
Abstract
The present review article strives to compile the latest synthetic approaches for the synthesis of triazolothiadiazine and its derivatives, along with their diverse pharmacological activities, viz. anticancer, antimicrobial, analgesic and anti-inflammatory, antioxidant, antiviral, enzyme inhibitors (carbonic anhydrase inhibitors, cholinesterase inhibitors, alkaline phosphatase inhibitors, anti-lipase activity, and aromatase inhibitors) and antitubercular agents. The review focuses particularly on the structure–activity relationship of biologically important 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines, which have profound importance in drug design, discovery and development. In silico pharmacokinetic and molecular modeling studies have also been summarized. It is hoped that this review article will be of help to researchers engaged in the development of new biologically active entities for the rational design and development of new target-oriented 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine-based drugs for the treatment of multifunctional diseases.
Graphical Abstract

Similar content being viewed by others

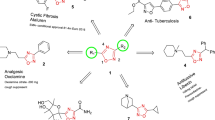
1 Introduction
Heterocyclic compounds are of prime importance due to their extensive therapeutic uses [1]. One of the outstanding structural features innate to heterocycles, which continues to be exploited to great advantage by the drug industry, lies in their ability to manifest substituents around a core scaffold in defined three-dimensional (3D) representations. Among the collection of heterocycles, triazolothiadiazine, a hybrid nucleus made by fusion of two pharmaceutically active moieties, i.e., triazole and thiadiazine, is one of the most essential nuclei owing to their wide range of applications as synthetic intermediates and promising pharmaceuticals. The ability of hydrogen bond accepting and donating characteristics makes this core a much précised pharmacophore with a bioactive profile, as it can make specific interactions with different target receptors. On the basis of fusion permutations of a five-membered triazole ring with a six-membered thiadiazine ring, four isomeric structural variants of triazolothiadiazine, namely 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (I), 1,2,4-triazolo[5,1-b][1,3,5]thiadiazines (II), 1,2,4-triazolo[1,5-c][1,3,5]thiadiazines (III), and 1,2,3-triazolo[5,1-b][1,3,4]thiadiazines (IV) may exist (Fig. 1).

Different possible isomers of triazolothiadiazine
As a class of privileged pharmacophores, the chemistry and synthesis of compounds possessing a 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine nucleus has attracted enormous attention. This review is focused on the synthetic protocols and biological potential, including structure–activity relationship (SAR), in silico pharmacokinetics, molecular modeling and mechanistic studies of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines since 2010.
2 Synthetic Approaches to the 1,2,4-Triazolo[3,4-b][1,3,4]thiadiazine Scaffold
The literature is enriched with several approaches for synthesis of the 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine nucleus. Synthetic design for 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine involves primarily four routes: a, b, c and d (Fig. 2). Route a involves the acid-catalyzed cyclization of 2-hydrazinyl-6H-1,3,4-thiadiazine with ortho esters, thereby annulation of the triazole ring on thiadiazole [2]. However, this method has limited substrate scope. Route b involves the cyclocondensation of 4-amino-3-mercaptotriazoles with various bielectrophiles such as α-bromopropenone [3], phenacyl bromides [4,5,6,7,8], α-tosyloxy ketones [9], ethyl bromoacetate [10], hydrazonoyl halides [11], α-chloroacetylacetone [12] and propargyl bromide in presence of heteropolyacids [13] under conventional heating conditions to access the triazolothiadiazine scaffold. This can be credited to mercapto and amino groups as easily accessible nucleophilic centers in 4-amino-3-mercaptotriazole for the synthesis of condensed heterocyclic rings. Route b offers vast diversity in terms of substitution at 5-position of 4-amino-3-mercaptotriazole as well as choices of bielectrophiles; therefore, this route is the most common approach for the synthesis of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines. Route c involves synthesis of triazolothiadiazine derivatives by intermolecular condensation of 3-mercapto-4-arylideneamino-1,2,4-triazoles and α-halo compounds with activated methylene group [14, 15]. Route d involves ring transformation of oxadiazole-3H-thione upon treatment with phenacyl bromide followed by reaction with hydrazine hydrate to furnish triazolothiadiazine ring [16, 17].

Synthetic approaches for the preparation of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine
The present review encapsulates and highlights the development of synthetic protocols in the last decade (2010–2021) for the synthesis of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines as illustrated in Schemes 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33 and 34.

Synthesis of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives

Synthesis of 6,7-dihydro-5H-[1,2,4]triazolo[3,4-b]1,3,4]thiadiazine derivatives

Heteropolyacid catalyzed synthesis of [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives

Iodine mediated synthesis of [1,2,4]triazolo[3,4-b][1,3,4]thiadiazines

Synthesis of trisubstituted 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives

Synthesis of 3-arylamino-6-phenyl[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives

Synthesis of coumarin substituted 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives

Synthetic pathway for the formation of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine hybrids

Synthesis of 6-substituted-3-[2-(4-phenylpiperazin-1-yl)lquinolin-3-yl]-7H-[1,2,4]triazolothiadiazines

Synthesis of 3,6-disubstituted[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives

Regioselective synthesis of 7-aroyl-6-methyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine analogs

Synthesis of bis(s-triazolo[3,4-b][1,3,4]thiadiazine) derivatives

Synthesis of pyrazole linked mono- and bis-1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives

Synthesis of bis(triazolo[3,4-b]thiadiazine) derivatives tagged to arene centres

Synthesis of bis(1,2,4-triazolo[3,4-b][1,3,4]thiadiazine) derivatives

Synthesis of bis(5,6-dihydro-s-triazolo[3,4-b]thiadiazines)

Synthesis of benzofuran or benzo[d]thiazole linked mono- and bis-1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives

Synthesis of naphthoquinone fused 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives

Multicomponent assemblage for synthesis of 3-(pyrazolyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)-2H-chromen-2-ones

Multicomponent synthesis of 3-aryl-1-(6-aryl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl)-1H-pyrazol-5-amines and 3-(3-(5-amino-3-aryl-1H-pyrazol-1-yl)-7H-[1,2,4]triazolo[3,4-b] [1,3,4]thiadiazin-6-yl)-8-ethoxy-2H-chromen-2-ones

Multicomponent synthesis of 4-(arylydrazono)-3-methyl-1-(6-(coumarin-3-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl)-1H-pyrazol-5(4H)-ones

Synthesis of phthalazinedione based 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines

Multicomponent synthesis of [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives

One-pot multicomponent synthesis of substituted [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives

Synthesis of 3-ethyl-6-methyl-7-(2-arylhydrazono)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives

Synthesis of (7Z)-7-[2-(aryl)hydrazinylidene]-6-methyl-3-(pyridine-4-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines

Synthesis of pyrimidine clubbed 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives

Synthesis of pyrimido[5,4-e][1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives

Synthesis of 7-benzylidene-3-methyl-6,7-dihydro-5H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine

Synthesis of highly functionalized 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines

Synthesis of 5-substituted [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine) derivatives

