
Abstract
Precision oncology aims to tailor clinical decisions specifically to patients with the objective of improving treatment outcomes. This can be achieved by leveraging omics information for accurate molecular characterization of tumors. Tumor tissue biopsies are currently the main source of information for molecular profiling. However, biopsies are invasive and limited in resolving spatiotemporal heterogeneity in tumor tissues. Alternative non-invasive liquid biopsies can exploit patient’s body fluids to access multiple layers of tumor-specific biological information (genomes, epigenomes, transcriptomes, proteomes, metabolomes, circulating tumor cells, and exosomes). Analysis and integration of these large and diverse datasets using statistical and machine learning approaches can yield important insights into tumor biology and lead to discovery of new diagnostic, predictive, and prognostic biomarkers. Translation of these new diagnostic tools into standard clinical practice could transform oncology, as demonstrated by a number of liquid biopsy assays already entering clinical use. In this review, we highlight successes and challenges facing the rapidly evolving field of cancer biomarker research.
Lay Summary
Precision oncology aims to tailor clinical decisions specifically to patients with the objective of improving treatment outcomes. The discovery of biomarkers for precision oncology has been accelerated by high-throughput experimental and computational methods, which can inform fine-grained characterization of tumors for clinical decision-making. Moreover, advances in the liquid biopsy field allow non-invasive sampling of patient’s body fluids with the aim of analyzing circulating biomarkers, obviating the need for invasive tumor tissue biopsies. In this review, we highlight successes and challenges facing the rapidly evolving field of liquid biopsy cancer biomarker research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cancer is a class of complex diseases characterized by abnormal cellular growth and the potential to invade healthy tissues and organs. The incidence of cancer and cancer-related death rates has been on the rise globally [1]. With more than 18 million new cases, and more than 9 million deaths per year, cancer is the first or the second cause of death before the age of 70 in 91 out of 172 countries [1].
Cancer survival rates vary substantially between different types of cancer, where diagnosis at late stage worsens prognosis even for treatable cancers. Metastatic spread to distal sites, which is the definition of late-stage cancer, accounts for 90% of cancer-related deaths [2]. There is a case to be made that population-wide screening and early cancer detection might have a substantial positive impact on cancer morbidity and mortality [3]. Despite this need for earlier detection of cancer, screening tests with proven clinical utility are uncommon. The advent of high-throughput technologies and computational tools is likely to facilitate early diagnosis in years to come. However, adoption of new biomarkers for early cancer diagnosis requires careful consideration of available evidence for associated benefits, costs, and potential harms [4].
For cancers in which early diagnosis is possible and treatment options exist, favorable outcomes are often impeded by our limited understanding of patient stratification to guide treatment decisions. The current clinical practice for diagnosis and treatment decisions is commonly based on methods like tissue biopsy, imaging techniques (CT, MRI, or PET), and cytology. The information gained from these approaches is coarse-grained, because they provide little detail at the molecular level about the underlying cancer. This can complicate treatment decisions, because the interpatient tumor heterogeneity often dictates the response to the available therapies [5, 6].
Rapid advances in omics technologies, such as genomics, transcriptomics, epigenomics, proteomics, and metabolomics, can be used to profile biopsied tumor samples at great detail, enabling precision oncology [7, 8]. However, even such detailed analyses render only static snapshots of the tumor tissue. Rather than being homogeneous, tumors exhibit spatial heterogeneity and undergo Darwinian evolution [9,10,11,12], which can confound prognosis or render treatment decisions ineffective. Unfortunately, solid tumors can only be repeatedly biopsied with invasive procedures, necessitating alternative non-invasive diagnostic strategies.
Precision oncology is turning towards liquid biopsy as an approach for non-invasive and risk-free detection and monitoring of cancer. Liquid biopsy relies on deriving diagnostic information about cancer by detecting and measuring tumor-related biomarkers in non-solid biological tissues (most commonly blood, urine, and stool) [13,14,15,16]. Liquid biopsy can target diverse classes of biomarkers, such as circulating tumor cells (CTC), circulating free and tumor DNA (cfDNA and ctDNA, respectively), RNA, exosomes with their corresponding biological cargo, circulating proteins, or metabolites [16,17,18]. Integration of these omics technologies enables interrogation of clinical samples for early diagnosis, prediction of therapy response for patient stratification [18], and longitudinal monitoring in cancer patients. For cancers which can be treated, precise localization, burden quantification, and knowledge of molecular signatures can be used to longitudinally tailor treatments. Furthermore, accurate cancer profiling can readily identify non-aggressive cancers and help patients avoid overtreatment.
Biomarkers are defined as a “characteristic that is measured as an indicator of normal biological processes, pathogenic processes, or a response to an exposure or intervention” [19]. Therefore, effective implementation of personalized medicine depends on identifying substances, patterns, or activities which can be reliably assayed as indicators for differential diagnosis (diagnostic), classifying tumors based on the probable outcome in the absence of treatment (prognostic) or assessing the probability that a patient will respond positively to a particular treatment (predictive). The goal of the cancer biomarker research is to develop robust, sensitive, specific, and cost-effective strategies for these clinical uses.
A survey of cancer biomarker literature shows a steady increase of interest in the field (Fig. 1). However, only a fraction of putative cancer biomarkers has made it to clinical trials, and only precious few validated in clinical trials (Fig. 1). This discrepancy has been a topic of several reviews [20, 21] and will be discussed in later sections. Briefly, in order to have clinical utility, performance of cancer biomarkers should conform to a set of analytical and clinical requirements [22]. Analytical requirements, such as precision, trueness, limit of detection and quantitation, linearity range, and specificity, while necessary, are not sufficient evidence of clinical validity or utility. Clinical validity of a biomarker is the ability of the biomarker to accurately identify patients with the targeted pathological state [23], while the utility measures the benefit (such as reduced mortality) of using a biomarker in clinical settings [24]. Evaluating the clinical performance [24] (intended use, clinical specificity and sensitivity, ROC analysis, positive and negative predictive values, cost-effectives, fast turn-around) of a biomarker requires carefully designed studies with large cohorts. Adopting biomarkers into standard clinical use without evidence of their clinical utility can be highly problematic. For example, population-wide screening based on biomarkers with poor specificity or low positive predictive values can translate into large numbers of patients undergoing unnecessary, expensive, and potentially harmful procedures. However, validation and utility studies are lengthy and expensive, and often out of reach for most laboratories conducting basic research. On the other hand, investment of time and resources into rigorous validation of biomarkers and molecular-level profiling of tumors might become more valuable than histopathological information for therapeutic decisions and drug approval in the future [25].

Cancer biomarker studies reported in PubMed. Grey bars show the percentage of biomarker articles among all journal articles and letters indexed in PubMed in English (articles with substance name “Biomarkers, Tumor” MeSH term “Humans,” excluding MeSH terms “Tumor Cells, Cultured, and Cell Line”). The orange bars show the percentage of PubMed articles on circulating biomarkers (biomarker articles and letters with MeSH terms “blood”, “plasma”, “serum”, “urine”, or “saliva”). Cyan bars show the subset of all biomarker articles and letters from the trial, multicenter, validation, clinical, or evaluation studies
This review discusses current standard and emerging approaches in the space of liquid biopsy. We categorize cancer biomarkers by omics layers (Fig. 2) and for each omics, we first evaluate the scientific rationale for its relevance in cancer. Second, we review progress and challenges associated with established or commercially exploited emerging technologies. And lastly, we examine and highlight some of the recent success stories and challenges in the clinical translation of cancer biomarkers. Some of the recently developed technologies still far from producing evidence of clinical validity are outside of the scope of this paper and reviewed elsewhere [17].

Liquid biopsy and biomarkers for precision medicine. Liquid biopsy is used to collect body fluid samples from individual patients. Samples typically contain multiple sources of biomarkers, which can in turn be analyzed using modern high-throughput omics technologies. High dimensional and heterogeneous data can be integrated into biological networks with interacting biochemical circuits and pathways. This complex multi-dimensional data can be analyzed and reduced using statistical and machine learning algorithms, with the end goal of producing robust and accurate classifiers for diagnosis, prediction of response to treatment and prognosis. Finally, the same scheme can be used for longitudinal monitoring of patients, obviating the need for repeated invasive biopsies
Sources of Biomarkers
Genome
Scientific Rationale
The onset and progression of cancer is dependent of alterations to DNA sequence of the genomes of cancer cells. The illumination of these alterations and their functional consequences can help improve clinical management of cancer patients. The investigation of genetic underpinnings of cancer led to the discovery of genetic and genomic cancer risk factors and biomarkers, some of which are well established in cancer research and clinical use. A classic example of genetic risk assessments is based on identifying inherited alleles of BRCA1 and BRCA2 genes associated with increased risk for breast cancer [26]. Genotyping cancer biopsy samples by PCR, Sanger, and next-generation sequencing is becoming standard clinical practice [27], a trend that is likely to continue as the whole genome sequencing (WGS) costs approach $1,000 per genome [28] (however, see refs [29, 30] for a critical evaluation of costs for NGS in clinical practice). Unfortunately, sample collection for tissue genotyping requires invasive procedures and returns only static information about a disease which is ultimately spatiotemporally dynamic. Thus, circulating DNA [31,32,33] is being investigated extensively as an emerging biomarker for personalized medicine.
Circulating or cell free DNA (cfDNA) is released from healthy as well as cancer tissues. cfDNA probably originates from apoptotic and necrotic cells, but the exact origin and mechanism of release are still a topic of investigation [34, 35]. Primary tumors, circulating tumor cells, occult and overt metastases can release DNA, increasing concentrations of cell-free DNA (cfDNA) in bodily fluids of cancer patients, compared to healthy people [31, 36]. Circulating tumor DNA (ctDNA) has been found in blood, urine, stool, and saliva. In people with cancer, about 1% of circulating DNA is ctDNA [35, 37], but this percentage can vary substantially between patients and is affected by stage and tumor volume [34]. Because ctDNA displays mutations characteristic of the progenitor tumor, it can serve as a biomarker for diagnosis, prognosis, and prediction.
The half-life of ctDNA is short (2–5 h in blood and urine [33, 34, 38]) and concentrations can decrease rapidly in response to treatment [33]. ctDNA levels are therefore useful as a monitoring biomarker [33] for estimating cancer burden in patients and assessing treatment responses.
Current and Emerging Technologies
Current and upcoming genomics technologies to detect and quantify ctDNA range from targeted to unbiased genome-wide methods and vary dramatically in their analytical sensitivity, specificity, costs, and throughput. Despite the technological progress of the field, reliable detection of low ctDNA amounts and variants with low mutant allele frequencies (MAF) can be challenging. Because ctDNA typically makes up only a small fraction of cfDNA, detection of rare tumor alleles might require large amounts of input materials and prohibitive sample volumes. Genomics technologies and their limitations have been a topic of a recent comprehensive review [34] and we will only cover them briefly in the following paragraphs.
Detection and quantification of predetermined alleles from ctDNA by Sanger sequencing, single and multi-locus PCRs [39], and qPCR [40, 41] is established in clinical oncology (e.g., cobas assay [40, 42, 43]). However, these methods are typically limited to detecting MAFs well above 1% [34, 44, 45], which would leave many cancers smaller than 10 cm3 undetected because their expected plasma MAFs can be 0.1% or lower [17, 46]. Advances such as digital droplet PCR (ddPCR) [47] and BEAMing [48, 49] allow absolute quantification of allele frequencies as low as 0.01%.
Next-generation sequencing (NGS) methods can be used for both targeted and unbiased (the whole genome (WGS) approach) identification of patient-specific structural aberrations [50], copy number variations, and SNPs. However, NGS approaches are limited by the low abundance of ctDNA fraction in total cfDNA, short ctDNA fragment length, as well as by high error rates that preclude reliable detection of rare variants. The sensitivity of targeted NGS methods can be boosted by identifying cancer-specific mutations (from patient-specific tumor tissue samples or known cancer-related alleles), and then applying this information as a selector in liquid biopsy samples. This approach is exemplified by tagged amplicon deep sequencing (TAm-Seq) [51], and enrichment by hybrid capture and molecular barcoding methods such as Safe-Seq [52] and CAPP-Seq [53] to detect MAF as low as 0.00025%. The record analytical sensitivity of detecting 4 in 105 cfDNA molecules was achieved by combining CAPP-Seq with molecular barcoding and in silico digital error suppression [54]. Sensitivity of unbiased methods can be improved by using methods such as whole genome amplification (WGA) [55].
Whole exome sequencing (WES) from liquid biopsy samples offers the ability to longitudinally follow cancer patients under treatment and monitor the appearance of resistance-conferring mutations. For example, in a proof-of-principle study, WES from plasma has been used to monitor six patients with metastatic breast, ovarian, and lung cancers for up to 2 years, revealing resistance-conferring mutations evolving in response to treatment [56]. However, such unbiased approaches have low sensitivity and can only be used on patients with advanced cancer.
Short-read massively parallel sequencing is now being increasingly used in the biomarker discovery and routine clinical applications. However, the aforementioned short read technologies suffer from GC bias, difficulty resolving complex and repeating sequences and phasing alleles. Genotyping liquid biopsy samples using long read single molecule sequencing offers a new approach that mitigates some of these limitations. There are currently two commercially available single molecule sequencing platforms with long reads commercialized by Pacific Biosciences and Oxford Nanopore Technologies. These two sequencing platforms enable phasing of nucleotide variants [57], sequencing of complex repetitive regions, and structural rearrangements [58], and epigenetic modifications [59, 60]. Furthermore, they allow rapid sequencing runs, which is desirable in diagnostic settings [61].
Single molecule real-time sequencing (SMRT) [62] platform by Pacific Biosciences is the more mature of the two platforms. SMRT technology derives sequence data from real time recording of incorporation of fluorescent nucleotides by polymerases immobilized on solid phase [62]. The polymerase reads the circularized template, resulting in long reads (> 10 kb on average, > 80 kb maximal read length [63]), albeit with a relatively high error rate (~ 13%) [63]. Where shorter (< 10 kb) reads suffice, templates can be read multiple times, allowing computation of highly accurate circular consensus sequences (CCS) from individual single passes (subreads). CCS-based sequencing has already been used to detect mutations at frequencies below 0.5% in the stool of CRC patients [64].
The long-read platform commercialized by Oxford Nanopore Technologies (ONT) [65] is a relative newcomer to the market. Nanopore sequencing platform derives sequence information from nucleotide-specific ionic current changes observed as ssDNA passes through a membrane-bound nanopore [66]. Standard ONT protocols result in DNA fragments of ~ 8 kb; however, there is no technical limitation to read length, apart from the length of DNA templates in the sample [63]. The biggest drawback of the ONT platform is its high error rate (~ 15%) [67], with no possibility of sequencing the same strand multiple times. The accuracy can be improved through 2D nanopore sequencing, where the two strands of a dsDNA template are linked by a hairpin and sequenced consecutively as ssDNA, allowing a consensus sequence to be computed.
The potential of SMRT and ONT platforms for advancing liquid biopsy is big, but long read technologies suffer from several drawbacks that currently limit more widespread use and implementation into routine clinical settings. Notably, per-base costs are considerably higher than for short read sequencing and currently available bioinformatics algorithms are less mature than for short read sequence analysis [68]. However, recent developments in base-calling software [69, 70] and future improvements in sequencing chemistries are expected to facilitate adoption of long read sequencing platforms into biomarker development and clinical use.
Translational Status
Molecular profiling of tumor tissues by genomics techniques has a critical role in advancing cancer research and is gradually becoming standard clinical practice. Given the wealth of knowledge these approaches accumulated about cancer molecular biology and technical advances in sequencing, it is not surprising that ctDNA from liquid biopsies are already yielding biomarkers validated in clinical trials. Furthermore, some of the assays have already been FDA-approved and are moving into clinical practice.
The first FDA-approved liquid biopsy test on the market is cobas EGFR Mutation Test v2 (Roche Diagnostics Inc) [42, 43]. The assay relies on detecting cancer-specific mutations in cfDNA and is used as a companion diagnostic test for detection of mutations in the epidermal growth factor receptor (EGFR) gene to identify patients with metastatic non-small cell lung cancer (NSCLC) eligible for treatment with erlotinib.
One of the standing problems in clinical practice is identifying patients with high risk of recurrence after surgery. The fact that plasma ctDNA can correlate with minimal residual disease, cancer-specific MAFs can serve as proxies for recurrence risk. Patient-specific cancer alleles can be specified by analyzing the cancer tissue at loci known to be recurrently mutated in cancer. The locus with the highest MAF can be chosen, and its abundance in plasma ctDNA can be used as a prognostic biomarker [71]. This approach was used to identify high-risk colon cancer patients. Specifically, quantification of patient-specific mutated alleles in ctDNA by dPCR in post-operative samples showed that ctDNA-positive patients were at a higher risk for recurrence in a prospective multicenter cohort of patients with resected stage II colon cancer (HR = 18; N = 230) [71].
In addition to revealing cancer-specific SNPs, ctDNA reflects other tumor-specific changes, such as chromosomal rearrangements. The potential of combining patient-specific somatic mutations and structural variants (first identified in tumor samples by SNP [72] arrays and WGS [72, 73], and then assayed in ctDNA by ddPCR) to estimate the risk of recurrence has been tested in two studies on patients with colorectal cancer [72, 73]. The first study investigated the ability of ctDNA to predict recurrence in retrospectively selected six relapsing and five non-relapsing patients following colorectal surgery [72]. Somatic structural variants in ctDNA were shown to be valuable prognostic biomarkers in terms of detecting post-surgery relapse with 100% sensitivity and 100% specificity. Importantly, relapse was detected on average 10 months earlier than when using conventional follow-up. The second study [73] was a part of a single-site prospective observational cohort with colorectal cancer patients undergoing surgical treatment, split into two cohorts. The investigation off the first cohort (N = 27 retrospectively selected with 1:1 ratio of relapsing and non-relapsing patients) showed that patients treated with curative intent for localized disease who were ctDNA-positive within 3 months of surgery had a very high risk (100%) of relapsing (HR 37.7). The ctDNA analysis of an independent validation cohort (retrospectively selected 18 CRC patients with liver metastases) showed that ctDNA within the first 3 months was associated with a high relapse risk (HR 4.9). These estimates should be interpreted with caution given the relatively low sample size. Indeed, patients are being recruited for a multi-site prospective observational cohort (N = 1800, NCT03637686) to validate the ctDNA as a biomarker for detection of subclinical residual disease and precisely estimate the risk of recurrence from colorectal cancer.
In a single-center retrospective study of patients with non-metastatic breast cancer, the abundance of rearrangements in ctDNA was used to identify patients who will eventually develop metastases [74]. Authors first carried out whole genome sequencing of primary tumors to identify patient-specific chromosomal rearrangements. Next, they analyzed plasma ctDNA from follow-up blood samples and quantified tumor-specific rearrangements using ddPCR. The assay could discriminate patients with eventual metastasis from those with long-term disease-free survival with 93% sensitivity and 100% specificity (AUC = 0.98, N = 20) [74]. This exploratory study shows the potential of ctDNA to detect occult metastases. SAGA Diagnostics is deploying two ultrasensitive ddPCR assays (IBSAFE and KROMA) based on these results.
A similar approach has been successfully implemented for personalized cancer profiling by deep sequencing (CAPP-Seq) in NSCLC patients (5 healthy controls and 13 patients undergoing treatment for newly diagnosed or recurrent NSCLC) [53]. The development of a personalized profile is a multistep process. The first step is the identification of recurrently mutated cancer-specific regions in The Cancer Genome Atlas (TCGA) [75]. The next step is to design a library of biotinylated DNA oligonucleotides (CAPP-Seq “selector” library) that can selectively enrich identified targets in the sample. CAPP-Seq library is then used to identify patient-specific aberrations in tumor tissue samples. In the last step, CAPP-Seq is applied to plasma cfDNA to enrich and quantify ctDNA [75]. The assay had a maximal sensitivity 85% and maximal specificity of 96% to discriminate between NSCLC patients and healthy controls. CAPP-Seq has been adopted by Roche in AVENIO ctDNA NGS liquid biopsy kit.
