
Abstract
The past 20 years have seen major advances in the diagnosis and management of pulmonary hypertension, a disease associated with significant morbidity and mortality. The 6th World Symposium in Pulmonary Hypertension (WSPH) took place in February 2018 and attempted to consolidate the current knowledge in the field into practical recommendations to help prioritize an action plan to improve patient outcomes and identify future research directions. In this review, we will summarize the highlights of the 6th WSPH proceedings, including revisions to the hemodynamic definitions and classification of the various types of pulmonary hypertension, genetic advances, approaches to risk stratification, and updated treatment algorithms.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The hemodynamic definition of pulmonary hypertension (PH) has been revised to a mean pulmonary artery pressure (mPAP) > 20 mmHg. |
Genetic studies have continued to reveal novel genetic risk factors for pulmonary arterial hypertension and pulmonary veno-occlusive disease. |
Patients with group 2 and 3 PH should not be treated with PH specific therapy due to high risk of complications. |
Patients with chronic thromboembolic disease should be referred early for thromboendarterectomy. For patients who are not candidates for surgery, balloon angioplasty and/or medical therapy should be considered. |
Introduction
Pulmonary hypertension (PH) continues to be a rapidly evolving field with a myriad of diagnostic and therapeutic challenges for clinicians and patients alike. Since the first World Symposium in Pulmonary Hypertension (WSPH) in 1973, there has been a concerted effort by the clinical and scientific community to review the most important advances in the field and propose strategies to improve the quality of care and accelerate research discovery in the field of PH. In 2018, the 6th WSPH took place in Nice, France, and major revisions were proposed to the hemodynamic definitions and classification of the various types of PH, genetics, and risk stratification. This review will summarize the major highlights from the 6th WSPH proceedings and will inform practitioners of major changes to diagnosis and management of PH aimed at improving patient outcomes and quality of life. This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Revisions to the Hemodynamic Definition of PH
While many aspects of classification, diagnosis, and treatment of PH have been updated, perhaps the most impactful change was the updated hemodynamic definition. Per the proceedings, it was suggested that PH be now defined as a mean pulmonary artery pressure (mPAP) of > 20 mmHg, a decrease from the previous definition of mPAP ≥ 25 mmHg [1].
The authors cited a number of reasons for the change. They felt that the previous cutoffs were chosen arbitrarily and were not based on physiologic data. Previous studies have demonstrated that a mPAP of 25 was greater than two standard deviations above the norm for the population. In a 2009 review of all published data on pulmonary arterial pressures in healthy individuals, a normal mPAP was identified as 14 ± 3.3 mmHg [2], which led the authors to suggest that a mPAP ≥ 25 mmHg was not necessarily inclusive of all individuals with abnormally elevated pulmonary pressures. This was also pointed out in the 5th WSPH but the switch was not made mainly due to concern about the consequences of over-diagnosing PH, as well as the lack of evidence that patients in the previously considered “borderline” range (mPAP 21–24 mmHg) suffered worse outcomes.
The update to the hemodynamic definition of PH was made at the 6th WSPH because four studies now exist to suggest that patients with pulmonary pressures > 20 mmHg do suffer from worse clinical outcomes. One study of patients with systemic sclerosis found that 42% of participants with mPAP 21–24 mmHg developed overt PH (mPAP ≥ 25 mmHg) over ~ 2 years of follow-up with ~ 14% suffering deaths despite therapy. The authors argue that this data suggest that PA pressures > 2 standard deviations above the mean may be associated with risk of future PH and increased mortality, at least in the population with systemic sclerosis [3]. Moreover, several larger studies of the general population have recently demonstrated worse survival in patients with borderline PH [4, 5]. Given this new data, the task force agreed that the evidence now exists to include patients with mPAP > 20 under the diagnosis of PH.
The change has not been made without resistance from experts in the field. Gibbs and Torbicki published a rebuttal to the updated proceedings in the European Respiratory Journal in February of 2019, arguing that the data to support the change in the hemodynamic definition of PH are lacking [6]. They point out that the addition of a category for “borderline PH” has been addressed at past symposia and that the previous concern for overdiagnosis, which led to its removal from the guidelines, remains valid. They also argue that making this change without data to suggest that treatment of patients with a mPAP 21–24 mmHg improves outcomes is premature. Their position that the selected trials cited in the updated definition tell us nothing about the vast majority of patients of interest is not without merit as the majority of the studies included populations known to be at high risk for development of pulmonary arterial hypertension (PAH). Additionally, Gibbs and Torbicki argue that the two standard deviations argument is not consistent, since the pulmonary artery wedge pressure (PAWP) and pulmonary vascular resistance (PVR) cutoff values do not meet these same criteria. In fact, some experts have argued for also revising the PVR cutoff from ≥ 3 Wood units (WU) to 2 WU or even 1.5 WU, which would potentially create another cohort of patients who are being “missed” by both the old and new hemodynamic definitions.
Since the publication of the 6th WSPH proceedings, several retrospective studies have looked at how many patients were “missed” in previous cohorts. Jaafar et al. looked retrospectively at a cohort of patients at high risk for PH who underwent right heart catheterization (RHC) between 2005 and 2019 [7]. Of the 268 patients who underwent RHC, 137 (51%) were originally diagnosed with various etiologies of PH. When the new definition was applied (mPAP > 20 mmHg) retrospectively, seven out of the 131 patients who previously did not meet criteria for PH were retroactively diagnosed with PH. These included one patient with PAH, three patients with group 2 disease, and three patients with group 3 disease. The authors noted that the one PAH patient who was “missed” with the prior definition had stable disease without signs or symptoms of progression at 7 years after the RHC. Interestingly, of the 124 who still did not meet criteria for PH, 76 had a mPAP > 20 mmHg, but had PAWP ≤ 15 and a PVR < 3 WU.