One-step synthesis of triazolo[3,4-b][1,3,4]thiadiazines

Synthesis of spirocyclic triazolo[3,4-b][1,3,4]thiadiazine derivatives

Synthesis of 3-alkyl/aryl-7/9-methyl-10,10a-dihydroindeno[1,2-e][1,2,4]triazolo[3,4-b][1,3,4]thiadiazines
2.1 Synthetic Strategies Using α-Halocarbonyl Compounds
Foroughifar et al. [14] reported an efficient route for the synthesis of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (5) by treating Schiff’s bases (3), obtained by reaction of 5-substituted 4-amino-1,2,4-triazole-3-thiols (1) with appropriate benzaldehydes (2) in acetic acid, with ethyl chloroacetate (4) in the presence of sodium hydride (Scheme 1).
Al-Etaibi et al. [15] used this method for stereoselective synthesis of 6,7-dihydro-5H-[1,2,4]triazolo[3,4-b]1,3,4]thiadiazine derivatives (9). Several 4-arylideneamino-1,2,4-triazole-3(2H)-thiones (3) (generated by the reaction of 3-aryl-4-amino-5-mercapto-1,2,4-triazoles (1) with benzaldehydes (2) on condensation with ethyl bromoacetate or phenacyl bromide (6) in presence of triethylamine in refluxing ethanol gave trans-6,7-dihydro-5H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (8), which isomerized gradually in CDCl3 and rapidly in DMSO-d6 into corresponding cis-stereoisomers (9) (Scheme 2).
Motamedi et al. [18] developed a heteropolyacid (HPA)-catalyzed one-pot synthesis of [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives (11) and (13) in excellent yields via cyclocondensation reaction of 5-substituted 4-amino-1,2,4-triazole-3-thiols (1) with α-chloroacetone (10) or α-chloroacetonitrile (12) in refluxing acetic acid (Scheme 3).
Jakhar and Makrandi [19] described one-pot synthesis of [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives (15) by iodine-mediated condensation of 3-alkyl/aryl-4-amino-5-mercapto-1,2,4-triazoles (1) with 3-acetyl-6-substituted coumarin (14) in dimethylformamide (DMF) under microwave (MW) irradiation (Scheme 4). This protocol avoids the use of highly lachrymatory molecular bromine and its main advantage is good yields (78–92%) in a very short time.
Knak et al. [20] reported the synthesis of 3,6,7-trisubstituted 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (17) in excellent yields by stirring 4-amino-3-mercapto-l,2,4-triazoles (1) with 2-bromo-1,2-diphenylethan-1-ones (16) initially in ethanol at 20 °C and subsequently at refluxing for 4 h (Scheme 5).
Marathe et al. [21] reported an efficient synthesis of 3-arylamino-6-phenyl[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines derivatives (19) by heating 4-amino-3-arylamino-5-mercapto-1,2,4-triazoles (18) with α-bromoacetophenone (6) in dry DMF, initially in presence of potassium carbonate for 2 h, and subsequently with p-TsOH (Scheme 6).
A literature survey revealed that manifestation of the coumarin nucleus in heterocycles is found to have broad pharmacological activities [22, 23]. Vaarla et al. [24] designed a series of coumarin substituted 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (20) via cyclocondensation of 5-substituted 4-amino-1,2,4-triazole-3-thiols (1) with differently substituted 3-(2-bromoacetyl)coumarins (6) in ethanol under reflux (Scheme 7). Substitution of coumarin with electron-withdrawing groups at R1 and/or R2 provided higher yields of products than electron rich groups.
Utilizing a conventional approach, Lechani et al. [25] synthesized [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine hybrids bearing aryl-, pyran-2-one-, or coumarin moieties (21) in moderate to good yields by acetic acid catalyzed cyclocondensation reaction of 2-(4-amino-5-mercapto-4H-[1,2,4]triazol-3-yl)phenol (1) with aromatic or heterocyclic α-bromoacetyl derivatives (6) in refluxing EtOH (Scheme 8). Simple reaction conditions, short reaction times, good yields and synthesis of highly functionalized hybrids bearing oxygen-, nitrogen-, and sulfur-based heterocycles make this method potentially valuable.
Tang et al. [26] synthesized a new series of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (22) via reaction of 4-amino-5-[2-(2-piperazinyl-1-yl)quinolin-3-yl]-4H-1,2,4-triazole-3-thiol (1) with various phenacyl bromides (6) in refluxing ethanol/DMF mixture (10:1) (Scheme 9).
Kattimani et al. [27] successfully synthesized 3,6-disubstituted[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives (23) via reaction of phenacyl bromide (6) with various 4-(4-amino-5-mercapto-4H-1,2,4-triazol-3-yl)methyl)-2-aryl-2H-1,2,4-triazol-3(4H)-ones (1) in refluxing ethanol (Scheme 10).
In recent years, organic reactions under visible-light irradiation are grabbing intense attention as a powerful strategy involving the concept of ‘Green Chemistry’ to reduce hazardous ecological imprints of organic synthesis. In order to achieve a green protocol, Aggarwal et al. [28] developed first visible-light mediated regioselective synthesis of novel acyl derivatives of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines 26 involving reaction of binucleophilic 4-amino-3-mercapto-1,2,4-triazoles (1) with in situ generated α-bromodiketones (25) from unsymmetrical β-diketones (24). In principle, reaction of unsymmetrical α-bromodiketones (25) (having two carbonyl carbon of different electrophilicity) with 4-amino-3-mercapto-1,2,4-triazoles (1) may lead to the formation of two regioisomers- 7-aroyl-6-methyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (26) and 7-acetyl-6-aryl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (27). However, the reaction exhibited high level of regioselectivity yielding only (26), not the other possible isomer, 7-acetyl-6-aryl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (27). The structure of the regioisomer formed has been established unambiguously by heteronuclear 2D-NMR [(1H–13C) and (1H–15N) HMBC, (1H–13C) HMQC] spectroscopic and X-ray crystallographic studies. The substrate scope of the reaction was established by taking differently substituted 4-amino-3-mercapto-1,2,4-triazoles (1) and unsymmetrical diketones (24) under optimized conditions. All combinations resulted in exclusive formation of (26) with high regioselectivity. This rapid and efficient method involves use of simple and readily available starting materials, environment benign conditions, and provided excellent yields of the products (85–97%) in a fairly short time (Scheme 11). A solvent-free protocol using p-TsOH as a catalyst was also investigated to achieve the synthesis of (26); however, yields were comparatively lower (72–85%).
Bis-heterocycles with a flexible linker reported to possess a wide array of pharmaceutical and biological activities [29]. In this context, Ginidi et al. [30] introduced a novel series of bis(s-triazolo[3,4-b][1,3,4]thiadiazines) (29) tethered via phenoxymethyl spacers. Piperidine catalyzed cyclocondensation reaction of 4-amino-3-mercapto-1,2,4-triazole derivatives (1) with bis(α-bromoketones) (28) in EtOH-DMF mixture under reflux afforded corresponding bis(s-triazolo[3,4-b][1,3,4]thiadiazines) (29) in good yields (Scheme 12).
Synthesis of novel mono- and bis-1,2,4-triazolo[3,4-b][1,3,4]thiadiazines linked with pyrazole moiety (32) and (34) was described by Salem et al. [31]. The reaction of 4-amino-3-mercapto-1,2,4-triazole derivatives (1) with 2-bromo-1-(5-methyl-1-phenyl-1H-pyrazol-4-yl)ethanone (30) in refluxing ethanol in the presence of triethylamine as catalyst, afforded an intermediate (31), which underwent cyclization in refluxing acetic acid to access pyrazolyl(5,6-dihydro-s-triazolo[3,4-b]thiadiazines) (32) in good yields (70–75%) (Scheme 13). Bis(6-pyrazolyl-s-triazolo[3,4-b][1,3,4]thiadiazines) linked to the alkyl spacer (34) were synthesized in one-step by cyclocondensation of bis(4-amino-5-mercapto-1,2,4-triazol-3-yl)alkanes (33) with 2 equivalents of (30) in refluxing EtOH containing a few drops of triethylamine.
Alternatively, bis(triazolo[3,4-b]thiadiazine) derivatives (35) linked to arene centres via alkyl/ether/sulfanylmethylene insertions were prepared by treating bis(4-amino-5-mercapto-s-triazolo-3-y1) derivatives (33) with two equivalents of phenacyl bromide (6) in DMF/ethanol mixture under reflux (Scheme 14) [32].
Shaaban and Elwahy [33] reported the synthesis of bis(5,6-dihydro-s-triazolo[3,4-b]thiadiazines) (36) in good-to-excellent yields by the reaction of bis(α-bromoketones) (28) with 4-amino-3-mercapto-1,2,4-triazole derivatives (1) in ethanol under refluxing conditions (Scheme 15), which, upon reduction with sodium borohydride in refluxing methanol, afforded bis 4-(6,7-dihydro-3-substituted-5H-1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (37) in good yields.
Sayed et al. [34] have reported the synthesis of bis(5,6-dihydro-s-triazolo[3,4-b]thiadiazines) (38) in moderate yields (60–66%) via reaction of bis(α-bromoketones) (28) with 4-amino-3-mercapto-1,2,4-triazole derivatives (1) in catalytic amount of triethylamine in refluxing ethanol-DMF mixture (Scheme 16).
Recently, the synthesis of mono- and bis(triazolothiadiazine) derivatives coupled with benzofuran or benzo[d]thiazole moieties (39) and (40) in good yields (69–79%) has been achieved by the reaction of bromoacetylbenzothiazole/bromoacetylbenzofuran (6) with each of 4-amino-3-mercapto-1,2,4-triazoles (1) and bis([1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl)alkanes (33) in catalytic amount of triethylamine in refluxing ethanol (Scheme 17) [35].
In view of the significant biological potential of fused naphthoquinones [36], Khalafy et al. [37] reported the synthesis of a new series of 10-substituted-5H-naphtho[1,2-e][1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-5-ones (43) from the reaction of 4-amino-5-aryl/alkyl-3-mercapto-1,2,4-triazoles (1) with 2-bromo-1,4-naphthoquinone (41) in ethanol (Scheme 18). This two-step procedure involves the intermediacy of 2-[(4-amino-5-aryl-4H-1,2,4-triazol-3-yl)thio]naphthalene-1,4-diones (42), which underwent intramolecular cyclization in EtOH-HCl under reflux to afford products (43) in 56–82% yields.
2.2 Multicomponent Reaction Approach
Recently, multicomponent reactions (MCRs) due to their high efficiency, atom economy, and reduced time are being explored to access functionalized triazolothiadiazines in a one-pot one-step fashion. In general, construction of triazolothiadiazine scaffold using MCR involves condensation of two components, namely, α-halocarbonyl compounds and 4-amino-5-hydrazinyl-3-mercapto-1,2,4-triazole and the third component is being employed for preparing derivatives or variations at hydrazinyl group.
Pavurala and Vedula [38] reported a one-pot multicomponent approach for synthesis of 3-(pyrazolyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)-2H-chromen-2-ones (46) in excellent yields. Reaction of 4-amino-5-hydrazinyl-3-mercapto-1,2,4-triazole (44), substituted 3-(2-bromo-acetyl)coumarins (6) and acetyl acetone (45) in absolute ethanol under reflux afforded (46) (Scheme 19). The method has the merits of operational simplicity, good yields and accomplished the synthesis of two heterocycles in one stride.
Keeping in view the importance of pyrazole-linked heterocyclic compounds, Sujata et al. [39] reported one-pot multicomponent synthesis of 3-aryl-1-(6-aryl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl)-1H-pyrazol-5-amines (48) and 3-(3-(5-amino-3-aryl-1H-pyrazol-1-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)-8-ethoxy-2H-chromen-2-ones (49). Acetic acid catalyzed condensation of (44), various phenacyl bromides (6) or 3-(2-bromoacetyl)coumarins (6′) and benzoylacetonitriles (47) in refluxing ethanol yielded 48 and 49 in excellent yields (Scheme 20).
Aychiluhim et al. [40] described one-pot multicomponent reaction of 3-(2-bromo acetyl)coumarins (6), (44) and ethyl 2-(2-(aryl)hydrazono)-3-oxobutanoates (50) to produce novel 4-(arylhydrazono)-3-methyl-1-(6-(coumarin-3-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl)-1H-pyrazol-5(4H)-ones (51) (Scheme 21). Short reaction period, higher yields, mild conditions and one-pot synthesis are some of the key features of this methodology.
Keeping the importance of triazolo[3,4-b][1,3,4]thiadiazines and phthalazine-1,4-dione, Jilloju et al. [41] explored multicomponent synthesis of 2-(6-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl)-2,3-dihydrophthalazine-1,4-dione derivatives (53). Three-component reactions of (44) with phenacyl bromides (6) and phthalic anhydride (52) in acetic acid yielded (53) in good to excellent yields (Scheme 22).
A simple, multicomponent efficient synthesis of [1,2,4]triazolo[3,4-b]thiadiazines (54) was reported by Sujatha et al. [42] involving pseudo four component condensation of (44), aromatic aldehydes (2), and various phenacyl bromides (6) in presence of triethyl amine in absolute ethanol to afford novel [1,2,4]triazolo[3,4-b]thiadiazine derivatives in excellent yields (Scheme 23). Shorter reaction time, good yields, operational simplicity, mild and clean reaction profiles are some key features of this method.
Thirupaiah et al. [43] have described the synthesis of a pyran and phthalazine substituted [1,2,4]triazolo[3,4-b][1,3,4]-thiadiazine derivative (55) by multicomponent reaction of equimolar 3-(2-bromoacetyl)-4-hydroxy-6-methyl-2H-pyran-2-one (6), (44), and phthalic anhydrides (52) in acetic acid under refluxing conditions (Scheme 24). Similarly, the three-component reaction of 44, aldehydes (2) and 3-(2-bromoacetyl)-4-hydroxy-6-methyl-2H-pyran-2-one (6), with few drops of acetic acid in absolute ethanol afforded substituted 3-[3-(N1-benzylidene-hydrazino)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl]-4-hydroxy-6-methyl-pyran-2-ones (56) in good yields.
2.3 Synthesis from Hydrazonoyl Chlorides
Products of the reactions between 4-amino-3-mercaptotriazoles and hydrazonoyl halides are [1,2,4]triazolo[3,4-b][1,3,4]thiadiazines bearing 2-arylhydrazono group at the 7-position. Abdelrazek et al. [44] reported the facile triethylamine-catalyzed synthesis of 3-ethyl-6-methyl-7-(2-arylhydrazono)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives (59) from the reaction of 4-amino-5-ethyl-3-mercapto-1,2,4-triazole (1) with variously substituted hydrazonoyl chlorides (57) in refluxing dioxane. S-alkylated intermediates (58), generated in situ, are cyclized to afford the desired products (59) in good yields (80–88%) (Scheme 25).
4-Amino-3-(4-pyridyl)-5-mercapto-1,2,4-triazole (1) couples with substituted aryl hydrazonoyl chlorides (57) in a similar manner to yield 7-(2-arylhydrazono)[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives (60) (Scheme 26) [45].
2.4 Miscellaneous Reactions
Ebrahimpour et al. [46] synthesized pyrimido[4,5-e][1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives (64) in good yields via S/N type Smiles rearrangement. Initially, the reaction of 5-bromo-2,6-dichloropyrimidine (61) with 4-amino-5-methyl-3-mercapto-1,2,4-triazole 1 in CH3CN afforded intermediate 3-((5-bromo-6-chloropyrimidin-4-yl)thio)-5-methyl-4H-1,2,4-triazol-4-amine (62) and subsequently nucleophilic substitution with secondary amines gave corresponding heteroaryl thioethers (63). Treatment of (63) in refluxing CH3CN in the presence of NaNH2 afforded the regioisomer 64 instead of expected regioisomer 65 by an intramolecular SNAr type reaction (Scheme 27). X-ray structural analysis of the product having NR2 = morpholine group demonstrated the formation of 64 through Smiles rearrangement.
A series of pyrimido[5,4-e][1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives (69) was synthesized by Afrough et al. [47] through the condensation reaction of 5-bromo-4-[1-(5-bromo-2,6-dichloropyrimidin-4-yl)ethyl]-2-chloro-6-methylpyrimidine (66) with 4-amino-5-methyl-3-mercapto-1,2,4-triazole (1) (Scheme 28). This three-step reaction involves the formation of intermediate (67), which underwent nucleophilic substitution with secondary amine to give corresponding diheteroaryl sulfide intermediates (68). Intramolecular SNAr reaction of compounds 68 in the presence of NaNH2 afforded the desired products (69). Interestingly, nucleophilic substitution with piperidine provided the desired products in good yields (73–75%) whereas substitution with piperazines resulted in lower yields of products (62–68%).
Skryl'nikova et al. [48] reported the synthesis of 7-benzylidene-3-methyl-6,7-dihydro-5H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine (74) by intramolecular cyclization of 5-methyl-4-(3-phenylprop-2-yn-1-yl)amino-4H-1,2,4-triazole-3-thiol (72) in DMF at 100 °C in the presence of catalytic amounts of a 40% aqueous solution of KOH (Scheme 29). Compound 72, in turn, was formed by p-toluenesulfonic acid (p-TsOH)-catalyzed reaction of 4-amino-3-mercapto-1,2,4-triazole (1) with phenylpropiolaldehyde (70), involving synthesis of 5-methyl-4-[(3-phenylprop-2-yn-1-ylidene)amino]-4H-1,2,4-triazole-3-thiol intermediate (71), followed by hydrogenation with NaBH4. The hydrogenation with NaBH4 excluded the possibility of formation of side product, 3-methyl-8-phenyl-5,6-dihydro[1,2,4]triazolo[3,4-b][1,3,4]thiadiazepine (73).
An efficient synthesis of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (76) in excellent yields (> 90%) has been presented via a one-pot metal-free protocol by nucleophilic addition of 4-amino-3-mercapto-1,2,4-triazole derivatives (1) to dibenzoylacetylene (DBA) (75) under stirring in acetonitrile at room temperature (Scheme 30) by Shah et al. [49]. The structure of product 76 having R = Ph was unambiguously confirmed by X-ray crystallographic data.
Pandey et al. [50] have synthesized a series of novel 5-[(3-aralkyl amido/imidoalkyl) phenyl]-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazine derivatives (78) in good yields by condensation reaction of 4-amino-5-mercapto-3-[(3-aralkyl amido/imidoalkyl)phenyl]-1,2,4-triazoles (1) with benzoin (77) in polyphosphoric acid under reflux (Scheme 31).
The reaction of 4-amino-3-mercapto-5-phenyl-s-triazole (1) with aromatic/aliphatic/cyclic ketones 79 and 81 in refluxing acetic acid afforded the corresponding triazolo[3,4-b][1,3,4]thiadiazines 80 and 82 in a single step (Scheme 32) [51]. This method is better than classical methods in terms of synthesis of 6- and 7-substituted derivatives (rare), and avoidance of use of highly toxic and irritating halo compounds.
A base catalyzed condensation reaction of 4-amino-3-[(4-nitrobenzyl)thio]-4H-1,2,4-triazoles (1) with alkylisatins (83) in ethanol was described by Gaponenko et al. [52]. Spirocyclic triazolo[3,4-b][1,3,4]thiadiazine derivatives (85) were obtained by the formation of new C–C bond in the final step of diastereospecific heterocyclization of intermediate 84 (Scheme 33).
Prakash et al. [53] synthesized a series of 3-alkyl/aryl-7/9-methyl-10,10a-dihydroindeno[1,2-e][1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (87) (67–93% yields) by the cycloconsensation of 4/6-methyl-2-tosyloxy-1-indanones (86), generated by oxidation of 4/6-methyl-1-indanones with [(hydroxy)tosyloxyiodo]benzene (HTIB) in acetonitrile, and 3-alkyl/aryl-4-amino-5-mercapto-1,2,4-s-triazoles (1) in refluxing ethanol (Scheme 34).
3 Pharmacological Properties of 1,2,4-Triazolo[3,4-b][1,3,4]thiadiazines
Reported literature protocols have provoked researchers to discover new biological active compounds consisting of triazolothiadiazine as a core motif. 1,2,4-Triazolo[3,4-b][1,3,4]thiadiazine derivatives have been reported to possess significant pharmacological activity as anticancer, antimicrobial, analgesic and anti-inflammatory, antioxidant, antiviral, enzyme inhibitors and antitubercular agents through different mechanisms of action, such as enzyme inhibition, interaction with intracellular targets, etc. Triazolothiadiazines also find applications in other fields, such as intermediates for photographical couplers [54, 55], PDE4 inhibitors [56], and corrosion inhibitors [57, 58].
3.1 Anticancer Agents
Aytaç et al. [59] designed and synthesized a series of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (88) linked with structural motifs of nonsteroidal analgesic anti-inflammatory drugs (ibuprofen, naproxen and flurbiprofen) at the 3-position. All synthesized derivatives were screened for their in vitro cytotoxic potential against liver (Huh7), breast (MCF-7) and colon (HCT116) cancer cell lines. Preliminary results indicated that all triazolothiadiazine derivatives exhibited significant cytotoxic potential (IC50 = 1.1–18.8 µM) than synthon, 4-amino-1,2,4-triazole-5-thiones (IC50 = 20.2–67.9 µM), thereby implying that fusion of triazole with thiadiazine is crucial for cytotoxic potential. Among triazolothiadiazines, compounds 88a–j with lower IC50 values were further evaluated against hepatocellular carcinoma cell lines (HepG2, Hep-3B, Mahlavu, FOCUS and Snu475) and were found to display substantial cytotoxic activities (IC50 = 1.5–10.1 µM) (Fig. 3). According to fluorescent microscopy and flow cytometry analysis, the compounds 88a–c, 88f and 88j were found to induce apoptosis and SubG1 arrest in Huh7 cells. Mechanistic study revealed that compounds 88b and 88c initiated oxidative stress induced apoptosis through ASK-1 protein activation and Akt protein inhibition.

Structure–activity relationship (SAR) studies of non-steroidal drugs linked to 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines as anticancer agents
Ahmad et al. [60] synthesized a series of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (89) having an alkenyl/hydroxyalkenyl fatty acid chain and evaluated them for their in vitro anticancer activity against three different human cancer cell lines, namely Hep3 B, MCF-7, HeLa (human cervical carcinoma) and toxicity against PBMCs (peripheral blood mononuclear cells). All the synthesized compounds were found to be non-toxic against PBMCs. SAR studies revealed that compounds 89c and 89d containing a hydroxyl group on a fatty acid chain exhibited maximum cytotoxic potential (IC50 = 6.0–8.8 µM) against all the three tested cancer cells, as depicted in Fig. 4.

SAR analysis of fatty-acid-chain-substituted 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines as anticancer agents
Xu et al. [61] synthesized novel 3,6-diaryl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives (90) as anti-proliferative agents against three human cancer cell lines: gastric adenocarcinoma (SGC-7901), lung adenocarcinoma (A549) and fibrosarcoma (HT-1080) cells. From the series, compounds 90a–h exhibited excellent potency against SGC-7901, A549 and HT-1080 cell lines with IC50 values at the nanomolar level (IC50 = 0.011–0.079 µM). A SAR study revealed the significance of substitution at position-3 of the triazolothiadiazine scaffold as shown in Fig. 5. Compound 90g displayed the most promising antiproliferative activity against the three human cancer cell lines, with an IC50 value of 0.011–0.015 μM and effectively induced cell cycle arrest at G2/M phase in SGC-7901 cells. This compound effectively inhibited tubulin polymerization, with an IC50 value of 1.6 μM. Docking studies revealed that compound 90g binds to the tubulin perfectly at the colchicine binding site.

SAR exploration of 3,6-diaryl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines as anticancer agents
Khan et al. [62] synthesized a series of 6-aryl-3-(pyridin-4-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (91) and screened their anticancer activity against kidney fibroblast (BHK-21) and lung carcinoma (H157) cell lines at four different concentrations (100, 10, 1 and 0.1 µM) by cell growth inhibition. Among the tested series, compound 91c showed the highest potency, with 78.6% inhibition (100 µM) against H157 cells, which was better than the standard drug vincristine (74.5%). Compounds 91a and 91d exhibited excellent cytotoxicity against BHK-21 cells with 73.3% and 72.6% inhibition (100 µM). SAR studies presented the significance of aryl substituents at position-6 of triazolothiadiazine for anticancer activity and substitution of C6 by biphenyl group significantly decreased anticancer activity (Fig. 6).

SAR analysis and anticancer activity of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines against BHK-21 and H157 cell lines
In another work, Khan et al. [63] synthesized 6-aryl-3-(pyridin-3-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives (92) and screened their cytotoxic potential against lung carcinoma (H157) and vero cell lines. Compound 92 h (R = pyridin-3-yl, Ar = 3,4-diClPh) presented excellent antiproliferative activity against H157 cell lines with an IC50 value of 0.96 ± 0.43 µM at 1 µM concentration as compared with the standard drug vincristine (IC50 = 1.03 ± 0.04 µM) (Fig. 6).
Kamel and Abdo [64] accomplished the synthesis of pyridine linked 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (93) and investigated in vitro anticancer activity against six human cancer cell lines, namely human gastric cancer (NUGC), human colon cancer (DLD1), human liver cancer (HA22T and HEPG2), nasopharyngeal carcinoma (HONE1), human breast cancer (MCF) and normal fibroblast cells (WI38). Among them, compound 93a displayed significant cytotoxic potential (IC50 = 59–469 nM) against all the tested cancer cell lines except the cell line DLD1(IC50 = 2350 nM) (Fig. 7).