Signatera RUO (Natera) is a multiplex-PCR NGS ctDNA test for monitoring and minimal residual disease assessment in NSCLC patients. The Signatera RUO technology is being validated in an observational prospective cohort trial called TRACERx (Tracking Non-Small-Cell Lung Cancer Evolution Through Therapy (Rx)) study (NCT01888601) with the goal of defining the relationship between intratumor heterogeneity over 5 years and clinical outcome following surgery and adjuvant therapy. The analysis of ctDNA samples of the first 96 participants with NSCLC [46] shows evidence of adjuvant chemotherapy resistance. Furthermore, the assay was able to identify patients with recurrence with sensitivity of 93% and specificity of 90% in the sub-group of 24 patients from the TRACERx cohort (retrospectively selected 10 control cases and 14 confirmed relapses).
Similar efforts have been undertaken by other companies. For example, Guardant Health has just completed a trial comparing a liquid cfDNA assay Guardant360 to tissue biopsy for detecting predictive and prognostic genetic markers in NSCLC patients (NCT03615443). Similarly, Foundation Medicine has established a companion diagnostics test called FoundationOne Liquid based on profiling more than 70 genes and genomic biomarkers for microsatellite instability.
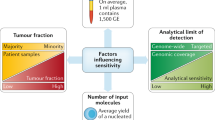
The aforementioned approaches are applied once the cancer has been diagnosed and treatment initiated. However, most cancers present symptomatically and are only diagnosed at difficult to treat stages. Arguably, pre-symptomatic population wide screening for early cancer detection is one of the most attractive applications of liquid biopsies and could in principle improve clinical outcomes [3]. However, this application comes with a unique set of challenges that will need to be addressed before clinical adoption. First and foremost, in a setting with asymptomatic individuals, liquid biopsy biomarkers are likely to be the only source of diagnostic information. Second, already present tumors will tend to be small in size and at early stages, which means that the concentration of cancer biomarkers might be close to the methods’ limit of detection. Third, mutational analysis will need to account for non-tumor somatic mutations. Finally, establishing true clinical sensitivity and specificity of the assay will be of utmost importance, which will require large cohorts of healthy individuals with longitudinal follow-up.
The genomics company GRAIL is setting out to tackle these challenges, by leveraging cfDNA sequencing for early detection in one of the largest genomics medicine studies to date. Currently, GRAIL is conducting two clinical studies. The first, called STRIVE (NCT03085888), is a prospective observational cohort study with enrollment of approximately 100,000 women undergoing mammography. Participants will be followed for five years to record clinical outcome data, with the ultimate goal of validating GRAIL’s blood-based assay for early breast cancer detection. The second study (NCT02889978), called the Circulating Cell-Free Genome Atlas (CCGA), is a prospective observational case-control study at the recruitment stage. CCGA is enrolling more than 10,000 subjects to characterize the landscape of cfDNA found in the blood of cancer patients and healthy individuals. During 2019, GRAIL also plans to deploy a clinical study (SUMMIT) to evaluate the blood assay and sequence cfDNA in 50,000 participants with no cancer diagnosis at the time of enrolment. This study will follow up patients for 3 years, and then track them through medical registries for additional five to evaluate clinical outcomes. Planned sample sizes will be sufficient for showing clinical validity of the screening approach. However, adoption of population-wide screening will require evidence for clinical utility, which might require additional large cohorts with long follow-ups.
Circulating Tumor Cells
Scientific Rationale
Circulating tumor cells (CTCs) were first discovered in 1869 [76]. CTCs have reemerged as a topic of interest in the last couple of decades because of their role in tumor biology and potential to serve as biomarkers for liquid biopsy. CTCs are cells that enter the circulatory system from the primary tumor site during the growth and metastasis [77, 78]. The biology of CTC release into the bloodstream involves at least two independent mechanisms. The first mechanism requires epithelial-mesenchymal transition (EMT), where tumor cells lose their epithelial phenotype and acquire mesenchymal traits [79, 80]. The second mechanism does not require EMT and could depend on external forces, such as surgery [79, 81]. How these two mechanisms of CTCs release contribute to metastatic spread of cancer is still a topic of intensive investigation.
The potential of CTCs for liquid biopsy is manifold. CTC enumeration has to this point been most intensively investigated as a biomarker. Typically, CTCs are low in abundance (fewer than 10 cells per milliliter of blood [79, 82, 83]), and the abundance of CTCs correlates with reduced progression-free and overall survival [84], and can therefore be used as liquid biopsy biomarker for determining the cancer burden. CTCs can retain some of the markers of the tissue of origin, allowing localization of the tumor of origin [85]. Furthermore, the advent of microfluidic and single-cell omics methodologies [86] allows the analysis of CTCs by technologies that were earlier limited to bulk tissue samples [87]. However, the effective use CTCs in clinical practice is mainly hampered by methodological and standardization challenges related to enrichment and selection of CTCs, as outlined below.
Current and Emerging Technologies
The main technological challenge for analysis of CTCs for basic research and clinical use is their low abundance in blood. Therefore, the first step in any detection and analysis procedure is enrichment and selection. Enrichment methods (reviewed in [79, 88]) take advantage of CTCs biological properties (expression of surface cell proteins [89, 90]) or physical properties (size/shape [91, 92], electric charge [93, 94]). Once enriched, CTCs can be separated from normal cells on the basis of cell-surface antigen expression.
To complicate things further, CTCs exist as either apoptotic or viable populations, each in turn harboring subpopulations with different phenotypical characteristics [79, 95]. Thus, different enrichment procedures bias the enriched CTC pool towards specific subpopulations. Depending on the intended use of CTCs for liquid biopsy, this methodological challenge will need to be addressed differently [79]. For example, the early detection of cancer requires extreme sensitivity to the lowly abundant CTCs. Prognostic use of CTCs requires comprehensive enrichment of all CTC subpopulations, such that all the relevant tumor heterogeneity can be captured. Using CTCs as predictive and monitoring biomarkers requires enrichment of all living CTCs as well as their characterization to determine the tumor’s sensitivity or resistance to therapeutic interventions.
Despite this challenge, there are many attempts to use CTCs for liquid biopsy. To date, most progress has been made using CTC enumeration as biomarker prognostic biomarker in patients treated for metastatic breast [96], colorectal (CRC) [97], or prostate cancer (PCa) [98].
In addition to CTC enumeration, single-cell omics methodologies are being leveraged to characterize tumors and extract clinically meaningful information. Whole genome amplification (WGA) can be used to facilitate the genomic analysis of CTCs [99,100,101]. Multi-gene panel sequencing of CTCs from patients with stage IV CRC uncovered genotypes that were subsequently identified at subclonal levels in primary tumors and metastases of the same patients [102]. Whole genome sequencing of CTCs has been used in patients with metastatic breast cancer [103]. The long-read technology enabled identification of driver mutations as well as the primary tissue of origin. A similar study based on the whole exome sequencing for metastatic prostate cancer [104] uncovered concordance between mutations found in CTCs genomes and tumor tissues (primary and metastatic), demonstrating that CTC genomics profiling could be a valuable diagnostic and prognostic tool [103, 104].
Genome-wide epigenomic analysis of DNA from liquid biopsy samples might not be possible at the moment, but there are already attempts to probe the methylation status of targeted genes [105] and promoters in CTCs [106]. Single cell RNA sequencing has already been used to investigate CTC transcriptomes in hepatocellular [87] cancer, melanoma [107], pancreatic [108], and breast cancer [109]. Established CTCs protein analysis includes identification of CTCs via antibody staining [96]. However, higher throughput and coverage will advance through recent improvements in microfluidics [110]. Microfluidics devices are already enabling the analysis of proteomics [111] and secretomics [112], as well as metabolic [113] biomarkers from CTCs.
Translational Status
Despite the promise of CTC omics assays for personalized oncology, more clinical trials are needed to establish clinical validity and utility. As we mentioned earlier, the field is still hampered by technical and reproducibility issues related to CTC enrichment.
To date, the only FDA-approved CTC assay on the market is CellSearch (Menarini Silicon Biosystems, Inc, acquired from Janssen Diagnostics LLC/Veridex LLC). The assay exploits the epithelial cell adhesion molecule (EpCAM), expressed by many carcinoma cells to isolate CTC cells [82, 114]. Because EpCAM expression is linked to poor survival, CellSearch enumeration of EpCAM positive CTCs can serve as a prognostic biomarker in combination with other clinical information. In a pivotal prospective, double-blind, multi-center clinical trial a total of 177 patients with metastatic breast cancer were recruited to investigate CTC counts as predictors of progression-free survival and overall survival [96]. The results show that patients with higher CTC counts (≥ 5 CTC/mL) had significantly shorter OS and PFS. Later three prospective multi-center cohort studies showed similar results for metastatic breast, colorectal, and prostate cancer [115].
The aforementioned trials demonstrate the clinical validity of the CellSearch-based CTC enumeration as predictive and prognostic biomarker. However, the utility of CTC liquid biopsy in specific clinical contexts is still unclear and a subject of ongoing clinical trials (reviewed in ref [116]). For example, HER2 status of CTC cells enriched by CellSearch in patients with metastatic breast cancer showed potential as a prognostic and predictive biomarker for clinical response to therapies targeting HER2 [117, 118]. One therapy for HER2-positive metastatic breast cancer is trastuzumab emtansine (trastuzumab-DM1 or T-DM1), a conjugate of a tumor-activated prodrug and humanized anti-HER2 monoclonal antibody [119]. An interventional multi-site trial (NCT01975142) was carried out to test if patients with metastatic HER2-negative breast cancer but with HER2-positive CTC cells respond to treatment with T-DM1. The study found that HER2-positive CTCs can be detected in a subpopulation of HER2-negative metastatic breast cancer, but there was an overall low response to anti-HER2 therapy [120].
CellMaxLife is currently conducting clinical trials on using an automated liquid biopsy platform CellMax CTC (CMx) [85, 121] for early detection of colorectal (NCT03476122), prostate (NCT03488706), and breast cancer (NCT03511859).
In addition to CTC enumeration, omics profiling is also being investigated in clinical trials. Oncotype DX AR-V7 is an assay that detects AT-V7 proteins in nuclei of CTCs. The assay has shown efficacy in identifying metastatic prostate cancer patients who will not respond to androgen receptor (AR)-targeted therapies. Clinical studies show that Oncotype DX AR-V7 can be used to guiding the choice of treatment between taxanes and androgen receptor signaling inhibitors [122,123,124]. In a cross-sectional single-site cohort study of histologically confirmed mCRPC undergoing a change in therapy, it was investigated if pretherapy nuclear AR-V7 in CTCs is a treatment-specific marker. The results showed that taxanes result in improved OS compared to ARS inhibitors in patients with AR-V7-positive CTCs (HR 0.24) [122]. These findings were later validated in an independent, multi-site, blinded, cross-sectional cohort (N = 225) [124].
Epigenome
Scientific Rationale
Epigenetic modifications, such as DNA and histone methylation, play a critical role in regulating core cellular processes, such as transcription, DNA repair, and replication. Because dysregulation of DNA-templated processes is a crucial step in neoplastic progression, proteins and complexes involved in epigenetic modifications are often found mutated across different cancer types [125]. Changes in epigenomic regulators during tumorigenesis in turn lead to changes in methylation patterns compared to normal cells [125, 126].
Exactly how epigenomics changes modulate cellular processes in normal and cancer cells is a field of intense research. This research has established a causal relationship between epigenetic changes and some of the hallmarks of cancer [125, 126]. DNA methylation is the most widely researched epigenetic modification. Repression of tumor suppressor genes can be readily achieved through epigenetic changes [127, 128]. Specifically, methylation patterns at CpG sites in promoter regions can regulate expression of downstream genes. Tumor suppressor specific hypermethylation is a process where cancer cells methylate CpG islands in promoters of tumor suppressor genes, thus downregulating their expression. Furthermore, genome-wide hypomethylation has also been observed in cancer [129, 130]. Hypomethylation of promoters that are methylated in normal cells can dysregulate gene expression, which can promote tumorigenesis when targets of dysregulation are proto-oncogenes. In combination with hypomethylation of repetitive sequences [131], this can elevate mutation rates, cause genomic instability, and promote tumor formation [126, 132]. Because these processes are crucial for cancer progress, key players are being investigated as therapeutic targets [125, 133,134,135] (reviewed recently in ref [136])
The epigenomic aberrations characteristic of cancer cells can readily distinguish healthy from cancer tissues with high accuracy [137]. Therefore, epigenomics changes such as DNA methylation patterns have been recognized as potential diagnostic, prognostic, and predictive biomarkers. Furthermore, there is evidence of concordance between epigenetic changes found in ctDNA and its tissue of origin [138,139,140], making epigenomic biomarkers promising candidates for liquid biopsy.
DNA methylation patters are highly tissue-specific [141], enabling detection of tissue of origin for ctDNA [142]. A recently published atlas of human cfDNA methylation patterns [143] could bolster efforts for early cancer detection and help pinpoint the tissue of origin.
Current and Emerging Technology and Biomarkers
Epigenomic biomarkers can be assessed in cfDNA, exosomes and CTCs. It is possible to approach epigenomic biomarkers by looking at specific changes at predetermined loci (for example at recurring cancer-specific epigenetic changes or at patient-specific methylation patterns), or to probe genome-wide epigenomic patterns characteristic of cancer.
5-Methylcytosine (5mC) is the most extensively studied DNA methylation in epigenomics [125]. Epigenomics techniques to assess the methylation patterns of DNA rely on bisulfite treatment, which converts unmethylated cytosine to uracil, but leaves 5mC unaffected. Bisulfite-treated DNA is then subject to different methods that attempt to distinguish uracil from cytosine in the template [126, 144,145,146].
Bisulfite PCR methods to assess methylation in liquid biopsy biomarkers include single [147] or multiplex [144, 148] quantitative real-time PCR, bisulfite dPCR (methyl-BEAMing) [146], and methylation-specific PCR [145]. While it is possible to make diagnostic decisions by assaying the methylation status of one marker or small gene panels [144], the accurate identification of individual epigenomic biomarkers is typically problematic because of low sensitivity due to background plasma DNA noise [149]. Furthermore, DNA methylation profiles are strongly influenced by gender, ethnicity, and individual differences and spatial heterogeneity within tumors [149, 150], necessitating the use of large biomarker panels to increase accuracy, as well as large and diverse cohorts to validate clinical utility of epigenomics biomarkers for liquid biopsy.
Aforementioned approaches rely of quantifying changes in methylation at one or more predetermined loci. An alternative is to assay methylation patterns in cfDNA in a highly parallel manner, for example, by using methylation microarrays [151]. Similarly, massively parallel bisulfite sequencing offers an unbiased way to assay genome-wide methylation patterns [129] in circulating DNA. A recent study shows that an unbiased DNA methylome can predict the outcome in patients with juvenile myelomonocytic leukemia in small discovery and validation cohorts [152]. A modification of the general method, involving methylated CpG tandems amplification and sequencing (MCTA-Seq) [153] can allow detection of methylated alleles at frequencies as low as 0.25%.
The major limitation of bisulfite-based approaches is that the initial step of bisulfite conversion degrades most of the input DNA [154]. To address this issue, a new bisulfite-free method for epigenomic profiling was developed [155]. This cell-free methylated DNA immunoprecipitation and high-throughput sequencing (cfMeDIP-seq) is an unbiased method that captures and enriches methylated cfDNA fragments to assess genome-wide methylation patterns from lowly abundant DNA samples, albeit at a lower resolution (100–300 bp) [156].
Another type of DNA methylation is 5-hydroxymethylcytosine (5-hmC). The regulation of 5-hmC is known to be affected in some human cancer types, making 5-hmC a potential biomarker for cancer [157,158,159]. A recently developed bisulfite-free method targeting 5-hmC takes advantage of the fact that the hydroxymethyl group in 5-hmC can be selectively chemically labeled [160]. This reduces the background noise and allows higher analytical sensitivity in samples with low input DNA [160].
Given that dramatic changes in epigenomes are universal to all cancers, it might be possible to go beyond cancer type-specific biomarkers and develop pan-cancer methylation biomarkers in the future. An innovative approach along these lines investigates how the methylation landscape (“Methylscape” [161]) affects physicochemical properties of DNA. Specifically, authors exploited changes in solvation and gold affinity of plasma cfDNA caused by cancer-specific methylation, and developed an innovative and rapid electrochemical and colorimetric assays for the presence of breast and colorectal cancer (case-control study with 45 healthy individuals and combined 100 breast and CRC cancer patients, AUC = 0.887 for discrimination of healthy vs cancer patients) [161].
In general, epigenomics biomarkers have a lot of potential for liquid biopsy. However, major technological limitations are low sensitivity, high costs, and the required expert knowledge. Furthermore, different protocol for isolating cfDNA show significant variation in the amount of recovered DNA, necessitating analytical validation and standardization of new methods before clinical implementation.
Translational Status
Despite being a field in its infancy, epigenomics is already yielding biomarkers and assays for liquid biopsy in clinical practice. Assays based on even a single epigenetic marker can serve as powerful diagnostic tools. The first FDA-approved blood test for CRC diagnosis, Epi proColon (Epigenomics, Inc), uses qualitative real-time PCR to detect methylated Septin 9 DNA in plasma. Epi proColon has shown similarly sensitivity to the fecal immunochemical test (FIT), albeit at a lower specificity in a multicenter observational case control study (NCT01580540) [147]. The study with the primary goal of establishing non-inferiority of Epi proColon compared to FIT was conducted on two cohorts: the first had subjects with diagnosed CRC (N = 102) and the second had subjects scheduled to undergo colonoscopy (N = 199). Sensitivity to discriminate between CRC and non-CRC was 73%, and specificity was 82%. Interestingly, a randomized controlled two-site trial with average-risk adults overdue for screening (NCT02251782) showed that Epi proColon boasts better patient adherence to screening than FIT (N = 413, 99.5% vs 88.1%) [162].
Another FDA-approved assay for liquid biopsy is Cologuard assay (Exact Sciences). Cologuard is multitargeted stool DNA test that can detect colorectal cancer. Aberrant methylation patterns in NDRG4 and BMP3 are detected through allele-specific real-time PCR and employed as diagnostic biomarkers in combination with hemoglobin and the allelic status of KRAS gene [148]. The sensitivity and specificity of the assay were determined in a large observational case-only prospective study involving asymptomatic persons at average risk for colorectal cancer scheduled to undergo screening colonoscopy (N = 9989). The sensitivity was 92.3% (versus 73.8% for the FIT assay) for discriminating between CRC and non-CRC patients, including those with negative findings on colonoscopy, precancerous lesions, and non-advanced adenomas. Cologuard had a lower specificity than FIT (89.8% for Cologuard vs 96.4% for FIT) for patients with negative results on colonoscopy [148].
Nucleix has recently published a study detailing the clinical performance of their Bladder EpiCheck. The test assays methylation patterns in a urine panel of 15 DNA methylation biomarkers [163] for follow-up of patients with non-muscle-invasive bladder cancer (blinded, single-arm, prospective multicenter study, N = 353, AUC = 0.82 for cancers including and AUC = 0.94 for excluding LG Ta tumors). With a high NPV (99.3%), EpiCheck could be used in follow-up to reduce the burden of repeated cystoscopies, which is the standard of care to diagnose bladder cancer progression.