In addition to this major shift in mPAP cut-off for PH diagnosis, PVR is now included in the diagnostic criteria of all forms of PH. Previously, PVR was only included in the definition of group 1 PH. In addition, diagnosis of pre-capillary PH, post-capillary PH, and combined pre- and post-capillary PH has been revised and all include a PVR cutoff in their definitions. Additionally, other forms of pre-capillary PH (group 3 and group 4) also include a PVR cutoff of ≥ 3 WU in their definition. The details of the hemodynamic diagnosis of these forms of PH will be discussed later in this review.
While the implications of the new hemodynamic definition are still beginning to unfold, there continues to be uncertainty about how to treat these patients. At the very least, they should be followed closely at an expert center, as they are at high risk for progression. Further studies are needed to better define the new population of patients with mPAP 21–24 and to elucidate which patients will benefit from treatment.
Advances in the Genetic Basis of PAH and PVOD
With the development of cutting-edge genetic research methods, the scientific community’s understanding of the complex genetic underpinnings of PH has grown exponentially. High-throughput sequencing in recent years has improved understanding beyond the well-described BMPR2 mutation to include new potential pathologic mechanisms for heritable PAH (HPAH). As technology continues to identify new heritable pathways and therapeutic targets, an age of personalized genetic-based therapies for PH is on the horizon.
The identification of BMPR2 in 2000 by two independent groups was the opening salvo for the age of genetic medicine in PH [8]. Almost 20 years later, it is estimated that BMPR2 mutations account for ~ 80% of heritable and ~ 20% of idiopathic PAH (IPAH) [9]. Several proteins within the BMPR2 signaling pathway have also been sequenced after pedigree construction in affected families and linked to PAH, including ALK1/ACVRL1 (a heterodimeric partner of BMPR2), BMP9 (a BMPR2 ligand), ENG (a coreceptor for BMPR2 signaling) and SMAD1, 4, and 9 (downstream BMP signaling molecules) [10,11,12,13,14]. Whole exome sequencing (WES) in families with PH has led to the discovery of CAV1 a protein involved in membrane localization of BMPR2 and nitric oxide (NO) metabolism [15]. Interestingly, low protein expression of BMPR2 and CAV1 is found in the absence of genetic mutation in patients with PH without heritable PAH, implying processes beyond simple genetic makeup, which are at play in differential protein expression in PAH [16, 17]. It is unclear if wild-type BMPR2 is expressed in the setting of heterozygosity which may confer a milder phenotype.
WES was also used to identify KCNK3, a potassium channel responsible for regulating resting membrane potential and pulmonary vascular tone in PAH patients. More recently, mutations in TBX4 (a gene linked to small patella syndrome) has been identified as a cause of PAH in pediatric patients with hereditary and IPAH [18, 19]. Recently, WES in children demonstrated that after BMPR2 mutations, TBX4 variants confer the highest degree of genetic risk in childhood-onset PAH [20]. Finally, two groups in 2014 independently identified mutations in EIF2AK4 as causative mutation of pulmonary capillary hemangiomatosis (PCH) and pulmonary veno-occlusive disease (PVOD) [21, 22]. If present and biallelic, this mutation is 100% penetrant and is sufficient for diagnosis of these conditions without the necessity of a lung biopsy.
Beyond WES, whole genome sequencing (WGS) has recently been applied to screen for pathogenic genetic variants in large populations of PAH patients. A study of 1048 patients found novel mutations in GDF2 (which codes for BMP9), ATP13A3, AQP1, and SOX17, though the specific pathogenic mechanism of the last three of these genes in HPAH is not yet understood [23]. In addition to genomic DNA, a recent study has now shown that mitochondrial DNA of certain haplogroups shows a proclivity for protection or propensity for the development of PAH [24]. As sequencing technology becomes faster, inexpensive and readily available new mechanisms for the development of PAH will reveal themselves and hopefully provide researchers with novel therapeutic targets.
Many processes associated with translation and post-translational modifications have been implicated in the genetics of PH. Micro (mi) RNA expression is known to play a role in cardiovascular disease, and there are known miRNAs which play a role in BMPR2 mRNA degradation, NFAT activation, and PASMC proliferation currently under investigation [25]. Type I HDACs have been shown to be associated with PASMC proliferation as well, though DNA acetylation and methylation remain areas of active preclinical studies [26, 27].
The importance of discussing the results of genetic screens with patients and families is critical. The psychosocial concerns of genetic screening for a disease for which there is no prevention and no cure presents unique challenges to patient counseling and calls into question the utility of testing outside of specific circumstances. Given the intricacies of the heritable nature of certain types of PAH, it is recommended that genetic screening be done with a multidisciplinary team involving a geneticist as well as PH expert to discuss the benefits of testing to the patient and the choice of gene panel (e.g., commercially available panels, WES or WGS).
Risk Stratification of PH Patients
There are several well-validated risk stratification tools for patients with PAH that have been developed through retrospective analysis of large patient registries (Table 1). These tools use a variety of data points including demographics, functional status, laboratory values, and hemodynamic data to sort patients into low-, intermediate-, or high-risk categories. These categories serve as a framework for choosing an initial treatment strategy and can be measured longitudinally to assess response to treatment as well as disease progression.
