
Abstract
Numerous cellular functions occur in spatially and temporally confined regions. Recent studies have shown that membrane-less organelles and compartments in the cell are assembled via liquid–liquid phase separation (LLPS). In vitro LLPS assays using recombinant expressed and purified proteins are necessary for us to further understand how the assembly of phase-separated compartments is regulated in cells. However, uniform standards and protocols are lacking for these in vitro studies. Here, we describe a step-by-step protocol commonly used to investigate in vitro LLPS using purified proteins. This protocol includes expression and purification of the studied proteins, induction of LLPS of the purified proteins, and studies of the biophysical properties of the liquid droplets formed by LLPS. These protocols can be easily followed by researchers to investigate the LLPS behaviors of proteins of interest.
Similar content being viewed by others


Introduction
One of the most important questions in cell biology is how the numerous biochemical reactions in the cell are spatiotemporally coordinated. Tightly controlled assembly of membrane-less organelles and compartments by liquid–liquid phase separation (LLPS) in cells is a key event for such coordination (Aguzzi and Altmeyer 2016; Banani et al. 2017). LLPS is the conversion of a homogeneous liquid phase into inhomogeneous phases. Unlike organelles, which are encapsulated by membranes, no physical barriers confine the nonmembrane-bound compartments formed by LLPS. The membrane-less compartments have defined boundaries and remain dynamic by exchanging materials with the surrounding environment (Shin et al. 2017). These membrane-less compartments are largely composed of proteins and RNAs, and are maintained by molecular interactions (Banani et al. 2017; Bergeron-Sandoval et al. 2016).
Protein LLPS is triggered by multivalent interactions conferred by interaction of modular domains and/or intrinsically disordered regions (LDRs) in proteins (Aguzzi and Altmeyer 2016; Banani et al. 2017). The multivalent interactions between proteins and RNAs also contribute to LLPS-mediated formation of RNP granules such as stress granules and P bodies (Aguzzi and Altmeyer 2016; Banani et al. 2017). Phase separation is regulated by factors affecting multivalent interactions, including protein concentration, temperature, pH, salt concentration, and posttranslational modifications (such as phosphorylation, methylation, and ubiquitination) (Dao et al. 2018; Nott et al. 2015; Su et al. 2016), by the presence of other molecules (such as another protein and RNA) (Saha et al. 2016; Zeng et al. 2016), and also by the tag used for protein expression. A standardized protocol for protein LLPS is urgently needed.
In this study, we present a step-by-step protocol, which summarizes some recently used assays to induce and study protein LLPS. This protocol includes the purification of proteins, the in vitro phase-separation assays and the assays for studying the biophysical properties of the phase-separated liquid droplets. We hope the proposed protocol will enable users to easily and efficiently conduct protein LLPS experiments.
Materials and equipment
Reagents
Buffers
-
Binding buffer: 50 mmol/L Tris–Cl pH 7.9, 500 mmol/L NaCl, and 10 mmol/L imidazole
-
Ni–NTA elution buffer: 50 mmol/L Tris–Cl pH 7.9, 500 mmol/L NaCl, and 500 mmol/L imidazole
-
GST elution buffer: 50 mmol/L Tris–Cl pH 7.9, 500 mmol/L NaCl, and 10 mmol/L GSH
-
MBP elution buffer: 50 mmol/L Tris–Cl pH 7.9, 500 mmol/L NaCl, and 10 mmol/L maltose
-
HS (high salt) buffer: 25 mmol/L HEPES pH 7.5, 500 mmol/L NaCl, and 1 mmol/L DTT
-
LS (low salt) buffer: 25 mmol/L HEPES pH 7.5 and 1 mmol/L DTT
-
LLPS buffer: 25 mmol/L HEPES pH 7.5, 150 mmol/L NaCl, and 1 mmol/L DTT
Equipment
-
HiPrep 26/60 Sephacryl S-200 HR columns (GE Healthcare, 17-1195-01)
-
AKTA purifier (GE Healthcare)
-
Glass-bottom cell culture dishes (Nest)
-
Glass slides and cover glasses (Citoglas)
-
Confocal microscope (LSM 880 Meta plus Zeiss Axiovert zoom, Carl Zeiss)
-
Centrifugal filters (Millipore)
Software
-
Imager M2 (Carl Zeiss)
-
GraphPad Prism (GraphPad Software)
Overview of experimental design
This protocol includes four sections. The first section provides the detailed procedure for the expression and purification of the studied proteins. The second section summarizes the methods to induce LLPS of the purified proteins. The third section describes the standards to detect and determine whether the purified proteins undergo LLPS. The fourth section introduces the techniques to study the biophysical properties of liquid droplets formed by LLPS.
