). Wherever required, the phase identification was also carried out using the Philips X-ray diffractometer using CuKα radiation diffraction, which was necessary for determining the reaction mechanism. During electro-reduction of TiO2, the partially reduced pellets were also removed after different time intervals between 0.5 h and 4 h of reaction at 900 °C for the verification of phases formed during complete metallization. The results of X-ray powder diffraction, SEM, and EDX analyses are discussed for explaining the reaction mechanism of electro-reduction of TiO2 to metallic titanium.
3 Results and Discussion
The current–time plots for the electro-reduction of TiO2 with carbon and inert anodes are compared in Fig. 1a and b, respectively. It is evident that in case of each anode, there is a fast transient before 5 h after which the rate of either rise or fall in current slows down significantly. Note that the rate of change in current with time in the electro-reduction experiments with inert anode differs significantly from that observed for carbon anodes, when both the anodes were used for the electro-reduction of TiO2:K2O = 1:0.5 mixed pellet. Since the cell potential is fixed at 3.1 V, the trends in the rise and fall in current with time represent the decreasing and increasing cell resistance, respectively. The reason for the decreasing current in carbon anode, as reported previously, is due to the formation of the dominant perovskite (CaTiO3) phase6,7,8. By comparison, the rising current in case of inert anode is due to increased metallization via the electro-reduction of TiO2 which increases the overall conductivity with time. It was also observed that when the TiO2 was not mixed with K+ -ions in the TiO2 pellet, the formation of perovskite was observed with both types of anodes, leading to almost cessation of the overall electro-reduction.

The current–time diagram for the electro-reduction of TiO2 pellet via cathodic dissociation of TiO2 mixed with K2O (as KHCO3) in a ratio of TiO2:K2O = 1:0.5. a with inert metallic anode; b with carbon anode. Temperature of electro-reduction was 900 °C in KCl-LiCl (9:1 weight ratio) molten salt bath. Atmosphere of argon gas was maintained at a rate of 500 ml/min.
For analysing the mechanism of the electro-reduction reaction and its distinct characteristics of rising current with time, the phase changes with time were characterised using the X-ray powder diffraction technique. The partially reduced samples were extracted at 0.5, 1, and 4 h of reduction and washed and dried before X-ray diffraction. In Fig. 2, the diffraction pattern of the partially reduced pellets at 0.5, 1, and 4 h of time intervals are compared, from which the diminishing diffraction intensities of oxides of titanium (Ti3O5, TiO), perovskite, and CaTi2O4 phases confirm that in the presence of K+-ion in the pellet the reducibility of TiO2 increases significantly. Although the Ti-metal starts forming from a time interval of 0.5 h during the electro-reduction, the phase intensities of perovskite and CaTi2O4 diminish rapidly in 4 h of reduction. The metallisation of Ti metal in the CaCl2-LiCl bath follows the thermodynamic equilibrium condition which we have verified using the predominance area diagram in the Ti-Ca-O-Cl diagram in Fig. 3. No potassium complexes or salts were observed in the diffraction data, which may be explained because of high solubility of alkali and CaCl2 in water.

X-ray diffraction pattern of TiO2 pellet electrolysed for different time intervals using inert anode and TiO2:K2O = 1:0.5. Temperature: 900 °C. Atmosphere of electro-reduction argon gas maintained at 0.5 l min−1.

A phase predominance area diagram constituting phases in the Ti–Ca–O–Cl system at 900 °C19.