The Registry to Evaluate Early and Long-Term PAH Disease Management (REVEAL) risk stratification tool uses 12–14 variables to calculate risk of 1-year mortality. It has been shown to be predictive of survival at baseline [28], at 1-year follow-up [29], and at 5 years [30]. The recently published REVEAL 2.0 calculator has been shown to predict clinical worsening and mortality in patients with PAH who have survived at least 1 year from initial enrollment in the cohort [31]. A European group has also developed a risk-stratification tool using a large PH registry called Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA) [32]. The COMPERA tool uses fewer data points [8] than REVEAL, but it classifies patients into low-, intermediate-, and high-risk categories with similar efficacy as the REVEAL score [33]. The Swedish PAH Registry (SPAHR) [34] and French Pulmonary Hypertension Network (FPHN) [35] have developed similar tools by using data from large registries.
All of these risk stratification tools have similar efficacy in stratifying patients into low- (< 5%), intermediate- (5–10%), and high-risk (> 10%) of mortality at 1 year. Regardless of which tool a clinician chooses, they all seem to have comparable efficacy for identifying patients at high risk. Along with experience and clinical judgement, these tools can provide valuable insight into prognosis and treatment decisions. However, these risk stratifications have a number of limitations, including the use of retrospective data, non-modifiable risk factors, and data points that are not routinely collected in PAH patients [33]. Independent of which tool is chosen, their application will help the practitioner identify patients at high risk of 1-year mortality and should help prioritize therapy and transplant referral when appropriate.
Treatment Algorithm: Group 1 PAH
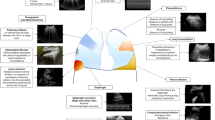
The 6th WSPH provided updated guidance on treatment strategies for group 1 PAH (Fig. 1). The task force recommends vasoreactivity testing be performed only in patients with IPAH, HPAH, and PAH associated with drugs and toxins. Vasoreactive patients should be treated with calcium channel blocker (CCB) monotherapy, progressively titrated to high doses under the supervision of a PH expert. For non-vasoreactive low- and intermediate-risk patients, initial oral combination therapy with an endothelin receptor antagonist (ERA) and a phosphodiesterase-5 inhibitor (PDE5i) is the treatment of choice. The AMBITION trial randomized 500 patients to receive either up-front combination therapy with ambrisentan and tadalafil vs. monotherapy with either drug plus placebo. The patients who received the combination therapy were shown to have significantly decreased rates of clinical failure events [36] (a composite endpoint of death from any cause, hospitalization for worsening PAH, disease progression, and unsatisfactory clinical response). Sitbon et al. [37] looked retrospectively at a cohort of 97 patients who were treated with up-front combination therapy (bosentan or ambrisentan plus sildenafil or tadalafil), and found that all regimens were associated with improved functional class, 6MWD, and hemodynamic variables at 4-month follow-up. At further follow-up (mean time 30 months), these patients had much higher survival rates that were initially calculated by the FPHN risk-stratification score [37]. For high-risk patients, first-line therapy is an intravenous prostacyclin analogue (PCA) (alone or in combination), which has been shown to have a mortality benefit. Finally, the task force recommends that monotherapy should only be used under certain circumstances [33] and should not be a substitute for combination therapy when appropriate.

Treatment algorithm for group 1 pulmonary arterial hypertension
Patients should be re-evaluated 3–6 months after initiation of therapy. Achieving a low-risk status is ideal and these patients should continue on the chosen therapy. For patients who are at intermediate risk after the initial treatment period, escalation to double or triple combination therapy is recommended. For patients who are high risk after the initial treatment period, escalation to maximal medical therapy (including intravenous PCA) is warranted. Any patient who is on maximal medical therapy should be referred for lung transplant evaluation.
Update on Group 2 PH
It is well known that patients with left heart disease (LHD) commonly have comorbid elevations in their right-sided pressures [38]. Usually, these elevations in mPAP are secondary to pulmonary venous congestion secondary to elevated left-sided pressures and do not represent a separate disease entity. However, there is a subset of LHD patients that exhibit significant PH in the setting of severe right ventricular failure and elevated PVR, which has led to the term combined pre- and post-capillary pulmonary hypertension (Cpc-PH). In heart transplant patients, elevations in pre-transplant PVR have been independently associated with increased 30-day post-transplant mortality [39]. Interestingly, increased mortality in transplant patients with elevated pre-transplant PVR was similar in patients with mPAP 20–24 mmHg. Regarding the pathology of Cpc-PH, one autopsy study that looked at the pulmonary vasculature of patients with heart failure reported that the severity of PH correlated most strongly with pulmonary venous and small indeterminate vessel intimal thickening [40]. Interestingly, this was very similar to findings seen in patients with PVOD, though notably less severe.
Based on available evidence, the 6th WSPH task force updated the hemodynamic definition of group 2 PH to further define the distinct clinical phenotypes of PH due to LHD (PH-LHD).
-
Isolated post-capillary PH (IpcPH) (i.e., pulmonary venous hypertension)
-
mPAP > 20 mmHg
-
PAWP > 15 mmHg
-
PVR < 3 WU
-
-
Combined post-capillary and pre-capillary PH (CpcPH) (i.e., out of proportion PH)
-
mPAP > 20 mmHg
-
PAWP > 15 mmHg
-
PVR ≥ 3 WU
-
Importantly, the committee no longer recommends using diastolic pressure gradient (DPG) [DPG = diastolic pulmonary artery pressure (dPAP) − PAWP] to help define PH-LHD and advices on relying on PVR and the transpulmonary gradient (TPG, mPAP-PAWP).