Pyridine linked 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines as anticancer agents
Zhang et al. [65] synthesized novel 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (94) bearing furan and thiophene nucleus and evaluated them for their antiproliferative activities against four human cancer cell lines, namely PC-3 (prostate cancer cells), HepG2 (hepatoma cells), A549 (alveolar epithelial cells), and MCF-7 (mammary adenocarcinoma cells). Results indicated that most of the tested compounds showed good-to-excellent antiproliferative activities against PC-3 and A549 cell lines. A SAR study revealed that thiophene derivatives displayed higher cytotoxic potential against four tested cell lines than furan derivatives (Fig. 8). Compound 94b exhibited 2- to 11-fold higher activity against PC-3, HepG2 and A549 cells (IC50 = 5.09–12.74 µM) than the standard drug fluorouracil (IC50 = 22.28–46.83 µM) and arrested G2/M cell-cycle in PC-3 cells in a dose-dependent manner.

SAR of thiophene/furan linked 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines as anticancer agents
Farghaly et al. [66] carried out the synthesis and anticancer evaluation of 3,6,7-trisubstituted-1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (95). Among them, compound 95a exhibited potential activity against both HEPG-2 and HCT cell lines, with IC50 values of 8.63 and 8.33 µg ml−1, respectively, whereas compound 95h presented excellent activity against HEPG-2 cell line with an IC50 value of 5.63 µg ml−1 (Fig. 9).

SAR evaluation of 3,6,7-trisubstituted-1,2,4-triazolo[3,4-b][1,3,4]thiadiazines as anticancer agents
Through an HTS process, LaPorte et al. [67] identified a series of pyrazole-linked 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (96) as potent and selective STAT3 inhibitors. STAT3 is an attractive target for anticancer agents. Among the tested series, compounds 96a and 96b displayed significant antiproliferative activity against several head and neck squamous cell carcinoma (HNSCC) cell lines, namely 686LN, Cal33, FaDu, and OSC19 (GI50 = 14–44 µM) by reduction of phosphorylated STAT3 levels and downstream effects on STAT3 gene expression targets. Compounds 96a and 96b proved to be selective inhibitors STAT3 with IC50 values of 6.8 and 9.6 µM, respectively, over STAT1 (IC50: > 50 µM for each). SAR studies revealed the significance of pyrazole group and specific aryl substitution on the thiadiazine for inhibition of STAT3 pathway as shown in Fig. 10. Further, improvements in potency and metabolic stability were accomplished by the introduction of α-methyl group on the thiadiazine (96c, GI50 = 1.9 µM, IC50 = 8.2 µM).

SAR analysis and anti-proliferative activities of pyrazole-containing 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines as STAT3 inhibitors
Sever et al. [68] synthesized a series of indomethacin based 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (97) and evaluated their cytotoxic potential against the human glioma cell line T98. Among the evaluated series, compound 97h (Fig. 11) exhibited the most potent anti-tumor activity via inhibition of COX-2 mRNA levels in dose-dependent manner. The results of flow cytometric analysis demonstrated that apoptosis stimulating percentages of compound 97h at 50 μM and 100 μM doses were calculated as 11% and 12%, respectively, in comparison with the control group (1.1%). Docking analysis revealed compound 97h and indomethacin were found to bind similarly to the active site of the COX-2 enzyme.

Anti-tumor activity of indomethacin based 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines
Poojary et al. [69] synthesized a series of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (98) possessing arylfurfuryl, 2,4-dichloro-5-fluorophenyl and aryloxymethyl/anilinomethyl moieties (98a–h) and screened compounds 98d and 98h (Fig. 12) for their anticancer potential initially against NCI-H460 (lung), MCF-7 (breast) and SF 268 (CNS) cell lines. Further evaluation of compounds 98d and 98h against a full panel of 60 cell lines indicated that compound 98d showed good activity against SW-620 (colon cancer), MALME-3 M (melanoma) and T-47D (breast cancer) cell lines with GI50 values of 3.1, 3.8 and 7.9 µM, respectively, whereas compound 98h showed good activity against MMOLT-4 (leukemia), EKVX (non-small cell lung cancer) and NCI/ADR-RES (breast cancer) cell lines with GI50 values of 3.3, 7.5 and 6.4 µM, respectively.

Anticancer activities of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines
A series 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (99) was prepared by Fan et al. [70] and evaluated for their in vitro anticancer activity against two prostate cancer cell lines PC-3 and DU-145 using SRB (sulforhodamine B) and MTT (3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide) assay. All the synthesized compounds exhibited remarkable cytotoxicity against targeted cell lines with GI50 values ranging from 12.0 to 32.5 μg ml−1 and 16.5–27.7 μg ml−1 against DU-145 and PC-3 cell lines, respectively. SAR studies revealed that compounds 99a, 99d and 99e combined with benzimidazole and quinoline moieties exhibited the most promising activity, with 63, 71 and 66% cytotoxicity, respectively (Fig. 13).

SAR examination of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines with different heteroaromatics as anticancer agents
Ibrar et al. [71] designed several coumarin-triazolothiadiazine hybrids (100) and screened their in vitro anticancer activity against kidney fibroblast (BHK-21) and lung carcinoma (H-157) cell lines. Among the series, compounds 100d and 100h showed the highest inhibition against BHK-21 cell lines (IC50=1.24 and 1.39 µM), while compound 100i showed the highest rate of inhibition against the H-157 cell line (IC50=1.01 µM), greater than the standard drugs vincristine (IC50 = 1.03 µM) and cisplatin (IC50 = 1.31 µM). A SAR study revealed the significance of substituents as shown in Fig. 14.

SAR studies and anticancer evaluation of coumarin-linked 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines
Yakaiah et al. [72] carried out synthesis of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (101) containing coumarin and benzotriazole moieties and evaluated their anticancer against a human lung cancer (HCC 827) cell line. Bioassay results indicated that compound 101k exhibited promising activity, with an IC50 value of 0.821 µM. SAR studies indicated that the presence of an electron-rich group on the coumarin ring was responsible for better activity, as shown in Fig. 15.

Benzotriazole and coumarin-combined triazolothiadiazine as anticancer agent
Iqbal et al. [73] reported the synthesis of coumarin-triazolothiadiazine conjugates (102) and explored them for their cytotoxic potential against three different cancer cell lines: breast cancer cells (MCF-7), bone-marrow cancer cells (K-562), and cervical cancer cells (HeLa). Among the tested series, compounds 102a and 102g exhibited a significant anti-proliferative effect in cancer cells (IC50 = 2.21–21.2 µM). Moreover, compound 102a with an IC50 value of 2.21 µM was found to be more potent than the standard drug carboplatin (IC50 = 3.91 µM) against MCF-7 cell lines, and this compound induced 50.1% apoptosis in via G2/M phase arrest in MCF-7 cells. A SAR study was carried out to correlate the importance of substituents with the observed activity (Fig. 16).

Coumarin-triazolothiadiazines with anticancer activity
Arandkar and Vedula [74] introduced a series of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (103) and screened them for anticancer activity at a concentration 10–5 M against 60 cancer cell lines comprising leukemia, non-small cell lung cancer, colon cancer, CNS cancer, melanoma, ovarian, renal, prostate cancer and breast cancer lines. A SAR study revealed that compounds containing halo groups exhibited higher potency than other groups (Fig. 17). From the tested compounds 103d, 103e, 103f and 103l exhibited significant activity against renal cancer OU-31 cell line with 47.42%, 54.64%, 46.76 and 48.14% growth, respectively. Compound 103f also exhibited significant activity on leukemia MOLT-4 cell line with 49.82% growth.

SAR inspection and anticancer activities of triazolothiadiazine analogs
Mohamady et al. [75] designed a series of naphthoquinone-hydrazino[1,2,4]triazolo[3,4-b]-1,3,4-thiadiazine derivatives (104) and screened their cytotoxicity against breast cancer cell lines (MCF-7) and hepatocellular carcinoma cell lines (HepG2) via MTT assay. Among the tested compounds, compound 104a (Fig. 18) exhibited maximum cytotoxicity against HepG2 cell lines (IC50 = 0.07 µM) and significantly upregulated caspase 3, with induction of apoptosis at pre-G1 phase and cell death at G2/M phase. Compounds 104a–c exhibited significant inhibition of Topoisomerase-IIβ (0.55–0.64 μM). Docking analysis explained the potential interactions of 104a–c within the ATP catalytic binding domain of Topo-IIβ.

SAR study of naphthoquinone-hydrazinotriazolothiadiazines as anticancer agents
Recently, Çevik et al. [76] synthesized a series of benzimidazole clubbed 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (105) and tested them for their in vitro anticancer activity against MCF-7 cancer cell lines, among which compounds 105c, 105e, and 105k–l exhibited significant anticancer activity (IC50 = 0.016–0.119 µM). Moreover, compounds 105e and 105l were found to be more active (IC50 = 0.016 and 0.018 µM, respectively) than the standard drug cisplatin (IC50 = 0.020 µM). A SAR study explained the effect of substituents (Fig. 19).

SAR of naphthoquinone-hydrazinotriazolothiadiazines as anticancer agents
Puthiyapurayil et al. [77] synthesized several 6-arylsubstituted-3-[2-(4-substitutedphenyl)propan-2-yl]-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (106) and evaluated them for their in vitro cytotoxicity against MCF-7 and A549. Among the screened series, compound 106h and 106i showed most promising cytotoxicity, with an IC50 value 10.54 and 14.28 µM, respectively, against the MCF-7 cell line. A SAR study showed the significance of substituents for favorable activity (Fig. 20).

SAR and anticancer potential of 6-arylsubstituted-3-[2-(4-substitutedphenyl)propan-2-yl]-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines
Several 6-ferrocenyl-3-subsituted-7H-1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (107) were synthesized and evaluated biologically against human fibrosarcoma cells (HT1080) and human foreskin fibroblasts (hTERT-BJ) cell lines by Miao et al. [78]. Compounds 107a and 107b (Fig. 21) exhibited a higher inhibitory effect on HT1080 cells than hTERT-BJ cells. Bioassay results revealed that compounds 107a and 107b suppressed the viability of HT1080 cells by inducing cell cycle arrest in G1 phase, apoptosis, decreasing mitochondrial membrane potential and increasing the ratio of Bax/Bcl-2.

Anti-tumor activity of 6-ferrocenyl-3-subsituted-7H-1,2,4-triazolo[3,4-b][1,3,4]thiadiazines
Ansari et al. [79] synthesized 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (108) bearing the 3,4,5-trimethoxyphenyl moiety and screened their antiproliferative potential against three human cancer cell lines, namely adenocarcinomic human alveolar basal epithelial cells (A549), breast cancer cells (MCF7), and human ovarian carcinoma cells (SKOV3). Among them, three compounds 108a (IC50 = 0.70 µM), 108m (IC50 = 0.30 µM) and 108 (IC50 = 0.60 µM) were significantly more potent than the reference drug etoposide (IC50 = 1.89–2.99 µM) against MCF7, SKOV3 and A549 cells, respectively. SAR studies revealed the significance of trimethoxy substitution for anti-tumor activity as shown in Fig. 22. Moreover, compounds 108a and 108m exhibited inhibitory potential on tubulin polymerization with IC50 values of 1.03 and 1.61 µM, respectively. Docking studies also indicated that compounds 108a, 108m, 108o inhibited tubulin polymerization through binding to the colchicine-binding site.

SAR and anticancer activities of 3,4,5-trimethoxyphenyl linked 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines
Radwan et al. [80] evaluated anticancer activity 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives 109a and 109b against liver human cancer cell lines (HEPG2) in vitro (Fig. 23). Compounds 109a and 109b with IC50 values of 31.1 μM and 19.6 μM, respectively, were more effective than reference drug doxorubicin (IC50 = 36.3 μM). The most active compound 109b exhibited in vivo anti-hepatocellular carcinoma efficacy in albino mice via regulating tumor angiogenesis by reducing the level of vascular endothelial growth factor (VEGF) by 30% and restoring the activities of hepatic marker enzymes, hepatic tyrosine kinase (HTK), aspartate and alanine-aminotransferase (AST and ALT) as well as improved the liver levels of lipid peroxides (by 62%) and glutathione (GSH) (by 99%).

SAR study of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives as potent anticancer agents
Liu et al. [81] synthesized and screened 3‐(3‐methoxyphenyl)‐6‐(3‐amino‐4‐methoxyphenyl)‐7H‐[1,2,4]triazolo[3,4‐b][1,3,4]thiadiazine 110 for its cytotoxic potential against fibroblast (L929), alveolar basal epithelial (A549), cervical carcinoma (HeLa), fibrosarcoma (HT‐1080), breast (MCF-7) and gastric (SGC‐7901) cell lines. Compound 110 exhibited significant cytotoxicity against HeLa and SGC‐7901 cells, with IC50 values of 0.11 and 0.12 μM, respectively (Fig. 24). Immunofluorescence and flow cytometry studies revealed that compound 110 induced cell cycle arrest in G2/M phase. Western blot analysis and examination of mitochondrial membrane potential revealed that compound 110 induced apoptosis in SGC‐7901 cells through mitochondria‐mediated and a death receptor‐mediated pathway, whereas in HeLa cells induction was mainly via a mitochondria‐mediated pathway.

3‐(3‐Methoxyphenyl)‐6‐(3‐amino‐4‐methoxyphenyl)‐7H‐[1,2,4]triazolo[3,4‐b][1,3,4]thiadiazine as potent anticancer agent
Ma et al. [82] synthesized a series of [1,2,4]triazolo[3,4‐b][1,3,4]thiadiazine derivatives (111) and studied in vitro anticancer activity against four human cancer cell lines: colorectal adenocarcinoma (HT-29), cervical carcinoma (HeLa), lung adenocarcinoma (A549) and breast (MCF-7) cell lines. Among them, compound 111j was found to have significant cytotoxic (IC50 = 0.004–0.098 μM) and anti-tubulin (IC50 = 1.69 μM) properties and also exhibited excellent selectivity with no cytotoxicity over the normal human embryonic kidney (HEK-293) cell line (IC50 > 100 μM). SAR studies revealed the significance of substitution on the thiadiazine for better potency as shown in Fig. 25. Compound 111j induced cell cycle arrest in A549 cells at G2/M phase by regulation of p-cdc2 and cyclin B1 expressions, and subsequently apoptosis via up-regulating cleaved PARP, activation of caspase-3 and down-regulating Bcl-2 expression. Molecular modeling studies revealed that compound 111j significantly blocked tubulin polymerization through binding to the colchicine site of tubulin. Moreover, compound 111j displayed promising in vivo activity in an A549 lung cancer xenograft mouse model.

SAR study of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives as potent anticancer agents
Ji et al. [83] introduced spirooxindole derivatives of [1, 2, 4]triazolo[3,4‐b][1,3,4]thiadiazine (112) and evaluated their cytotoxic potential. Among the evaluated series, compound 112d was found to exhibit 3.0, 1.6, 2.7 and 1.3 times more potency than the standard drug 5-fluorouracil, with IC50 values of 8.02, 6.62, 9.49 and 17.65 μM against prostate (DU145), esophageal (EC109), gastric carcinoma (MGC803) and breast (MCF-7) cell lines, respectively. A SAR study revealed the significance of substituents, as shown in Fig. 26.

SAR study of spirooxindoles incorporating 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives as potent anticancer agents
Ismail et al. [84] synthesized 6-aryl-3-(aryl/hetarylbenzylidene)hydrazineyl-7H-[1,2,4]triazolo[3,4‑b][1,3,4]thiadiazines (113) and studied their anticancer potential against full NCI-60 cancer cell lines. Compound 113h was found to show the highest percent inhibition over the 60 cell lines, with mean growth inhibition percentage of 45.44%. SAR studies revealed that compounds with p-cholorophenyl substitution at C-6 postion of triazolothiadiazine nucleus had better activity than analogs with an unsubstituted phenyl ring (Fig. 27). Mechanistic studies demonstrated that compound 113h selectively inhibited the glycogen synthase kinase-3β (GSK-3β), with IC50 = 0.883 μM and increased active caspase-3 levels. Further, compound 113h induced cell cycle arrest in G2/M phase and apoptosis in PC-3 prostate cancer-treated cells.