The phases identified during the course of electro-reduction were found to be in good agreement with the predicted equilibrium phase composition, as shown in Fig. 3. It is evident from the equilibrium analysis that the electro-reduction bath after 1 h of reaction must be reaching oxygen potential below log10 (PO2) = − 26 atm, at which the Magnelli oxides of Ti (TinO2n−1) start complexing with CaO by forming, say CaTi2O4 spinel-like phase in equilibrium with Ti3O5. From the phase analysis, it is also evident that the formation TiO concurs with the Ti–O interstitial solid-solution. The reference red lines in Fig. 2 for TiO and Ti–O solid solutions are drawn between 2θ = 37.5° and 43.8°, in which range the strongest diffraction peaks of these two phases are present. By considering the vertical red lines in Fig. 2 as a reference for the Ti–O and TiO phases, the apparent shifts in the peak positions of these two phases with time for electro-reduction suggest that the resulting change in the d-spacing (or the lattice dimensions) of TiO and Ti–O solid-solution phases which form as intermediate phases during the electro-reduction of TiO2.
Note that during electrolysis, the potassium bicarbonate (KHCO3) mixed with TiO2 pellet decomposes to K2O via \({\text{2KHCO}}_{{3}} {\text{ = K}}_{{2}} {\text{O + H}}_{{2}} {\text{O + 2CO}}_{{2}}\). The decomposition reaction occurs rapidly and combines with the TiO2 by forming potassium titanate complexes, as shown in Eqs. (3a) and (3b). Equation (3c) shows the anion-cation exchange reactions. The Gibbs energy change for these reactions are large and negative at 900 °C.
The potassium titanate, formed in situ, melts at 900 °C and breaks down the perovskite barrier and allows a greater surface area for electro-reduction. This means that the liquid rich in K4TiO4 becomes the main source of the O2− anion transport which is mediated by the presence of reciprocal salt mixture, shown in reaction 3c. This is possible because under the applied potential of 3.1 V, the K4TiO4 liquid continues to dissociate and generate K2O which then follows equilibrium conditions in Eqs. (3a) and (3b). Under these thermodynamic conditions, the perovskite phase does remain stable, and decomposes readily which is why the electro-reduction reaction in the first 5 h accelerates for both types of anodes.
As shown in Fig. 4a, the dense metallic microstructure formed as a result of the initial stage of electro-reduction is a consequence of the presence of complex titanate liquid. Once the dense metallic layer forms, there is a volumetric change resulting in the shrinkage which appears to form discreet pores on the peripheral surface resulting. Since the weight ratio of K2O:TiO2 is 1:2, the overall reaction suffers from the paucity of K+-rich liquid inside the pores of unreduced TiO2 and its oxides. The resulting reaction slows down after 5 h with each type of anode and then progresses at much reduced rate, as shown in Fig. 1a and b.
In Fig. 5a the high magnification image of the dendritic region in Fig. 4a is shown and compared. The dendritic structure of titanium metal is evident and also dominated by metallic titanium. There is little evidence for the presence of any perovskite phase in Figs. 4a, b and 5a, b.

a The scanning electron microscopic image (see 10 µm) with dendritic and dense layer of Ti metal in a TiO2:K2O = 1:0.5 mixed pellet. b The energy dispersive X-ray spectrum of the analysed area in the region identified with the red arrow.

The microstructure of the fully metallized TiO2 cathode, a showing clearly the dendritic and solidified Ti metal morphologies in a TiO2:K2O = 1:0.5 mixed pellet, and b EDX spectrum of the solidified region shows traces of oxygen, calcium, chlorine, and aluminium (from alumina crucible).
The effect of increased K2O:TiO2 = 1:1 ratio on the reduction is shown by presenting the microstructural evidence in Fig. 6 in which the formation of dense outer layer is evident and this layer seems more continuous around the peripheral surface than that in Fig. 4. Since the Gibbs energy changes for (3a)–(3c) reactions at 900 °C are large and negative, which suggest that in the presence of freshly formed K2O from the decomposition of KHCO3, the perovskite surrounding the unreacted TiO2 is removed by the formation of potassium titanate (K4TiO4)-rich liquid via reaction (3a). The solid K2TiO3 may form via reaction (3b) only below 500 °C19, as it decomposes to form K4TiO4 and K8Ti5O14 phase mixture. It should be noted that since the Gibbs energy change for the formation of K2TiO3 is much smaller than that for K4TiO4, which is why it is the liquid rich in K4TiO4 is likely to be more prevalent than K2TiO3.