The 6th WSPH recognizes that PH-LHD patients are a heterogenous population with a variety of comorbidities and risk factors. As such, the committee recommends a three-step approach to diagnose patients with PH-LHD (or alternative etiologies of PH). First, clinicians should classify PH-LHD patients into three distinct clinical phenotypes: PH due to heart failure with preserved ejection fraction (PH-HFpEF), PH due to heart failure with reduced ejection fraction (PH-HFrEF), and PH due to valvular heart disease (PH-VHD) [38]. Second, clinicians should determine the pre-test probability of a patient having PH-LHD, by looking at a variety of risk factors that increase the risk of having LHD. These risk factors include older age, elements of the metabolic syndrome, atrial fibrillation, and results from cardiac testing including EKG, echocardiogram, and cardiac MRI. Third, in patients with intermediate and high probability of PH-LHD, clinicians should proceed with hemodynamic evaluation prior to any attempts at using PH-specific therapies.
RHC continues to be the gold standard for diagnosing PH. In patients with LHD, there can be a number of confounding variables (such as atrial fibrillation and large V waves), which can make the interpretation of RHC data difficult. As such, the task force recommends that RHCs be done at expert centers. The task force also recognizes that RHC results can be equivocal in some cases. In these cases, a fluid loading challenge could be useful (500 ml over 5 min), since a PAWP > 18 mmHg immediately after fluid administration is considered abnormal.
Regarding treatment options, the 6th WSPH task force reviewed all available literature on PH specific therapy in PH-LHD, and found no definitive evidence that PH specific therapies provide benefit in PH-LHD [38]. In fact, multiple studies have shown that PH-specific therapies could be harmful in PH-LHD. The MELODY-1 trial randomized patients with Cpc-PH to receive macitentan or placebo, and found that patients in the macitentan arm were more likely to retain fluid and be hospitalized than patients in the placebo arm [41]. The SIOVAC trial randomized patients who had corrected valvular heart disease (VHD) and persistent PH to receive either sildenafil or placebo [42]. Patients in the sildenafil arm had worse clinical outcomes compared to placebo. Based on these trials and others, the 6th WSPH task force issued a strong recommendation against all PH specific therapies for group 2 PH [38].
Update on Group 3 PH
PH is a common comorbid condition in patients with many forms of chronic lung disease (CLD) [43]. CLD-associated pulmonary hypertension (CLD-PH) has been shown to be associated with reduced functional status and worse outcomes [44, 45]. The most well characterized CLD-PH entities are chronic obstructive pulmonary disease (COPD), the idiopathic interstitial pneumonias (IIP) including idiopathic pulmonary fibrosis (IPF), and sarcoidosis. In one retrospective study of patients with Global Initiative for Chronic Obstructive Lung Disease stage IV (GOLD IV) COPD [46], many patients had a mean pulmonary artery pressure (mPAP) > 20 mmHg, but only 2.7% had mPAP > 40 mmHg. Many of these patients with severe PH (mPAP > 40 mmHg) had other identifiable causes of PH, such as pulmonary embolism (PE). PH is also common in patients with IPF and other IIPs. Prevalence in this population ranges from 8% to 45% depending on severity of the CLD [47,48,49]. Severe PH (mPAP > 40 mmHg) is relatively uncommon, occurring in < 10% of patients with IPF referred for lung transplant [47]. PH is also common in patients with combined pulmonary fibrosis and emphysema (CFPE)[50] and sarcoidosis [51]. It has also been described in patients with lymphangioleiomyomatosis (LAM), pulmonary Langerhans cell histiocytosis (PLCH), bronchopulmonary dysplasia, cystic fibrosis (CF), and lung cancer [43].
The 6th WSPH recommends the following hemodynamic definitions of CLD-PH [43]:
-
1.
CLD without PH (mPAP < 21 mmHg, or mPAP 21–24 with PVR < 3 WU).
-
2.
CLD with PH (mPAP 21–24 mmHg with PVR ≥ 3 WU, or mPAP 25–34) (CLD-PH).
-
3.
CLD with severe PH [mPAP ≥ 35 mmHg, or mPAP ≥ 25 mmHg and low cardiac index (CI) (< 2.0 l/min/m2)] (CLD-severe PH).
The 6th WSPH committee recognizes that distinguishing between group 1 and group 3 PH can be very challenging. Certain features favor a diagnosis of group 1 PAH, including mild lung disease (FEV1 > 60%, FVC > 70%) and more severe PH. Conversely, more severe lung disease (FEV1 < 60%, FVC < 70%) and milder PH favors a diagnosis of group 3 PAH. As with other types of PH, if there is diagnostic uncertainly or severe disease, the patient should be referred to an expert center. Echocardiography remains the best non-invasive measure to screen for CLD-PH. Other tests, including plasma levels of brain natriuretic peptide (BNP) or N-terminal pro-BNP (NT-proBNP), and increased ratio of the main pulmonary artery to ascending aorta diameter [52], can be suggestive of CLD-PH. RHC is recommended in patients with CLD when significant PH is suspected and the clinician suspects that the diagnosis will change management [43].
In all patients with CLD-PH, the lung disease should be treated according to current guidelines. Oxygen should be given to patients when indicated (i.e., resting oxygen saturation ≤ 88%). With regards to PH-specific treatment, the data is quite heterogenous. While there are some data that suggest PH-specific therapy may improve hemodynamics [53], symptoms and 6MWD [54] in COPD-PH patients with a mPAP ≥ 35 mmHg, the 6th WSPH does not make specific treatment recommendations. There is similar heterogeneity of treatment data in patients with IIPs, and the 6th WSPH does not make specific recommendations for treatment.