Transcriptome
Scientific Rationale
The pool of all expressed RNA species is collectively called the transcriptome. The transcriptome plays a pivotal role in cellular processes. The human genome encodes approximately 20,000 genes that are transcribed into mRNA, rRNA, and tRNA. The importance of these RNA species for regulation and protein synthesis has been recognized since the inception of molecular biology. The non-coding RNAs (ncRNA) have been discovered in the last couple of decades, expanding the concept of transcriptome to include microRNA (miRNA) [164], piRNA [165], tiRNA [166], snoRNA [167], Y RNA [168], PASR [169], TSS-RNA [170], snRNA [171], and lncRNA [169, 172, 173].
While DNA content is mostly identical in different cells of an organism, transcriptional profiles can vary dramatically across cell types, space, and time. Therefore, changes in the transcriptome offer an opportunity to associate cellular phenotypes to underlying molecular mechanisms and potential genotypic changes. The experimental methods, algorithms, and the underlying domain knowledge have been developing more than four decades [174]. With the advent of RNA-Seq, transcriptomics has arguably become the most mature omics approach in the functional genomics toolset.
The role of transcriptional changes in healthy and diseased states, carcinogenesis in particular, have been the focus of intense investigation. Notably, aberrant mRNA expression levels are associated with dysregulation in cancer. Comprehensive profiling of gene expression patterns across many tissues and cancers have yielded molecular classifications of cancer (sub)types [75, 175]. Furthermore, unbiased sequencing of transcripts has enabled detection of cancer- and patient-specific somatic mutations [176, 177] and fusions/rearrangements, spearheading the discovery of novel mRNA biomarkers [178].
Methodological advances in transcriptomics have also helped uncover the role of non-coding RNAs in health and disease. Small ncRNAs have been implicated in the regulation of transcription, post-transcriptional processing pathways, gene silencing, epigenetic processes, translation, and protein activity [179, 180]. The most widely studied class of ncRNAs in cancer are miRNAs, known to regulate tumor suppressors and oncogenes [181]. Aberrant miRNA expression has been implicated in the occurrence and progression of tumors [182]. More importantly, miRNA expression patterns are cancer-specific and can accurately identify tissue of origin in metastatic cancer [183]. Because miRNAs are dysregulated in all stages of cancer, they can be used as biomarkers for early detection, prognosis, or treatment selection. Similarly, lncRNA have a wide range of regulatory functions in cancer and normal cells [174, 184]. The functions of other ncRNAs in carcinogenesis are still poorly understood.
The existence of the circulating transcriptome as a liquid biopsy biomarker was first recognized when cell free RNA (cfRNA) from Epstein-Barr virus was discovered in the blood of nasopharyngeal carcinoma patients [185] and circulating miRNAs were found in the serum of B-cell lymphoma patients [186]. cfRNAs can be released into body fluids passively through processes such as cell death, or actively secreted in exosomes or in complexes with proteins [187].
miRNAs are remarkably stable and abundant in fluids like blood, making them most widely investigated transcriptomics biomarkers for liquid biopsy [187, 188]. The major impediment for discovery and clinical use of other cfRNA biomarkers, mRNA specifically, is their poor stability in body fluids, mostly due to degradation by RNase [187]. Recent studies uncovered that longer circulating RNA species can be found in exosomes or complexes with (lipo)proteins, both of which increase the stability and the resistance to RNase [184, 188].
Current and Emerging Technology and Biomarkers
Quantification methods include qRT-PCR, dPCR, microarrays, and RNA-Seq (for both miRNA and lncRNA). PCR-based methods tend to be quick, sensitive, and easy to interpret, but they lack throughput and can only analyze a small panel of predetermined RNAs [189]. Microarrays have the advantage of analyzing many biomarkers in parallel [190, 191]. However, they are characterized by lower analytical sensitivity and specificity [192]. RNA-Seq allows detection of high-throughput analysis, the capacity to identify novel fusions, but at a cost of higher complexity of analysis and the larger amount of sample input.
There are a number of studies exploring the potential of circulating RNAs as diagnostic, predictive, and prognostic biomarkers. Plasma [193, 194] and serum [195] miRNAs have shown potential as biomarkers for early detection of NSCLC. Furthermore, patterns of miRNA and their precursors (pri-miRNA) can be used to detect NSCLC and enable discrimination between squamous cell carcinoma and adenocarcinoma NSCLC subtypes [196]. Similarly, circulating miRNA have been proposed as markers for guiding therapy by identifying triple negative breast cancer [197]. Finally, miRNA species in blood show potential as metastatic biomarkers in osteosarcoma [198], bladder cancer [199], and ovarian cancer [200]. However, one of the major drawbacks of miRNA as cancer biomarkers is that most species exist in both healthy and cancer patients, and their expression differences can be rather small [187].
The number of studies investigating potential miRNAs biomarkers is high, but there are recent examples that show the potential of using other RNA species. For example, different mRNAs from liquid biopsies can be potential biomarkers for predicting sensitivity to chemotherapy in gastric cancer [201,202,203]. Currently, one of the most promising mRNA biomarkers is telomerase reverse transcriptase (TERT), showing potential as a diagnostic, prognostic, and predictive biomarker in gastric [204], prostate [205], hepatocellular cancer [206], CRC [207], and breast cancer [208].
A recent study [188] looked at combined patterns of altered expression of different RNA classes, using a combination of NGS and validation by qPCR and ddPCR. The study uncovered a number of novel microRNA, mRNA, and YRNA in plasma of melanoma patients compared to healthy control. However, these novel biomarkers will need to be validated in larger studies [188].
The choice of RNA quantification technology needs to be accompanied by other analytical considerations. The first step in any transcriptomics protocol is the isolation and purification or RNA. Extraction from body fluids often results in samples with low RNA quality and quantity. Furthermore, the use of different isolation protocols can affect purity and quality of isolated cfRNA [184]. To complicate matters further, cfRNA concentrations and fractions can vary drastically between different body fluids [184], tissues, organs, and individuals [184]. In addition to these technical challenges, data analysis procedures protocols are not standardized [209, 210].
Translational Status
The published results on transcriptomics-based liquid biopsy highlight an abundance of potential (mostly miRNA) biomarkers. However, many proposed circulating RNA biomarkers were validated only in underpowered retrospective single-center studies with small cohorts [187], often leading to contradictory results [211]. Nevertheless, RNA-based assays are being validated in clinical trials, and there are already tests ready for commercial and clinical use.
A large validation study was conducted to validate miR-Test, a screening blood assay based on signatures of 13 serum miRNAs quantified by microarrays [212]. Specifically, high-risk individuals enrolled in a single-center non-randomized lung cancer screening trial (COSMOS study) [213] were selected, split into four cohorts, and screened with the miR-Test [212]. The first, calibration cohort (N = 24) was screened using an extended panel of 34 miRNAs. The initial miRNA set was then reduced to the most informative 13 miRNAs, whose discriminatory power was validated in a second cohort (N = 1008 from the COSMOS trial, where 36 patients had low-dose computed tomography (LDCT)-detected lung cancer). miR-Test showed the sensitivity of 77.8% to detect all tumors and the specificity of 74.8%. In the third cohort, the miR-Test was assayed for the ability to discriminate non-malignant from malignant diseases (N = 83 patients who never developed cancer during a 5-year follow-up), showing specificity of 86.7%. The fourth cohort was a clinical validation set (N = 74 patients diagnosed with stage I-III lung cancer outside of the COSMOS study) where sensitivity to detect cancer was 70.3%, with similar performance across different stages [212]. A similar result was obtained in a randomized multi-center prospective trial (N = 939) carried out to test the clinical utility of qRT-PCR-based miRNA signatures in plasma [214]. The test showed good diagnostic performance with NPV = 99%, as well as the ability to identify malignancy and the aggressiveness by predicting death as a result of the disease with sensitivity of 95% and an NPV of 100% [214]. Cumulatively, these two examples show that miRNA-based liquid biopsy assays could be used as biomarkers for early detection of lung cancers with similar sensitivity and specificity to low-dose computed tomography (LDCT), and could potentially be used to reduce false positive rates associated with LDCT screening alone [212, 214].
Another example is Progensa PCA3 [215] (Gen-Probe), a prostate-specific lncRNA overexpressed in primary and metastatic prostate adenocarcinoma [216]. A urine-based test specific for PCA3 [217] has shown good analytical [218] and clinical performance in a number of clinical studies, a greater specificity but lower sensitivity than the classical PSA biomarker [216]. In a multicenter prospective cohort of men scheduled for repeat prostate biopsy, the assay showed 77.5% sensitivity and 57.1% specificity for repeat biopsy outcome (N = 441) [215]. The PCA3 test has been approved by FDA for clinical decisions about repeat biopsy of prostate cancer [217].
SelectMDx (MDxHealth, Inc) is a liquid biopsy test based on detecting HOXC6 and DLX1 mRNA levels in urine for prediction of clinically significant prostate cancer prior to prostate biopsy. The performance of the test has been investigated in a trial with two multicenter cohorts, a discovery cohort (N = 519), and validation cohort (N = 386). The results show that the test performs with AUC = 0.90 for a classifier that combines mRNA levels with PSA levels and clinical risk factors, excluding DRE results [219]. The clinical use of SelectMDx has shown potential to increase health outcomes and reduce overall treatment costs through elimination of unnecessary biopsies in two simulations of cost-effectiveness [220, 221].
Cxbladder is a urine-based multiplex RNA test for detecting, monitoring, and stratification in bladder cancer [222, 223]. The test derives its score from increased expression levels of five genes, MDK, HOXA13, CDC2, IGFBP5, and CXCR2. In a multi-center study with prospectively recruited patients with hematuria, Cxbladder detected 82% cases (at specificity of 85%) of urothelial carcinoma from urine samples taken prior to cystoscopy, outperforming other tests and cytology (N = 485) [224]. Furthermore, it distinguished between low-grade Ta tumors and other detected urothelial carcinoma with a sensitivity of 91% and a specificity of 90% in the same cohort [224].
Proteome
Scientific Rationale
The proteome is the entire set of proteins expressed in a cell, tissue, or an organism. Unlike the genome, the composition of the proteome can change with time and in response to varying intracellular and extracellular conditions. Furthermore, because expression of a single gene can involve alternative splicing, recoding events, and a wide range of post-translational modifications, the number of expressed proteins and proteoforms vastly outnumbers the number of genes. The identification and quantification of expressed proteoforms can illuminate molecular pathways, interactions, and events underlying cellular phenotypes in healthy and disease contexts.
Oncoproteomics investigates proteins involved in the carcinogenic processes with the aim of understanding the underlying molecular mechanisms, deriving novel targets for therapy, and identifying proteins that can serve as diagnostic, prognostic, and predictive biomarkers. Historically, the proteome has been the major source of circulating biomarkers for clinical use. Due to analytical and clinical limitations of these early markers, the field of oncoproteomics has been focused on discovering new protein molecules and signatures that can inform clinical decisions.
The advancement in proteomics methods and instrumentation, paired with other omics methodologies, raised hopes for the development of protocols to aid the rapid discovery of novel biomarkers and to implement personalized approaches in clinical practice. The promise of proteomics to advance basic biology and cancer research has culminated in projects to elucidate the maps of the human (tissue) proteome [225,226,227,228] and the cancer proteome [229]. However, despite the advances in methods and the accumulation of domain knowledge, much of the current oncoproteomics research for biomarkers remains in the discovery phase. The introduction of novel protein biomarkers and proteomic technologies in the clinic is still hampered by technical challenges, poor analytical performance, reproducibility issues, a lack of standards, and the lack of validation in large and rigorously designed clinical studies. The careful analysis of past success and failures has been critical for shaping the guidelines and regulations for advancing the field of biomarker discovery and for bringing new biomarkers into standard clinical practice [20, 21, 230,231,232,233].
Current and Emerging Technology and Biomarkers
Of all the sources of biomolecular information that can be used to diagnose and characterize cancers through liquid biopsy, protein biomarkers have the longest standing tradition in clinical practice. These circulating protein biomarkers include CEA [234], PSA [235], β-hGC [236], AFP [237, 238], FDP [239, 240], HE4 [241], ALT/AST [242], LDH [243, 244], CA 125 [245, 246], CA 15-3 [247], CA 19-9 [248], CA 27.29 [247] (reviewed in [230, 249,250,251,252]). Standard clinical protein biomarker assays typically target a single or a small number of prespecified tumor-associated antigens using immune-based methods [253]. These methods have a high analytical sensitivity and assays can easily be automated using liquid handling robots [254]. The limitation is that antibodies can detect multiple proteoforms [255] and non-specifically interact with interfering compounds generating false positives [256, 257]. Limited dynamic range is another technical issue affecting immunoassays [254].
High-throughput proteomics techniques have emerged as a viable alternative to address some of these issues. Importantly, newer proteomics methods can go beyond detecting predetermined panels of biomarkers, complementing genomic and transcriptomic approaches in probing the molecular signature of the underlying cancer. Furthermore, proteomics can provide additional information about concentrations of expressed proteins and their post-translational modifications, offering unique approaches to analyze and stratify cancer types.
The most widely used proteomics approach is mass-spectrometry (MS) [254]. Mass-spectrometry is an umbrella term that describes a wide array of methods that ionize analytes and then detect and analyze ions in the gas phase. Different ionization methods (electrospray ionization (ESI) [258] or matrix-assisted laser desorption/ionization (MALDI) [259], surface-enhanced laser desorption/ionization (SELDI) [259, 260], etc.) can be coupled with different analyzers (quadrupoles, time-of-flight, orbitrap analyzers, etc.) to achieve different analytical characteristics [254]. Furthermore, MS instruments can be combined in tandem (MS/MS) to afford structural information on analyzed ions. This setup can allow implementation of selected and multiple reaction monitoring (SRM [261] and MRM [262], respectively) in order to detect and quantify pre-specified ions. This mode of analysis is highly specific and can be used to multiplex quantification of approximately 200 proteins [263, 264]. In general, MS-based proteomic methods can yield large targeted or unbiased datasets with high precision and resolution [265]. In clinical laboratory, they offer lower routine costs, higher specificity and throughput, as well as the possibility to multiplex assays. Limitations of MS-based proteomics include reproducibility issues, analytical sensitivity to proteoforms with low abundance, method validation and standardization, instrument cost, and the need for expert interpretation.
An emerging platform for proteomics biomarker discovery is the protein array technology [266]. Protein (micro)arrays or chips are solid surfaces that immobilize up to thousands of purified or synthesized proteins at high densities [266,267,268,269]. Protein arrays are used to quantify large predetermined panels of proteins by capturing a wide range of protein binding activities [270]. The technology is characterized by high-throughput, high sensitivity, and robustness. The discovery of protein biomarkers for liquid biopsy is an important application of protein arrays. Arrays with immobilized antibodies are a low-cost method to profile the expression of many proteins in parallel [269], but they rely on using pre-existing antibodies for targets of interest. Functional protein arrays [271] contain complements of purified protein and can be used for unbiased assays of the entire circulating proteome. While it is difficult and costly to fabricate functional protein arrays, there are commercial variants available on the market [266]. An interesting variation of the technology are reverse-phase lysate microarrays, which allow lysates (e.g., from liquid biopsy) to be printed on a micro-array and quantified by immunochemical methods in a massively parallel fashion [272, 273]. These arrays are typically cheaper than functional ones but are limited by the availability of antibodies.
Another high-throughput proteomics technology with potential use in biomarker discovery is an aptamer-based platform called SOMAscan (Somalogic Inc.) [274]. SOMAscan is a multiplex quantitative affinity-based assay where immobilized aptamers bind to proteins with high specificity and affinity [274].
Proteomics technologies for liquid biopsy are being improved at a rapid rate, and some of the technical challenges are being addressed. Further improvements in this field will benefit from organized and institutionalized efforts to characterize cancers by integrating proteogenomic technologies and to establish standards for translation of the research in cancer biology. An example of such effort is the National Cancer Institute’s Clinical Proteomic Tumor Analysis Consortium (CPTAC), which is advancing integrated proteogenomic analysis for illumination of molecular bases of colorectal, breast and ovarian cancer [275,276,277]. The CPTAC initiative led to the creation of two additional programs, The Applied Proteogenomics OrganizationaL Learning and Outcomes (APOLLO) [278] network and the International Cancer Proteogenome Consortium (ICPC) [279]. These two networks are advancing the translation of proteogenomic research into routine clinical practice and enhancement of precision oncology through international data sharing.
Translational status
Assays for classic circulating protein biomarkers have been in clinical use for a couple of decades. Systematic reviews call in question the indiscriminate use of some of these biomarkers for population-wide screening [280,281,282,283], with the major limitation being a lack of specificity to a single cancer type, and high false positive rates associated with non-cancerous conditions [284]. These issues can translate into unnecessary treatments that harm patients and increase healthcare costs.
Despite the general issues with classical protein biomarkers, they show utility in appropriately defined clinical settings, especially when used in combination with other clinical variables. CEA has limited specificity and sensitivity for routine follow-up of early-stage CRC patients [285], but in combination with clinical, radiological, and histological findings, it can predict recurrence of CRC after resection [286, 287]. Likewise, PSA-based screening is associated with false positive rates and overdiagnosis [288, 289], but it was shown to reduce mortality from PCa when screening high-risk patients between 55 and 69 years of age in conjunction with digital rectal examination [288], it can detect PCa recurrence after resection, as well as predict response to treatment [290]. Similarly, while LDH is unsuitable as a biomarker for diagnosis of early-stage melanoma, it is a validated prognostic biomarker in metastatic melanoma (and currently the only serologic marker for melanoma) [244, 291].
New proteomics technologies have been used extensively in biomarker discovery in the last two decades. However, these methods are not easily translated into routine clinical use due to technical complexity, low throughput, and low reproducibility [292]. There are efforts to address these issues in a systematic fashion. A multi-site study has been implemented to standardize protocols and limit variability and irreproducibility in multiple reaction monitoring for plasma proteomics [293]. Attempts to translate proteomics-based biomarkers into clinical trials can be improved by following fit-for-purpose recommendations for designing analytical validation experiments and reporting findings [294].
Poorly designed and underpowered trials have hampered the translation of proteomic biomarkers into the clinic [20]. Some of the known examples of biomarker failures include the use of exoprotease-generated peptidome patterns in serum for detection of various cancer types [295]. In the original study, MALDI-TOF MS analysis of peptidomic patterns yielded a panel of biomarkers for accurate discrimination of three cancer types form healthy controls [295]. However, subsequent reanalysis of the study design revealed a number of flaws in the study design, namely improper case-control matching (age and sex brackets different in case and control patients) and inappropriately selected patient population (cohort including mostly patients with late-stage cancer, where intended purpose was early cancer detection) [20]. Later validation study found no evidence that serum peptidomic patterns can be used as cancer biomarkers [296]. Another example of a flawed design is a study that suggested a four biomarker panel yielding 95% sensitivity and 95% specificity, thus outperforming the standard CA125 biomarker for detection of ovarian cancer [297]. A subsequent study found that expanding the biomarker panel to six analytes could further improve specificity (99%) to OvCa [298]. However, an independent, blinded attempt to validate these findings showed that none of the biomarkers outperformed CA125 in sensitivity and specificity to OvCa [20, 299]. Reanalysis of the original study design revealed that authors used partially overlapping training and validation cohorts, causing overfitting [300]. Furthermore, cases and control patients came from different sources, where sample collection procedure was not standardized, introducing potential biases into the analysis [300]. Similar study design flaws prevented OvaCheck, an MS-based serum test to identify ovarian cancer patients [301], to be marketed for public use. Namely, differential handling of samples from different sources created batch effects that confounded the signal [302,303,304].