SAR and anticancer study of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives
Hu et al. [85] synthesized [1,2,4]triazolo[3,4‑b][1,3,4]thiadiazine derivatives (114) linked with antibacterial fluoroquinolones (FQ) at the 3-position. All synthesized derivatives were screened for their in vitro cytotoxic potential against murine leukemia (L1210) and Chinese hamster ovary (CHO) cell lines. Compound 114b having cyclopropyl substituted fluoroquinolones displayed better activity against L1210 cell lines, with an IC50 value of 0.14 µmol/L (Fig. 28).

SAR study of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives as potent anticancer agents
Gomha et al. [86] explored bis-triazolothiadiazines (115) as potent anticancer agents against human hepatocellular carcinoma (HepG-2) cell lines using harmine as a reference. Bioassay results showed that compounds 115i–k exhibited promising potency with IC50 value of 24.8, 28.3, and 31 μM, respectively. SAR studies demonstrated that compound substituted with naphthocoumarin (IC50 = 24.8) showed higher activity than a coumarin (IC50 = 28.3 μM), pyridine (IC50 = 31 μM) and pyrazole (IC50 = 59.0 μM) substituted analog (Fig. 29). Substitution of the phenyl ring with electron-donating (CH3 or OCH3) as well as electron-withdrawing groups (Cl, F or NO2) diminished the cytotoxic activity compared with the unsubstituted phenyl group. In docking studies, potential interactions of 115i and 115j within the active site of DYRK1A are observed.

SAR study of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives as potent anticancer agents
3.2 Antimicrobial Agents
Deohate et al. [87] accomplished the synthesis of 6,7-disubstituted phenyl-3-pyridin-4-yl-5H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (116) and screened them for antimicrobial activity. Screening results indicated that compounds 116b, 116e, f showed highest inhibition against bacterial strains Escherichia coli, Staphylococcus aureus, and Salmonella typhi (zone of inhibition = > 21 mm) and moderate activity against Aerobacter aerogenes (zone of inhibition = 17–20 mm). Most of the tested compounds were found inactive against Bacillus subtilis (zone of inhibition = 13–16 mm). A SAR study showed that compounds containing electron donating substituents on the phenyl ring at position-6 and -7 of triazolothiadiazine enhanced the biological response markedly in comparison with electron-withdrawing groups, as depicted in Fig. 30.

SAR study and antimicrobial properties of 6,7-disubstituted phenyl-3-pyridin-4-yl-5H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines
Penta et al. [88] synthesized pyrazolyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)-2H-pyran-2-one derivatives (117) and evaluated their in vitro antibacterial activity against S. aureus, B. subtilis, E. coli and Klebsiella pneumonia and antifungal activity against Candida albicans. Among the tested compounds, compound 117e showed better inhibition (zone of inhibition = 27–35 mm) than standard drugs (zone of inhibition = 25–32 mm). SAR study revealed that derivative containing electron-withdrawing group on phenyl ring exhibited most potent antimicrobial activity (Fig. 31).

Structure and antimicrobial properties of pyrazolyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)-2H-pyran-2-one derivatives
Mekala et al. [89] reported indole-based 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (118) and screened them for antifungal activity against Aspergillus fumigatus, C. albicans,Trichophyton rubrum and Trichophyton mentagrophytes. Amongst the tested compounds, compound 118c was highly potent against T. rubrum and T. mentagrophytes (MIC = 3.12 µg ml−1) while compound 118e showed high activity against C. albicans (MIC = 3.12 µg ml−1) even better than standard amphotericin-B (MIC = 6.25 µg ml−1). SAR studies revealed that the electron-withdrawing group enhanced the biological impact (Fig. 32).

SAR studies and antimicrobial properties of triazolothiadiazine analogs
Jadhav et al. [90] synthesized a series of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (119) linked with chromeno[4,3-b]pyridin-5-one moiety and screened for antibacterial potency against S. aureus, E. coli, B. cereus and antifungal activity against C. albicans and Aspergillus clavatus. Compound 119c showed significant antifungal effect against C. albicans (MIC = 5 µg ml−1), comparable to that of standard ciprofloxacin (MIC = 5 µg ml−1). Compound 119d showed good inhibitory activity against the bacteria B. cereus (MIC = 5 µg ml−1) and fungus A. clavatus (MIC = 5 µg ml−1 which was nearly the same as the standard ciprofloxacin and ketokonazole (MIC = 5 µg ml−1. SAR studies revealed that compounds containing chloro or methoxy substituent displayed high potency (Fig. 33).

SAR evaluation and antimicrobial properties of triazolothiadiazines containing chromeno[4,3-b]pyridin-5-one moiety
Iradyan et al. [91] carried out synthesis and antibacterial evaluation of 6-(2′-alkoxyphenyl)-3-aryl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (120) against S. aureus, Shigella flexneri and E. coli. Of the screened compounds, 120a and 120b showed the most promising results (zone of inhibition = 15.6–23.6 mm). SAR studies showed that propoxy group at ortho-position of the phenyl ring was accountable for promoting the activity (Fig. 34).

SAR appraisal and antibacterial properties of triazolothiadiazines derivatives
Ghosh et al. [92] introduced a series of triazolothiadiazines (121) bearing substituted phenylquinolin-2-one moiety and evaluated them for their antibacterial activity against Pseudomonas aeruginosa, E. coli, S. aureus, and antifungal activity against A. fumigatus, A. niger and C. albicans. SAR study revealed that triazolothiadiazine derivatives bearing phenyl quinoline moiety possessed moderate to good antimicrobial activities and halogen substituted compounds 121d, 121g and 121h showed the promising antifunfal activity against C. albicans with zones of inhibition of 20 mm, 23 mm and 19 mm, respectively (Fig. 35).

SAR and antimicrobial (inhibition zone) activities of triazolothiadiazine derivatives
El-Sayed et al. [93] reported synthesis of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (122) bearing a 1,2,3-triazole moiety and screened them for antimicrobial activity against bacterial and fungal strains. Out of the tested series, the most potent compound 122b exhibited moderate activity against bacterial strains of E. coli, P. aeruginosa, S. aureus, and B. subtilis (zone of inhibition = 14–18 mm) and fungal strains of C. albicans, and Aspergillus flavus (zone of inhibition = 17 and 19 mm) as compared with standard drugs. SAR study revealed the effect of different substituents on 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine scaffold for antimicrobial activity as depicted in Fig. 36.

SAR evaluation and antimicribial properties of triazolothiadiazine derivatives
Bhat et al. [45] reported the synthesis of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (123) and in vitro antifungal potential against various Candida species, namely C. albicans, C. tropicalis, C. parapsilosis and other Candida isolates (Candida sp. [HVS] 13,184, Candida sp. [HVS] 11,972, Candida sp. [HVS] 178, Candida sp. [urine] 300, Candida sp. [urine] 12,341, Candida sp. [urine] 12,485, Candida sp. [blood] 12,810). Among the screened derivatives, compounds 123a, 123c, 123e and 123g exhibited higher antifungal activity against some Candida species (MIC = 6.25–12.5 µg ml−1) than the standard drug ketoconazole (MIC = 7–30 µg ml−1). SAR studies indicated that p-chlorophenyl substitution on triazolothiadiazine ring positively influenced the anti-Candidal activity (Fig. 37).

SAR and anti-Candidal active triazolothiadiazines derivatives
Altintop et al. [94] synthesized new 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (124) and screened for their in vitro anti-Candidal activity. Among all compounds, 124i (R = cyclohexyl, R1 = Cl) exhibited more potent activity against C. albicans (ATCC 90,028) with a MIC value of 0.03 mg ml−1 in comparison to ketoconazole (MIC = 0.04 mg ml−1). SAR studies revealed the significance of cyclohexyl group and p-chlorophenyl substituent on triazolothiadiazine ring for anti-Candidal activity (Fig. 38).

SAR appraisal and anti-Candidal activities of triazolothiadiazines derivatives
Prakash et al. [95] synthesized two series of dihydroindeno (125) and indeno-clubbed 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (126) and evaluated their antimicrobial effects against S. aureus, B. subtilis, E. coli, P. aeruginosa, A. niger, A. flavus and Penicillium species. Bioassay results showed that dihydroindeno derivatives are more potent than indeno derivatives in terms of antimicrobial activity, and the tested compounds showed more potency towards Gram positive bacteria than towards Gram negative bacteria. Dihydroindeno compounds 125d–e, 125g–i and indeno compounds 126d, 126g, 126j exhibited noticeable antibacterial activities (zone of inhibition = 14.3–28.6 mm). In antifungal screening, compounds 125h, 126g and 126j exhibited highest potential (mycelial growth inhibition = 51.1–68.8%) compared with standard drug fluconazole. A brief SAR study is presented in Fig. 39.

SAR and activity values of dihydroindeno and indeno clubbed 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines
Badr et al. [96] accomplished the synthesis of 6-(4-substituted phenyl)-3-(5-nitrofuran-2-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine analogs (127) and screened their antibacterial potential against S. aureus and E. coli. Results revealed that the tested compounds showed good activity against S. aureus. Compounds 127a and 127b were found to be as effective (MIC = 25 µg ml−1) as the reference drug ampicillin (MIC = 25 µg ml−1) against S. aureus. From the SAR, it was deduced that halogen groups on the aryl ring are essential for biological response (Fig. 40).

SAR of 6-(4-substituted phenyl)-3-(5-nitrofuran-2-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines as antibacterial agents
Sumangala et al. [97] synthesized 6-aryl-3-[4-(methylsulfonyl)benzyl]-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (128) and evaluated their antibacterial activity against S. aureus, E. coli, P. aeruginosa, B. cereus and antifungal activity against C. albicans. Compounds 128d, 128i and 128h showed excellent antibacterial activity against E. coli (MIC = 3.125 mg ml−1), P. aeruginosa (MIC = 3.125 mg ml−1) and B. cereus (MIC = 3.125 mg ml−1), respectively. Compounds 128b, 128e, 128f and 128h exhibited good antifungal activity (MIC = 1.562–3.125 mg ml−1). A brief SAR of most active compounds is explained in Fig. 41.

SAR and antimicrobial potential of 6-aryl-3-[4-(methylsulfonyl)benzyl]-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines
Pundeer et al. [98] synthesized 7H-7-alkoxy-3-alkyl/phenyl-6-aryl-s-triazolo[3,4-b][1,3,4]thiadiazine derivatives (129) and tested for antimicrobial potency against S. aureus, B. subtilis, E. coli, P. aeruginosa, Saccharomyces cerevisiae and C. albicans. Compounds 129f and 129l exhibited maximum inhibition against S. aureus, with MIC = 16 and 16 µg ml−1, respectively, while compound 129i generated good inhibition against B. subtilis with MIC = 32 µg ml−1. In antifungal screening, compounds 129c, 129f and 129i exhibited potent activity (MIC = 32–64 µg ml−1) in comparison with the standard drug amphotericin-B (MIC = 100 µg ml−1). A SAR study revealed the effect of substitution on the scaffold (Fig. 42).

SAR examination and antimicrobial screening results of 7H-7-alkoxy-3-alkyl/phenyl-6-aryl-s-triazolo[3,4-b][1,3,4]thiadiazines
Puthiyapurayil et al. [77] developed a novel 6-arylsubstituted-3-[2-(4-substituted phenyl)propan-2-yl]-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (130) and evaluated antimicrobial potential against E. coli, S. aureus, P. aeruginosa, B. subtilis, Rhizopus oryzae, A. fumigatus, A. niger and T. mentagrophytes. Bioassay results revealed that, out of the evaluated derivatives, compounds 130e, 130j and 130m showed equipotent antibacterial and antifungal activity to the standard streptomycin and amphotericin-B (MIC = 6.25 µg ml−1). SAR studies explained that, due to their increased lipophilicity, highly electronegative substituents (F and Cl) facilitate hydrophobic interactions of the compounds with specific binding sites on either receptor or enzymes (Fig. 43).

SAR assesment and antimicrobial assay of 6-arylsubstituted-3-[2-(4-substitutedphenyl)propan-2-yl]-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines
Purohit et al. [99] reported a new series of 3-(3-chlorophenyl)-6-aryl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (131) and evaluated their antimicrobial effects against B. megaterium, S. aureus, E. coli, P. aeruginosa, and A. niger. Among the tested series, compounds 131g and 131i showed significant activity against bacterial and fungal strains (zone of inhibition = 18–24 mm) while 131c and 131f exhibited promising antibacterial activity against B. megaterium, P. aeruginosa and E. coli (zone of inhibition = 16–20 mm). The pharmacological assessment and SAR of active compounds are given in Fig. 44.

SAR and antimicrobial studies of triazolothiadiazine derivatives
Sim et al. [100] synthesized and screened pyrimidine linked 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (132) for their antibacterial potential against B. subtilis, B. cereus, S. aureus, P. aeruginosa, S. typhimurium, and E. coli. Screening results indicated that compound having 4-flurophenyl ring 132f demonstrated moderate activity against P. aeruginosa with MIC = 62.5 μg ml−1 (Fig. 45).

SAR appraisal and antibacterial studies of pyrimidine linked triazolothiadiazine derivatives
Almajan et al. [101] accomplished the synthesis of phenylsulfonylphenyl containing 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (133) and assessed their in vitro antibacterial potential against the Gram-positive (E. faecalis, S. aureus, S. epidermidis, B. cereus) and Gram-negative (A. baumanii, C. freundii, E. clocae, E. coli, P. aeruginosa) bacterial strains through the broth dilution method. Compound 133a was found to be most potent derivative against S. epidermidis with MIC = 32 µg ml−1 (Fig. 46). Bioassay results demonstrated that screened derivatives exerted better inhibition against Gram-positive bacterial strains than Gram-negative bacterial strains.

Antibacterial evaluation and SAR of triazolothiadiazine derivatives
Reddy et al. [102] synthesized 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (134) and evaluated their in vitro antibacterial activity. Among them, compound 134c exhibited promising antibacterial activity against E. coli, K. pneumoniae, S. dysentriae, and S. flexnei with zone of inhibition value of 30 mm, 29 mm, 29 mm, and 27 mm, respectively, which was more active than standard neomysin (inhibition zone = 20 mm) and equipotent to streptomycin (inhibition zone = 30 mm) (Fig. 47).

SAR and antibacterial studies of triazolothiadiazine derivatives
Kumar et al. [103] prepared a series of 3-(4-isopropylthiazol-2-yl)-6-substituted phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (135) and evaluated them for antimicrobial activity against S. aureus, S. faecalis, B. subtilis, K. pneumoniae, E. coli and P. aeruginosa and antifungal activity against S. cerevisiae, C. tropicalis and A. niger. Compound 135b revealed significant antimicrobial potential against tested bacterial and fungal strains (MIC = 8, 4, 8, 16, 16, 8, 16, 31.25, 16 µg ml−1 against S. aureus, S. faecalis, B. subtilis, K. pneumoniae, E. coli, P. aeruginosa, S. cerevisiae, C. tropicalis and A. niger, respectively) (Fig. 48).

SAR and antibacterial evaluation of isopropylthiazole clubbed derivatives of triazolothiadiazine
Indole clubbed derivatives of triazolothiadiazine (136) were evaluated for their antibacterial potential against B. subtilis, B. cereus, S. aureus, M. luteus, E. coli, P. aeruginosa, S. typhimurium and P. vulgaris by Sim et al. [104]. Compound 136d was found to show significant inhibition against B. subtilis, B. cereus and S. aureus with MIC = 3.91 μg ml−1. SAR studies revealed the importance of chlorine on indole moiety and p-methylphenyl group at C-6 position of triazolothiadiazine ring (Fig. 49).

SAR and antibacterial studies of indole clubbed triazolothiadiazines as potential antibacterial agents
3.3 Analgesic and Anti-inflammatory Agents
Puthiyapurayil et al. [105] introduced amide linked 3,6-disubstituted-7H-1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (137) and screened them for their in vivo analgesic and anti-inflammatory potential using acetic acid-induced writhing test and carregeenan-induced rat hind paw edema, respectively. From the tested series, compounds 137a, 137b and 137c exhibited potent and consistent analgesic activity in both 100 and 200 mg kg−1 po−1 doses with less ulcerogenic risk (inhibition = 28–43%) as well as excellent anti-inflammatory activity (at a dose of 50 mg kg−1) (edema inhibition = 38–61%). SAR studies demonstrated that derivatives with N-methyl-[4-(trifluoromethyl)phenyl]pyrazole moiety at the position-3 of the fused ring are more potent than the other derivatives bearing [1-(4-chlorophenyl)-1-methylethyl] as shown in Fig. 50. Compound 137b also displayed promising cytotoxic effect against human breast cancer (MCF-7) cell line with IC50 value 14.24 µM.