In the first section of this protocol, we will provide a guideline for constructing the expression plasmids, expressing recombinant proteins, and carrying out purification. This guideline describes a generally used procedure in our lab, but it is recommended that readers optimize this procedure according to their studied proteins.
In the second section of this protocol, we provide four methods to induce LLPS of the purified proteins, including decreasing the ionic strength of the buffer, addition of precipitators, co-addition of interacting partners, and removal of the recombinant tag by protease. Different proteins undergo LLPS under different conditions according to their properties.
The third section of this protocol guides readers to recognize the phenomenon of LLPS. Several assays and standards are recommended to determine whether the proteins undergo LLPS, including assessing the turbidity of the solution, microscopic examination of liquid droplets, monitoring the fusion and fission of liquid droplets, assessing their wetting properties, and subjecting the mixture to sedimentation to analyze the partitioning of proteins between the different phases.
The fourth section of this protocol provides some techniques to study the biophysical properties of liquid droplets formed by LLPS, including fluorescence recovery after photobleaching (FRAP) analysis and assays for salt resistance and droplet aging. Using these assays, the biophysical properties of liquid droplets, such as mobility, solubility, and degree of gelation, can be determined.
Summarized procedure
-
(1)
Amplified cDNA fragments encoding the studied proteins are cloned into appropriate expression vectors.
-
(2)
The studied proteins are expressed in E. coli BL21-CodonPlus (DE3) cells.
-
(3)
The BL21 cells expressing the studied proteins are collected, lysed, and centrifuged to remove heavy debris.
-
(4)
The studied proteins in the supernatants of cell lysates are purified to homogeneity by means of various purification methods.
-
(5)
The purified proteins are induced to phase separate by means of various methods. No priority should be given to any method. Method i is performed with proteins dissolved in HS buffer; whereas methods ii–iii are performed with proteins dissolved in LLPS buffer.
-
(i)
Decreasing the ionic strength of HS buffer to that of the LLPS buffer.
-
(ii)
Co-adding a precipitator to the studied proteins in LLPS buffer.
-
(iii)
Removing the recombinant tag from the studied proteins with a specific protease in LLPS buffer.
-
(iv)
Mixing the studied proteins with interacting biomacromolecules in LLPS buffer.
-
(i)
-
(6)
Various methods are used to detect whether the proteins undergo LLPS. The methods are not listed in order of priority.
-
(i)
Detecting the turbidity of the solution.
-
(ii)
Detecting the morphological of liquid droplets formed by LLPS by examination using a differential interference contrast (DIC) microscope.
-
(iii)
Detecting the fusion and fission events of liquid droplets formed by LLPS using DIC microscopy.
-
(iv)
Examining the wetting properties of liquid droplets on the surface of a glass slide using a wetting assay.
-
(v)
Examining the composition and ratio of proteins in liquid droplets during sedimentation experiments.
-
(i)
-
(7)
The biophysical properties of liquid droplets formed by LLPS are examined using various assays. The assays are not listed in order of priority.
-
(i)
Examining the mobility of liquid droplets with fluorescence recovery after photobleaching (FRAP) analysis.
-
(ii)
Examining the solubility of liquid droplets using a salt-resistance assay.
-
(iii)
Examining the degree of gelation and speed of phase transition of liquid droplets using a droplet-aging assay.
-
(i)
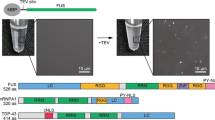
Protein expression and purification
The procedure for protein expression and purification contains several steps, as illustrated in Fig. 1.

Flowchart of protein expression and purification
Construction of plasmids and expression of recombinant proteins
-
(1)
The DNA fragments encoding the studied proteins are PCR amplified from cDNA libraries of the appropriate species, and inserted into bacterial vectors. To express recombinant proteins, the amplified cDNA fragments are transferred into various modified pET-32a vectors, resulting in recombinant proteins with N-terminal His6, GFP-His6, GST-His6, or MBP-His6 tags. All these recombinant tags can be cleaved with specific proteases, such as PreScission or TEV proteases. The constructed plasmids should be confirmed by DNA sequencing. The studied proteins can also be expressed using commercial vectors, such as pET-28, pGEX, and pMal vectors to obtain His6-, GST-, and MBP-fused recombinant proteins, respectively.