a Micrograph of electro-reduced TiO2:KHCO3 = 1:1 pellet at 900 °C in a CaCl2-LiCl bath. Note the difference in the sheet-like and dendritic Ti metal, surrounded by pores. b Isothermal ternary section at 800 °C K-Ti–O (oxygen rich section) showing the evidence for the presence of K4TiO4 liquid in equilibrium with other potassium titanate phases, using the FACT Sage 6.0 software19.

Elemental maps of the cross section of a partially reduced TiO2 pellet (TiO2:K2O = 1:0.2) with inert metallic anode at 900 °C. The right-hand side of the micrograph shows the interfacial microstructure of TiO2 pellet in contact with CaCl2. The topochemical progression of electro-reduction is apparent from the cross-section examination.

A comparison of change in mass (Δm,g) with time (h, hour) during electro-reduction of TiO2 present in TiO2:K2O = 1:0.5 mixed powder mixture pressed into a pellet. Reaction under consideration for mass lost was: TiO2 = Ti + 2O2 + 4e−.
The presence of porosity, which may arise as a result of the solidified microstructure during the phase equilibrium condition of electro-reduction with simultaneous evolution of CO2 from the decomposition of KHCO3, may be responsible for maintaining the porosity on the surface of the pellet. It is these microscopic pathways through which the CaCl2 liquid may reach the inner core of unreduced pellet for removing O2− ion for maintaining the steady state of ion transport during the electro-reduction process. On the other hand, where the CaCl2 liquid remains in contact with more impervious metallic Ti, the interstitial diffusion of atomic oxygen might be the viable transport for the O2− ions transport: \(\left[ {O^{2 - } } \right] \leftrightarrow \left[ O \right]_{{{\text{atomic}}}} + 2e\) either across the metallic layer or through the interdendritic pores of titanium metal. This is likely because the values of the atomic diffusivities of oxygen in α and β forms of Ti metal are of the order of \(\sim\) 1 × 10–6 cm2 s−1, when compared with the 1.323 × 10–6–4.033 × 10–5 cm2s−1 for O2− anion diffusion in CaTiO39,10 under high and low partial pressures of O2. Since at a later stage of electro-reduction, the overall oxygen potential of the cell reduces, when compared at the start of the electro-reduction, the apparent diffusivity O2− anion also increases by three times at 900 °C9. The slow atomic and anionic diffusion in metallic Ti and perovskite phase, respectively, may become the rate governing step in achieving near completion of electro-reduction for near 100% metallization. A clear transition in the morphological distinction between the sheet-like and dendritic titanium metal may be the result of K2O-rich liquid assisted anion transport between time, t = 0 and t = 5 h on the surface of the pellet at the start of the electro-reduction. The surface reaction is then taken over by the anion transport in combination with interstitial diffusion of atomic oxygen in oxygenated β-Ti at later stage beyond 5 h of electroreduction. Direct evidence for the presence of oxygenated β-Ti may be confirmed by the presence oxygen peak in the EDX spectrum in Fig. 5b. Based on the anionic and atomic diffusivity data, given above, the estimated thickness of metallic layer growth may be of the order of 60–70 μm h−1 of the reduced pellet when using the inert anode. For comparing the effect of current on the reaction mechanism, a carbon anode was also used during electrolysis in the presence of KHCO3 in the pellet, as shown in Fig. 1b. However, only partial metallization was achieved in this case, which is consistent with the experimental observations made by Schwandt et al. 20 and Alexander et al. 21. In this study, we show that the low current is a manifestation of much slower metallization of TiO2 in spite of the use of TiO2 mixed with KHCO3. To support this point of view, in Fig. 7 we show the elemental maps of a cross-section of partially reduced TiO2 sample which contained TiO2:KHCO3 = 1:0.2. Although high current was observed at the start, as shown in Fig. 1b, the formation metallic layer was only confined to 500 μm. We also confirmed the formation of KCl via reaction (3c), which also supports other accompanying reactions in 3a and 3b, for example. It should be mentioned that no K2TiO3 was found in the partially reduced sample.