SAR assessment and pharmacological evaluation results of triazolothiadiazines derivatives
Shehry et al. [106] reported synthesis and anti-inflammatory activity of 3-(2,4-dichlorophenoxy)methyl)-1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (138). Compounds 138a and 138b exhibited potent anti-inflammatory activity (edema inhibition = 59–60% at 3 h). The percent change in paw volume compared with base line measurement was recorded after interval of 1 h. SAR studies demonstrated that 2,4-dichlorophenoxy at the 3-position of triazolothidiazine ring was found crucial for biological effect (Fig. 51).

SAR study and anti-inflammatory potential of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives
Hussein et al. [107] synthesized and evaluated 4-substituted phenyl-3-(pyridin-4-yl)-7H-(1,2,4)triazolo[3,4-b]-[1,3,4]thiadiazine derivatives (139) for their analgesic and anti-inflammatory effects. Among them, compounds 139d, 139e and 139f (0.03 mmol kg−1) displayed maximum anti-inflammatory effect at 5 h, with inflammatory inhibition rates of 75% and 84.85 and 75.31%, respectively. Moreover, compounds 139d–f (0.02 mmol kg−1) exhibited potent analgesic effects with 79–145% activity compared to indomethacinin at the 5 h interval. Figure 52 illustrates a brief SAR and activity profile of most active compounds.

SAR and analgesic/anti-inflammatory evaluation of triazolothiadiazines derivatives
Koksal et al. [17] reported a series of 3,6-diaryl-1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (140) for their anti-inflammatory activity. The outcome showed that compounds 140c–h and 140j were found to have better inhibitory potency (50.2–62.8%) at different time interval with less ulcerogenic risk than the standard indomethacin (49.2% in 120 min, 54.9% in 240 min). SAR studies were carried out to determine the active functional groups at position-6 of triazolthiadiazine ring in order to enhance potency (Fig. 53).

SAR study and anti-inflammatory activity of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives
Naveena et al. [108] reported the synthesis and anti-inflammatory activity evaluation of novel 6-substituted-3-(2-(aryloxymethyl)phenyl)[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives (141). Bioassay results showed that the tested compounds displayed anti-inflammatory activity ranging from 40.52% to 70.88%. Compounds 141a–e exhibited significant anti-inflammatory activity with 68.65–70.88% inhibition. A SAR study demonstrated that substitution with 5-chloro-2-sulfanamido-3-thienyl and 4-nitrophenyl groups at position-6 of triazolothiadiazine scaffold enhanced the anti-inflammatory activity (Fig. 54).

SAR studies and anti-inflammatory data of active triazolothiadiazines derivatives
3.4 Antioxidant Agents
Ranganatha et al. [109] synthesized diarylmethanone linked 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (142) and evaluated their in vitro antioxidant potential by DPPH, nitric oxide and hydrogen peroxide scavenging assay. Among the tested series, compounds 142b, 142c and 142d exhibited significant antioxidant effect in DPPH (2,2-diphenyl-1-picryl-hydrazyl-hydrate) test with the IC50 values of 18.33, 16.32, 16.55 µg ml−1, respectively, in comparison with standard ascorbic acid (IC50 = 14.98 μg ml−1). SAR studies were carried out to determine the active functional groups in order to enhance the potency (Fig. 55).

SAR and antioxidant potential of active triazolothiadiazines derivatives using the DPPH (2,2-diphenyl-1-picryl-hydrazyl-hydrate) method
Patrao et al. [110] synthesized and screened antioxidant activity of a cluster of α and β-naphthoxy methyl linked 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (143) using the DPPH assay. The results indicated that, among the tested derivatives, compound 143b exhibited highest antioxidant potential, with inhibition of 60% at a concentration of 100 μg ml−1. SAR studies demonstrated that derivatives linked with α-naphthoxy group exhibited enhanced activity in comparison with β-naphthoxy derivatives (Fig. 56).

SAR and radical scavenging activity of triazolothiadiazines derivatives
Kaushik et al. [111] prepared 3-substituted phenyl-6-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives (144) and screened their antioxidant activity on the basis of the scavenging activity of the stable DPPH free radical. Compounds 144a and 144e possess significant antioxidant potential with IC50 values of 17.27 and 17.05 µg ml−1, respectively. SAR studies revealed that antioxidant activity of compounds may depend on the basic skeleton of the molecule as well as on the nature of substituents (Fig. 57).

SAR and antioxidant activities of triazolothiadiazines derivatives
Shakir et al. [112] reported synthesis and antioxidant activity evaluation of 6-amino-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl derivatives (145) bearing 2,6-dimethoxy-4-(methoxymethyl)phenol moiety by DPPH and ferric reducing antioxidant power (FRAP) assays. Among the screened compounds, compound 145b exhibited remarkable antioxidant ability in both assays (DPPH inhibition = 92%; IC50 = 56 µg ml−1 which is higher than standard butylated hydroxytoluene (BHT) (DPPH inhibition = 66%; IC50 = 78 µg ml−1). SAR studies revealed that compounds 145a–c exhibited higher antioxidant potential with inhibition in the range 79–83% due to presence of thiourea moiety than compounds 145d–f containing amide group with 68–75% inhibition (Fig. 58).

SAR and antioxidant profile of active triazolothiadiazines derivatives
Sathyanarayana et al. [113] designed and synthesized benzimidazole linked 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (146) and assessed their in vitro antioxidant potential using DPPH, nitric oxide and superoxide radical scavenging methods. Among them, compound 146c showed more activity in both DPPH and nitric oxide radical assay, with IC50 values of 13.57 and 18.55 μg ml−1 compared with standard (IC50 = 23.75 and 23.14 μg ml−1, respectively). SAR studies demonstrated that derivatives substituted with electron-donating groups exhibited enhanced activity in comparison with electron-withdrawing groups (Fig. 59).

SAR and antioxidant studies of triazolothiadiazines derivatives
3.5 Antiviral Agents
Pavurala et al. [114] synthesized a series of bis coumarinyl bis triazolothiadiazinyl ethane derivatives (147) and screened their antiviral activity against a broad spectrum of RNA and DNA viruses (herpes simplex virus-1 (KOS), herpes simplex virus-2 (G), vaccinia virus, vesicular stomatitis virus, herpes simplex virus-1TKKOSACVr, adenovirus-2, vesicular stomatitis virus, coxsackie virus B4, respiratory syncytial virus, para-influenza-3 virus, reovirus-1, sindbis virus, punta torovirus, feline coronavirus (FIPV), feline herpes virus, influenza A H1N1 subtype, influenza A H3N2 subtype, and influenza B) in HEL, HeLa, Vero, and CRFK cell medium. Out of 11 tested compounds, 147k exhibited good activity against influenza A virus. In a cytopathic effect (CPE) reduction assay, compound 147k exerted inhibitory effects against influenza virus infected Madin–Darby canine kidney (MDCK) cells, with EC50 values of 20–72 μM. Docking results revealed that compound 147k (MoleDock score = −173 kcal mol−1) inhibits the H1N1 neuraminidase enzyme via hydrogen bonding, electrophilic and hydrophobic interactions in the catalytic unit of H1N1 neuraminidase (Fig. 60). SAR studies revealed that compounds having an aromatic ring on a coumarin nucleus exhibit better antiviral activity than other substituents such as chloro, bromo or methoxy substituents.

Anti-influenza viral activity of bis coumarinyl bis triazolothiadiazinyl ethane derivatives
Pandey et al. [50] introduced a series of 5-[(3′-aralkyl amido/imidoalkyl) phenyl]-1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (148) and screened their in vitro antiviral potential against two animal viruses, namely Japanese encephalitis virus (JEV) strain P20778 and Herpes simplex virus-1 (HSV-1) strain 753,166. Screening results revealed that compound 148c, with a 2-phenyl-3-methyl-quinazolin-(3H)-4-one group, is mainly responsible for exerting antiviral activity against JEV (EC50 = 7.8 µg ml−1 inhibition = 50%; therapeutic index = 32) while compounds 148a, 148b and 148d containing phthalimido, phthalimidomethyl, and nicotinamido substituents, respectively, exhibited weak activity (10–20%) against JEV and HSV-I (Fig. 61).

Antiviral activity of 5-[(3′-aralkyl amido/imidoalkyl) phenyl]-1,2,4-triazolo[3,4-b][1,3,4]thiadiazines
Khan et al. [115] synthesized 6-aryl-3-(furan-2-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives (149) and assessed their in vitro anti-HIV-1 (strain IIIB) and anti-HIV-2 (strain ROD) activity by monitoring the inhibition of the virus-induced cytopathic effect in MT-4 cells. Bioassay results indicated that compound 149f exerted inhibitory effects against replication of human immunodeficiency virus (HIV)-1 and -2 in cell cultures with EC50 of >0.67 μm, but no selectivity was witnessed (SI < 1) (Fig. 62). Compound 149 g with a nitro group at 4 position of the phenyl ring exhibited less anti-HIV activity (EC50 = > 83.20 μm).

SAR estimation and anti-HIV activity of furan linked triazolothiadiazines derivatives
3.6 Enzyme Inhibitors
SitaRam et al. [116] synthesized 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (150) containing benzenesulfonamide moieties and screened their inhibitory potential against four isomers of the α-carbonic anhydrases (CAs) comprising hCAs I and II (cytosolic, ubiquitous isozymes) and hCAs IX and XII (transmembrane, tumor-associated isozymes). Bioassay results revealed that compound 150a exhibited excellent inhibitory potential against all four isozymes (Ki = 3.0–89 nM), 150b inhibited isozymes hCA I, IX and XII (Ki = 84, 6.5 and 3.8 nM, respectively) while 150c inhibited isozymes hCA IX and XII (Ki = 25 and 18 nM, respectively). Compounds 150d–e selectively inhibited hCA IX (Ki = 21–25 nM) even better than the standard drug acetazolamide (Ki = 25 nM). SAR revealed the effect of different substituents on 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine for hCA inhibition (Fig. 63).

SAR and carbonic anhydrases inhibition of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives
Alafeefy et al. [117] introduced a series of benzenesulfonamides incorporating aroylhydrazone-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazinyl group (151) and examined their inhibitory potential against four CAs, the human (h) isoforms hCA I, II, IX and XII. Bioassay results revealed that compound 151a presented significant inhibitory effects against hCA I (Ki = 86.4 nM) (threefold more effective than the standard acetazolamide; Ki = 250 nM)) and hCA XII (Ki = 4.3 nM) while 151b exhibited inhibition against hCA II and hCA XII (Ki = 0.93 and 0.85 nM, respectively) (Fig. 64).

SAR and pharmacokinetics of triazolothiadiazine derivatives as carbonic anhydrase inhibitors
Khan et al. [62] screened 3-(pyridin-4-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives (152) for cholinesterase inhibition against electric eel acetylcholinesterase (EeAChE) and horse serum butyrylcholinesterase (hBChE) as well as for alkaline phosphatase inhibition. Results indicated that compound 152f (IC50 = 13.66 ± 0.31 µM) exhibited remarkable inhibition against EeAChE, while compounds 152g (IC50 = 16.6 ± 2.72 µM) and 152h (IC50 = 12.2 ± 1.67 µM) showed greater inhibitory potential against BChE more than the standard drug neostigmine. Compound 152h also exhibited significant inhibitory effect on alkaline phosphatase with IC50 = 1.32 ± 0.20 µM. SAR studies revealed the role of different functional groups on the triazolothiadiazine nucleus as shown in Fig. 65. In another work, evaluation of 3-(pyridin-3-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (Fig. 65) for cholinesterase inhibitory activity by Khan et al. [63] revealed that compound 153g (IC50 = 15.1 ± 0.213 µM) and 153h (IC50 = 1.09 ± 0.156 µM) exerted strong inhibitory effects on EeAChE and hBChE, respectively.

SAR, cholinesterase and alkaline phosphatase inhibitory activities of triazolothiadiazines
Ozil et al. [118] synthesized triazole-linked bis([1,2,4]triazolo[3,4-b][1,3,4]thiadiazine) derivatives (154) and assessed anti-lipase activity with regard to pancreatic lipase inhibition. Among them, compounds 154a–c showed considerable anti-lipase activity at various concentrations (inhibition = 90–94%; IC50 = 0.11–0.42 µg ml−1). SAR studies identified the significance of R groups on triazolothiadiazine ring for inhibition of pancreatic lipase (Fig. 66).

SAR and anti-lipase activity of triazolothiadiazine derivatives
Recently, Çevik et al. [119] introduced benzimidazole-triazolothiadiazine derivatives (155) as aromatase inhibitors (IC50 = 0.037 ± 0.001 μM to 1.345 ± 0.061) using fluorimetric assay kit with letrozole as the reference drug. Among tested compounds, compound 155g was found to be most promising inhibitor against aromatase enzyme (IC50 = 0.037 μM), which was slightly less potent than standard drug letrozole (IC50 = 0.024 μM) (Fig. 67).

SAR analysis and effect of benzimidazole linked triazolothiadiazine derivatives as aromatase inhibitors
A series of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines (156) was explored as dual inhibitors of cholinesterase (ChE) and monoamine oxidase (MAO) by Khan et al. [120]. Bioassay results revealed that compounds 156e and 156h exhibited high selectivity towards EeAChE, with IC50 values of 0.065 and 0.075 μM, respectively, which is around 250- and 218-fold stronger inhibition than the standard drug neostigmine (IC50 = 16.3 μM). Compound 156c showed ~330-fold more inhibition than the standard drug clorgyline (IC50 = 3.64 μM) towards MAO-A, with IC50 values of 0.011 μM, while compound 156n exhibited remarkable selectivity for MAO-B (IC50 = 0.08 μM). SAR revealed the effect of different substituents on 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine on ChE inhibition and AChE/ BChE selectivity (Fig. 68).

SAR and exploration of triazolothiadiazine derivatives as cholinesterase (ChE) and monoamine oxidase (MAO) inhibitors
Baeeri et al. [121] synthesized 3,6-diaryl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines (157) as potential phosphodiesterase-4 inhibitors. Among the tested derivatives, compound 157d exerted remarkable specificity for PDE-4 by enhancing the concentration of cAMP (ED50 = 13 µM), however the level of cGMP remains unaffected (ED50 = 49.8 µM) (Fig. 69). Docking studies also validated the selective binding of these compounds in cAMP-specific PDE enzymes binding site.

SAR and evaluation of triazolothiadiazine derivatives as phosphodiesterase-4 inhibitors
3.7 Antitubercular Agents
Li et al. [122] reported new analogs of 3,6-disubstituted 1,2,4-triazolo[3,4‐b][1,3,4]thiadiazine (158) and examined their antitubercular activities against Mycobacterium tuberculosis (Mtb) H37Rv, multi-drug-resistant (MDR)-TB and RDRTB (rifampin-resistant strains) by the BacT/ALERT 3D liquid culture technology using rifampin and isoniazid as the standard drugs. According to the results of antitubercular determination, compounds 158a, 158i, 158k, 158r, 158t and 158o exhibited most potent activity (MIC = 0.25–4 µg ml−1; IC50 = 28–252 µg ml−1). A SAR study was carried out to correlate the effect on activity of aromatic or hetero-aromatic substituents at the 3- and 6-position of triazolothiadiazine, which is presented in Fig. 70.

SAR and antitubercular activity of 3,6-disubstituted 1,2,4-triazolo[3,4‐b][1,3,4]thiadiazine derivatives
Kincaid et al. [123] identified 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives (159) as antimycobacterial agents via inhibition of uridine 5′-diphosphate-galactopyranose mutase (UGM) by means of virtual screening (docking and X-ray crystallography studies). Bioassay outcomes showed that compounds 159g–j (Kd = 5–15 µM; inhibition = 97–100%) possessed the most promising activity a,nd compound 159j fully blocked the K. pneumoniae UGM activity at 50 μM. These derivatives also contributed antimicrobial potency against Mtb and Mycobacterium smegmatis. SAR studies of the derivatives revealed that compounds with larger or more polarizable phenyl ring on the position-6 and larger side-chains at position-3 of triazolothiadiazine ring exhibited effective inhibition (Fig. 71). Docking and X-ray structure showed consistency with the observed SAR.