There are two important trials which show harm for PH-specific therapies in CLD-PH. The ARTEMIS trial was stopped early after patients in the ambrisentan group were more likely to have disease progression [48]. In the RISE-IIP trial, there was increased mortality and serious adverse events in the riociguat group [55]. Based on the results of these two trials, ambrisentan and riociguat are contraindicated in IPF and IIP, respectively.
There are minimal data on efficacy of PH-specific treatment in sarcoidosis and the other CLDs. The general recommendations for patients with CLD-PH by the 6th WSPH are as follows [43]:
-
1.
For patients with mild lung disease and severe PH, the distinction between group 1 and group 3 disease is critical, and these patients should be referred to an expert center.
-
2.
No PH-specific therapy is recommended in patients with severe lung disease and less severe PH (mPAP 21–24 with PVR ≥ 3 WU, or mPAP 25–34 mmHg).
-
3.
Patients with severe lung disease and severe PH (mPAP ≥ 35 mmHg) should be referred to an expert center for “individualized patient care”.
-
4.
Patients with “end-stage” lung disease and PH should also be referred to an expert center for care. Extraordinary measures such as extracorporeal membrane oxygenation (ECMO) and mechanical ventilation should only be used as a bridge to lung transplantation.
PH has also been observed in patients with hypoventilation and hypoxia. PH in patients with obstructive sleep apnea (OSA) is rare and usually mild. Interestingly, PH is much more common in patients with obesity hypoventilation syndrome (OHS) and is often severe, marked by chronic right ventricular failure and poor outcomes [56]. The treatment for both OSA and OHS associated PH is non-invasive ventilation.
Update on Group 4 PH
Chronic thromboembolic pulmonary hypertension (CTEPH) is a possible sequelae of acute PE, with an estimated incidence between 0.4% and 6.2% [57]. Interestingly, not all patients found to have CTEPH have a history of acute PE. While the exact pathogenesis of CTEPH is unknown, it is characterized by acute thromboembolism progressing to chronically organized thromboembolic material and associated vascular remodeling [57]. There are various factors that likely contribute to the progression to CTEPH, including disturbances in angiogenesis (which is an important step in recanalization), fibrinolysis, and the endothelium [58].
Ventilation/perfusion (V/Q) scan remains the diagnostic test of choice for CTEPH. Once the diagnosis of CTEPH has been made, computed tomography of the pulmonary arteries (CTPA) and pulmonary angiography can be useful for surgical planning. Patients with CTEPH require lifelong anticoagulation. Vitamin K antagonists (such as warfarin) have historically been the mainstay of treatment; however, the 6th WSPH task force recognizes that there is minimal evidence to suggest that one class of anticoagulant is better than another (i.e., the newer oral anticoagulants).
All patients with a diagnosis of CTEPH should be referred to an expert center for evaluation for pulmonary endarterectomy, which is the recommended first-line treatment. Exact criteria for operability are institution-specific, but clear disease concordance on imaging, bilateral lower lobe disease, better functional class, absent signs of right heart failure and lack of significant medical comorbidities are all signs of a favorable risk–benefit assessment for pulmonary endarterectomy [57].
About 40% of patients with CTEPH are deemed to be non-operable [59]. These patients do have options for PH-directed medical therapy. The CHEST-1 trial randomized patients with non-operable CTEPH or persistent PH after PEA to receive either riociguat or placebo [60]. Patients in the riociguat arm had significant improvement in exercise capacity and PVR. Riociguat is currently the only FDA-approved medication for the treatment of CTEPH. Other randomized controlled trials have looked at bosentan [61] and macitentan [62] in non-operative CTEPH, with some benefits in hemodynamics and 6MWD, but these medications are not routinely used.
Balloon pulmonary angioplasty (BPA) is an emerging therapy for CTEPH. This procedure involves the endovascular insertion of a balloon into the CTEPH lesions, with dilation leading to recanalization of the pulmonary arteries. The procedure is technically challenging, and should only be performed at high-volume centers, where it remains an option for some patients with non-operable CTEPH.
Chronic thromboembolic disease (CTED) is an emerging clinical entity which has created some confusion in the PH community. CTED is defined as perfusion defects on V/Q scan accompanied by symptoms (dyspnea, exercise limitation, etc.), but without hemodynamic evidence of PH at rest [57]. Some possible etiologies of the symptoms patients with CTED experience include abnormal pulmonary vascular response to exercise [63] and increased dead space ventilation [64]. There is currently no evidence that CTED evolves into CTEPH, but there is some data that suggests that selected patients with CTED may have improved exercise capacity after PEA [65]. However, the 6th WSPH task force warns that the CTEPH treatment guidelines should not be applied to CTED.
Update on Group 5 PH
The 6th WSPH again considered group 5 PH to be an area in need of further study. The group, classified as “Pulmonary Hypertension with unclear and or multi-factorial mechanisms” since the 4th WSPH represents a group of diseases in which affected individuals are at increased risk for PH compared to the general population. Noteworthy changes following the 6th WSPH mainly revolve around the removal of certain clinical conditions as potential causes of PH. Specifically, splenectomy and thyroid dysfunction have both been removed as clinical conditions that are associated with PH in isolation and are now considered risk factors for PH. The committee recommends maintaining adequate control of an individual’s thyroid dysfunction when treating PH. LAM has also been removed from group 5 and has been re-classified in group 3. Further studies in the LAM population have suggested that severity of PH is usually mild and that it correlates with the degree of lung dysfunction [66].