To address aforementioned study design issues, recommendations have been put in place for the phased design in biomarker discovery [305] aimed at better evaluation of classification accuracy in the intended clinical context. A novel candidate protein biomarker for pancreatic ductal adenocarcinoma (PDAC) has been put forth using the recommended study design [306, 307]. Authors first carried out an experiment where they reprogrammed human PDAC cells into an induced pluripotent stem cell-like line (10-22 cell line) [308]. A proteomic analysis of media from 10-22 cell-derived precursor pancreatic intraepithelial neoplasia cultured as organoids revealed 107 candidate proteins. Authors cross-referenced this candidate set against human plasma proteins [308] and focused on proteins with low abundance in healthy human plasma [306, 309]. They further reduced the set to three candidates, matrix metalloproteinase 2 (MMP2), MMP10, and thrombospondin-2 (THBS2), and assayed these in human plasma samples from a biospecimen repository (N = 10 cancer cases and N = 10 controls). MMP2 and MMP10 were uninformative, but THBS2 could discriminate pancreatic cancer from healthy controls (AUC = 0.76), and resectable and locally advanced PDAC against healthy controls (AUC = 0.886) in this small discovery cohort. A combined panel with THBS2 and CA19-9 performed well across all stages on PDAC in a larger validation cohort (AUC = 0.970, N = 161, 81 cancer and 80 healthy controls). The authors then validated the two-marker panel in an independent cohort (N = 337) and found that it can discriminate between cancer and healthy subjects with AUC = 0.97 [306].
In 2009, FDA has approved OVA1, the first in vitro Diagnostic Multivariate Index Assay (IVDMIA) for Ovarian Cancer [231]. OVA1 combines results of assays for CA-125 II, prealbumin, apolipoprotein A-1, β2-microglobulin, and transferrin into a single score that can distinguish malignant ovarian tumors from non-malignant forms that do not warrant referral to surgery. The assay was initially developed as a SELDI-TOF assay, but due to reproducibility issues, it was finally implemented as an immunoassay [310]. The OVA1 test was first validated in combination with physician assessment to predict malignancy in a prospective multicenter cohort of women scheduled for surgery of an ovarian tumor (N = 516, sensitivity 96%, specificity 35%) [311]. In a second study, the assay was validated in the context of its intended use, for risk stratification of ovarian malignancy after enrollment by non-gynecologic oncology providers. This prospective multicenter trial assayed pre-operative serum of 494 women and correlated the results with surgical pathology results. In combination with clinical variables, the assay could distinguish malignant from benign adnexal masses with the sensitivity of 95.7% and specificity 50.7% [312]. A recent improvement of the assay, OVA2, showed increased specificity (69%) to OvCa in a multi-site prospective cohort (N = 493) [313].
Another successful example of a commercialized liquid biopsy test is Veristrat (Biodesix). Veristrat classifies NSCLC patients as “Good” or “Poor” based on a multivariate MALDI-TOF proteomics blood test via detection of inflammatory states associated with aggressive lung cancer [314, 315]. Veristrat was used for patient stratification in a randomized phase III multi-center clinical trial with the goal of measuring survival and response to EGFR-TK inhibitor (erlotinib) or chemotherapy [316]. In this cohort of 285 confirmed, second-line, stage IIIB or IV NSCLC patients [316], those with a Veristrat classification of “Poor” had shorter overall survival on erlotinib than on chemotherapy (3 vs 6.4 months, HR = 1.72). Biodesix has now partnered with MRM Proteomics Inc. in an effort to implement the iMALDI platform for proteomic biomarkers to further enhance diagnosis and prognosis for lung cancer.
In 2018, Integrated Diagnostics announced the results of its multi-site prospective PANOPTIC (Pulmonary Nodule Plasma Proteomic Classifier) Trial [317] (NCT01752114). PANOPTIC was designed to validate Xpresys Lung 2, a liquid biopsy assay that integrates clinical data and MRM quantification of two plasma proteins, LG3BP and C163A, to distinguish benign from malignant lung nodules. The assay showed high sensitivity (97%), specificity of 44%, NPV of 98% and could in principle reduce procedures carried out on benign nodules by 40% (N = 392 patients with pulmonary nodules) [317].
Array-based proteomics technologies are also showing their potential for translation. IMMray™ PanCan-d (Immunovia) is a blood test based on machine learning to derive a diagnostic classifier from antibody microarray data. The test could classify samples from pancreatic cancer patients or healthy controls accurately with AUC > 0.95 in six retrospective cohorts to date [318,319,320,321,322]. IMMray™ PanCan-d is currently undergoing a clinical trial to investigate its diagnostic accuracy for detection of pancreatic cancer in high-risk groups (NCT03693378). The antibody microarray technology has potential to be used in diagnostics of other types of cancer and has already been tested to longitudinally monitor sera of patients with breast cancer [323] and to classify patients based on risk for developing prostate cancer [324].
The potential of SOMAscan assay to bridge biomarker discovery and validation phases was tested in three multi-site prospectively designed case/control studies. These studies used archived samples to discover and validate protein biomarkers for NSCLC detection in high-risk patient populations [325]. The first cohort (N = 363) was used to discover a robust panel of protein NSCLC biomarkers from 1033 SOMAscan analytes. The analysis resulted in a 7-marker panel with an AUC of 0.85 for all cases of NSCLC vs benign nodule controls. The histopathological sensitivity of the 7-protein panel was validated in a second cohort and showed similar discrimination between cancer and healthy subjects (AUC = 0.81, N = 138), and AUC = 0.89 for squamous cell carcinoma. Authors performed an additional validation study on an EDRN multicenter reference set for validating biomarkers for detection of lung cancer (N = 135). In this cohort, they found that the biomarker panel could detect NSCLC vs healthy samples with AUC = 0.77 and squamous cell carcinoma with AUC = 0.87[325].
Metabolome
Scientific Rationale
Cancer metabolism dramatically differs from that of a normal tissue. This phenomenon, dubbed metabolic reprogramming, is recognized as one of the hallmarks of cancer [326]. The first known example of metabolic reprogramming in cancer cells, the Warburg effect, was discovered 90 years ago [327]. The Warburg effect describes a metabolic phenotype where cancer cells display higher glycolytic flux and produce lactate at a higher rate than normal cells despite oxygen availability.
The acquisition and maintenance of neoplastic processes such as abnormal cellular proliferation and metastasis generally increases the demand for energy and biosynthetic building blocks, and changes the redox balance [328, 329]. Consequently, the cellular metabolism changes to accommodate those requirements and increase the fitness of cancer cells. The exact mechanisms of metabolic reprogramming and how they contribute to malignant phenotypes are active topics of investigation.
Oncometabolites are small molecules whose abundance is drastically increased as a consequence of cancer-associated metabolic reprogramming or because of somatic mutations in specific enzymes [328]. The most commonly known oncometabolite is D-2-hydroxyglutarate (D2HG), a reduced form α-ketoglutarate. The abundance of D2HG is low in normal tissues but increases in tumors harboring mutations in isocitrate dehydrogenase 1 or 2 (IDH1 or IDH2) [328, 330, 331].
The discovery of oncometabolites prompted the search for metabolomic biomarkers. However, looking for a single oncometabolite to serve as an accurate diagnostic, prognostic, or predictive biomarker for complex diseases like cancer might prove to be futile. A more promising perspective might be to rely on metabolite panels or signatures. This better reflects the reality of dysregulated pathways, as well as improves statistical robustness of the biomarker-informed decision-making.
Discovery in cancer metabolism can proceed with two different pathways, either by quantifying metabolites (metabolomics) or by measuring activities of metabolic pathways (e.g., by metabolic flux analysis) [332, 333]. These two approaches are not interchangeable and provide complementary information. A combination of both can yield important insights into metabolic phenotypes in cancer and uncover oncometabolite biomarkers for precision medicine.
Current and Emerging Technology and Biomarkers
Metabolomic experiments typically use analytical platforms such as nuclear magnetic resonance (NMR) [334], liquid-chromatography mass spectrometry (LC-MS) [335, 336], gas chromatography mass spectrometry (GC-MS) [335], and capillary electrophoresis mass spectrometry (CE-MS) [335, 337] to analyze and quantify metabolites in a sample [338]. These experiments could be targeted to a specific metabolite, a class of metabolites, or attempt to comprehensively assess all metabolites in an unbiased manner. Targeted approaches focus on up to a hundred metabolites, while untargeted analyses can cover hundreds or even thousands in a single experiment.
Different platforms are suitable for different applications or the analysis of different metabolites. GC-MS is typically used to assay metabolites smaller than 1,000 Da that are volatile or can be made volatiles via chemical derivatization [338]. In a single acquisition, GC-MS can resolve a couple of hundred metabolites with different properties (such as sugars and their derivates, amino and organic acids, amines, sterols and fatty acids) [339]. The advantage of GC-MS is reproducibility across different platforms, the existence of comprehensive spectral libraries, robustness, and relatively low costs. LC-MS is mostly used for larger metabolites that are non-volatile [339]. LC-MS can generally resolve a larger number of molecular species than GC-MS. CE-MS is ideal for the analysis of polar and ionic compounds, especially from low volumes. However, the sensitivity is generally lower, and variability is higher than that of GC-MS and LC-MS [338]. NMR has an advantage over MS methods by being a non-destructive analytical technique. Furthermore, in addition to quantification, NMR can be used to unambiguously resolve structures of unknown metabolites. Results of a 1H-NMR analyses are highly reproducible, but they typically have lower sensitivity than MS-based approaches [338].
Targeted approaches are empowered by known changes in cancer metabolism. In some cases, it might be possible to associate cancer-specific genetic alterations to changes in metabolite concentration and devise a scheme to discriminate between different types of cancer. For example, a mutation in isocitrate dehydrogenase (IDH) is a driver event in malignant gliomas [330]. The accumulation of 2-hydroxyglutarate (2HG) is observed in glioma cells with IDH1 mutation [340]. Plasma and urine levels of 2-hydroxyglutarate can be used to predict response to treatment [341].
Targeted metabolomics can also focus on oncometabolite concentration changes caused by dysregulation in metabolic pathways. Known examples are changes to concentrations of prostaglandins resulting from reprogramming of the eicosanoid pathway, which was shown to promote tumor growth [342]. These changes can in turn create distinct metabolic signatures in urine, which can be detected by targeted methods such as SRM. This approach showed the potential of using urine prostaglandins as biomarkers for detecting patients with high risk of developing pancreatic cancer [343]. Similar studies highlighted prostaglandins as biomarkers for identifying patients at high risk of breast cancer [344] and for prognosis of lung metastases in breast cancer patients [345]. Another example looks at polyamines as circulating biomarkers. Dysregulation of intracellular polyamine metabolism is a hallmark of most cancers [346] and a potential target for therapy. Furthermore, urine and plasma polyamine content in cancer patients can mirror intracellular levels of these metabolites, presenting an opportunity to use them as liquid biopsy biomarkers. The biomarker potential of polyamines was recently investigated using targeted analysis with LC MS/MS in colorectal [347, 348], ovarian [349], and prostate cancer [350].
The second approach uses untargeted/unbiased analysis of circulating metabolites to find differences between cancer patients and healthy individuals. Unsurprisingly, the findings of these untargeted approaches can sometimes recapitulate known cancer-related metabolic changes. For example, dysregulation of the citric acid cycle can alter plasma and urine concentration of pathway intermediates, such as succinate and fumarate [328]. The potential of these two biomarkers for diagnosing and staging renal cell carcinoma was demonstrated using untargeted 1H-NMR and GC-MS [351]. Similarly, extracellular protein breakdown is a hallmark of pancreatic cancer [352] and leads to release of branched-chain amino acids (BCAA). A recent study relied on LC-MS metabolic profiling of individuals from four prospective cohorts uncovered that three BCAAs (isoleucine, leucine, and valine) whose elevated levels in plasma can indicate twofold increased risk of PDAC future diagnosis [353].
An innovative unbiased metabolomic approach bypassing urine and plasma sampling relies on analyzing volatile organic compounds (VOC) from breath and breath condensate. The analytical platform can be MS or a proprietary Field Asymmetric Ion Mobility Spectrometry (FAIMS) technology (Owlstone Inc). This approach is being investigated to uncover novel metabolic biomarkers of lung cancer [354, 355]. A systematic review of VOC studies for early detection of lung cancer found that 2-butanone and 1-propanol are commonly reported as best discriminators between healthy and lung cancer subjects [356].
Different metabolites can differ drastically in their size and physicochemical properties. Because no single method can be used to separate, detect, and quantify a wide range of molecular species, multiple sample preparation and analytical methods can be used to capture the metabolome. Using multiplatform metabolomic approaches is a powerful strategy to identify biomarkers signatures. For example, combined metabolomics and lipidomics approaches using 1H-NMR, GC-MS, and LC-MS have uncovered potential biomarkers originating from alterations in lipid metabolism that could help identifying breast cancer [357]. Similarly, a panel of Fourier transform ion cyclotron resonance mass spectrometry (FTICR-MS), liquid chromatography (LC) MS/MS, and NMR was used for discovery of a set of biomarkers from altered lipid metabolism with the potential for early detection of colorectal cancer [358].
One of the examples of discovering cancer-specific metabolomic signatures by first computationally identifying dysregulated pathways, and then confirming these predictions using metabolomics comes from our group. We explored the integration of multiomics data with genome-scale metabolic models and showed that genetic alterations in clear cell renal cell carcinoma (ccRCC) were associated to ccRCC-specific metabolic reprogramming [359, 360]. The detailed computational analysis of ccRCC metabolism showed altered regulation of glycosaminoglycan (GAG) biosynthesis [360]. CE-based plasma and urine analysis of samples from ccRCC patients showed that a GAG panel (19 metabolic species) can be used to derive a GAG signature with diagnostic and prognostic potential [360,361,362].
Studies cited above demonstrate that advancements in instrumentation and computational methods are adequate to allow metabolomics biomarker discovery. However, the field is still hindered by different analytical challenges. Most importantly, experimental procedures and materials, as well as data handling and statistical analyses will need to be standardized to ensure analytical validation of biomarkers.
Translational Status
Arguably, the translational efforts in oncometabolomics are still in its infancy. There are currently no FDA-approved metabolomic liquid biopsy tests on the market. There are many challenges that will need to be addressed before metabolic biomarkers in the discovery phase can be validated and translated into clinical practice. Typically, only a small number of samples is collected during clinical trials, making the subsequent identification and validation of discovered metabolites challenging. Another challenge is found in the difficulty of measuring small differences in metabolites between healthy and cancer patients, masked by high inter-individual variation due to genetic and environmental factors [363]. Some of these challenges can partially be addressed by the advent of the Human Metabolome Database (HMDB) [364]. The database contains information on ranges of specific metabolites in human populations and their link to cancer pathways and disease phenotypes. However, like with other classes of biomarkers, the conclusive evidence of biomarker’s validity can only be established through clinical trials with large and diverse cohorts.
Some of the mentioned issued with metabolomic biomarkers are exemplified by sarcosine. Plasma sarcosine has shown promise as a biomarker for prostate cancer [365]. However, its validity and utility as a viable biomarker were questioned when it failed in later validation trials [366,367,368], and then resurfaced as a biomarker in a recent study [369]. The definite decision about the clinical utility will require large and rigorously designed validation trials.
The sensitivity of circulating metabolites to external factors means that biomarker validation studies need to account for ethnographic and dietary habits. Aminoindex Cancer Screening (AICS, Ajinomoto Co., Inc.) system is a blood test based on multivariate analysis of plasma-free amino-acids (PFAA) [370, 371]. The AICS assay has been validated in a series of clinical trials covering approximately 2,500 patients with 7 different types of cancer and 15,000 healthy controls [372,373,374]. The AICS test is currently being used to screen for early detection of lung cancer in Japan [372, 375]. Ajinomoto is sponsoring a single-site observational prospective case-control study to investigate performance characteristics of the AICS test for gynecological cancers in the US population (NCT02178462).
Our group has completed a number of prospective and retrospective studies to establish clinical validity of a GAGs as biomarkers for liquid biopsy in ccRCC. We found that plasma and urine GAG scores readily distinguish cancer patients from healthy controls (100% specificity and 100% sensitivity in a discovery cohort with 34 mccRCC patients and 16 healthy individuals, and a validation cohort with 18 mccRCC and 9 healthy subjects) [360]. Furthermore, the diagnostic and prognostic value of GAG was investigated in two additional studies [361, 362]. The first study explored the association between urine and plasma GAG scores with progression free and overall survival in a prospective cohort of 31 patients diagnosed with ccRCC [361]. The results show that urine GAG score was a predictor of PFS and OS (hazard ratio (HR) 4.62 and HR 10.13, respectively). The second study [362] investigated plasma GAG score as a biomarker for pre-operative detection of early-stage RCC and prediction of recurrence and death after RCC surgery. This retrospective case-control study consisted of a consecutive series of surgically treated 175 RCC patients and 19 healthy controls [362]. We found that the GAG score could correctly classify RCC from healthy subjects with AUC = 0.999 in the discovery part of the cohort (N = 67). In the validation set (N = 108), the GAG score achieved an AUC of 0.991, and achieved 93.5% sensitivity at the predetermined cutoff [362]. This test will be validated in the multicenter prospective clinical study AURORAX-087A for detection of post-surgical recurrence in ccRCC (NCT04006405).
A systematic review of VOC studies for early detection of lung cancer found that 2-butanone and 1-propanol are commonly reported as best discriminators between healthy and lung cancer subjects [356]. However, there is some discordance between relevant VOCs, as one study points to n-dodecane as having the highest discriminatory power between patients with histologically proven lung cancer and healthy controls (sensitivity 76%, specificity of 100%, when using a decision tree based on n-dodecane and 9 other peaks; N = 50 cancer patients and N = 39 healthy subjects) [354]. Instead of relying on identification of individual VOC, entire VOC signatures have potential for lung cancer detection. For example, one pilot study compared VOC patterns of 32 patients with cytological or histological diagnosis of lung cancer and 54 healthy controls. Combinations of VOC peaks could discriminate cancer from healthy subjects with leave-on-out cross-validation accuracy of 100% [355]. Owlstone Ltd. is currently sponsoring a multi-center case-control study on patients suspected to have lung cancer aimed at evaluating VOC analysis using their breath biopsy technology for early detection of lung cancer (NCT02612532).
‘Breathomics’ are not limited to lung cancer detection. An observational cohort trial on patients referred to CRC surgery or for diagnostic colonoscopy was completed (NCT02332213), where breath biopsy samples were collected prior to surgery or colonoscopy. The GC-MS analysis of VOC revealed that acetone and ethyl acetate were elevated in CRC patients, compared to healthy controls. A discriminant function analysis of breath VOC patterns could discriminate CRC vs healthy controls with 85% sensitivity and a 94% specificity (N = 209) [376]. Two recently published studies shown that VOCs have the potential to discriminate esophagogastric cancer patients from heathy controls [377, 378]. Another study shows that VOCs can be used for early detection of pancreatic cancer [379]. Owlstone Ltd. is currently recruiting patients for a pan-cancer prospective cross-sectional observational case-control study to evaluate if breath biopsy can differentiate between healthy subjects and patients with gastric, esophageal, pancreatic, renal, prostate, and bladder cancer from matched controls (NCT03756597). Breath biopsies are a promising source of biomarkers for precision medicine. The ongoing trials will help address some of the challenges in the field, such as low reproducibility and low VOC concordance between different studies.