SAR and uridine 5′-diphosphate-galactopyranose mutase (UGM) inhibitory potential of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine derivatives
4 Rational Design Based on SAR of Triazolothiadiazine Derivatives
The present review captures the diverse biological activities exhibited by triazolothiadiazines reported in the literature. After careful analysis, general structural features have been identified that can be used in the design and development of novel triazolothiadiazine deivatives having clinical efficiency. Unfused 5-membered triazole ring systems were found to be biologically inactive; however, fusion of the 5-membered triazole ring with 6-membered thiadiazine ring is important for bioactivity [59, 84]. Unsaturation in the 6-membered thiadiazine ring fueled bioactivity, in comparison with saturated derivatives [84]. At position-3 of the triazole ring, aromatic substitution connected through a spacer (0–3 atoms) exhibited better activity. Aromatic substituents at position-6 of the triazolothiadiazine ring displayed improved potency over the aliphatic analogs. Halogen-substituted aryl groups, specifically chloro-substituted aryl results in better activity than electron-donating groups, as aliphatic substitution bring down the activity. Substitution at position-7 of the triazolothiadiazine ring can be important for activity, but is not essential. Figure 72 summarizes the structural modifications in terms of different substituents at position-3, 6 and 7 (R1, R2 and R3) with respect to various biological activities, namely anticancer, antiviral, antimicrobial, antioxidant, antitubercular, enzyme inhibitiors, analgesic and anti-inflammatory activities.

Structural modifications on triazolothiadiazine scaffold and their effect on bioactivities
5 Pharmacokinetic Profile
Pharmacokinetics is the study of processes by which drugs are absorbed, distributed, metabolized and eliminated from the body. Performing standard experimental procedures for absorption, distribution, metabolism and excretion (ADME) profiling for a vast number of compounds would have been time-consuming and economically unfeasible. Most drug molecules fail to enter the market due to their adverse pharmacokinetic properties. Recently, computer simulation platforms are gradually becoming popular as they can accelerate the pharmacokinetic development of the drug molecule [124, 125]. In the primary phase of drug discovery, in silico approaches for the prediction of pharmacokinetics of drug candidates play an essential role in terms of cost effectiveness, time factors and avoidance of the ethical problems that arise from a large number of animals required for experiments. A successful lead should obey Lipinski's rule of five, which states that the drug should be easily absorbed, distributed to its target site, not easily metabolized, and excreted easily.
The drug-likeness of a compound was assessed according to Jorgensen’s rule of three [126] and Lipinski’s rule of five [127], which indicates that a chemical substance can be orally active in humans by estimating structural or physio-chemical properties (descriptors) which are important for drug pharmacokinetics. Jorgensen’s rule of three states that Caco-2 cell permeability (PCaco) > 22 nm/s, predicted aqueous solubility (PlogS) > − 5.7 and number of primary metabolites < 7. Lipinski’s rule of five states that orally active drug has no more than one violation of the following criteria: molecular weight < 500 Da; hydrogen bond donors ≤ 5; hydrogen bond acceptors ≤ 10; and octanol/water partition coefficient (log P) ≤ 5 (or MlogP not greater than 4.15) [128, 129]. Topological polar surface area (TPSA) ≤ 140 Å2 and the number of rotatable bonds ≤ 10 are also considered additional criteria used to determine oral bioavailability [130].
Triazolo[3,4-b][1,3,4]thiadiazine derivatives have been identified as compounds with broad-spectrum activity, high safety, and excellent pharmacokinetic properties in a variety of studies. Therefore, analysis of arrangement of substituents around triazolothiadiazine scaffold (Fig. 73) might provide useful information to construct novel triazolothiadiazine derivatives with better biological proficiency.

Structural variations on triazolothiadiazine moiety and their predicted pharmacokinetic profile
Çevik et al. [76] calculated ADME parameters of benzimidazole-triazolothiadiazine derivatives 105 by QikProp 4.8 software for comparing with those of 95% of known drugs. All compounds 105 were found in accordance with the Jorgensen’s rule of three and Lipinski’s rule of five for drug-likeness properties by causing no more than one violation. According to predictions of ADME properties for compounds 105e and 105l, including molecular weight 433.489 and 467.934, respectively, the number of rotatable bond values (1), only one hydrogen bond donor group, hydrogen bond acceptors 6 and logP 4.468 and 4.96, respectively, it can be suggested that the active compounds 105e and 105l have desirable pharmacokinetic properties.
Sathyanarayana et al. [113] reported that novel [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives 146 bearing benzimidazole moiety obeyed most of the ADME parameters. Most of these compounds showed TPSA < 80 Å2, indicating their good intestinal absorption. The most potent antioxidant 146c showed excellent percentage human oral absorption (100%). Instead of having molecular weight ≥ 500 Da, these compounds have high percentage of human oral absorption and a violation of Lipinski’s rule of five will not affect oral absorption [131]. QikProp predicted the number of possible metabolic reactions of these compounds in the range 2–5 (recommended range 1–8), indicating the compounds can easily reach the target site. Moreover, the LogKp values for compounds 146 range from −1.35 to −3.26 (acceptable range −8.0 to −10.0), which indicates that compounds 146 can be administrated dermally.
In silico ADME properties of the most selective triazolothiadiazine derivatives 160a and 160b as a new oral anti-inflammatory agent were accessed using the SwissADME web tool by Abdellatif et al. [132], which revealed that both compounds violated Lipinski’s rule of five by two represented by molecular weight (589.94 and 663.61 Da, respectively) and MlogP values (6.46 and 4.55, respectively). Consequently, these triazolothiadiazine derivatives have lower bioavailability scores (0.17) compared with that of the reference drug celecoxib (0.55).
Alsaedi et al. [133] synthesized triazolo[3,4-b][1,3,4]thiadiazine derivative 161 as potent VEGFR2 inhibitor with anti-hepatocellular carcinoma activity and predicted pharmacokinetic properties using SwissADME tool. Compound 161 showed good gastrointestinal (GI) absorption, high oral bioavailability, and fitted well in the Lipiniski’s rule. ADME prediction revealed that compound 161 has a desirable pharmacokinetic profile and can be considered as a hopeful drug candidate.
Sever et al. [134] predicted pharmacokinetic profiles of triazolothiadiazine derivatives using QikProp program of Schrodinger’s Maestro molecular modelling package. Compounds exhibited human oral absorption percentages ranging from 91.010% to 100.000%. All compounds comply with Lipinski’s rule of five and Jorgensen’s rule of three. On the basis of these findings, they are expected to possess favorable oral bioavailability and drug-like properties. According to in silico ADME studies for triazolothiadiazine 162, the total solvent accessible surface area (SASA) was 622.356 Å2, the predicted octanol/water partition coefficient (logPo/w) was 5.224, logarithm of predicted binding constant to human serum albumin, logKHSA value was 0.957 and the predicted apparent Caco-2 cell permeability PCaco-2 was 1763.718. These results were in agreement with recommended range, thus indicating compound 162 to possess a favorable ADME profile.
6 Molecular Modeling
In receptor-based drug design, the 3D structure of the binding site is modelled on the target with the aid of molecular simulations. Docking experiments are carried out to identify the binding sites through computationally model ligand–receptor interactions as well as lead candidates from the binding energy scores [135]. A literature survey reflected the prominence of the 1,2,4-triazolo[3,4‐b][1,3,4]thiadiazine structural motif in the drug designing aimed at diverse pharmacological activities by its robust interactions with various receptors. Extensive research has focused on structural attributes of 1,2,4-triazolo[3,4‐b][1,3,4]thiadiazines responsible for its bioactivity on particular targets via computational studies (docking and molecular dynamics). Structural variations on triazolothiadiazine scaffold and their interactions with different biological receptors are shown in Fig. 74. 1,2,4-Triazolo[3,4-b][1,3,4]thiadiazine derivative 111j exhibited promising anticancer potential, which is supported by several interactions of its core nucleus with amino acids Lys352, Ala250, Lys254, Leu248, Asn258 and the H-bond between oxygen atom of its trimethoxyphenyl ring and the thiol group of Cys241 in the colchicine binding site of tubulin polymerase [82]. The methylpropylphenyl moiety of triazolothiadiazines 88b–d inserted into a hydrophobic basin formed by Val349, Leu352, Phe518 and Ile523 amino acids of the COX-1 protein to form a hydrophobic interaction [59]. In compound 113h, the N2 of the triazole ring and NH at position-3 interacted with Val135 via two H-bonds inside the ATP binding pocket of GSK-3β, one of the targets of anticancer activity of this compound [84]. Compound 104a displayed H-bond interactions with Asp479 and Lys456 present at ATP catalytic binding domain of Topo-Iiβ [75]. Docking studies displayed that compound 102a formed strong H-bond interactions with Ser93, His321 and His154 in active site of tissue-nonspecific alkaline phosphatase (h-TNAP), while compound 102d formed important interactions with the active site of intestinal alkaline phosphatase (h-IAP), which included two H-bonds between two nitrogen atoms of triazole ring and Arg166 and metal interaction of carbonyl group of coumarin core with Zn2+ [73]. Docking studies indicated that compound 105e could inhibit aromatase enzyme through an H-bond interaction of CN group at the para-position of the phenyl ring with Ser314. Besides, benzimidazole ring formed two π–π interactions with Arg115 and Phe134 and the triazolothiadiazine ring could interact by a salt bridge with the Hem600 molecule and Cys437 [76].