Conclusions
In this review, we discussed the recent updates to the hemodynamic definition of PH, recent advances in genetics, risk stratification strategies, and general updates in the classification and treatment of all five groups of PH. As the field of PH is constantly growing, we anticipate that discoveries will continue at an advanced pace. Therefore, practitioners should remain attentive as studies aimed at elucidating the impact of the new hemodynamic definition PH are being conducted to inform on the approach to take when dealing with this new subset of patients with mPAP > 20 but < 25 mmHg. Regardless of the outcome of these studies, practitioners have a toolkit at their disposal to help treat PH patients and ensure appropriate care at all stages of the disease.
References
Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, Krowka M, et al. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J. 2019;53(1):1801913.
Kovacs G, Berghold A, Scheidl S, Olschewski H. Pulmonary arterial pressure during rest and exercise in healthy subjects: a systematic review. Eur Respir J. 2009;34(4):888–94.
Coghlan JG, Wolf M, Distler O, Denton CP, Doelberg M, Harutyunova S, et al. Incidence of pulmonary hypertension and determining factors in patients with systemic sclerosis. Eur Respir J. 2018;51(4):1701197.
Douschan P, Kovacs G, Avian A, Foris V, Gruber F, Olschewski A, et al. Mild elevation of pulmonary arterial pressure as a predictor of mortality. Am J Respir Crit Care Med. 2018;197(4):509–16.
Maron BA, Hess E, Maddox TM, Opotowsky AR, Tedford RJ, Lahm T, et al. Association of borderline pulmonary hypertension with mortality and hospitalization in a large patient cohort: insights from the veterans affairs clinical assessment, reporting, and tracking program. Circulation. 2016;133(13):1240–8.
Gibbs JSR, Torbicki A. Proposed new pulmonary hypertension definition: is 4 mm (Hg) worth re-writing medical textbooks? Eur Respir J. 2019;53(3):1900197.
Jaafar S, Visovatti S, Young A, Huang S, Cronin P, Vummidi D, et al. Impact of the revised haemodynamic definition on the diagnosis of pulmonary hypertension in patients with systemic sclerosis. Eur Respir J. 2019;54(2):1900586.
Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA, Loyd JE, et al. Heterozygous germline mutations in BMPR2, encoding a TGF-β receptor, cause familial primary pulmonary hypertension. Nat Genet. 2000;26(1):81–4.
Evans JDW, Girerd B, Montani D, Wang X-J, Galiè N, Austin ED, et al. BMPR2 mutations and survival in pulmonary arterial hypertension: an individual participant data meta-analysis. Lancet Respir Med. 2016;4(2):129–37.
Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med. 2001;345(5):325–34.
Chaouat A. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax. 2004;59(5):446–8.
Nasim MDT, Ogo T, Ahmed M, Randall R, Chowdhury HM, Snape KM, et al. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat. 2011;32(12):1385–9.
Wang X-J, Lian T-Y, Jiang X, Liu S-F, Li S-Q, Jiang R, et al. Germline BMP9 mutation causes idiopathic pulmonary arterial hypertension. Eur Respir J. 2019;53(3):1801609.
Harrison RE, Berger R, Haworth SG, Tulloh R, Mache CJ, Morrell NW, et al. Transforming growth factor-β receptor mutations and pulmonary arterial hypertension in childhood. Circulation. 2005;111(4):435–41.
Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, Phillips JA, et al. Whole exome sequencing to identify a novel gene (Caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet. 2012;5(3):336–43.
Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105(14):1672–8.
Achcar ROD, Demura Y, Rai PR, Taraseviciene-Stewart L, Kasper M, Voelkel NF, et al. Loss of caveolin and heme oxygenase expression in severe pulmonary hypertension. Chest. 2006;129(3):696–705.
Ma L, Roman-Campos D, Austin ED, Eyries M, Sampson KS, Soubrier F, et al. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med. 2013;369(4):351–61.
Kerstjens-Frederikse WS, Bongers EMHF, Roofthooft MTR, Leter EM, Douwes JM, Van Dijk A, et al. TBX4 mutations (small patella syndrome) are associated with childhood-onset pulmonary arterial hypertension. J Med Genet. 2013;50(8):500–6.
Zhu N, Gonzaga-Jauregui C, Welch CL, Ma L, Qi H, King AK, et al. Exome sequencing in children with pulmonary arterial hypertension demonstrates differences compared with adults. Circ Genom Precis Med [Internet]. 2018. https://doi.org/10.1161/CIRCGEN.117.001887.
Best DH, Sumner KL, Austin ED, Chung WK, Brown LM, Borczuk AC, et al. EIF2AK4 mutations in pulmonary capillary hemangiomatosis. Chest. 2014;145(2):231–6.
Eyries M, Montani D, Girerd B, Perret C, Leroy A, Lonjou C, et al. EIF2AK4 mutations cause pulmonary veno-occlusive disease, a recessive form of pulmonary hypertension. Nat Genet. 2014;46(1):65–9.
Gräf S, Haimel M, Bleda M, Hadinnapola C, Southgate L, Li W, et al. Identification of rare sequence variation underlying heritable pulmonary arterial hypertension. Nat Commun. 2018;9(1):1416.
Farha S, Hu B, Comhair S, Zein J, Dweik R, Erzurum SC, et al. Mitochondrial haplogroups and risk of pulmonary arterial hypertension. West J, editor. PLOS One. 2016;11(5):e0156042.