Exosomes
Scientific Rationale
Exosomes are bioactive nanovesicles (30–150 nm in size) enclosed in lipid bilayer membranes [380,381,382]. Exosomes are released from endosomes of almost all cell types. The molecular cargo of exosomes includes diverse classes of biomolecules (proteins, DNA, various RNA species, lipids, and other metabolites). The exact composition of exosomes can be very heterogeneous, and likely reflects the composition and the phenotypic state of the cell of origin. The exact mechanisms of exosome biogenesis and function are still under investigation, but there is evidence showing that exosomes have an important role in inter-cellular communication, both in healthy and diseased states [383].
Exosomes are being investigated as important factors in carcinogenesis, with potential to both promote tumor growth and restrain it [384]. Studies show that tumor-derived exosomes can modulate tumor progression, angiogenesis, and metastasis [385]. Given their role in cancer biology, exosomes are being studied as targets for anticancer-therapy [386], potential drug delivery vectors [387], and as biomarkers [383].
Exosomes are rich in biomolecules that reflect the state and the composition of progenitor cells; they are a rich source of biomarkers [383]. In fact, genomics [388], transcriptomics [388], proteomics [389], and metabolomics [390] analyses can be applied to exosome-derived biomarkers. Importantly, because exosomes can be readily isolated from blood, plasma, saliva, urine, breast milk, semen, ascites fluid, amniotic fluid, and cerebrospinal fluid, they are an ideal target for liquid biopsy. Finally, exosomes can obviate the need for repeated tumor biopsies. Because exosome heterogeneity can be associated with intra-tumor heterogeneity, the entire phenotypic and genotypic tumor landscape can be captured with a single liquid biopsy.
Current and Emerging Technologies and Biomarkers
To fully exploit exosomes as a source of robust and accurate biomarkers, it is important to carefully consider methods for sample handling, isolation, and enrichment. The main methods for isolation of exosomes are differential ultracentrifugation [391, 392], density-gradient ultracentrifugation [391, 392], polymer-facilitated precipitation [393, 394], immunoaffinity capture [391, 395], and size-exclusion chromatography [396]. The most commonly used method is differential centrifugation [392]. However, the method can cause vesicle aggregation and co-isolation of protein contaminants [397]. Density-gradient ultracentrifugation can produce samples of higher purity, but it is laborious and lengthy, making it unsuitable for clinical applications. Size exclusion chromatography can produce similarly pure samples, and minimally affect exosome characteristics [398]. Exosome purification using commercial polymer-facilitated precipitation is rapid and easy to implement, but results can be poor [399]. Isolation using immunoaffinity can be used to enrich specific subpopulations of exosomes based on surface antigens [391].
One of the major challenges in the field of exosome biology is the lack of standardization of protocols used for exosome enrichment and characterization. Biased isolation of exosome components can introduce variability in results [400]. Differences in experimental procedures can affect results across different studies. As a way of tackling this issue and establishing experimental guidelines, a crowdsourcing knowledgebase resource called EV-TRACK has been established [401]. Once exosomes are isolated, they can be used as starting materials for further characterization. Exosomes can be characterized by electron and atomic force microscopy methods [402,403,404,405], but here we focus on molecular characterization drawing on omics technologies described earlier.
Exosomal nucleic acid content represents the genetic variants of the originating cancer, as could therefore be used to tailor therapeutic decisions and monitor response to therapy. Given the high abundance and the stability of exosomes, compared to cfDNA and CTCs in patients who have undergone therapy, exosome-derived DNA (exoDNA) is a robust source of genetic biomarkers to guide therapy. Indeed, driver mutations such as BRAF V600E mutation in melanoma and EGFR L858R and T790M mutations in lung cancer have been readily detected in exoDNA [406]. On the other hand, results of exosomal liquid biopsies in population-wide screening should be carefully interpreted, because driver mutations, such as KRAS and TP53, can be found in exosomes of healthy individuals [407, 408].
In addition to DNA, exosomes are a rich source of variety of RNA species [184, 409]. Of all exosomal RNA (exoRNA), miRNAs have been the main focus of exosomal biomarker discovery [410]. On the other hand, long RNAs, such as lncRNA [411] and mRNA [412], are also informative and can be used to identify somatic mutations and changes in gene expression characteristic of cancers. Exosomal hTERT mRNA can be detected in exosomes of patients with different malignancies [412]. The utility of this potential pan-cancer marker will need to be validated in larger cohorts.
Exosomes are significantly more abundant than CTCs in body fluids. This means that exosomally derived proteins are more amenable to proteomics analyses. Exosome proteomes can be probed for biomarkers in an unbiased way, using untargeted MS-proteomics approaches [413, 414], or using affinity-based approaches, such as binding to aptamer-based SOMAscan arrays [415]. Indeed, studies have shown that single protein biomarkers and protein panels from exosomes have the potential to be used as diagnostic and prognostic biomarkers in pancreatic cancer [416, 417], melanoma [418], lung cancer [389], and colorectal cancer [419].
Combining multiple omics approaches can yield more robust biomarkers than relying on a single method. For example, exoDNA and exoRNA can be investigated in tandem to detect oncogenic fusion transcripts [420]. This approach can be very useful for combined genomics and transcriptomics profiling of cancers that are not amenable to solid tissue biopsies [420]. Similarly, improvements in sensitivity and specificity can be achieved by simultaneous investigation of protein and miRNA panels derived from exosomes [421].
Recent developments in the field use various strategies to bypass purification steps. In one such strategy, called ExoScreen, authors used two types of antibodies and photosensitizer-beads to directly capture and detect cancer-derived circulating EVs [422]. ExoScreen uses antibodies against CD9 and CD147 antigens to capture CD9/CD147 double-positive EVs which were enriched in the serum of stage I colorectal cancer patients [422]. Similarly, protein microarrays with cocktails of antibodies against exosome-specific tetraspanins can be used to ensure specific capture of all exosomes. This approach called Extracellular Vesicle Array (EV Array) [423] enabled the detection of exosomes from a crude biofluid sample in a high-throughput manner.
Microfluidics platforms [424,425,426] are a new avenue towards integrated isolation, detection, and multi-omics characterization of exosomes for liquid biopsies. Microfluidic devices obviate the need for lengthy and laborious protocols, enable working with smaller sample volumes, and offer higher throughput. Clinical validity of microfluidic devices is an active area of research, but early efforts have showed promise for liquid biopsies. Microfluidic devices like ExoChip [424] can offer integrated quantification of exosome levels in biofluids. ExoChip relies on immuno-isolation of exosomes by CD63 [424, 427], followed by fluorescence staining and detection/quantification using a standard plate readers. A similar platform, ExoSearch [426], uses continuous flow to isolate CD9-positive exosomes, which are then stained with fluorescently labeled antibodies against exosomal tumor markers (CA125, EpCAM, and CD24), followed by multiplex fluorescence imaging. Another innovative method for exosome capture and characterization, called nano-plasmonic exosome (nPLEX) sensor, uses an array of periodic nanoholes embedded in a gold film [419]. nPLEX arrays are functionalized with exosome-specific affinity ligands, where exosome capture causes changes in the local refractive index proportional to the target protein levels. Importantly, captured exosomes can be released, facilitating analysis of mRNA cargo by qRT-PCR [419]. Microfluidic exosome isolation can also be carried out via size selection. An example of this approach is Exodisc lab-on-a-chip [428]. Exodisc uses tandem nano-filters to enrich exosome subpopulations in the range of 20–600 nm. An integrated immunoassay can then be used to quantify and characterize isolated exosomes.
These emerging technologies have a higher throughput and require much lower volume than standard methods for exosome isolation. Whether this will promote wider adoption of exosome-derived biomarkers for clinical decision-making will remain to be seen after some of the methods are validated in independent prospective clinical trials.
Translational Status
The clinical use of exosome-based biomarkers is fraught with challenges. Exosomes can vary in size and concentrations in different biological samples [429]. Moreover, external factors such as physical activities undertaken prior to sampling or the time of sampling can influence the composition of exosomes in liquid biopsy samples [397]. Once samples are taken, it is critical to consider rapid sample processing and storage because circulating cells inside the sample can continue producing exosomes [397]. Additionally, any downstream biomarker analysis is reliant on costly specialized instrumentation and kits for exosome isolation [430]. Moreover, isolation techniques are often matrix-specific and lack standardization necessary for clinical use. Future improvements in standardization, scalability, and turnover time for exosome isolation will pave way for routine use of this important source of biomarkers in the clinic.
Despite these challenges, exosome-based liquid biopsy tests might make it to the clinic in the near future. Exosome Diagnostics has two assays on the market. ExoDx Prostate (IntelliScore) is a urine-based liquid biopsy test that uses exosomal RNA to quantify expression of three genes and predict the aggressiveness of prostate cancer [431]. A multi-site prospective cohort study of patients undergoing prostate biopsy was used to validate the ExoDx assay for discrimination between aggressive (Gleason grade 7 and higher) versus Gleason grade 6 or benign prostate cancer. In a training cohort (N = 255), the exosome assay score in combination with PSA and clinical variables could detect GS7 or higher prostate cancer with AUC = 0.77 [431]. In the validation cohort (N = 519), the performance was similar, with AUC = 0.73 to detect GS7 cancer or higher, outperforming the PSA-based standard of care test (AUC = 0.63) [431]. The test is available for clinical use and Exosome Diagnostics is currently conducting a trial to investigate its utility and evaluate its potential to reduce the number of initial prostate biopsies (NCT03235687). Their second test, ExoDx Lung (ALK), currently available for research only, is a qPCR test that detects EML4-ALK fusion transcripts in plasma exosomes to inform therapy selection for lung cancer. ExoDx Lung (ALK) was tested for longitudinal monitoring in response to treatment in a prospective cohort of ALK-positive patients (N = 52, total 144 longitudinal samples) [432]. The assay detected exoRNA ALK-fusions in 50% of patients at baseline. Furthermore, 98% of samples from patients who showed objective response or stable disease were tested negative, showing that ExoDx Lung (ALK) has potential as a monitoring biomarker for NSCLC [432].
Systems Biomarkers
Scientific Rationale
The biology of cancer is characterized by complex phenotypes such as genomic instability, metabolic reprogramming, changes in proliferative signaling, evasion of apoptosis and immune response, induction of angiogenesis, invasion and metastasis, collectively known as cancer hallmarks [433]. To understand, identify, and target these neoplastic processes, we need systems-level integration and understanding of information from multiple layers of biological activity (Fig. 2).
The first application of systems biology for precision oncology is the discovery of new (multiomics) biomarkers. Systems biology provides a framework to investigate complex cancer phenotypes in terms of pathways and networks. The state-of-the-art statistical and computational algorithms can be applied to the accumulated multi-layered biological data and integrated with known cancer-related biochemical pathways to guide the discovery of new biomarker panels [434]. The biomarker candidates can then be analytically and clinically validated in clinical trials.
The second application addresses the problem of intra- and inter-patient cancer heterogeneity. Despite general commonalities [433], cancers are defined by distinct background genotypes and molecular signatures. To implement successful personalized treatment protocols, we need to account for genetic and environmental differences between individuals, as well as temporal and spatial heterogeneity of cancer cells within patients. Multiomics patient data collection and assays for known biomarkers can be combined with machine learning approaches [435] for precise patient stratification. The complexity and costs associated with these approaches are still a barrier to implementing systems biology into clinical oncology. However, as the costs of omics analyses keeps declining, and as analysis tools become more powerful and easier to use, multiomics strategies might approach routine use in the next decade.
Current and Emerging Technology
The main drivers of systems biology are big data sets generated through omics technologies described in previous sections. Comprehensive multiomics profiling over thousands of cancer patients has resulted in large databases of cancer-related biological data. Arguably, the most important publicly available resource for cancer systems biology is The Cancer Genome Atlas (TCGA) [75]. TCGA contains petabytes of data from (epi)genomics, transcriptomics, and proteomics experiments and clinicopathologic annotation data describing 33 cancer types from 11,160 patients and has been instrumental for translational cancer research [436]. Another similar resource is The International Cancer Genome Consortium (ICGC) cancer data portal that contains multiomics data from 84 cancer projects and more than 20,000 patients worldwide [437].
The availability of big multiomics dataset is not enough to produce biological or clinical insight on its own. In fact, our ability to generate big data sets is vastly greater than the ability to analyze and integrate them [438]. Combining the results of biological assays with imaging, biopsy, and clinical data is used routinely in clinical practice. Extracting useful information from high-dimensional and heterogeneous biological data sets requires a different approach. The data can be used in combination with models of cellular processes and pathways [18] to reduce the dimensionality and generate candidate list of features. Another approach is to directly combine biological information from different omics platforms (multianalyte approach) to derive classifiers and diagnostic scores uses sophisticated computational methods [435]. These methods include various network topology analyses, dimensionality reduction methods, anomaly detection, supervised and unsupervised machine learning algorithms, as well as summarization and visualization techniques for complex high-dimensional data [439]. These methods have been used for feature selection on big data sets directly [440, 441].
The sensitivity, specificity, and confidence of clinical decision-making can be boosted by leveraging orthogonal multianalyte panels. However, using multi-layered information and multiple markers runs the risk of over-fitting predictive models [439]. The importance of identified multiomics biomarkers (genes, transcripts, proteins, and metabolites from the candidate list) needs to be investigated by targeted assays and omics experiments and be validated in retrospective clinical studies. Robust statistical methods can then be used to remove biomarkers with the minimal impact on accuracy, to identify meaningful correlations, and devise predictive models [439]. The final set of biomarkers, in combination with appropriate statistical methods, can then be validated in larger cohorts [439].
Multi-omics technologies and their integration with diverse clinical data will become even more important for robust patient stratification and cancer diagnostics with the advent of artificial intelligence and deep learning (DL) algorithms [442]. Deep learning is a subclass of machine learning algorithms that use neural networks, multi-layered data processing networks capable of feature extraction and pattern recognition in large and diverse data sets [443]. While the successful applications of DL algorithms in medicine are mostly focused on automated classification of medical imaging data [444, 445], there are promising examples of applications to (multi)-omics in precision medicine. In one study, authors used an artificial neural network (ANN) to distinguish multiple myeloma patients from healthy subjects with 95% sensitivity at 95% specificity (N = 84, case-control study) based on MALDI TOF MS low mass spectral fingerprint/metabolomic analysis of peripheral plasma samples [446]. In another study, miRNA sequence data from serum samples of epithelial ovarian cancer (EOC) patients was used to train a neural network, resulting in a diagnostic algorithm that outperformed CA125 in distinguishing cancer patients from healthy controls and benign tumors (AUC = 0.9, N = 179, case-control studies) [447]. The authors then reduced the set of diagnostic miRNAs to only seven that could be detected via qPCR, adapted the neural network to the reduced set, and validated the classifier on 51 pre-operative clinical samples to achieve an AUC = 0.85 [447].
The power of deep learning is that it can readily integrate disparate data, such as multi-omics data, medical images, and clinical information to enhance prediction accuracy [443]. However, further progress is limited by small data sets. The full realization of DL potential will have to wait on the availability of sufficiently large, matched, and carefully annotated datasets. However, even with perfect datasets of sufficient size, validation and assurance of proper use might require interpretability of predictions before DL is adopted into routine clinical use [448, 449].
Translational Status
Translational use of systems biology and multi-analyte biomarkers is a relatively new addition to the field. However, there have been some notable studies yielding new biomarker panels for liquid biopsy. Some of these biomarkers show promise in early clinical trials and await validation in larger cohorts, while others are on their way to being commercialized and used in clinical practice.
CancerSEEK is a promising multianalyte blood test, with potential for pan-cancer diagnosis [450]. The initial case-control study (N = 1005) on patients with clinically detected stage I-III cancers shows that the test could detect a median of 70% over eight common cancer types by quantifying levels of protein markers in plasma and cancer-specific mutations in cfDNA. Importantly, CancerSEEK protein markers were useful in detecting the candidate tissue of origin, which is a critical feature in a population-wide screens for early pan-cancer detection. However, the specificity needs to be assessed and validated on large prospective cohorts [450].
Large biomarker panels can in some cases be replaced by only a few biomarkers. For example, combining digital droplet PCR to determine KRAS mutant allele fraction (MAF) in cfDNA and exoDNA can help devise a classifier to predict liver cancer. This classifier was tested for clinical utility in a longitudinal prospective cohort of 194 patients undergoing treatment for clinically and histologically confirmed localized or metastatic pancreatic adenocarcinoma [32]. The baseline multianalyte analysis of the cohort showed that the ctDNA and exoDNA MAFs ≥ 5% to be a significant predictor of OS (HR, 7.73). Furthermore, longitudinal multianalyte monitoring of exoDNA showed that MAF peak above 1% is associated with radiologic progression (sensitivity 79% and specificity 100%) [32]. This study shows that longitudinal monitoring of circulating nucleic acids can provide useful predictive and prognostic information.
A larger multianalyte test has been investigated for prediction of pancreatic cancer in a population with risk for familial pancreatic cancer [451]. The panel used multiple analytes across different sample matrices: tissue (miRNA: miR-196b), serum (snRNA: RNU2-1f; protein: LCN2, TIMP1, Glypican-1, and CA 19-9), duodenal juice exosomes (protein: Glypican-1), and duodenal cfDNA (KRAS mutations). The validation in a small cohort showed that the entire panel could be reduced to the three plasma analytes (miR-196b, TIMP1, and LCN2), and distinguish stage I PDAC (N = 5) from healthy individuals (N = 20) with an AUC = 1, and sensitivity, and specificity at 100% [451]. Validating the specificity of this multi-analyte panel towards early stage PDAC is necessary. However, it might be challenging to do so, because clinically validated stage I PDAC samples are extremely rare [451].
A successful case of a multianalyte test that also incorporates clinical data is the Stockholm 3 model (STHLM3). This test combines plasma protein biomarkers (PSA, free PSA, intact PSA, hK2, MSMB, MIC1), 232 genetic polymorphisms associated with prostate cancer in earlier studies, and clinical variables to identify high risk prostate cancer at biopsy [324]. STHLM3 was validated in an independent multi-center community cohort of 533 patients scheduled for prostate biopsy [452]. Blood samples drawn prior to biopsy and analyzed to compare STHL3 to PSA-based diagnosis of clinically significant prostate cancer (ISUP Grade Group (GG) 2 or higher). STHLM3 showed better diagnostic performance than PSA alone (AUC = 0.859 vs 0.642 for PSA and 0.748 for PSA density) for detection of Gleason grade group ≥ 2 vs benign and Gleason grade group = 1 PCa, with the potential to reduce the total number of biopsies by 38% [452]. The Stockholm3 test is now entering clinical use in Sweden, Norway, and Finland.
An upcoming AI Genomics start-up Freenome is recruiting patients for an observational study focused on colorectal cancer screening. Their approach will be to analyze all cfDNA (most of which originates from immune cells [453]), cfRNA and circulating protein as potential circulating biomarkers. Their first report, on sequencing cfDNA for early CRC detection, was available as a preprint at the time of writing this review [454]. The study was performed on retrospectively collected 871 plasma samples from international institutions and commercial biobanks (from 546 predominantly early stage CRC cases, and 271 non-cancer controls). The authors estimated ctDNA fraction in plasma cfDNA from copy number variation and used machine learning (logistic regression and support vector machine) to discriminate between healthy and CRC samples. Using k-fold cross validation, the procedure showed sensitivity of 85% at 85% specificity for CRC versus healthy subjects.
Conclusions and Future Directions
The molecular characterization of tumor tissue biopsy samples is currently the gold standard of precision and personalized medicine. However, the invasiveness of (repeated) biopsies is one of the main drivers of research on liquid biopsy biomarkers for clinical decision making. Moreover, liquid biopsies can offer insights into biological phenomena and clinically important information about spatiotemporal heterogeneity of tumors that is not readily accessible through tissue biopsy.