-
(2)
To express recombinant proteins, the plasmids expressing target proteins are transformed into E. coli BL21-CodonPlus (DE3) cells, which are further cultured on LB medium plates at 37 °C overnight. A single colony from each transformation plate is grown in 10 mL LB medium for 4 h in a 37 °C shaker with a rotation speed of 220 r/min. Then the bacterial cultures are inoculated into 1 L LB medium and grown for about 2 h to OD600 = 0.5–0.6 before induction. The culture bottles are cooled with ice for 10 min, supplemented with 0.3 mmol/L IPTG and subjected to shaking at 220 r/min at 18 °C to express recombinant proteins. After 16–20-h shaking, the BL21 cells are collected by sedimentation for 15 min at 5000 r/min. Different proteins require different growing temperatures and induction times. The detailed procedure for protein expression should be optimized.
Purification of recombinant proteins
-
(1)
The BL21 cells collected from 1–2 L bacterial culture are resuspended in 30 mL binding buffer (50 mmol/L Tris–Cl pH 7.9, 500 mmol/L NaCl, and 10 mmol/L imidazole), and lysed with a high-pressure homogenizer.
-
(2)
The lysates are centrifuged at 18,000 r/min for 30 min at 4 °C to remove the heavy debris.
-
(3)
To purify His6-, GST-, and MBP-tagged proteins, the supernatants of lysates are loaded onto columns containing Ni–NTA, GST, and MBP agarose beads, respectively, and incubated at 4 °C for 30 min under frequent stirring. After washing 2 or 3 times with binding buffer, the bound proteins are eluted with Ni–NTA elution buffer (50 mmol/L Tris–Cl pH 7.9, 500 mmol/L NaCl, and 500 mmol/L imidazole), GST elution buffer (50 mmol/L Tris–Cl pH 7.9, 500 mmol/L NaCl, and 10 mmol/L GSH), and MBP elution buffer (50 mmol/L Tris–Cl pH 7.9, 500 mmol/L NaCl, and 10 mmol/L maltose), respectively.
-
(4)
Recombinant-expressed tags often affect the behavior of the protein. It is therefore recommended that the tags are removed by specific proteases.
-
(5)
The proteins are further purified by ion-exchange chromatography to remove cleaved tags, followed by size-exclusion chromatography (GE Healthcare) on an AKTA purifier with HS buffer (25 mmol/L HEPES pH 7.5, 500 mmol/L NaCl, and 1 mmol/L DTT) or LLPS buffer (25 mmol/L HEPES pH 7.5, 150 mmol/L NaCl, and 1 mmol/L DTT).
-
(6)
The purified proteins are concentrated by centrifugal filtration (Millipore). The protein concentration is then measured. The quality of the proteins is affected by repeated freezing and thawing, so it is recommended that the purified proteins are stored in small aliquots at −80 °C. All proteins are centrifuged at 13,000 r/min for 10 min at 4 °C before being used.
In vitro phase-separation assays
Several methods are used to induce LLPS according to the properties of the studied proteins.
Induction of LLPS by decreasing the ionic strength
Proteins dissolved in high-salt (HS) buffer containing 25 mmol/L HEPES pH 7.5, 500 mmol/L NaCl, and 1 mmol/L DTT are further concentrated to the desired concentration, and mixed with low-salt (LS) buffer containing 25 mmol/L HEPES pH 7.5 and 1 mmol/L DTT to adjust the concentration of NaCl to 150 mmol/L (for example, 30 μL purified protein in HS buffer is mixed with 70 μL LS buffer). Upon induction, the system is subjected to microscopic examination and other analyses to detect whether the studied protein undergoes LLPS. These methods will be detailed in the next section.
[?TROUBLESHOOTING No. 1]
Induction of LLPS by addition of a precipitator
If LLPS fails to be induced by the method described above, co-addition of molecular-crowding agents such as dextran may facilitate protein LLPS.
To induce LLPS, various concentrations of dextran are added into the LLPS system. For reference, 30 μL of studied protein with the desired concentration dissolved in HS buffer is mixed with 70 μL of LP buffer containing 7.14%–28.57% (w/v) dextran, to make the final buffer of 25 mmol/L HEPES pH 7.5, 150 mmol/L NaCl, 1 mmol/L DTT, and 5%–20% (w/v) dextran. A sample of the mixture is then removed by pipette for microscopic examination (see below).