Courboulin A, Paulin R, Giguère NJ, Saksouk N, Perreault T, Meloche J, et al. Role for miR-204 in human pulmonary arterial hypertension. J Exp Med. 2011;208(3):535–48.
Cavasin MA, Demos-Davies K, Horn TR, Walker LA, Lemon DD, Birdsey N, et al. Selective class i histone deacetylase inhibition suppresses hypoxia-induced cardiopulmonary remodeling through an antiproliferative mechanism. Circ Res. 2012;110(5):739–48.
Liu D, Yan Y, Chen J-W, Yuan P, Wang X-J, Jiang R, et al. Hypermethylation of BMPR2 promoter occurs in patients with heritable pulmonary arterial hypertension and inhibits BMPR2 expression. Am J Respir Crit Care Med. 2017;196(7):925–8.
Benza RL, Gomberg-Maitland M, Miller DP, Frost A, Frantz RP, Foreman AJ, et al. The REVEAL Registry Risk Score calculator in patients newly diagnosed with pulmonary arterial hypertension. Chest. 2012;141(2):354–62.
Benza RL, Miller DP, Foreman AJ, Frost AE, Badesch DB, Benton WW, et al. Prognostic implications of serial risk score assessments in patients with pulmonary arterial hypertension: a registry to evaluate early and long-term pulmonary arterial hypertension disease management (REVEAL) analysis. J Heart Lung Transplant. 2015;34(3):356–61.
Farber HW, Miller DP, Poms AD, Badesch DB, Frost AE, Rouzic EM-L, et al. Five-year outcomes of patients enrolled in the REVEAL registry. Chest. 2015;148(4):1043–54.
Benza RL, Gomberg-Maitland M, Elliott CG, Farber HW, Foreman AJ, Frost AE, et al. Predicting survival in patients with pulmonary arterial hypertension. Chest. 2019;156(2):323–37.
Hoeper MM, Kramer T, Pan Z, Eichstaedt CA, Spiesshoefer J, Benjamin N, et al. Mortality in pulmonary arterial hypertension: prediction by the 2015 European pulmonary hypertension guidelines risk stratification model. Eur Respir J. 2017;50(2):1700740.
Galiè N, Channick RN, Frantz RP, Grünig E, Jing ZC, Moiseeva O, et al. Risk stratification and medical therapy of pulmonary arterial hypertension. Eur Respir J. 2019;53(1):1801889.
Kylhammar D, Kjellström B, Hjalmarsson C, Jansson K, Nisell M, Söderberg S, et al. A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. Eur Heart J. 2018;39(47):4175–81.
Boucly A, Weatherald J, Savale L, Jaïs X, Cottin V, Prevot G, et al. Risk assessment, prognosis and guideline implementation in pulmonary arterial hypertension. Eur Respir J. 2017;50(2):1700889.
Galiè N, Barberà JA, Frost AE, Ghofrani H-A, Hoeper MM, McLaughlin VV, et al. Initial use of ambrisentan plus tadalafil in pulmonary arterial hypertension. N Engl J Med. 2015;373(9):834–44.
Sitbon O, Sattler C, Bertoletti L, Savale L, Cottin V, Jaïs X, et al. Initial dual oral combination therapy in pulmonary arterial hypertension. Eur Respir J. 2016;47(6):1727–36.
Vachiéry J-L, Tedford RJ, Rosenkranz S, Palazzini M, Lang I, Guazzi M, et al. Pulmonary hypertension due to left heart disease. Eur Respir J. 2019;53(1):1801897.
Crawford TC, Leary PJ, Fraser CD, Suarez-Pierre A, Magruder JT, Baumgartner WA, et al. Impact of the new pulmonary hypertension definition on heart transplant outcomes. Chest. 2019. https://doi.org/10.1016/j.chest.2019.07.028.
Fayyaz AU, Edwards WD, Maleszewski JJ, Konik EA, DuBrock HM, Borlaug BA, et al. Global pulmonary vascular remodeling in pulmonary hypertension associated with heart failure and preserved or reduced ejection fraction. Circulation. 2018;137(17):1796–810.
Vachiéry J-L, Delcroix M, Al-Hiti H, Efficace M, Hutyra M, Lack G, et al. Macitentan in pulmonary hypertension due to left ventricular dysfunction. Eur Respir J. 2018;51(2):1701886.
Bermejo J, Yotti R, García-Orta R, Sánchez-Fernández PL, Castaño M, Segovia-Cubero J, et al. Sildenafil for improving outcomes in patients with corrected valvular heart disease and persistent pulmonary hypertension: a multicenter, double-blind, randomized clinical trial. Eur Heart J. 2018;39(15):1255–64.
Nathan SD, Barbera JA, Gaine SP, Harari S, Martinez FJ, Olschewski H, et al. Pulmonary hypertension in chronic lung disease and hypoxia. Eur Respir J. 2019;53(1):1801914.
Hayes D, Black SM, Tobias JD, Kirkby S, Mansour HM, Whitson BA. Influence of pulmonary hypertension on patients with idiopathic pulmonary fibrosis awaiting lung transplantation. Ann Thorac Surg. 2016;101(1):246–52.
Medrek SK, Sharafkhaneh A, Spiegelman AM, Kak A, Pandit LM. Admission for COPD exacerbation is associated with the clinical diagnosis of pulmonary hypertension: results from a retrospective longitudinal study of a veteran population. COPD J Chronic Obstr Pulm Dis. 2017;14(5):484–9.