Towards Best Practices for Discovery and Validation of Biomarkers
The accumulation of understanding of cancer biology and advances in omics technologies have already yielded many potential circulating biomarkers. The declining costs of high-throughput assays and propagation of efficient computational methods have enabled both targeted and unbiased genome-wide studies on biomarkers for clinical applications. The emerging liquid biopsy tests are increasingly focused on multiparametric assays, involving multiple analytes from a single layer of biological information or multi-omics analytes. These kinds of studies are propelled by advances in statistical and machine learning methods for analyzing big data. However, while current research highlights the promise of liquid biopsy biomarkers for precision oncology, the majority of studies are still in an early proof-of-concept phase.
There are many challenges that will need to be overcome before many of the new omics biomarkers can enter into standard clinical practice. One of the reasons for the discrepancy between the number of biomarkers in the primary literature and clinical practice is the gulf between the experimental evidence needed to establish a finding in basic science and the requirements for a robust diagnostic assay [230]. Moreover, even when analytical validity is established, assays need to be clinically validated in well-designed trials. Currently, there are many biomarkers that show promise in retrospective and case-control pilot studies, but there is a general lack of large prospective studies demonstrating—at the very least—clinical validity. For example, a recent systematic assessment of clinical proteomics literature [455] revealed that only 10–20% studies mention potential clinical application, while the rest focus mainly on development of technical aspects of an assay or sample preparation. Furthermore, even where reviewed studies included clinical validation, it was found to be underpowered for the specific context of use (low sample size), the potential biomarker was not tested against current methods, the study was not performed on the population of interest (e.g., screening biomarker tested in a case-control study with different prevalence than in reality), or tested in the intended context of use [455]. Moreover, the evidence of clinical utility is lacking for the majority of liquid biopsy biomarkers.
Demonstrating clinical validity and utility is perhaps even more challenging for circulating biomarkers that would enable population-wide screening and early detection of tumors. While the early data may show promising diagnostic performance, caution is necessary because of large numbers of false positives even when clinical specificity is high (> 99%). Because the incidence and the mortality from any specific cancer is low, clinical utility studies need to be long and cover a sufficiently large population.
The weight of evidence and the scope of clinical trials needed to successfully complete a phased biomarker research for screening applications is well outside of reach for most research groups. In fact, multidisciplinary collaborations are often needed to successfully complete the process of biomarker discovery and validation through multiple phases that require a diverse and orthogonal sets of expertise: biological, analytical, statistical, clinical, ethical, and regulatory [456]. Inadequate attention to one or more of these facets of biomarker development has led to failures in validation studies. Specifically, past biomarker failures have been linked to poor selection of patients, low-quality sample acquisition, processing and storage, insufficient statistical power, lacking standards for biomarker profiling, and problems with reporting and analysis [20, 21, 456, 457].
To address common problems in biomarker development, various authors and organizations have devised sets of standards and guidelines for study design and reporting. Some of the important resources that provide guidelines for phased biomarker development, study design, and reporting standards for protocols and results include: guidelines for phased development of biomarkers for early detection of cancer [307], guidelines for case-control study design [305], validation steps for omics biomarkers [304], the Biospecimen Reporting for Improved Study Quality (BRISQ) [458], PRospective-specimen-collection, retrospective-Blinded-Evaluation (PRoBE) [305], design strategies for identification of predictive biomarkers [459], REporting recommendations for tumor MARKer prognostic studies (REMARK) [460], Consolidated Standards of Reporting Trials (CONSORT) [461], and standards for reporting of diagnostic accuracy (STARD) [462]. The National Biomarker Development Alliance (NBDA) set forth to establish standards and point out phases of the systems-based, end-to-end biomarker development that still need to be standardized [463]. Some critical points along the biomarker development trajectory are addressed below.
Many research and clinical ambiguities can be avoided by establishing the clinical question early at the inception of the biomarker research program [464]. This should be done by consulting with clinicians and taking unmet clinical needs into account (see the following subsection). Prespecifying the context of use enables early and productive engagement with regulatory bodies. Moreover, predetermined intended use of the biomarker dictates the details of all downstream phases. Specifically, it assures that patient selection, sample sources, quality, and adequate size/number are relevant to the intended clinical utility [299].
The importance of a large sample size is pronounced where multi-omics data are analyzed for biomarker discovery—small N leads to overfitting and false positives [463]. Equally important are independent confirmatory studies, where independent training and blinded test sets can increase confidence in the validity of a biomarker. In cases where biomarkers are being developed for a cancer (sub)type with low prevalence, it might be necessary to use biobanks and samples from multiple research centers to reach requisite numbers for validation. Furthermore, access to all clinical data is necessary to rule out patient characteristics other than the disease state as the source of biomarker level variation.
Prior to clinical validation, it is necessary to carry out the analytical validation of the biomarker assay. The assurance of the analytical validity of the test assay requires the use reference standards and can be assessed by measuring different parameters: accuracy, trueness, precision, reproducibility, robustness, linearity, analytical sensitivity and specificity, the limit of detection, and interfering substances. Notably, using multiomics procedures necessitates adherence to strict quality control standards [304, 463]. As the last step for ensuring the validity of the assay, it is important to establish multi-site assay precision and reproducibility. Adoption of FDA Good Laboratory Practice [465] and CLIA laboratory proficiency testing [466] can enhance analytical and statistical rigor.
Protocols for all pre-analytical (processing, handling, transport, and storage), analytical (assay methods and instrumentation) and post-analytical (statistical/computational pipeline for data analysis and interpretation) procedures need to be standardized and strictly defined (“locked down”) at this point in order to mitigate reproducibility issues downstream [463].
Data collection, storage, and analysis algorithms are another critical area where special care is necessary [463]. Increasingly complex multi-omics approaches require validation and application of new analytical and statistical approaches, the development of analytical standards, and robust open-source classification algorithms. Collection, storage, and sharing of high-quality data and metadata is important throughout the development process. Importantly, data sharing and publication of negative and contradictory results can simplify investigation of reasons for biomarker failure. Researchers need to adhere to data quality standards, use established ontologies, vocabularies, minimum reporting standards, and utilize accepted exchange formats. FDA and NIH jointly developed BEST (Biomarkers, EndpointS, and other Tools) Resource [19] to facilitate this process.
Clinical validation of the biomarker requires careful study design according to the intended use of the biomarker. Prospective randomized trials are considered to be a gold standard for biomarker validation; however, some cancer biomarkers currently in use were validated using retrospective analyses of clinical trials [459] (e.g., KRAS [467, 468] and BRCA mutations [469]). Critically, statistical procedures and threshold values for evaluating biomarker utility need to be prespecified. Depending on the intended use, biomarker characteristics will be estimated by receiver operating characteristic (ROC) curve analysis, estimated as clinical sensitivity and specificity, positive and negative predictive value (PPV and NPV, respectively).
As the final goal of biomarker development, it is necessary to establish clinical utility. In this phase, it is necessary to demonstrate the improvement of patient outcomes through the use of the biomarker (e.g., through overall or progression-free survival, mortality), show economical utility, and compare it to the established biomarkers and the standard of care.
Even if the number of aforementioned aspects to take in consideration for a successful translation of newly discovered biomarkers in the clinic may seem overwhelming and beyond the capacity of academic research, in our experience, biomarker validation is an iterative process (Fig. 3). Each iteration redefines the level of readiness of the liquid biopsy technology. Through each “cycle” of validation, evidence is accumulated on the clinical validity/utility of the biomarker, before the ultimate decision can be made about the commercialization of the assay. Noteworthy, reaching the market does not mark the end of the process, because biomarker performance needs to be monitored even after it has been approved for commercialization [463].

The process of biomarker validation in practice. Biomarker validation can be seen as an iterative process where the liquid biopsy assay at an increasing level of analytical validation is tested for clinical validity for an intended purpose and on a fit-for-purpose patient population. The study design and an endpoint need to be predetermined, and sample and clinical data collection need to be carried out in accordance with clinical and biomarker development guidelines. Upon completion of the study, assay performance characteristics need to be reported to requisite regulatory bodies. Upon enhancement of the analytical performance all protocols need to be locked down and validated again
Towards Unmet Clinical Needs
Despite all the challenges, translational efforts in liquid biopsy have resulted in validated and commercialized biomarkers, some of which are already in clinical use (Table 1). In particular, diagnostic, prognostic, and predictive circulating biomarker options exist for lung, breast, colorectal, bladder, ovarian, cervical, and prostate cancer. Additionally, the first chronic myeloid leukemia liquid biopsy assay for monitoring treatment response, QXDx AutoDG ddPCR System (Bio-Rad Laboratories), received FDA clearance at the time of writing this review. However, management of patients with solid tumors like gastric, esophageal, liver, pancreatic, endometrial, brain, thyroid, head and neck, melanoma, and renal cancers still has no validated and established liquid biopsy biomarkers.
Gastric (GC) and esophageal cancer are estimated to cause death in more than a 1.2 million people worldwide in 2019 [1]. There is an unmet need for diagnostic and prognostic liquid biopsy biomarkers for these two types of cancer. However, all available studies have been carried out in basic studies or retrospective and prospective studies with small cohorts.
Hepatocellular carcinoma is another example of a common and deadly cancer with an urgent need for liquid biopsy biomarkers [470]. Many liver cancer patients are diagnosed at late stage, where curative treatment is no longer an option. Importantly, sorafenib, a drug used to treat advanced hepatocellular carcinoma, currently has no clinically validated biomarkers for response prediction.
Similarly, pancreatic cancer, one of the most aggressive tumors, is asymptomatic at an early stage and most diagnosis are made at late stage where there are limited options for treatment [471]. The established biomarker for pancreatic cancer, CA19-9, is not suitable for screening and diagnosis. Moreover, pancreatic cancer has four different subtypes with complex molecular signatures, which are impossible to resolve using current diagnostic procedures [471].
Another example is endometrial cancer, typically diagnosed at an early stage when it is treatable. However, about 20% of the cases are diagnosed at late stage where 5-year survival is drastically lower [472]. Late stage endometrial cancer is treated with surgery, radiotherapy, and chemotherapy. However, chemotherapy is less effective than with other cancers, and different cancer subtypes require different therapeutic decisions, warranting development and validation of biomarkers for monitoring recurrence and response to therapy [472].
Perhaps the most compelling reason for development of liquid biopsy biomarkers are tumors of the central nervous system. Clinical decision-making, including monitoring response to treatment, is heavily reliant of neuroimaging. However, chemoradiation and antiangiogenic therapy can alter contrast enhancement and confound imaging results by affecting the permeability of the blood-brain barrier and the tumor vasculature [473]. Furthermore, brain tumor tissue biopsy carries significant risk for the patient, creating difficulties for diagnosis, prediction, and prognosis. Thus, there is an unmet clinical need for non-invasive liquid biopsy biomarkers for brain tumors.
Precision and personalized medicine will benefit greatly when analytical and clinical challenges affecting circulating biomarker development are addressed. While it might take a long time before clinical validity and utility are demonstrated for early detection, liquid biopsy biomarkers are becoming crucial for patient stratification and therapy response prediction. We believe that these trends will continue, and that liquid biopsy will play an increasingly important role in personalized cancer patient management in the future.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424. https://doi.org/10.3322/caac.21492.
Chaffer CL, Weinberg RA. A perspective on cancer cell metastasis. Science. 2011;331(6024):1559–64. https://doi.org/10.1126/science.1203543.
Ahlquist DA. Universal cancer screening: revolutionary, rational, and realizable. npj Precision Oncol. 2018;2(1). https://doi.org/10.1038/s41698-018-0066-x.
Schiffman JD, Fisher PG, Gibbs P. Early detection of cancer: past, present, and future. Am Soc Clin Oncol Educ Book. 2015:57–65. https://doi.org/10.14694/EdBook_AM.2015.35.57.
Bedard PL, Hansen AR, Ratain MJ, Siu LL. Tumour heterogeneity in the clinic. Nature. 2013;501(7467):355–64. https://doi.org/10.1038/nature12627.
Jamal-Hanjani M, Quezada SA, Larkin J, Swanton C. Translational implications of tumor heterogeneity. Clin Cancer Res. 2015;21(6):1258–66. https://doi.org/10.1158/1078-0432.Ccr-14-1429.
Stewart E, McEvoy J, Wang H, Chen X, Honnell V, Ocarz M, et al. Identification of therapeutic targets in rhabdomyosarcoma through integrated genomic, epigenomic, and proteomic analyses. Cancer Cell. 2018;34(3):411–26.e19. https://doi.org/10.1016/j.ccell.2018.07.012.
Yu KH, Snyder M. Omics profiling in precision oncology. Mol Cell Proteomics. 2016;15(8):2525–36. https://doi.org/10.1074/mcp.O116.059253.
Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. New Engl J Med. 2012;366(10):883–92. https://doi.org/10.1056/NEJMoa1113205.
Shi H, Hugo W, Kong X, Hong A, Koya RC, Moriceau G, et al. Acquired resistance and clonal evolution in melanoma during BRAF inhibitor therapy. Cancer Discov. 2014;4(1):80–93. https://doi.org/10.1158/2159-8290.CD-13-0642.
Johnson BE, Mazor T, Hong C, Barnes M, Aihara K, McLean CY, et al. Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science. 2014;343(6167):189–93. https://doi.org/10.1126/science.1239947.
Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21(7):751–9. https://doi.org/10.1038/nm.3886.
Duffy MJ. Role of tumor markers in patients with solid cancers: a critical review. Eur J Intern Med. 2007;18(3):175–84. https://doi.org/10.1016/j.ejim.2006.12.001.
Siravegna G, Marsoni S, Siena S, Bardelli A. Integrating liquid biopsies into the management of cancer. Nat Rev Clin Oncol. 2017. https://doi.org/10.1038/nrclinonc.2017.14.
Bardelli A, Pantel K. Liquid biopsies, what we do not know (Yet). Cancer Cell. 2017;31(2):172–9. https://doi.org/10.1016/j.ccell.2017.01.002.
Marrugo-Ramirez J, Mir M, Samitier J. Blood-based cancer biomarkers in liquid biopsy: a promising non-invasive alternative to tissue biopsy. Int J Mol Sci. 2018;19(10):E2877. https://doi.org/10.3390/ijms19102877.
Heitzer E, Haque IS, Roberts CES, Speicher MR. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat Rev Genet. 2019;20(2):71–88. https://doi.org/10.1038/s41576-018-0071-5.
Nielsen J. Systems biology of metabolism: a driver for developing personalized and precision medicine. Cell Metab. 2017;25(3):572–9. https://doi.org/10.1016/j.cmet.2017.02.002.
FDA-NIH Biomarker Working Group. BEST (biomarkers, endpoints, and other tools) resource [Internet]. BEST (Biomarkers, EndpointS, and other Tools) Resource. Silver Spring (MD) 2016.
Diamandis EP. Cancer biomarkers: can we turn recent failures into success? J Natl Cancer I. 2010;102(19):1462–7. https://doi.org/10.1093/jnci/djq306.
Ioannidis JP. Biomarker failures. Clin Chem. 2013;59(1):202–4. https://doi.org/10.1373/clinchem.2012.185801.
Sturgeon C, Hill R, Hortin GL, Thompson D. Taking a new biomarker into routine use - a perspective from the routine clinical biochemistry laboratory. Proteomics Clin Appl. 2010;4(12):892–903. https://doi.org/10.1002/prca.201000073.
Bossuyt PMM. Clinical validity: defining biomarker performance. Scand J Clin Lab Invest. 2010;70:46–52. https://doi.org/10.3109/00365513.2010.493383.
Pletcher MJ, Pignone M. Evaluating the clinical utility of a biomarker a review of methods for estimating health impact. Circulation. 2011;123(10):1116–U261. https://doi.org/10.1161/Circulationaha.110.943860.
Prasad V, Kaestner V, Mailankody S. Cancer drugs approved based on biomarkers and not tumor type-FDA approval of pembrolizumab for mismatch repair-deficient solid cancers. JAMA Oncol. 2018;4(2):157–8. https://doi.org/10.1001/jamaoncol.2017.4182.
Antoniou A, Pharoah PDP, Narod S, Risch HA, Eyfjord JE, Hopper JL, et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case series unselected for family history: a combined analysis of 22 studies. Am J Hum Genet. 2003;72(5):1117–30. https://doi.org/10.1086/375033.
Ross JS, Cronin M. Whole cancer genome sequencing by next-generation methods. Am J Clin Pathol. 2011;136(4):527–39. https://doi.org/10.1309/Ajcpr1svt1vhugxw.
Hayden EC. Technology: The $1,000 genome. Nature. 2014;507(7492):294–5. https://doi.org/10.1038/507294a.
Plothner M, Frank M, von der Schulenburg JG. Cost analysis of whole genome sequencing in German clinical practice. Eur J Health Econ. 2017;18(5):623–33. https://doi.org/10.1007/s10198-016-0815-0.
Schwarze K, Buchanan J, Taylor JC, Wordsworth S. Are whole-exome and whole-genome sequencing approaches cost-effective? A systematic review of the literature. Genet Med. 2018;20(10):1122–30. https://doi.org/10.1038/gim.2017.247.
Aravanis AM, Lee M, Klausner RD. Next-generation sequencing of circulating tumor DNA for early cancer detection. Cell. 2017;168(4):571–4. https://doi.org/10.1016/j.cell.2017.01.030.
Bernard V, Kim DU, San Lucas FA, Castillo J, Allenson K, Mulu FC, et al. Circulating nucleic acids are associated with outcomes of patients with pancreatic cancer. Gastroenterology. 2019;156(1):108–18.e4. https://doi.org/10.1053/j.gastro.2018.09.022.
Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nat Med. 2008;14(9):985–90. https://doi.org/10.1038/nm.1789.
Volckmar AL, Sultmann H, Riediger A, Fioretos T, Schirmacher P, Endris V, et al. A field guide for cancer diagnostics using cell-free DNA: from principles to practice and clinical applications. Genes Chromosom Cancer. 2018;57(3):123–39. https://doi.org/10.1002/gcc.22517.
Ossandon MR, Agrawal L, Bernhard EJ, Conley BA, Dey SM, Divi RL, et al. Circulating tumor DNA assays in clinical cancer research. Jnci-J Natl Cancer I. 2018;110(9):929–34. https://doi.org/10.1093/jnci/djy105.
Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6(224):224ra24. https://doi.org/10.1126/scitranslmed.3007094.
Kahlert C, Melo SA, Protopopov A, Tang JB, Seth S, Koch M, et al. Identification of double-stranded genomic DNA spanning all chromosomes with mutated KRAS and p53 DNA in the serum exosomes of patients with pancreatic cancer. J Biol Chem. 2014;289(7):3869–75. https://doi.org/10.1074/jbc.C113.532267.
Cheng THT, Jiang PY, Tam JCW, Sun X, Lee WS, Yu SCY, et al. Genomewide bisulfite sequencing reveals the origin and time-dependent fragmentation of urinary cfDNA. Clin Biochem. 2017;50(9):496–501. https://doi.org/10.1016/j.clinbiochem.2017.02.017.
Katseli A, Maragos H, Nezos A, Syrigos K, Koutsilieris M. Multiplex PCR-based detection of circulating tumor cells in lung cancer patients using CK19, PTHrP, and LUNX specific primers. Clinical Lung Cancer. 2013;14(5):513–20. https://doi.org/10.1016/j.cllc.2013.04.007.
Jenkins S, Yang JCH, Ramalingam SS, Yu K, Patel S, Weston S, et al. Plasma ctDNA Analysis for detection of the EGFR T790 M mutation in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2017;12(7):1061–70. https://doi.org/10.1016/j.jtho.2017.04.003.