Effect of structural modifications of triazolo[3,4‐b][1,3,4]thiadiazine on molecular docking simulations with various receptors
Further modifications and changes in substitution patterns at the C-3 and C-6 positions could result in higher affinity towards different receptors with additional interactions. This wide statistics will impart directions to researchers in innovating target selective, optimized and diverse 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine analogs for the treatment of multifactorial diseases.
7 Conclusion
In conclusion, the present review article deliberated the various synthetic protocols engaged for synthesis of diverse array of triazolothiadiazine analogs via multi-component reactions (MCRs), conventional heating and visible-light-mediated organic reactions. Moreover, the triazolothiadiazine scaffold is an important structural motif in biologically active compounds, exhibiting more potency than standard marketed drugs. This review presented an in-depth analysis of medicinal aspects of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines for different biological activities such as anticancer, antimicrobial, analgesic and anti-inflammatory, antioxidant, antiviral, enzyme inhibitors and antitubercular activities. The activity profile of this scaffold particularly offers structural modifications at the C3 and C6 positions of the triazolothiadiazine ring to achieve new analogues. This study also revealed the structural diversity, biological importance and SAR examination of heterocyclic compounds containing a triazolothiadiazine nucleus, which will help the practitioners in further exploration of biological efficacy.
References
Aggarwal R, Sumran G (2020) An insight on medicinal attributes of 1,2,4-triazoles. Eur J Med Chem 205:112652
Kulikov AS, Epishina MA, Fershtat LL, Makhova NN (2018) Effective synthesis of 7H–1,2,4-triazolo[3,4-b][1,3,4]thiadiazines. Chem Heterocycl Compd 54:669–672
Holla B, Kalluraya B, Sridhar K, Drake E, Thomas L, Bhandary K, Levine M (1994) Synthesis, structural characterization, crystallographic analysis and antibacterial properties of some nitrofuryl triazolo[3,4-b]-1,3,4-thiadiazines. Eur J Med Chem 29:301–308
Ergenc N, Ulusoy N, Çapan G, Sanis GÖ, Kiraz M (1996) Synthesis and antimicrobial properties of new 4-(alkylidene/arylidene)-amino-5-(2-furanyl)-2,4-dihydro-3H-1,2,4-triazole-3-thiones and 6-aryl-3-(2-furanyl)-7H-1,2,4-triazolo[3,4-b][1,3,4]thiadiazines. Arch Pharm 329:427–430
Molina P, Alajarín M, de Veg MJP, de la Concepción F-F, Cano FH, Claramunt RM, Elguero J (1987) Synthesis of 6,7-dihydro-5H-1,2,4-triazolo[3,4-b][1,3,4]thiadiazines by a C–C ring cyclization under mild conditions. J Chem Soc Perkin Trans 1:1853–1860
Karabasanagouda T, Adhikari AV, Shetty NS (2007) Synthesis and antimicrobial activities of some novel 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles and 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines carrying thioalkyl and sulphonyl phenoxy moieties. Eur J Med Chem 42:521–529
Hui XP, Zhang LM, Zhang ZY, Wang Q, Wang F (2000) Synthesis and antibacterial activity of s-triazoles, s-triazolo[3,4-b]-1,3,4-thiadiazines and s-triazolo[3,4-b]-1,3,4-thiadiazoles of 5-methylisoxazole. J Chin Chem Soc 47:535–539
Jin JY, Zhang LX, Chen XX, Zhang AJ, Zhang HI (2007) Syntheses and biological activities of 6-aryl-3-(3-hydroxy-propyl)-7H-1,2,4-triazolo[3,4-b][1,3,4]thiadiazines. Molecules 12:297–303
Singh SP, Naithani R, Aggarwal R, Prakash O (1998) A facile [hydroxy(tosyloxy)iodo]benzene mediated synthesis of symmetrical triazolo-[3,4-b]-1,3,4-thiadiazines. Synth Commun 28:3133–3141
Ibrahim YA, Elwahy AHM, El-Fiky AEM (1994) Stereospecific synthesis of 6,7-dihydro-5H-1,2,4-triazolo[3,4-b][1,3,4]thiadiazines. Heteroat Chem 5:321–325
Shawali AS, Zeid IF, Abdelkader MH, Elsherbini AA, Altalbawy FMA (2001) Synthesis, acidity constants and tautomeric structure of 7-arylhydrazono[1,2,4]triazolo[3,4-b][1,3,4] thiadiazines in ground and excited states. J Chin Chem Soc 48:65–72
Xiong Y, Zhang L, Zhang A, Xu D (2002) Synthesis of 6-aryl-3-(d-gluco-pentitol-1-yl)-7H-1, 2, 4-triazolo-[3,4-b][1,3,4]thiadiazines. Synth Commun 32:3455–3459
Motamedi R, Heravi MM, Bamoharram FF, Haeri A (2008) Facile routes to 1,2,4-triazolo[3, 4-b][1,3,4]thiadiazines and 1,2,4-triazino[3,4-b][1,3,4]thiadiazine by heteropolyacides. J Heterocycl Chem 45:1211–1214
Foroughifar N, Mobinikhaledi A, Ebrahimi S (2010) Efficient and convenient protocol for the synthesis of novel 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines. Synth Commun 40:2421–2428
Al-Etaibi A, John E, Ibrahim MR, Al-Awadi NA, Ibrahim YA (2011) Stereoselective synthesis of dihydrothiadiazinoazines and dihydrothiadiazinoazoles and their pyrolytic desulfurization ring contraction. Tetrahedron 67:6259–6274
Chunduru VSR, Rao VR (2013) Synthesis of coumarin substituted triazolothiadiazine derivatives via ring transformation reaction. J Heterocycl Chem 50:159–163
Koksal M, Ozkan I, Yarim M, Bilge SS, Bozkurt A, Erol DD (2011) Studies on synthesis and anti-inflammatory activities of novel 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines. Rev Chim 62:1069–1072
Motamedi R, Monfared A, Nezamabadi ZG, Bamoharram FF (2011) Facile one-pot synthesis of [1,2,4]triazolo[3,4-b][1,3,4]thiadiazines and 3,7-dimethyl-4H-[1,2,4]triazino[3,4-b][1,3,4]thiadiazin-6-one using heteropolyacid catalysts. J Heterocycl Chem 48:604–607
Jakhar A, Makrandi JK (2010) Molecular iodine mediated one step synthesis of 3-substituted-6-(6-substituted-2-oxochromen-3-yl)-7H [1,2,4]triazolo[3,4-b][1,3,4]thiadiazines. J Chem Res 34:238–240
Knak S, Pfeiffer WD, Dollinger H, Saghyan AS, Langer P (2015) Synthesis of imidazo[2,1-b][2H-1,3,4]thiadiazines and 1,2,4-triazolo[3,4-b][2H-1,3,4]thiadiazines. J Heterocycl Chem 52:463–467
Marathe S, Karnik AV (2017) Synthesis of 3-arylamino-6-phenyl[1,2,4]triazolo[3,4-b][1,3,4] thiadiazine derivatives as potential bioactive molecules. Chem Heterocycl Compd 53:604–607
Aggarwal R, Kumar S, Kaushik P, Kaushik D, Gupta GK (2013) Synthesis and pharmacological evaluation of some novel 2-(5-hydroxy-5-trifluoromethyl-4, 5-dihydropyrazol-1-yl)-4-(coumarin-3-yl) thiazoles. Eur J Med Chem 62:508–514
Gaudino EC, Tagliapietra S, Martina K, Palmisano G, Cravotto G (2016) Recent advances and perspectives in the synthesis of bioactive coumarins. RSC Adv 6:46394–46405
Vaarla K, Vedula RR (2016) Synthesis of novel 3-(3-(5-methylisoxazol-3-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)-2H-chromen-2-ones. J Heterocycl Chem 53:69–73
Lechani N, Hamdi M, Kheddis-Boutemeur B, Talhi O, Laichi Y, Bachari K, Silva AMS (2018) Synthetic approach toward heterocyclic hybrids of [1,2,4]triazolo[3,4-b][1,3,4] thiadiazines. Synlett 29:1502–1504
Tang Z, Peng Y, Liu F (2020) Design and synthesis of novel quinoline derivatives bearing oxadiazole, isoxazoline, triazolothiadiazole, triazolothiadiazine, and piperazine moieties. J Heterocycl Chem 57:2330–2338
Kattimani PP, Kamble RR, Dorababu A, Hunnur RK, Kamble AA, Devarajegowda HC (2017) C5-Alkyl-1,3,4-oxadiazol-2-ones undergo dealkylation upon nitrogen insertion to form 2H-1,2,4-triazol-3-ones: Synthesis of 1,2,4-triazol-3-one hybrids with triazolothiadiazoles and triazolothiadiazines. J Heterocycl Chem 54:2258–2265
Aggarwal R, Sharma S, Hooda M, Sanz D, Claramunt RM, Twamley B, Rozas I (2019) Visible-light mediated regioselective approach towards synthesis of 7-aroyl-6-methyl-[1,2,4] triazolo[3,4-b][1,3,4]thiadiazines. Tetrahedron 75:130728
Thurston DE, Bose DS, Thompson AS, Howard PW, Leoni A, Croker SJ, Jenkins TC, Neidle S, Hartley JA, Hurley LH (1996) Synthesis of sequence-selective C8-linked pyrrolo [2,1-c][1,4]benzodiazepine DNA interstrand cross-linking agents. J Org Chem 61:8141–8147
Ginidi ARS, Shaaban MR, Farag AM, Elwahy AHM (2015) Bis(α-bromo ketones): Versatile precursors for novel bis(s-triazolo[3,4-b][1,3,4]thiadiazines) and bis(thiazoles). J Heterocycl Chem 52:1421–1428
Salem ME, Darweesh AF, Farag AM, Elwahy AHM (2016) 2-Bromo-1-(1H-pyrazol-4-yl) ethanone: versatile precursors for novel mono-, bis- and poly 6-(1H-pyrazol-4-yl)-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines. Tetrahedron 72:712–719
Sarhan RM, Badawy MA, Elwahy AHM (2014) Efficient routes for the synthesis of novel bis(s-triazolo[3,4-b][1,3,4]thiadiazines). J Heterocycl Chem 51:e176–e182
Shaaban MR, Elwahy AHM (2012) Bis(α-bromo ketones): versatile precursors for novel bis(s-triazolo[3,4-b][1,3,4]thiadiazines) and bis(as-triazino[3,4-b][1,3,4]thiadiazines). J Heterocycl Chem 49:640–645
Sayed OM, Mekky AEM, Farag AM, Elwahy AHM (2016) 3,4-Bis(bromomethyl)thieno[2,3-b]thiophene: versatile precursors for novel bis(triazolothiadiazines), bis(quinoxalines), bis(dihydrooxadiazoles), and bis(dihydrothiadiazoles). J Heterocycl Chem 53:1113–1120
Salem ME, Darweesh AF, Elwahy AHM (2020) Synthesis of novel scaffolds based on thiazole or triazolothiadiazine linked to benzofuran or benzo[d]thiazole moieties as new hybrid molecules. Synth Commun 50:256–270
Kumagai Y, Shinkai Y, Miura T, Cho AK (2012) The chemical biology of naphthoquinones and its environmental implications. Annu Rev Pharmacol Toxicol 52:221–247
Khalafy J, Mohammadlou M, Mahmoody M, Salami F, Marjani AP (2015) Facile synthesis of new 10-substituted-5H-naphtho[1,2-e][1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-5-ones. Tetrahedron Lett 56:1528–1530
Pavurala S, Vedula RR (2015) An efficient, multicomponent synthesis of pyrazolyl triazolo thiadiazinyl chromen-2-ones. J Heterocycl Chem 52:306–309
Sujatha K, Deshpande RP, Kesharwani RK, Babu PP, Rao Vedula R (2019) An efficient one-pot expeditious synthesis of 3-phenyl-1-(6-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl)-1H-pyrazol-5-amines via multicomponent approach. Synth Commun 49:49–55
Aychiluhim TB, Rao VR (2014) Efficient, one-pot synthesis of triazolothiadiazinyl-pyrazolone and pyrazolyl-triazolothiadiazine derivatives via multicomponent reaction. Synth Commun 44:1422–1429
Jilloju PC, Srikanth M, Kumar SV, Vedula RR (2020) One-Pot, multi-component synthesis of substituted 2-(6-phenyl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl)-2, 3-dihydrophthalazine-1,4-diones. Polycycl Aromat Compd 41:2043–2054. https://doi.org/10.1080/10406638.2019.1709212
Sujatha K, Vedula RR (2019) Multicomponent efficient synthesis of new [1,2,4]triazolo[3,4]thiadiazines. J Heterocycl Chem 56:832–838
Thirupaiah B, Vedula RR (2014) Novel one-pot multicomponent synthesis of substituted 2,3-dihydro-2-(6-(4-hydroxy-6-methyl-2-oxo-2H-pyran-3-yl)-7H-[1,2,4]triazolo[3,4-b][1,3,4] thiadiazin-3-yl)phthalazine-1,4-diones and substituted 3-[3-(n′-benzylidene-hydrazino)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl]-4-hydroxy-6-methyl-pyran-2-ones. Synth Commun 44:513–519
Abdelrazek FM, Gomha SM, Shaaban MEB, Lotfi AI, El-Shemy HN (2017) Facile synthesis of some novel triazolo[3,4-b]thiadiazines and triazolo[4,3-b]tetrazines. Synth Commun 48:32–37
Bhat MA, Khan AA, Khan S, Al-Dhfyan A (2014) Synthesis of new [1,2,4]triazolo[3,4-b][1,3,4]thiadiazines and study of their anti-candidal and cytotoxic activities. J Chem 2014:897141. https://doi.org/10.1155/2014/897141
Ebrahimpour Z, Shiri A, Bakavoli M, Seyedi SM, Asghari T, Mague J (2017) Synthesis of new pyrimido[4,5-e][1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives via S/N smiles rearrangement. J Heterocycl Chem 54:235–241
Afrough T, Bakavoli M, Eshghi H (2017) Synthesis and characterisation of new 3-methyl-6-[1-(pyrimidin-4-yl)ethyl]-5H-pyrimido[5,4-e][1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives. J Chem Res 41:376–379
Skryl’nikova MA, Khramchikhin AV, Krivchun MN (2017) A new approach to the synthesis of 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines. Russ J Gen Chem 87:1321–1322
Shah TA, Ahmad Z, Mir NA, Muneer M, Rath NP, Ahmad M (2015) One step synthesis of highly functionalized thiazolo[3,2-b][1,2,4]triazole, triazolo[1,5-a]pyrimidine and triazolo[3,4-b][1,3,4]thiadiazine. RSC Adv 5:107931–107937
Pandey VK, Tusi Z, Tusi S, Joshi M (2012) Synthesis and biological evaluation of some novel 5-[(3-aralkyl amido/imidoalkyl)phenyl]-1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines as antiviral agents. ISRN Org Chem 2012:760517. https://doi.org/10.5402/2012/760517
El-Sherief HAH, Hozien ZA, El-Mahdy AFM, Sarhan AAO (2010) Novel method for the synthesis of s-triazolo[3,4-b][1,3,4] thiadiazines. Synthesis 15:2636–2642
Gaponenko NI, Kolodina AA, Lesin AV, Kurbatov SV, Starikova ZA, Nelyubina YuV (2010) Synthesis of spiro[indole-3,3′-[1,3,4]thiadiazino[3,2-a]benzimidazoles] and spiro[indole-3,6′-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines]. Russ Chem Bull 59:838–844
Prakash O, Aneja DK, Wadhwa D, Kumar R, Arora S (2012) A facile synthesis of novel dihydroindeno[1,2-e][1,2,4]triazolo[3,4-b][1,3,4]thiadiazines using HTIB. J Heterocycl Chem 49:566–570
Douglas P, Clarke D (1991) Substituent and solvent effects on the thermal isomerisation of 7H-pyrazolo[5,1-c]-1,2,4-triazole azomethine dyes. J Chem Soc Trans 2:1363–1367
Kim CK, Lamicela WN, Allen DL (1991) Desulfurization of triazolothiadiazines, Patent Number 5,055,586
Skoumbourdis AP, LeClair CA, Stefan E, Turjanski AG, Maguire W, Titus SA, Huang R, Auld DS, Inglese J, Austin CP, Michnick SW, Xia M, Thomas CJ (2009) Exploration and optimization of substituted triazolothiadiazines and triazolopyridazines as PDE4 inhibitors. Bioorg Med Chem Lett 19:3686–3692
Arshad N, Altaf F, Akram M, Ullah M (2019) Furan and phenyl substituted triazolothiadiazine derivatives as copper corrosion inhibitors: electrochemical and DFT studies. Prot Met Phys Chem Surf 55:770–780
Arshad N, Akram AR, Akram M, Rasheed I (2017) Triazolothiadiazine derivatives as corrosion inhibitors for copper, mild steel and aluminum surfaces: electrochemical and quantum investigations. Prot Met Phys Chem Surf 53:343–358
Aytac PS, Durmaz I, Houston DR, Çetin-Atalay R, Tozkoparan B (2016) Novel triazolothiadiazines act as potent anticancer agents in liver cancer cells through Akt and ASK-1 proteins. Bioorg Med Chem 24:858–872
Ahmad A, Varshney H, Rauf A, Sherwani A, Owais M (2017) Synthesis and anticancer activity of long chain substituted 1,3,4-oxadiazol-2-thione, 1,2,4-triazol-3-thione and 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazine derivatives. Arab J Chem 10:S3347–S3357
Xu Q, Bao K, Sun M, Xu J, Wang Y, Tian H, Zuo D, Guan Q, Wu Y, Zhang W (2017) Design, synthesis and structure–activity relationship of 3,6-diaryl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines as novel tubulin inhibitors. Sci Rep 7:1–12
Khan I, Zaib S, Ibrar A, Rama NH, Simpson J, Iqbal J (2014) Synthesis, crystal structure and biological evaluation of some novel 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazoles and 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazines. Eur J Med Chem 78:167–177
Khan I, Ibrar A, Zaib S, Ahmad S, Furtmann N, Hameed S, Simpson J, Bajorath J, Iqbal J (2014) Active compounds from a diverse library of triazolothiadiazole and triazolothiadiazine scaffolds: synthesis, crystal structure determination, cytotoxicity, cholinesterase inhibitory activity, and binding mode analysis. Bioorg Med Chem 22:6163–6173
Kamel MM, Abdo NYM (2014) Synthesis of novel 1,2,4-triazoles, triazolothiadiazines and triazolothiadiazoles as potential anticancer agents. Eur J Med Chem 86:75–80
Zhang B, Li YH, Liu Y, Chen YR, Pan ES, You WW, Zhao PL (2015) Design, synthesis and biological evaluation of novel 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines bearing furan and thiophene nucleus. Eur J Med Chem 103:335–342
Farghaly TAER, Abdallah MA, Mmahmoud HK (2015) Synthesis of novel 1,2,4-triazoles and triazolo-thiadiazines as anticancer agents. Turk J Chem 39:955–969
LaPorte MG, Wang Z, Colombo R, Garzan A, Peshkov VA, Liang M, Johnston PA, Schurdak ME, Sen M, Camarco DP (2016) Optimization of pyrazole-containing 1,2,4-triazolo-[3,4-b]thiadiazines, a new class of STAT3 pathway inhibitors. Bioorg Med Chem Lett 26:3581–3585
Sever B, Altıntop MD, Kuş G, Özkurt M, Özdemir A, Kaplancikli ZA (2016) Indomethacin based new triazolothiadiazine derivatives: synthesis, evaluation of their anticancer effects on T98 human glioma cell line related to COX-2 inhibition and docking studies. Eur J Med Chem 113:179–186