Chaouat A, Bugnet A-S, Kadaoui N, Schott R, Enache I, Ducoloné A, et al. Severe pulmonary hypertension and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;172(2):189–94.
Shorr AF, Wainright JL, Cors CS, Lettieri CJ, Nathan SD. Pulmonary hypertension in patients with pulmonary fibrosis awaiting lung transplant. Eur Respir J. 2007;30(4):715–21.
Raghu G, Nathan SD, Behr J, Brown KK, Egan JJ, Kawut SM, et al. Pulmonary hypertension in idiopathic pulmonary fibrosis with mild-to-moderate restriction. Eur Respir J. 2015;46(5):1370–7.
Kimura M, Taniguchi H, Kondoh Y, Kimura T, Kataoka K, Nishiyama O, et al. Pulmonary hypertension as a prognostic indicator at the initial evaluation in idiopathic pulmonary fibrosis. Respiration. 2013;85(6):456–63.
Cottin V, Le Pavec J, Prevot G, Mal H, Humbert M, Simonneau G, et al. Pulmonary hypertension in patients with combined pulmonary fibrosis and emphysema syndrome. Eur Respir J. 2010;35(1):105–11.
Shorr AF. Pulmonary hypertension in advanced sarcoidosis: epidemiology and clinical characteristics. Eur Respir J. 2005;25(5):783–8.
Yagi M, Taniguchi H, Kondoh Y, Ando M, Kimura T, Kataoka K, et al. CT-determined pulmonary artery to aorta ratio as a predictor of elevated pulmonary artery pressure and survival in idiopathic pulmonary fibrosis: measurement of the PA:A ratio in IPF. Respirology. 2017;22(7):1393–9.
Vonbank K. Controlled prospective randomised trial on the effects on pulmonary haemodynamics of the ambulatory long term use of nitric oxide and oxygen in patients with severe COPD. Thorax. 2003;58(4):289–93.
Valerio G, Bracciale P, Grazia D’Agostino A. Effect of bosentan upon pulmonary hypertension in chronic obstructive pulmonary disease. Ther Adv Respir Dis. 2009;3(1):15–21.
Nathan SD, Behr J, Cottin V, Collard HR, Hoeper MM, Martinez FJ, et al. Idiopathic interstitial pneumonia-associated pulmonary hypertension: a target for therapy? Respir Med. 2017;122:S10–3.
Castro-Añón O, Golpe R, Pérez-De-Llano LA, López González MJ, Escalona Velasquez EJ, Pérez Fernández R, et al. Haemodynamic effects of non-invasive ventilation in patients with obesity-hypoventilation syndrome: haemodynamic changes in OHS. Respirology. 2012;17(8):1269–74.
Kim NH, Delcroix M, Jais X, Madani MM, Matsubara H, Mayer E, et al. Chronic thromboembolic pulmonary hypertension. Eur Respir J. 2019;53(1):1801915.
Lang IM, Pesavento R, Bonderman D, Yuan JXJ. Risk factors and basic mechanisms of chronic thromboembolic pulmonary hypertension: a current understanding. Eur Respir J. 2013;41(2):462–8.
Pepke-Zaba J, Delcroix M, Lang I, Mayer E, Jansa P, Ambroz D, et al. Chronic thromboembolic pulmonary hypertension (CTEPH): results from an international prospective registry. Circulation. 2011;124(18):1973–81.
Ghofrani H-A, D’Armini AM, Grimminger F, Hoeper MM, Jansa P, Kim NH, et al. Riociguat for the treatment of chronic thromboembolic pulmonary hypertension. N Engl J Med. 2013;369(4):319–29.
Jaïs X, D’Armini AM, Jansa P, Torbicki A, Delcroix M, Ghofrani HA, et al. Bosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension. J Am Coll Cardiol. 2008;52(25):2127–34.
Ghofrani H-A, Simonneau G, D’Armini AM, Fedullo P, Howard LS, Jaïs X, et al. Macitentan for the treatment of inoperable chronic thromboembolic pulmonary hypertension (MERIT-1): results from the multicentre, phase 2, randomised, double-blind, placebo-controlled study. Lancet Respir Med. 2017;5(10):785–94.
van Kan C, van der Plas MN, Reesink HJ, van Steenwijk RP, Kloek JJ, Tepaske R, et al. Hemodynamic and ventilatory responses during exercise in chronic thromboembolic disease. J Thorac Cardiovasc Surg. 2016;152(3):763–71.
Held M, Kolb P, Grün M, Jany B, Hübner G, Grgic A, et al. Functional characterization of patients with chronic thromboembolic disease. Respiration. 2016;91(6):503–9.
Taboada D, Pepke-Zaba J, Jenkins DP, Berman M, Treacy CM, Cannon JE, et al. Outcome of pulmonary endarterectomy in symptomatic chronic thromboembolic disease. Eur Respir J. 2014;44(6):1635–45.
Wu X, Xu W, Wang J, Tian X, Tian Z, Xu K. Clinical characteristics in lymphangioleiomyomatosis-related pulmonary hypertension: an observation on 50 patients. Front Med. 2019;13(2):259–66.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Christopher A Thomas, Ryan J Anderson, David F Condon, and Vinicio A de Jesus Perez have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.10304645.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any non-commercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Thomas, C.A., Anderson, R.J., Condon, D.F. et al. Diagnosis and Management of Pulmonary Hypertension in the Modern Era: Insights from the 6th World Symposium. Pulm Ther 6, 9–22 (2020). https://doi.org/10.1007/s41030-019-00105-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s41030-019-00105-5