Reck M, Hagiwara K, Han BH, Tjulandin S, Grohe C, Yokoi T, et al. ctDNA Determination of EGFR mutation status in European and Japanese patients with advanced NSCLC: the ASSESS study. J Thorac Oncol. 2016;11(10):1682–9. https://doi.org/10.1016/j.jtho.2016.05.036.
cobas EGFR Mutation Test v2. https://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm504540.htm. Accessed 05 Feb 2019
Wu YL, Lee V, Liam CK, Lu S, Park K, Srimuninnimit V, et al. Clinical utility of a blood-based EGFR mutation test in patients receiving first-line erlotinib therapy in the ENSURE, FASTACT-2, and ASPIRATION studies. Lung Cancer. 2018;126:1–8. https://doi.org/10.1016/j.lungcan.2018.10.004.
Fassunke J, Ihle MA, Lenze D, Lehmann A, Hummel M, Vollbrecht C, et al. EGFR T790 M mutation testing of non-small cell lung cancer tissue and blood samples artificially spiked with circulating cell-free tumor DNA: results of a round robin trial. Virchows Arch. 2017;471(4):509–20. https://doi.org/10.1007/s00428-017-2226-8.
Wu YL, Zhou C, Liam CK, Wu G, Liu X, Zhong Z, et al. First-line erlotinib versus gemcitabine/cisplatin in patients with advanced EGFR mutation-positive non-small-cell lung cancer: analyses from the phase III, randomized, open-label ENSURE study. Ann Oncol. 2015;26(9):1883–9. https://doi.org/10.1093/annonc/mdv270.
Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545(7655):446–51. https://doi.org/10.1038/nature22364.
Hindson BJ, Ness KD, Masquelier DA, Belgrader P, Heredia NJ, Makarewicz AJ, et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal Chem. 2011;83(22):8604–10. https://doi.org/10.1021/ac202028g.
Diehl F, Li M, Dressman D, He YP, Shen D, Szabo S, et al. Detection and quantification of mutations in the plasma of patients with colorectal tumors. P Natl Acad Sci USA. 2005;102(45):16368–73. https://doi.org/10.1073/pnas.0507904102.
Diehl F, Schmidt K, Durkee KH, Moore KJ, Goodman SN, Shuber AP, et al. Analysis of mutations in DNA isolated from plasma and stool of colorectal cancer patients. Gastroenterology. 2008;135(2):489–98. https://doi.org/10.1053/j.gastro.2008.05.039.
Leary RJ, Sausen M, Kinde I, Papadopoulos N, Carpten JD, Craig D, et al. Detection of chromosomal alterations in the circulation of cancer patients with whole-genome sequencing. Sci Transl Med. 2012;4(162):162ra154. https://doi.org/10.1126/scitranslmed.3004742.
Forshew T, Murtaza M, Parkinson C, Gale D, Tsui DWY, Kaper F, et al. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci Transl Med. 2012;4(136):136ra68. https://doi.org/10.1126/scitranslmed.3003726.
Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. P Natl Acad Sci USA. 2011;108(23):9530–5. https://doi.org/10.1073/pnas.1105422108.
Newman AM, Bratman SV, To J, Wynne JF, Eclov NCW, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20(5):552–8. https://doi.org/10.1038/nm.3519.
Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34(5):547–55. https://doi.org/10.1038/nbt.3520.
Gyanchandani R, Kvam E, Heller R, Finehout E, Smith N, Kota K, et al. Whole genome amplification of cell-free DNA enables detection of circulating tumor DNA mutations from fingerstick capillary blood. Sci Rep. 2018;8:17313. https://doi.org/10.1038/s41598-018-35470-9.
Murtaza M, Dawson SJ, Tsui DWY, Gale D, Forshew T, Piskorz AM, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013;497(7447):108–12. https://doi.org/10.1038/nature12065.
Suzuki A, Suzuki M, Mizushima-Sugano J, Frith MC, Makalowski W, Kohno T, et al. Sequencing and phasing cancer mutations in lung cancers using a long-read portable sequencer. DNA Res. 2017;24(6):585–96. https://doi.org/10.1093/dnares/dsx027.
Gong L, Wong CH, Cheng WC, Tjong H, Menghi F, Ngan CY, et al. Picky comprehensively detects high-resolution structural variants in nanopore long reads. Nat Methods. 2018;15(6):455–60. https://doi.org/10.1038/s41592-018-0002-6.
Jain M, Olsen HE, Paten B, Akeson M. The Oxford nanopore MinION: delivery of nanopore sequencing to the genomics community (vol 17, 239, 2016). Genome Biol. 2016;17:256. https://doi.org/10.1186/s13059-016-1122-x.
Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, et al. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods. 2010;7(6):461–U72. https://doi.org/10.1038/Nmeth.1459.
Ardui S, Ameur A, Vermeesch JR, Hestand MS. Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic Acids Res. 2018;46(5):2159–68. https://doi.org/10.1093/narlgkx066.
Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, et al. Real-Time DNA Sequencing from single polymerase molecules. Science. 2009;323(5910):133–8. https://doi.org/10.1126/science.1162986.
van Dijk EL, Jaszczyszyn Y, Naquin D, Thermes C. The third revolution in sequencing technology. Trends Genet. 2018;34(9):666–81. https://doi.org/10.1016/j.tig.2018.05.008.
Russo G, Patrignani A, Poveda L, Hoehn F, Scholtka B, Schlapbach R, et al. Highly sensitive, non-invasive detection of colorectal cancer mutations using single molecule, third generation sequencing. Appl Transl Genom. 2015;7:32–9. https://doi.org/10.1016/j.atg.2015.08.006.
Clarke J, Wu HC, Jayasinghe L, Patel A, Reid S, Bayley H. Continuous base identification for single-molecule nanopore DNA sequencing. Nat Nanotechnol. 2009;4(4):265–70. https://doi.org/10.1038/Nnano.2009.12.
Madoui MA, Engelen S, Cruaud C, Belser C, Bertrand L, Alberti A, et al. Genome assembly using nanopore-guided long and error-free DNA reads. BMC Genomics. 2015;16:327. https://doi.org/10.1186/s12864-015-1519-z.
Jain M, Tyson JR, Loose M, CLC I, Eccles DA, O’Grady J, et al. MinION analysis and reference consortium: phase 2 data release and analysis of R9.0 chemistry. F1000Res. 2017;6:760. https://doi.org/10.12688/f1000research.11354.1.
Mantere T, Kersten S, Hoischen A. Long-read sequencing emerging in medical genetics. Front Genet. 2019;10:426. https://doi.org/10.3389/fgene.2019.00426.
Teng HT, Cao MD, Hall MB, Duarte T, Wang S, Coin LJM. Chiron: translating nanopore raw signal directly into nucleotide sequence using deep learning. Gigascience. 2018;7(5). https://doi.org/10.1093/gigascience/giy037.
Wenger AM, Peluso P, Rowell WJ, Chang P-C, Hall RJ, Concepcion GT et al. Highly-accurate long-read sequencing improves variant detection and assembly of a human genome. bioRxiv. 2019:519025. https://doi.org/10.1101/519025.
Tie J, Wang YX, Tomasetti C, Li L, Springer S, Kinde I et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8(346). https://doi.org/10.1126/scitranslmed.aaf6219.
Reinert T, Scholer LV, Thomsen R, Tobiasen H, Vang SR, Nordentoft I, et al. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut. 2016;65(4):625–34. https://doi.org/10.1136/gutjnl-2014-308859.
Scholer LV, Reinert T, Orntoft MBW, Kassentoft CG, Arnadottir SS, Vang S, et al. Clinical implications of monitoring circulating tumor DNA in patients with colorectal cancer. Clin Cancer Res. 2017;23(18):5437–45. https://doi.org/10.1158/1078-0432.Ccr-17-0510.
Olsson E, Winter C, George A, Chen Y, Howlin J, Tang MH, et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Mol Med. 2015;7(8):1034–47. https://doi.org/10.15252/emmm.201404913.
TCGA Research Network. The Cancer Genome Atlas. http://cancergenome.nih.gov/. Accessed Feb 22 2019
Ashworth TR. A case of cancer in which cells similar to those in the tumors were seen in the blood after death. Australas Med J. 1869;14:146–9.
Cima I, Kong SL, Sengupta D, Tan IB, Phyo WM, Lee D, et al. Tumor-derived circulating endothelial cell clusters in colorectal cancer. Sci Transl Med. 2016;8(345):345ra89. https://doi.org/10.1126/scitranslmed.aad7369.
Alix-Panabieres C, Pantel K. Clinical applications of circulating tumor cells and circulating tumor DNA as liquid biopsy. Cancer Discov. 2016;6(5):479–91. https://doi.org/10.1158/2159-8290.Cd-15-1483.
Chen LC, Bode AM, Dong ZG. Circulating tumor cells: moving biological insights into detection. Theranostics. 2017;7(10):2606–19. https://doi.org/10.7150/thno.18588.
Rhim AD, Mirek ET, Aiello NM, Maitra A, Bailey JM, McAllister F, et al. EMT and dissemination precede pancreatic tumor formation. Cell. 2012;148(1-2):349–61. https://doi.org/10.1016/j.cell.2011.11.025.
Camara O, Kavallaris A, Noschel H, Rengsberger M, Jorke C, Pachmann K. Seeding of epithelial cells into circulation during surgery for breast cancer: the fate of malignant and benign mobilized cells. World J Surg Oncol. 2006;4:67. https://doi.org/10.1186/1477-7819-4-67.
Allard WJ, Matera J, Miller MC, Repollet M, Connelly MC, Rao C, et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin Cancer Res. 2004;10(20):6897–904. https://doi.org/10.1158/1078-0432.Ccr-04-0378.
Stott SL, Lee RJ, Nagrath S, Yu M, Miyamoto DT, Ulkus L, et al. Isolation and characterization of circulating tumor cells from patients with localized and metastatic prostate cancer. Sci Transl Med. 2010;2(25):25ra23. https://doi.org/10.1126/scitranslmed.3000403.
Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. New Engl J Med. 2004;351(8):781–91. https://doi.org/10.1056/NEJMoa040766.
Lu SH, Tsai WS, Chang YH, Chou TY, Pang ST, Lin PH, et al. Identifying cancer origin using circulating tumor cells. Cancer Biol Ther. 2016;17(4):430–8. https://doi.org/10.1080/15384047.2016.1141839.
Hou Y, Guo HH, Cao C, Li XL, Hu BQ, Zhu P, et al. Single-cell triple omics sequencing reveals genetic, epigenetic, and transcriptomic heterogeneity in hepatocellular carcinomas. Cell Res. 2016;26(3):304–19. https://doi.org/10.1038/cr.2016.23.
D'Avola D, Villacorta-Martin C, Martins SN, Craig A, Labgaa I, von Felden J, et al. High-density single cell mRNA sequencing to characterize circulating tumor cells in hepatocellular carcinoma. Sci Rep. 2018;8. https://doi.org/10.1038/s41598-018-30047-y.
Alix-Panabieres C, Pantel K. Challenges in circulating tumour cell research. Nat Rev Cancer. 2014;14(9):623–31. https://doi.org/10.1038/nrc3820.
Yokobori T, Iinuma H, Shimamura T, Imoto S, Sugimachi K, Ishii H, et al. Plastin3 Is a novel marker for circulating tumor cells undergoing the epithelial-mesenchymal transition and is associated with colorectal cancer prognosis. Cancer Res. 2013;73(7):2059–69. https://doi.org/10.1158/0008-5472.Can-12-0326.
Pecot CV, Bischoff FZ, Mayer JA, Wong KL, Pham T, Bottsford-Miller J, et al. A novel platform for detection of CK+ and CK- CTCs. Cancer Discov. 2011;1(7):580–6. https://doi.org/10.1158/2159-8290.Cd-11-0215.
Vona G, Sabile A, Louha M, Sitruk V, Romana S, Schutze K, et al. Isolation by size of epithelial tumor cells - a new method for the immunomorphological and molecular characterization of circulating tumor cells. Am J Pathol. 2000;156(1):57–63. https://doi.org/10.1016/S0002-9440(10)64706-2.
Sollier E, Go DE, Che J, Gossett DR, O'Byrne S, Weaver WM, et al. Size-selective collection of circulating tumor cells using Vortex technology. Lab Chip. 2014;14(1):63–77. https://doi.org/10.1039/c3lc50689d.
Polzer B, Medoro G, Pasch S, Fontana F, Zorzino L, Pestka A, et al. Molecular profiling of single circulating tumor cells with diagnostic intention. Embo Mol Med. 2014;6(11):1371–86. https://doi.org/10.15252/emmm.201404033.
Shim S, Stemke-Hale K, Tsimberidou AM, Noshari J, Anderson TE, Gascoyne PRC. Antibody-independent isolation of circulating tumor cells by continuous-flow dielectrophoresis. Biomicrofluidics. 2013;7(1):011807. https://doi.org/10.1063/1.4774304.
Chen JF, Ho H, Lichterman J, Lu YT, Zhang Y, Garcia MA, et al. Subclassification of prostate cancer circulating tumor cells by nuclear size reveals very small nuclear circulating tumor cells in patients with visceral metastases. Cancer. 2015;121(18):3240–51. https://doi.org/10.1002/cncr.29455.
Hayes DF, Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Miller MC, et al. Circulating tumor cells at each follow-up time point during therapy of metastatic breast cancer patients predict progression-free and overall survival. Clin Cancer Res. 2006;12(14):4218–24. https://doi.org/10.1158/1078-0432.Ccr-05-2821.
Cohen SJ, Punt CJA, Iannotti N, Saidman BH, Sabbath KD, Gabrail NY, et al. Relationship of circulating tumor cells to tumor response, progression-free survival, and overall survival in patients with metastatic colorectal cancer. J Clin Oncol. 2008;26(19):3213–21. https://doi.org/10.1200/Jco.2007.15.8923.
Danila DC, Heller G, Gignac GA, Gonzalez-Espinoza R, Anand A, Tanaka E, et al. Circulating tumor cell number and prognosis in progressive castration-resistant prostate cancer. Clin Cancer Res. 2007;13(23):7053–8. https://doi.org/10.1158/1078-0432.CCR-07-1506.
Liu HE, Triboulet M, Zia A, Vuppalapaty M, Kidess-Sigal E, Coller J, et al. Workflow optimization of whole genome amplification and targeted panel sequencing for CTC mutation detection. Npj Genom Med. 2017;2:34. https://doi.org/10.1038/s41525-017-0034-3.
Zhu ZY, Qiu S, Shao K, Hou Y. Progress and challenges of sequencing and analyzing circulating tumor cells. Cell Biol Toxicol. 2018;34(5):405–15. https://doi.org/10.1007/s10565-017-9418-5.
Ni XH, Zhuo ML, Su Z, Duan JC, Gao Y, Wang ZJ, et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. P Natl Acad Sci USA. 2013;110(52):21083–8. https://doi.org/10.1073/pnas.1320659110.
Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM, et al. Complex tumor genomes inferred from single circulating tumor cells by array-CGH and next-generation sequencing. Cancer Res. 2013;73(10):2965–75. https://doi.org/10.1158/0008-5472.Can-12-4140.
Gulbahce N, Magbanua MJM, Chin R, Agarwal MR, Luo XH, Liu J, et al. Quantitative whole genome sequencing of circulating tumor cells enables personalized combination therapy of metastatic cancer. Cancer Res. 2017;77(16):4530–41. https://doi.org/10.1158/0008-5472.Can-17-0688.
Lohr JG, Adalsteinsson VA, Cibulskis K, Choudhury AD, Rosenberg M, Cruz-Gordillo P, et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nat Biotechnol. 2014;32(5):479–84. https://doi.org/10.1038/nbt.2892.
Mastoraki S, Strati A, Tzanikou E, Chimonidou M, Politaki E, Voutsina A, et al. ESR1 methylation: a liquid biopsy-based epigenetic assay for the follow-up of patients with metastatic breast cancer receiving endocrine treatment. Clin Cancer Res. 2018;24(6):1500–10. https://doi.org/10.1158/1078-0432.Ccr-17-1181.
Pixberg CF, Raba K, Muller F, Behrens B, Honisch E, Niederacher D, et al. Analysis of DNA methylation in single circulating tumor cells. Oncogene. 2017;36(23):3223–31. https://doi.org/10.1038/onc.2016.480.
Ramskold D, Luo SJ, Wang YC, Li R, Deng QL, Faridani OR, et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat Biotechnol. 2012;30(8):777–82. https://doi.org/10.1038/nbt.2282.
Yu M, Ting DT, Stott SL, Wittner BS, Ozsolak F, Paul S, et al. RNA sequencing of pancreatic circulating tumour cells implicates WNT signalling in metastasis. Nature. 2012;487(7408):510–U130. https://doi.org/10.1038/nature11217.
Lang JE, Ring A, Porras T, Kaur P, Forte V, Tripathy D, et al. RNA Seq of circulating tumor cells in stage II-III breast cancer. Ann Surg Oncol. 2018;25(8):2261–70.
Hughes AJ, Herr AE. Microfluidic Western blotting. P Natl Acad Sci USA. 2012;109(52):21450–5. https://doi.org/10.1073/pnas.1207754110.
Sinkala E, Sollier-Christen E, Renier C, Rosas-Canyelles E, Che J, Heirich K, et al. Profiling protein expression in circulating tumour cells using microfluidic western blotting. Nat Commun. 2017;8:14622. https://doi.org/10.1038/ncomms14622.
Deng YL, Zhang Y, Sun S, Wang ZH, Wang MJ, Yu BQ, et al. An integrated microfluidic chip system for single-cell secretion profiling of rare circulating tumor cells. Sci Rep. 2014;4. https://doi.org/10.1038/srep07499.
Zhang Y, Tang Y, Sun S, Wang ZH, Wu WJ, Zhao XD, et al. Single-cell codetection of metabolic activity, intracellular functional proteins, and genetic mutations from rare circulating tumor cells. Anal Chem. 2015;87(19):9761–8. https://doi.org/10.1021/acs.analchem.5b01901.
de Wit S, van Dalum G, Lenferink ATM, Tibbe AGJ, Hiltermann TJN, Groen HJM, et al. The detection of EpCAM(+) and EpCAM(-) circulating tumor cells. Sci Rep. 2015;5:12270. https://doi.org/10.1038/srep12270.
Miller MC, Doyle GV, Terstappen LW. Significance of circulating tumor cells detected by the cellsearch system in patients with metastatic breast colorectal and prostate cancer. J Oncol. 2010;2010:617421. https://doi.org/10.1155/2010/617421.
Cabel L, Proudhon C, Gortais H, Loirat D, Coussy F, Pierga JY, et al. Circulating tumor cells: clinical validity and utility. Int J Clin Oncol. 2017;22(3):421–30. https://doi.org/10.1007/s10147-017-1105-2.
Fehm T, Muller V, Aktas B, Janni W, Schneeweiss A, Stickeler E, et al. HER2 status of circulating tumor cells in patients with metastatic breast cancer: a prospective, multicenter trial. Breast Cancer Res Tr. 2010;124(2):403–12. https://doi.org/10.1007/s10549-010-1163-x.
Helissey C, Berger F, Cottu P, Dieras V, Mignot L, Servois V, et al. Circulating tumor cell thresholds and survival scores in advanced metastatic breast cancer: the observational step of the CirCe01 phase III trial. Cancer Lett. 2015;360(2):213–8. https://doi.org/10.1016/j.canlet.2015.02.010.
Niculescu-Duvaz I. Trastuzumab emtansine, an antibody-drug conjugate for the treatment of HER2 + metastatic breast cancer. Curr Opin Mol Ther. 2010;12(3):350–60.