Using the data in the current–time diagrams in Fig. 1a and b, the rate of mass change (Δm,g) has been plotted against time duration for electro-reduction. The change in mass was estimated using the Faraday’s law. Based on the computed mass from the current–time plots, the apparent rates of reduction for the inert and carbon anodes were estimated and the derived data from the linear regression analysis (R2 ≥ 0.998) in Fig. 8 clearly show that the overall kinetic constant for electroreduction is much slower for carbon than that for the inert anode. The comparison of kinetic data derived from Fig. 1a and b and the evidence from the X-ray diffraction and SEM analyses help in deucing that the inert anode with potassium salt in electrolyte and pellet may be better choice for molten salt based electro-reduction of TiO2. Before we conclude this article, we compare the above-mentioned process of Ti production with other electrolysis methods in Table 2, in which different methods of TiO2 reduction are presented briefly with references.
4 Conclusions
When the inert anode was used for electro-reduction of pressed pellets of TiO2:K2O = 1:0.5 mixture at 900 °C, nearly 100% metallization of the TiO2 to metallic Ti was achieved in less than 16 h of electrolysis. The experimental evidence shows that more than 75% reduction was achieved within first 5 h when the decomposed KHCO3 yielded K2O by aiding the formation of K4TiO4-rich liquid at 900 °C. The results of phase and microstructural examination also demonstrate that the intermediate CaTiO3 phase decomposes under electro-reduction condition by exchanging anions via reactions (3a) and (3c), which then sustain the cathodic dissociation of TiO2 present. The presence of K4TiO4-rich liquid appears to be responsible for the formation of thick continuous layer of Ti-metal on the peripheral surface pressed pellets. Once the K4TiO4-rich liquid has exhausted, the remaining oxide proceeds via much slower combination of O2− ion diffusion in perovskite and atomic diffusion of oxygen in the β-Ti structure. There is also microanalytical evidence for the presence of larger concentrations of residual oxygen in the dendritic Ti-metal structure when compared with dense Ti-metal layer formed on the surface of the electro-reduced pellet. From the isothermal electro-reduction data, the rate constant was determined from the mass change versus time curves for the inert and carbon anode reduced pellets. It was found that the rate constant for carbon anode (kcarbon = 0.003 g h−1) was nearly an order of magnitude smaller in magnitude than that observed for inert anode (kinert = 0.04 g h−1). With carbon anode, the presence of K4TiO4 does not lead to complete reduction of TiO2 as the perovskite does not decompose spontaneously.
References
Bailera M, Lisbona P, Pena B, Romeo LM (2021) A review on CO2 mitigation in the iron and steel industry through power to X processes. J CO2 Utiliz 46:101456
International Energy Agency, Iron and Steel Technology Roadmap (2020)
Saevarsdottir G, Magnusson T, Kvande H (2021) J Sustain Metallurg 7:848
Fray DJ, Farthing TW, Chen Z (2000) Reducing the carbon footprint: primary production of aluminum and silicon with changing energy systems. Nature 407:361–363
Chen GZ, Fray DJ (2002) Voltammetric studies of the oxygen-titanium binary system in molten calcium chloride. J Electrochem Soc 149:E455. https://doi.org/10.1149/1.1513985
Dring K, Dashwood R, Inman D (2005) Voltammetry of titanium dioxide in molten calcium chloride at 900 C. J Electrochem Soc 152:E104. https://doi.org/10.1149/1.1860515