Induction of LLPS by removing the recombinant tag
Some proteins with a single His6 tag, such as His6–FUS, are expressed as inclusion bodies and cannot be purified. For these proteins, addition of a big tag such as MBP at the N-terminus may improve their solubility and simplify the purification process.
To induce LLPS, MBP-tagged proteins dissolved in LLPS buffer are treated with a specific protease to remove the MBP tag. The reaction system is incubated at the indicated temperature and subjected to microscopic examination.
Induction of LLPS by biomacromolecule interaction
Some proteins fail to undergo LLPS by themselves and only phase separate following the co-addition of specific binding partners. For example, a mixture of p62 and polyubiquitin, but neither p62 nor polyubiquitin alone, undergoes phase separation in vitro. The binding partners can also be other biomacromolecules, such as DNA and RNA.
To induce LLPS, the individual components of the complex dissolved in LLPS buffer are mixed and incubated at a range of temperatures, and then subjected to microscopic examination.
Detection of liquid droplets formed by LLPS
Detection of the turbidity of the solution
A solution undergoing protein phase separation will yield some liquid droplets, leading to the conversion of a single phase into multiple phases. This will change the turbidity of the solution. Therefore, if the protein undergoes phase separation, the solution will become turbid; otherwise, it will remain clear (Fig. 2A).

Assays used for examining in vitro phase-separated droplets. A The solution becomes turbid if it contains phase-separated proteins (3 μmol/L SEPA-1/PGL-1/-3), while it remains clear if the protein (3 μmol/L SEPA-1) fails to undergo LLPS. B, C Compared to the clear liquid containing protein that failed to phase separate (3 μmol/L SEPA-1) (B), spherical droplets are formed in the turbid liquid containing proteins that undergo LLPS (3 μmol/L SEPA-1/PGL-1/-3) (C). D and E show typical protein aggregates in the turbid liquid containing proteins that do not undergo LLPS but form aggregates. F Time-lapse analysis showing that phase-separated liquid droplets formed by 3 μmol/L SEPA-1/PGL-1/-3 fuse with each other (red arrowhead) and relax into a larger one. G Images showing that liquid droplets formed by 3 μmol/L SEPA-1/PGL-1/-3 exhibit a wetting phenotype when encountering the surface of a coverslip. The time refers to imaging time. Scale bars: 10 μm (B–G and inserts in B–E)
Morphological detection by microscopic examination
Pipette 10 μL of the solution onto a glass slide, place a cover glass on the solution and examine under a microscope. If the protein undergoes LLPS, spherical droplets should be seen in the solution; otherwise, the solution will remain clear (Fig. 2B, C). Protein aggregates might be formed by some proteins. The liquid droplets formed by LLPS are spherical and transparent, whereas protein aggregates are opaque and irregularly shaped (Fig. 2D, E).
[CRITICAL] Adjust the focal plane of the microscope to find the layer of liquid droplets. Don’t be confused by air bubbles. Under the DIC microscope, air bubbles are more refractile and have a blacker boundary than liquid droplets formed by LLPS in solution.
[CRITICAL] Some liquid droplets undergo sedimentation quickly and wet the surface of the slide in a few minutes, thus quickly losing their spherical shape. To detect spherical droplets, the sample should be examined by DIC microscopy immediately after being pipetted onto the glass slide.
[?TROUBLESHOOTING No. 2]
Liquid droplet fusion and fission assay
Pipette 10 μL of the solution onto a glass slide, place a cover glass on the solution and perform microscopic examination. If the protein undergoes LLPS, the spherical droplets in the solution will fuse when they encounter each other and then relax into a larger droplet. Droplets can also undergo a fission process. Protein aggregates do not undergo fusion and fission events (Fig. 2F).
[CRITICAL] As mentioned above, some liquid droplets undergo sedimentation quickly and no longer undergo fusion and fission even after lapse of a several minutes. To detect fusion and fission of the droplet, the sample should be examined by DIC microscopy immediately after being pipetted onto the glass slide.
[?TROUBLESHOOTING No. 3]
Wetting assay
Pipette 10 μL of the solution onto a glass-bottomed cell culture dish and perform microscope examination. Wetting is the ability of a liquid to spread across the surface of a solid, and liquids are much more dynamic than solids. If the protein phase separates, the spherical droplets in the liquid will wet the surface and form irregular shapes over time (Fig. 2G), whereas hydrogels and protein aggregates do not wet or change shape.