Poojary KN, Poojary B, Kumar V, Holla BS (2016) Synthesis, anticancer and antibacterial activities of triazolothiadiazines containing 2,4-dichloro-5-fluorophenyl moiety. J Appl Chem 5:331–339
Fan MC, Han GY, Zhang XJ, Xi HF (2015) Studies of in vitro anti-prostate cancer potential of newer 1,2,4-triazolo-1,3,4-thiadiazines with different heteroaromatics. Bangladesh J Pharmacol 10:308–315
Ibrar A, Zaib S, Jabeen F, Iqbal J, Saeed A (2016) Unraveling the alkaline phosphatase inhibition, anticancer, and antileishmanial potential of coumarin-triazolothiadiazine hybrids: design, synthesis, and molecular docking analysis. Arch Pharm Chem Life Sci 349:553–565
Yakaiah S, Durgaprasad K, Rao AKSB, Rani PB, Awantika S, Buchappa G, Aparna P (2016) Synthesis and cytotoxicity activity of novel benzotrizolyl methyl[1,2,4-triazolo][3,4-b] thiadiazinyl substituted chromene 2-ones. Asian J Chem 28:1381–1384
Iqbal J, Ejaz SA, Ibrar A, Umar MI, Lecka J, Sévigny J, Saeed A (2018) Expanding the alkaline phosphatase inhibition, cytotoxic and proapoptotic profile of biscoumarin-iminothiazole and coumarin-triazolothiadiazine conjugates. Chem Sel 3:13377–13386
Arandkar V, Vedula RR (2019) A facile one-pot expeditious synthesis of triazolothiadiazines and anticancer activity. Phosphorus Sulfur Silicon Relat Elem 194:533–539
Mohamady S, Gibriel AA, Ahmed MS, Hendy MS, Naguib BH (2020) Design and novel synthetic approach supported with molecular docking and biological evidence for naphthoquinone-hydrazinotriazolothiadiazine analogs as potential anticancer inhibiting topoisomerase-IIB. Bioorg Chem 96:103641
Çevik UA, Çavuşoğlu BK, Sağlık BN, Osmaniye D, Levent S, Llgin S, Özkay Y, Kaplancikli ZA (2020) Synthesis, docking studies and biological activity of new benzimidazole-triazolothiadiazine derivatives as aromatase inhibitor. Molecules 25:1642. https://doi.org/10.3390/molecules25071642
Puthiyapurayil P, Poojary B, Chikkanna C, Buridipad SK (2012) Synthesis, spectral characterization and biological evaluation of a novel series of 6-arylsubstituted-3-[2-(4-substitutedphenyl)propan-2-yl]-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines. Eur J Med Chem 57:407–416
Miao R, Wei J, Lv M, Cai Y, Du Y, Hui X, Wang Q (2011) Conjugation of substituted ferrocenyl to thiadiazine as apoptosis-inducing agents targeting the Bax/Bcl-2 pathway. Eur J Med Chem 46:5000–5009
Ansari M, Shokrzadeh M, Karima S, Rajaei S, Hashemi SM, Mirzaei H, Fallah M, Emami S (2019) Design, synthesis and biological evaluation of flexible and rigid analogs of 4H–1,2,4-triazoles bearing 3,4,5-trimethoxyphenyl moiety as new antiproliferative agents. Bioorg Chem 93:103300
Radwan RR, Zaher NH, El-Gazzar MG (2017) Novel 1,2,4-triazole derivatives as antitumor agents against hepatocellular carcinoma. Chem Biol Interact 274:68–79
Liu Z, Lang B, Gao M, Chang X, Guan Q, Xu Q, Wu D, Li Z, Zuo D, Zhang W (2020) 3-(3-Methoxyphenyl)-6-(3-amino-4-methoxyphenyl)-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine, a novel tubulin inhibitor, evokes G2/M cell cycle arrest and apoptosis in SGC-7901 and HeLa cells. J Cell Biochem 121:2184–2196
Ma W, Chen P, Huo X, Ma Y, Li Y, Diao P, Yang F, Zheng S, Hu M, You W (2020) Development of triazolothiadiazine derivatives as highly potent tubulin polymerization inhibitors: structure–activity relationship, in vitro and in vivo study. Eur J Med Chem 208:112847
Ji L, Zhou Y, Yu Q, Fang Y, Jiang Y, Zhao Y, Yuan C, Xie W (2021) Synthesis and anticancer activity of new spirooxindoles incorporating [1,2,4]triazolo[3,4-b][1,3,4] thiadiazine moiety. J Mol Struct 1227:129406
Ismail MI, Mohamady S, Samir N, Abouzid KA (2020) Design, synthesis, and biological evaluation of novel 7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazine inhibitors as antitumor agents. ACS Omega 5:20170–20186
Hu GQ, Hou LL, Yang Y, Yi L, Xie SQ, Wang GQ, Duan NN, Chao TY, Wen XY, Huang WL (2011) Synthesis and antitumor evaluation of fluoroquinolone C3 fused heterocycles (II): from triazolothiadiazines to pyrazolotriazoles. Chin Chem Lett 22:804–806
Gomha SM, Muhammad ZA, Ezz El-Arab E, Elmetwally AM, El-Sayed AA, Matar IK (2020) Design, synthesis, molecular docking study and anti-hepatocellular carcinoma evaluation of new bis-triazolothiadiazines. Mini Rev Med Chem 20:788–800
Deohate PP (2013) Synthesis, structural study and biological activity of bridgehead nitrogen containing triazolo-thiadiazine derivatives. Chem Sci Trans 2:556–560
Penta S, Gadidasu KK, Basavoju S, Rao VR (2013) An efficient one-pot synthesis of pyrazolyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-yl)-2H-pyran-2-one derivatives via multicomponent approach and their potential antimicrobial and nematicidal activities. Tetrahedron Lett 54:5663–5666
Yata MR, Mekala RVR, Talagadadivi RP (2014) Synthesis, characterization and biological screening of some novel indole based 1,2,4-triazolo 1,3,4-thiadiazines. J Drug Deliv Ther 4:43–46
Jadhav GR, Medhane VJ, Gaikwad VB, Deshmukh DG, Gaikwad SS, Bholay AD (2016) Synthesis and antimicrobial study of triazolo[3,4-b][1,3,4]thiadiazole and triazolo-[3,4-b][1,3,4] thiadiazine derivatives of chromeno[4,3-b]pyridin-5-one moiety. J Chem Pharm Res 8:854–859
Iradyan MA, Iradyan NS, Minasyan NS, Paronikyan RV, Stepanyan GM (2016) Synthesis and antibacterial activity of 3,6-diaryl-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines. Pharm Chem J 50:10–15
Ghosh S, Verma A, Mukerjee A, Mandal MK (2019) Synthesis, characterization and antimicrobial evaluation of some novel 1,2,4-triazolo[3,4-b][1,3,4]thiadiazine bearing substituted phenylquinolin-2-one moiety. Arab J Chem 12:3046–3053
El-Sayed HA, Assy MG, Mohamed AS (2020) An efficient synthesis and antimicrobial activity of N-bridged triazolo[3,4-b]thiadiazine and triazolo[3,4-b]thiadiazole derivatives under microwave irradiation. Synth Commun 50:997–1007
Altıntop MD, Kaplancikli ZA, Turan-Zitouni G, Özdemir A, İşcan G, Akalın G, Yildirim ŞU (2011) Synthesis and anticandidal activity of new triazolothiadiazine derivatives. Eur J Med Chem 46:5562–5566
Prakash O, Aneja DK, Hussain K, Lohan P, Ranjan P, Arora S, Sharma C, Aneja KR (2011) Synthesis and biological evaluation of dihydroindeno and indeno[1,2-e][1,2,4]triazolo[3,4-b][1,3,4]thiadiazines as antimicrobial agents. Eur J Med Chem 46:5065–5073
Badr SM, Barwa RM (2011) Synthesis of some new [1,2,4]triazolo[3,4-b][1,3,4]thiadiazines and [1,2,4]triazolo[3,4-b][1,3,4]thiadiazoles starting from 5-nitro-2-furoic acid and evaluation of their antimicrobial activity. Bioorg Med Chem 19:4506–4512
Sumangala V, Poojary B, Chidananda N, Arulmoli T, Shenoy S (2012) Facile synthesis, cytotoxic and antimicrobial activity studies of a new group of 6-aryl-3-[4-(methylsulfonyl) benzyl]-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines. Eur J Med Chem 54:59–64
Pundeer R, Kiran V, Prakash R, Bhatia SC, Sharma C, Aneja KR (2012) α, α-Dibromoacetophenones mediated synthesis of some new 7H-7-alkoxy-3-alkyl/phenyl-6-aryl-s-triazolo[3,4-b][1,3,4]thiadiazines and their antimicrobial evaluation. Med Chem Res 21:4043–4052
Purohit DH, Dodiya BL, Ghetiya RM, Vekariya PB, Joshi HS (2011) Synthesis and antimicrobial activity of some new 1,3,4-thiadiazoles and 1,3,4-thiadiazines containing 1,2,4-triazolo nucleus. Acta Chim Slov 58:53–59
Sim KM, Chan PQ, Boo XL, Heng KS, Lye KW, Teo KC (2018) Synthesis, characterization and antibacterial activity of some new 1,2,4-triazole Schiff bases and 1,2,4-triazolo[3, 4-b][1,3,4]thiadiazines. Lett Org Chem 15:575–582
Almajan GL, Barbuceanu SF, Saramet I, Draghici C (2010) New 6-amino-[1,2,4]triazolo [3,4-b][1,3,4]thiadiazines and [1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-6-ones: synthesis, characterization and antibacterial activity evaluation. Eur J Med Chem 45:3191–3195
Reddy CS, Rao LS, Sunitha B, Nagaraj A (2015) Synthesis and antibacterial activity of N-substituted-[1,2,4]triazoles and 1,2,4-triazole[3,4-b][1,3,4]thiadiazines. Indian J Chem 54B:1283–1289
Kumar GS, Prasad YR, Mallikarjuna B, Chandrashekar S (2010) Synthesis and pharmacological evaluation of clubbed isopropylthiazole derived triazolothiadiazoles, triazolothiadiazines and mannich bases as potential antimicrobial and antitubercular agents. Eur J Med Chem 45:5120–5129
Sim KM, Teo KC (2018) Synthesis, characterization and antibacterial evaluation of some new 1,2,4-triazolo[3,4-b][1,3,4]thiadiazines as potential antibacterial agents. Lett Drug Des Discov 15:733–743
Puthiyapurayil P, Poojary B, Buridipad SK (2014) Synthesis, characterization and biological evaluation of a novel series of 1,2,4-triazolo-[3,4-b]-1,3,4-thiadiazines containing an amide linkage. J Heterocycl Chem 51:E55–E67
Shehry MFE, Abu-Hashem AA, El-Telbani EM (2010) Synthesis of 3-((2,4-dichlorophenoxy) methyl)-1,2,4-triazolo(thiadiazoles and thiadiazines) as anti-inflammatory and molluscicidal agents. Eur J Med Chem 45:1906–1911
Hussein MA, Shaker RM, Ameen MA, Mohammed MF (2011) Synthesis, anti-inflammatory, analgesic, and antibacterial activities of some triazole, triazolothiadiazole, and triazolothiadiazine derivatives. Arch Pharm Res 34:1239–1250
Naveena CS, Poojary B, Chandrashekhar C, Kumari NS (2011) Synthesis and biological evaluation of some [1,2,4]triazolo[3,4-b][1,3,4]thiadiazoles and [1,2,4]triazolo[3,4-b][1,3,4] thiadiazines. Lett Drug Des Discov 8:189–200
Ranganatha L, Prashanth T, Naveen P, Begum B, Alghorbani M, Khanum SA (2013) Evaluation of antioxidant activity of diarylmethanone integrated thaiadiazine analogues. Indo Am J Pharm Res 3:4131–4137
Patrao P, Khader AMA, Kalluraya B, Vinayachandra (2013) Ring transformation of 1,3,4-oxadiazoles into novel [1,2,4]triazolo[3,4-b][1,3,4]thiadiazines and their antimicrobial and antioxidant properties. J Chem Pharm Res 5:326–334
Kaushik N, Kumar N, Kumar A (2015) Synthesis of triazolothiadiazine derivatives as antioxidant agents. Int J Pharm Pharm Sci 7:120–123
Shakir RM, Ali KF, Hussain DF (2017) Synthesis and antioxidant ability of some new 6-amino-7H-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazin-3-yl) derivatives bearing 2,6-dimethoxy-4-(methoxymethyl) phenol moiety. Oriental J Chem 33:2492–2500. https://doi.org/10.13005/ojc/330543
Sathyanarayana R, Poojary B, Chandrashekarappa RB, Kumar H, Merugumolu VK (2020) Novel [1,2,4]triazolo[3,4-b][1,3,4]thiadiazine derivatives embedded with benzimidazole moiety as potent antioxidants. J Chin Chem Soc 67:1501–1516
Pavurala S, Vaarla K, Kesharwani R, Naesens L, Liekens S, Vedula RR (2018) Bis coumarinyl bis triazolothiadiazinyl ethane derivatives: synthesis, antiviral activity evaluation, and molecular docking studies. Synth Commun 48:1494–1503
Khan I, Hameed S, Al-Masoudi NA, Abdul-Reda NA, Simpson J (2015) New triazolothiadiazole and triazolothiadiazine derivatives as kinesin Eg5 and HIV inhibitors: synthesis, QSAR and modeling studies. Zeitschrift für Naturforschung B 70:47–58
SitaRam CG, Khloya P, Vullo D, Supuran CT, Sharma PK (2014) Benzenesulfonamide bearing 1,2,4-triazole scaffolds as potent inhibitors of tumor associated carbonic anhydrase isoforms hCA IX and hCA XII. Bioorg Med Chem 22:1873–1882
Alafeefy AM, Abdel-Aziz HA, Vullo D, Al-Tamimi AMS, Awaad AS, Mohamed MA, Capasso C, Supuran CT (2015) Inhibition of human carbonic anhydrase isozymes I, II, IX and XII with a new series of sulfonamides incorporating aroylhydrazone-,[1,2,4]triazolo[3,4-b][1,3,4]thiadiazinyl-or 2-(cyanophenylmethylene)-1,3,4-thiadiazol-3(2H)-yl moieties. J Enzyme Inhib Med Chem 30:52–56
Özil M, Bodur O, Ülker S, Kahveci B (2015) Microwave-promoted synthesis and biological activity of some 2-hetarylmethyl-4-(4-hetarylphenyl)-5-methyl-2, 4-dihydro-3H-1,2,4-triazol-3-one derivatives. Chem Heterocycl Compd 51:88–96
Çevik UA, Sağlık BN, Osmaniye D, Levent S, Çavuşoğlu BK, Karaduman AB, Özkay Y, Kaplancikli ZA (2020) Synthesis and docking study of benzimidazole-triazolothiadiazine hybrids as aromatase inhibitors. Arch Pharm (Weinheim) 353:e2000008
Khan I, Bakht SM, Ibrar A, Abbas S, Hameed S, White JM, Rana UA, Zaib S, Shahid M, Iqbal J (2015) Exploration of a library of triazolothiadiazole and triazolothiadiazine compounds as a highly potent and selective family of cholinesterase and monoamine oxidase inhibitors: design, synthesis, X-ray diffraction analysis and molecular docking studies. RSC Adv 5:21249–21267
Baeeri M, Foroumadi A, Motamedi M, Yahya-Meymandi A, Firoozpour L, Ostad SN, Shafiee A, Souzangarzadeh S, Abdollahi M (2011) Safety and efficacy of new 3,6-diaryl-7H-[1, 2, 4]triazolo[3,4-b][1,3,4]thiadiazine analogs as potential phosphodiesterase-4 inhibitors in NIH-3T3 mouse fibroblastic cells. Chem Biol Drug Des 78:438–444
Li Z, Bai X, Deng Q, Zhang G, Zhou L, Liu Y, Wang J, Wang Y (2017) Preliminary SAR and biological evaluation of antitubercular triazolothiadiazine derivatives against drug-susceptible and drug-resistant Mtb strains. Bioorg Med Chem 25:213–220
Kincaid VA, London N, Wangkanont K, Wesener DA, Marcus SA, Héroux A, Nedyalkova L, Talaat AM, Forest KT, Shoichet BK (2015) Virtual screening for UDP-galactopyranose mutase ligands identifies a new class of antimycobacterial agents. ACS Chem Biol 10:2209–2218
Sliwoski G, Kothiwale S, Meiler J, Lowe EW Jr (2013) Computational methods in drug discovery. Pharmacol Rev 66:334–395. https://doi.org/10.1124/pr.112.007336
Daina A, Michielin O, Zoete V (2017) SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep 7:42717
Jorgensen WL, Duffy EM (2002) Prediction of drug solubility from structure. Adv Drug Deliv Rev 54:355–366
Lipinski CA, Lombardo F, Dominy BW, Feeney PJ (1997) Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliv Rev 23:3–25
Iyer VB, Gurupadayya B, Koganti VS, Inturi B, Chandan RS (2017) Design, synthesis and biological evaluation of 1,3,4-oxadiazoles as promising anti-inflammatory agents. Med Chem Res 26:190–204
Faidallah HM, Rostom SA (2017) Synthesis, anti-inflammatory activity, and COX-1/2 inhibition profile of some novel non-acidic polysubstituted pyrazoles and pyrano[2,3-c]pyrazoles. Arch Pharm 350:1700025
Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD (2002) Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem 45:2615–2623
Magda AA, Abdel-Aziz NI, Alaa AM, El-Azab AS, ElTahir KE (2012) Synthesis, biological evaluation and molecular modeling study of pyrazole and pyrazoline derivatives as selective COX-2 inhibitors and anti-inflammatory agents. Bioorg Med Chem 20:3306–3316
Abdellatif KRA, Abdelall EKA, Elshemy HAH, Philoppes JN, Hassanein EHM, Kahk NM (2021) Optimization of pyrazole-based compounds with 1,2,4-triazole-3-thiol moiety as selective COX-2 inhibitors cardioprotective drug candidates: design, synthesis, cyclooxygenase inhibition, anti-inflammatory, ulcerogenicity, cardiovascular evaluation, and molecular modeling studies. Bioorg Chem 114:105122
Alsaedi AM, Almehmadi SJ, Farghaly TA, Harras MF, Khalil KD (2022) VEGFR2 and hepatocellular carcinoma inhibitory activities of trisubstituted triazole derivatives. J Mol Struct 1250:131832
Sever B, Altıntop MD, Çiftçi GA, Özdemir A (2021) A new series of triazolothiadiazines as potential anticancer agents for targeted therapy of non-small cell lung and colorectal cancers: design, synthesis, in silico and in vitro studies providing mechanistic insight into their anticancer potencies. Med Chem 17:1104–1128
Sivakumar KC, Haixiao J, Naman CB, Sajeevan TP (2020) Prospects of multitarget drug designing strategies by linking molecular docking and molecular dynamics to explore the protein-ligand recognition process. Drug Dev Res 81:685–699
Acknowledgements
We are grateful to Council of Scientific and Industrial Research (CSIR), New Delhi, India for providing financial assistance (SRF) to Mona Hooda (Grant no. 09/105(0236)/2016-EMR-1) and JRF to Prince Kumar (Grant no. 09/105(0302)/2020-EMR-1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Aggarwal, R., Hooda, M., Kumar, P. et al. Vision on Synthetic and Medicinal Facets of 1,2,4-Triazolo[3,4-b][1,3,4]thiadiazine Scaffold. Top Curr Chem (Z) 380, 10 (2022). https://doi.org/10.1007/s41061-022-00365-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s41061-022-00365-x