Centeno-Sanchez RL, Fray DJ, Chen GZ (2007) Study on the reduction of highly porous TiO2 precursors and thin TiO2 layers by the FFC-Cambridge process. J Mater Sci 42:7494–7501. https://doi.org/10.1007/s10853-007-1588-8
Jiang K et al (2006) “Perovskitization”-assisted electrochemical reduction of solid TiO2 in molten CaCl2. Angew Chem Int Ed 45(3):428–432
Bak T, Nowotny J, Sorrel CC (2004) Chemical diffusion in calcium titanate. J Phys Chem Solids 65:1229–1241. https://doi.org/10.1016/j.jpcs.2004.01.015
Song MH, Han SM, Min DJ, Choi GS, Park JH (2008) Diffusion of oxygen in β-titanium. Scr Mater 59(6):623–626. https://doi.org/10.1016/j.scriptamat.2008.05.037
HSC Chemistry 10 (2020) http://www.chemistry-software.com/
Sanchez-Segado S, Makanyire T, Escudero-Castejon L, Hara Y, Jha A (2015) Reclamation of reactive metal oxides from complex minerals using alkali roasting and leaching–an improved approach to process engineering. Green Chem 17:2059–2080. https://doi.org/10.1039/C4GC02360A
Lahiri A, Jha A (2007) Kinetics and reaction mechanism of soda ash roasting of ilmenite ore for the extraction of titanium dioxide. Metall Mater Trans B 38(6):939–948. https://doi.org/10.1007/s11663-007-9095-5
Lahiri A, Jha A (2009) Selective separation of rare earths and impurities from ilmenite ore by addition of K+ and Al3+ ions. Hydrometallurgy 95:254–261. https://doi.org/10.1016/j.hydromet.2008.06.004
Ghambi S, Sanchez-Segado S, Chipakwe V, Jha A (2021) An investigation on hydrofluoric (HF) acid-free extraction for niobium oxide (Nb2O5) and tantalum oxide (Ta2O5) from columbite/tantalite concentrates using alkali reductive roasting. Miner Eng 173:107183. https://doi.org/10.1016/j.mineng.2021.107183
Abhishek L (2009) PhD Thesis on “physical and process chemistry of alkali roasting of titaniferous minerals”, pp 102, 128, 165
Chimupala Y (2015) Materials Science.https://www.semanticscholar.org/paper/Synthesis-and-characterization-of-the-TiO2(B)-phase-Chimupala/0b24c094cdf296d95a46dbe020329262ade6cc44
Yang X, Jha A (2005) 7th international symposium on molten salt and chemistry & technology, Toulouse France
Bale C et al (2020) FACTSAGE 5.5 and FACTSAGE 6.0: Ecole Polytechnique CRCT, Montreal, Quebec, Canada
Schwandt C, Fray DJ (2005) Electrochemical synthesis of titanium oxycarbide in a CaCl2 based molten salt. Electrochim Acta 51(1):66
Alexander DTL, Schwandt C, Fray DJ (2006) Microstructural kinetics of phase transformations during electrochemical reduction of titanium dioxide in molten calcium chloride. Acta Mater 54(11):2933
Robin A (2005) Influence of temperature on the reduction mechanism of Ti (III) ions on iron in the LiF–NaF–KF eutectic melt and on the electrochemical behavior of the resultant titanium coatings. Mater Chem Phys 89:438
Yan BCK (2016) Electrolysis of titanium oxide to titanium in molten cryolite salt, thesis, University of Toronto, Canada. http://hdl.handle.net/1807/72839
Jiao S, Zhu H (2006) Novel metallurgical process for titanium production. J Mater Res 21:2172
Acknowledgements
The authors wish to acknowledge the support for the EPSRC funded research grants (GR/T08074/01; EP/C007581/1), Millennium Inorganic Chemicals for the PhD Studentship of A Lahiri and Carbon Trust funding on Inert Anodes for metal extraction.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Lahiri, A., Jha, A. Accelerated Electro-Reduction of TiO2 to Metallic Ti in a CaCl2 Bath Using an Inert Intermetallic Anode. J Indian Inst Sci 102, 127–137 (2022). https://doi.org/10.1007/s41745-022-00296-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41745-022-00296-y