Sedimentation assay
A sedimentation assay is recommended to quantitatively examine protein phase separation. Following LLPS, 100 µL solution is centrifuged for 5 min at 13,000 r/min. The precipitate is resuspended in a volume of LLPS buffer equal to the supernatant, and the precipitate and supernatant fractions are denatured by adding 5× SDS loading buffer and boiled for 5 min. 1–10 µL supernatant and precipitate samples are loaded onto SDS–PAGE for Coomassie brilliant blue staining or western blotting analysis with the appropriate antibodies. The ratio of the total protein partitioned into phase-separated droplets is determined by measuring the intensity of the bands in the supernatant and precipitate samples.
[CRITICAL] Carefully pipette as much of the supernatant from the system as possible. Don’t touch the precipitate with the pipette tip.
Studying the biophysical properties of phase-separated liquid droplets
Fluorescence recovery after photobleaching (FRAP) analysis
Fluorescence recovery after photobleaching (FRAP) is a useful assay to determine the molecular dynamics and mobility of the phase-separated liquid droplets. FRAP experiments can be carried out on a confocal microscope equipped with the necessary laser. A region of interest is bleached with a laser, and then the fluorescence in this region is monitored as it recovers. The fluorescence recovery depends on the molecular dynamics of the phase-separated liquid droplets. If the phase-separated droplets are liquid-like, the fluorescence recovery is quick (Fig. 3A). The fluorescence recovery is slow in gel-like droplets, and the fluorescence signal fails to recover in protein aggregates after photobleaching.

Phase-separated liquid and gel-like droplets exhibiting distinct biophysical properties. A In phase-separated liquid droplets formed by 3 μmol/L SEPA-1/PGL-1::GFP/PGL-3, the fluorescent signal recovers after a portion of the droplet is photobleached. B Phase-separated liquid droplets formed by 3 μmol/L SEPA-1/PGL-1/-3 largely dissolve in a buffer containing a high concentration of salt. C The gel-like phase-separated droplets formed by 1 μmol/L EPG-2/SEPA-1/PGL-1/-3 remain largely undissolved in a buffer containing a high concentration of salt. D, E Phase-separated liquid droplets formed by 3 μmol/L SEPA-1/PGL-1/-3 undergo fusion and form larger irregularly shaped structures over time (D), while the size and shape of gel-like droplets formed by 1 μmol/L EPG-2/SEPA-1/PGL-1/-3 remain unchanged over time (E). Scale bars: 10 μm (B–E and inserts in B–E)
To perform the FRAP assay, fluorescence-tagged proteins are used to label the liquid droplets. Pipette 10 μL of the induced fluorescence-labeled liquid droplets on a glass-bottom cell culture dish, and then bleach a region of interest under a confocal microscope at room temperature. The fluorescence intensities in these regions are measured and collected by Mean ROI every several seconds, and the raw data are processed and further analyzed by GraphPad Prism. A method for processing the raw data is described by Carisey and colleagues (Carisey et al. 2011).
[CRITICAL] To perform FRAP experiments, select immobile droplets that are just attached to the glass-bottom cell culture dish for bleaching. Selection of mobile droplets for the experiments results in sudden discontinuous changes in the fluorescence recovery curve.
[CRITICAL] To compare the recovery rates of two different samples, select droplets with similar fluorescence intensity from the two different samples for the FRAP experiments. Also, the fluorescence intensity of the selected droplets should be bleached to similar levels.
[?TROUBLESHOOTING No. 4]
Salt-resistance assay
The salt-resistance assay is used to determine the solubility and degree of gelation of the formed liquid droplets. Pipette 10 μL of the induced liquid droplets (in buffer containing 25 mmol/L HEPES pH 7.5, 150 mmol/L NaCl, and 1 mmol/L DTT) into an Eppendorf tube, then adjust the concentration of NaCl to 500 mmol/L (typically add 7 μL buffer containing 25 mmol/L HEPES pH 7.5, 1 mol/L NaCl, and 1 mmol/L DTT to the induced liquid droplets), and examine the droplets under a microscope. Liquid droplets are dynamic, and they dissolve in high-salt solutions. Gel-like or solid droplets remain partially dissolved or undissolved in high-salt solutions (Fig. 3B, C).
Droplet-aging assay
The droplet-aging assay is used to study the phase-transition properties of liquid droplets formed by phase separation. After formation of liquid droplets for different lengths of time, pipette 10 μL of the liquid droplets onto a glass slide, place a cover glass over the liquid, and examine the shape under a microscope. Liquid droplets are very dynamic, and they fuse with each other. However, gel-like or solid droplets fuse at a much reduced rate, or not at all, and the shape remains unchanged (Fig. 3D, E). We recommend examining the morphology of liquid droplets assembled after 30 min, 1 h, 2 h, 4 h, 8 h, and 12 h.
The droplet-aging assay can be used in recombination with the FRAP and salt-resistance assays to obtain more rigorous conclusions.
[?TROUBLESHOOTING No. 5]
Future Perspectives
Protein LLPS assays using purified proteins have been widely performed to study the assembly of membrane-less organelles and compartments. In this paper, we provide a detailed and step-by-step protocol including protein expression and purification, in vitro phase-separation assays, and analysis of the biophysical properties of the liquid drops formed by LLPS. Protein LLPS is a burgeoning research field in cell biology, and many other techniques are being used to study this phenomenon, such as NMR, cryo-EM, and single-molecule fluorescence methods. Improvements in technology will drive developments in the field of protein LLPS. Finally, we recommend using a combination of different assays to investigate protein LLPS and determine the various properties of phase-separated structures. Ultimately, how the properties of droplets correlate with the biological function of membrane-less compartments assembled by LLPS should be investigated.
[?TROUBLESHOOTING]
Troubleshooting protocol is provided in Table 1.
References
Aguzzi A, Altmeyer M (2016) Phase separation: linking cellular compartmentalization to disease. Trends Cell Biol 26(7):547–558
Banani SF, Lee HO, Hyman AA, Rosen MK (2017) Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol 18(5):285–298
Bergeron-Sandoval LP, Safaee N, Michnick SW (2016) Mechanisms and consequences of macromolecular phase separation. Cell 165(5):1067–1079
Carisey A, Stroud M, Tsang R, Ballestrem C (2011) Fluorescence recovery after photobleaching. Methods Mol Biol 769:387–402
Dao TP, Kolaitis RM, Kim HJ, O’Donovan K, Martyniak B, Colicino E, Hehnly H, Taylor JP, Castaneda CA (2018) Ubiquitin modulates liquid-liquid phase separation of UBQLN2 via disruption of multivalent interactions. Mol Cell 69(6):965–978
Nott TJ, Petsalaki E, Farber P, Jervis D, Fussner E, Plochowietz A, Craggs TD, Bazett-Jones DP, Pawson T, Forman-Kay JD, Baldwin AJ (2015) Phase transition of a disordered nuage protein generates environmentally responsive membraneless organelles. Mol Cell 57(5):936–947
Saha S, Weber CA, Nousch M, Adame-Arana O, Hoege C, Hein MY, Osborne-Nishimura E, Mahamid J, Jahnel M, Jawerth L, Pozniakovski A, Eckmann CR, Julicher F, Hyman AA (2016) Polar positioning of phase-separated liquid compartments in cells regulated by an mRNA competition mechanism. Cell 166(6):1572–1584
Shin Y, Berry J, Pannucci N, Haataja MP, Toettcher JE, Brangwynne CP (2017) Spatiotemporal control of intracellular phase transitions using light-activated optodroplets. Cell 168(1–2):159–171
Su X, Ditlev JA, Hui E, Xing W, Banjade S, Okrut J, King DS, Taunton J, Rosen MK, Vale RD (2016) Phase separation of signaling molecules promotes T cell receptor signal transduction. Science 352(6285):595–599
Zeng M, Shang Y, Araki Y, Guo T, Huganir RL, Zhang M (2016) Phase transition in postsynaptic densities underlies formation of synaptic complexes and synaptic plasticity. Cell 166(5):1163–1175
Acknowledgements
They authors would like to thank the Strategic Priority Research Program of the Chinese Academy of Sciences (CAS) (XDB19000000), the National Natural Science Foundation of China (31561143001, 31630048, 31790403, and 31421002), the National Chinese Ministry of Science and Technology (2017YFA0503401), and the Key Research Program of Frontier Sciences, CAS (QYZDY-SSW-SMC006) for research funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Zheng Wang, Gangming Zhang, and Hong Zhang declare that they have no competing interests.
Human and animal rights and informed consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wang, Z., Zhang, G. & Zhang, H. Protocol for analyzing protein liquid–liquid phase separation. Biophys Rep 5, 1–9 (2019). https://doi.org/10.1007/s41048-018-0078-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41048-018-0078-7