Berger F, Bidard FC, Pierga JY, Sablin MP, Cottu P, Neffati S et al. 117PAnti-HER2 therapy efficacy in HER2-negative metastatic breast cancer with HER2-amplified circulating tumor cells: results of the CirCe T-DM1 trial. Annals of Oncology. 2017;28(suppl_5). https://doi.org/10.1093/annonc/mdx363.033.
Tsai WS, Chen JS, Shao HJ, Wu JC, Lai JM, Lu SH, et al. Circulating tumor cell count correlates with colorectal neoplasm progression and is a prognostic marker for distant metastasis in non-metastatic patients. Sci Rep. 2016;6:24517. https://doi.org/10.1038/srep24517.
Scher HI, Lu D, Schreiber NA. Association of AR-V7 on circulating tumor cells as a treatment-specific biomarker with outcomes and survival in castration-resistant prostate cancer. Jama Oncology. 2016;2(11):1441–9.
Scher HI, Graf RP, Schreiber NA, McLaughlin B, Lu D, Louw J, et al. Nuclear-specific AR-V7 protein localization is necessary to guide treatment selection in metastatic castration-resistant prostate cancer. Eur Urol. 2017;71(6):874–82. https://doi.org/10.1016/j.eururo.2016.11.024.
Scher HI, Graf RP, Schreiber NA, Winquist E, McLaughlin B, Lu D, et al. Validation of nuclear-localized AR-V7 on circulating tumor cells (CTC) as a treatment-selection biomarker for managing metastatic castration-resistant prostate cancer (mCRPC). J Clin Oncol. 2018;36(6):273. https://doi.org/10.1200/JCO.2018.36.6_suppl.273.
Dawson MA. The cancer epigenome: concepts, challenges, and therapeutic opportunities. Science. 2017;355(6330):1147–52. https://doi.org/10.1126/science.aam7304.
Ushijima T. Innovation - Detection and interpretation of altered methylation patterns in cancer cells. Nat Rev Cancer. 2005;5(3):223–31. https://doi.org/10.1038/nrc1571.
Suzuki H, Watkins DN, Jair KW, Schuebel KE, Markowitz SD, Chen WD, et al. Epigenetic inactivation of SFRP genes allows constitutive WNT signaling in colorectal cancer. Nat Genet. 2004;36(4):417–22. https://doi.org/10.1038/ng1330.
Chen WY, Zeng XB, Carter MG, Morrell CN, Yen RWC, Esteller M, et al. Heterozygous disruption of Hic1 predisposes mice to a gender-dependent spectrum of malignant tumors. Nat Genet. 2003;33(2):197–202. https://doi.org/10.1038/ng1077.
Chan KCA, Jiang PY, Chan CWM, Sun K, Wong J, Hui EP, et al. Noninvasive detection of cancer-associated genome-wide hypomethylation and copy number aberrations by plasma DNA bisulfite sequencing. P Natl Acad Sci USA. 2013;110(47):18761–8. https://doi.org/10.1073/pnas.1313995110.
Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301(5895):89–92. https://doi.org/10.1038/301089a0.
Ross JP, Rand KN, Molloy PL. Hypomethylation of repeated DNA sequences in cancer. Epigenomics. 2010;2(2):245–69. https://doi.org/10.2217/Epi.10.2.
Eden A, Gaudet F, Waghmare A, Jaenisch R. Chromosomal instability and tumors promoted by DNA hypomethylation. Science. 2003;300(5618):455. https://doi.org/10.1126/science.1083557.
Fenaux P, Mufti GJ, Hellstrom-Lindberg E, Santini V, Finelli C, Giagounidis A, et al. Efficacy of azacitidine compared with that of conventional care regimens in the treatment of higher-risk myelodysplastic syndromes: a randomised, open-label, phase III study. Lancet Oncol. 2009;10(3):223–32. https://doi.org/10.1016/S1470-2045(09)70003-8.
Lubbert M, Suciu S, Hagemeijer A, Ruter B, Platzbecker U, Giagounidis A, et al. Decitabine improves progression-free survival in older high-risk MDS patients with multiple autosomal monosomies: results of a subgroup analysis of the randomized phase III study 06011 of the EORTC Leukemia Cooperative Group and German MDS Study Group. Ann Hematol. 2016;95(2):191–9. https://doi.org/10.1007/s00277-015-2547-0.
San-Miguel JF, Hungria VTM, Yoon SS, Beksac M, Dimopoulos MA, Elghandour A, et al. Panobinostat plus bortezomib and dexamethasone versus placebo plus bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma: a multicentre, randomised, double-blind phase 3 trial. Lancet Oncol. 2014;15(11):1195–206. https://doi.org/10.1016/S1470-2045(14)70440-1.
Fardi M, Solali S, Hagh MF. Epigenetic mechanisms as a new approach in cancer treatment: an updated review. Genes Dis. 2018;5(4):304–11. https://doi.org/10.1016/j.gendis.2018.06.003.
Hao XK, Luo HY, Krawczyk M, Wei W, Wang WQ, Wang J, et al. DNA methylation markers for diagnosis and prognosis of common cancers. P Natl Acad Sci USA. 2017;114(28):7414–9. https://doi.org/10.1073/pnas.1703577114.
Wong IHN, Lo YMD, Zhang J, Liew CT, Ng MHL, Wong N, et al. Detection of aberrant p16 methylation in the plasma and serum of liver cancer patients. Cancer Res. 1999;59(1):71–3.
Gormally E, Caboux E, Vineis P, Hainaut P. Circulating free DNA in plasma or serum as biomarker of carcinogenesis: practical aspects and biological significance. Mutat Res-Rev Mutat. 2007;635(2-3):105–17. https://doi.org/10.1016/j.mrrev.2006.11.002.
Zhai RH, Zhao Y, Su L, Cassidy L, Liu G, Christiani DC. Genome-wide DNA methylation profiling of cell-free serum DNA in esophageal adenocarcinoma and Barrett esophagus. Neoplasia. 2012;14(1):29–U39. https://doi.org/10.1593/neo.111626.
Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518(7539):317–30. https://doi.org/10.1038/nature14248.
Sun K, Jiang PY, Chan KCA, Wong J, Cheng YKY, Liang RHS, et al. Plasma DNA tissue mapping by genome-wide methylation sequencing for noninvasive prenatal, cancer, and transplantation assessments. P Natl Acad Sci USA. 2015;112(40):E5503–E12. https://doi.org/10.1073/pnas.1508736112.
Moss J, Magenheim J, Neiman D, Zemmour H, Loyfer N, Korach A, et al. Comprehensive human cell-type methylation atlas reveals origins of circulating cell-free DNA in health and disease. Nat Commun. 2018;9(1):5068. https://doi.org/10.1038/s41467-018-07466-6.
Hoque MO, Topaloglu O, Begum S, Henrique R, Rosenbaum E, Van Criekinge W, et al. Quantitative methylation-specific polymerase chain reaction gene patterns in urine sediment distinguish prostate cancer patients from control subjects. J Clin Oncol. 2005;23(27):6569–75. https://doi.org/10.1200/Jco.2005.07.009.
Chimonidou M, Strati A, Malamos N, Georgoulias V, Lianidou ES. SOX17 Promoter methylation in circulating tumor cells and matched cell-free DNA isolated from plasma of patients with breast cancer. Clin Chem. 2013;59(1):270–9. https://doi.org/10.1373/clinchem.2012.191551.
Barault L, Amatu A, Bleeker FE, Moutinho C, Falcomata C, Fiano V, et al. Digital PCR quantification of MGMT methylation refines prediction of clinical benefit from alkylating agents in glioblastoma and metastatic colorectal cancer. Ann Oncol. 2015;26(9):1994–9. https://doi.org/10.1093/annonc/mdv272.
Johnson DA, Barclay RL, Mergener K, Weiss G, Konig T, Beck J, et al. Plasma septin9 versus fecal immunochemical testing for colorectal cancer screening: a prospective multicenter study. PLoS One. 2014;9(6):e98238. https://doi.org/10.1371/journal.pone.0098238.
Imperiale TF, Ransohoff DF, Itzkowitz SH, Levin TR, Lavin P, Lidgard GP, et al. Multitarget stool DNA testing for colorectal-cancer screening. New Engl J Med. 2014;370(14):1287–97. https://doi.org/10.1056/NEJMoa1311194.
Gai WX, Sun K. Epigenetic biomarkers in cell-free DNA and applications in liquid biopsy. Genes-Basel. 2019;10(1):E32. https://doi.org/10.3390/genes10010032.
Mazor T, Pankov A, Song JS, Costello JF. Intratumoral heterogeneity of the epigenome. Cancer Cell. 2016;29(4):440–51. https://doi.org/10.1016/j.ccell.2016.03.009.
Gordevicius J, Krisciunas A, Groot DE, Yip SM, Susic M, Kwan A, et al. Cell-Free DNA Modification dynamics in abiraterone acetate-treated prostate cancer patients. Clin Cancer Res. 2018;24(14):3317–24. https://doi.org/10.1158/1078-0432.Ccr-18-0101.
Stieglitz E, Mazor T, Olshen AB, Geng HM, Gelston LC, Akutagawa J, et al. Genome-wide DNA methylation is predictive of outcome in juvenile myelomonocytic leukemia. Nat Commun. 2017;8:2127. https://doi.org/10.1038/s41467-017-02178-9.
Wen L, Li JY, Guo HH, Liu XM, Zheng SM, Zhang DF, et al. Genome-scale detection of hypermethylated CpG islands in circulating cell-free DNA of hepatocellular carcinoma patients (vol 25, pg 1250, 2015). Cell Res. 2015;25(12):1376. https://doi.org/10.1038/cr.2015.141.
Grunau C, Clark SJ, Rosenthal A. Bisulfite genomic sequencing: systematic investigation of critical experimental parameters. Nucleic Acids Res. 2001;29(13):E65–5.
Shen SY, Singhania R, Fehringer G, Chakravarthy A, Roehrl MHA, Chadwick D, et al. Sensitive tumour detection and classification using plasma cell-free DNA methylomes. Nature. 2018;563(7732):579–83. https://doi.org/10.1038/s41586-018-0703-0.
Taiwo O, Wilson GA, Morris T, Seisenberger S, Reik W, Pearce D, et al. Methylome analysis using MeDIP-seq with low DNA concentrations. Nat Protoc. 2012;7(4):617–36. https://doi.org/10.1038/nprot.2012.012.
Buscarlet M, Tessier A, Provost S, Mollica L, Busque L. Human blood cell levels of 5-hydroxymethylcytosine (5hmC) decline with age, partly related to acquired mutations in TET2. Exp Hematol. 2016;44(11):1072–84. https://doi.org/10.1016/j.exphem.2016.07.009.
Li WS, Zhang X, Lu XY, You L, Song YQ, Luo ZG, et al. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res. 2017;27(10):1243–57. https://doi.org/10.1038/cr.2017.121.
Pfeifer GP, Xiong WY, Hahn MA, Jin SG. The role of 5-hydroxymethylcytosine in human cancer. Cell Tissue Res. 2014;356(3):631–41. https://doi.org/10.1007/s00441-014-1896-7.
Schutsky EK, DeNizio JE, Hu P, Liu MY, Nabel CS, Fabyanic EB, et al. Nondestructive, base-resolution sequencing of 5-hydroxymethylcytosine using a DNA deaminase. Nat Biotechnol. 2018;36(11):1083–90. https://doi.org/10.1038/nbt.4204.
Sina AA, Carrascosa LG, Liang Z, Grewal YS, Wardiana A, Shiddiky MJA, et al. Epigenetically reprogrammed methylation landscape drives the DNA self-assembly and serves as a universal cancer biomarker. Nat Commun. 2018;9(1):4915. https://doi.org/10.1038/s41467-018-07214-w.
Liles EG, Coronado GD, Perrin N, Harte AH, Nungesser R, Quigley N, et al. Uptake of a colorectal cancer screening blood test is higher than of a fecal test offered in clinic: a randomized trial. Cancer Treat Res Commun. 2017;10:27–31. https://doi.org/10.1016/j.ctarc.2016.12.004.
Witjes JA, Morote J, Cornel EB, Gakis G, Valenberg FJPV, Lozano F, et al. Performance of the Bladder EpiCheck™ methylation test for patients under surveillance for non–muscle-invasive bladder cancer: results of a multicenter, prospective, blinded clinical trial. Eur Urol Oncol. 2018;1(4):307–13. https://doi.org/10.1016/j.euo.2018.06.011.
Lee RC, Feinbaum RL, Ambros V. The C-elegans heterochronic gene Lin-4 encodes small Rnas with antisense complementarity to Lin-14. Cell. 1993;75(5):843–54. https://doi.org/10.1016/0092-8674(93)90529-Y.
Li XZG, Roy CK, Moore MJ, Zamore PD. Defining piRNA primary transcripts. Cell Cycle. 2013;12(11):1657–8. https://doi.org/10.4161/cc.24989.
Yamasaki S, Ivanov P, Hu GF, Anderson P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J Cell Biol. 2009;185(1):35–42. https://doi.org/10.1083/jcb.200811106.
Tyc K, Steitz JA. U3, U8 and U13 comprise a new class of mammalian Snrnps localized in the cell nucleolus. EMBO J. 1989;8(10):3113–9. https://doi.org/10.1002/j.1460-2075.1989.tb08463.x.
Lerner MR, Boyle JA, Hardin JA, Steitz JA. Two novel classes of small ribonucleoproteins detected by antibodies associated with lupus erythematosus. Science. 1981;211(4480):400–2. https://doi.org/10.1126/science.6164096.
Kapranov P, Cheng J, Dike S, Nix DA, Duttagupta R, Willingham AT, et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science. 2007;316(5830):1484–8. https://doi.org/10.1126/science.1138341.
Seila AC, Calabrese JM, Levine SS, Yeo GW, Rahl PB, Flynn RA, et al. Divergent transcription from active promoters. Science. 2008;322(5909):1849–51. https://doi.org/10.1126/science.1162253.
Hadjiolo AA, Venkov PV, Tsanev RG. Ribonucleic acids fractionation by density-gradient centrifugation and by agar gel electrophoresis - a comparison. Anal Biochem. 1966;17(2):263–7. https://doi.org/10.1016/0003-2697(66)90204-1.
Clark MB, Choudhary A, Smith MA, Taft RJ, Mattick JS. The dark matter rises: the expanding world of regulatory RNAs. Essays Biochem. 2013;54:1–16. https://doi.org/10.1042/Bse0540001.
Morris KV, Mattick JS. The rise of regulatory RNA. Nat Rev Genet. 2014;15(6):423–37. https://doi.org/10.1038/nrg3722.
Cieslik M, Chinnaiyan AM. Cancer transcriptome profiling at the juncture of clinical translation. Nat Rev Genet. 2018;19(2):93–109. https://doi.org/10.1038/nrg.2017.96.
Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The genotype-tissue expression (GTEx) project. Nat Genet. 2013;45(6):580–5. https://doi.org/10.1038/ng.2653.
Sheng QH, Zhao SL, Li CI, Shyr Y, Guo Y. Practicability of detecting somatic point mutation from RNA high throughput sequencing data. Genomics. 2016;107(5):163–9. https://doi.org/10.1016/j.ygeno.2016.03.006.
Piskol R, Ramaswami G, Li JB. Reliable identification of genomic variants from RNA-Seq data. Am J Hum Genet. 2013;93(4):641–51. https://doi.org/10.1016/j.ajhg.2013.08.008.
Mertens F, Johansson B, Fioretos T, Mitelman F. The emerging complexity of gene fusions in cancer. Nat Rev Cancer. 2015;15(6):371–81. https://doi.org/10.1038/nrc3947.
Holoch D, Moazed D. RNA-mediated epigenetic regulation of gene expression. Nat Rev Genet. 2015;16(2):71–84. https://doi.org/10.1038/nrg3863.
Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–74. https://doi.org/10.1038/nrg3074.
Zhou KC, Liu MX, Cao Y. New insight into microRNA functions in cancer: oncogene-microRNA-tumor suppressor gene network. Front Mol Biosci. 2017;4:46. https://doi.org/10.3389/fmolb.2017.00046.
Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6(11):857–66. https://doi.org/10.1038/nrc1997.
Rosenfeld N, Aharonov R, Meiri E, Rosenwald S, Spector Y, Zepeniuk M, et al. MicroRNAs accurately identify cancer tissue origin. Nat Biotechnol. 2008;26(4):462–9. https://doi.org/10.1038/nbt1392.
Anfossi S, Babayan A, Pantel K, Calin GA. Clinical utility of circulating non-coding RNAs - an update. Nat Rev Clin Oncol. 2018;15(9):541–63. https://doi.org/10.1038/s41571-018-0035-x.
Lo KW, Lo YM, Leung SF, Tsang YS, Chan LY, Johnson PJ, et al. Analysis of cell-free Epstein-Barr virus associated RNA in the plasma of patients with nasopharyngeal carcinoma. Clin Chem. 1999;45(8 Pt 1):1292–4.
Lawrie CH, Gal S, Dunlop HM, Pushkaran B, Liggins AP, Pulford K, et al. Detection of elevated levels of tumour-associated microRNAs in serum of patients with diffuse large B-cell lymphoma. Brit J Haematol. 2008;141(5):672–5. https://doi.org/10.1111/j.1365-2141.2008.07077.x.
Sole C, Arnaiz E, Manterola L, Otaegui D, Lawrie CH. The circulating transcriptome as a source of cancer liquid biopsy biomarkers. Semin Cancer Biol. 2019. https://doi.org/10.1016/j.semcancer.2019.01.003.
Sole C, Tramonti D, Schramm M, Goicoechea I, Armesto M, Hernandez LI, et al. The circulating transcriptome as a source of biomarkers for melanoma. Cancers (Basel). 2019;11(1):E70. https://doi.org/10.3390/cancers11010070.
Fernandez-Mercado M, Manterola L, Larrea E, Goicoechea I, Arestin M, Armesto M, et al. The circulating transcriptome as a source of non-invasive cancer biomarkers: concepts and controversies of non-coding and coding RNA in body fluids. J Cell Mol Med. 2015;19(10):2307–23. https://doi.org/10.1111/jcmm.12625.
Shen J, Liu ZL, Todd NW, Zhang H, Liao JP, Yu L, et al. Diagnosis of lung cancer in individuals with solitary pulmonary nodules by plasma microRNA biomarkers. BMC Cancer. 2011;11:374. https://doi.org/10.1186/1471-2407-11-374.
Baraniskin A, Nopel-Dunnebacke S, Ahrens M, Jensen SG, Zollner H, Maghnouj A, et al. Circulating U2 small nuclear RNA fragments as a novel diagnostic biomarker for pancreatic and colorectal adenocarcinoma. Int J Cancer. 2013;132(2):E48–57. https://doi.org/10.1002/ijc.27791.
Ono S, Lam S, Nagahara M, Hoon DSB. Circulating microRNA biomarkers as liquid biopsy for cancer patients: pros and cons of current assays. J Clin Med. 2015;4(10):1890–907. https://doi.org/10.3390/jcm4101890.
Zhang H, Mao F, Shen T, Luo Q, Ding Z, Qian L, et al. Plasma miR-145, miR-20a, miR-21 and miR-223 as novel biomarkers for screening early-stage non-small cell lung cancer. Oncol Lett. 2017;13(2):669–76. https://doi.org/10.3892/ol.2016.5462.
Geng Q, Fan T, Zhang BY, Wang W, Xu Y, Hu H. Five microRNAs in plasma as novel biomarkers for screening of early-stage non-small cell lung cancer. Respir Res. 2014;15:149. https://doi.org/10.1186/s12931-014-0149-3.
Zhu WY, Zhou KY, Zha Y, Chen DD, He JY, Ma HJ, et al. Diagnostic value of serum miR-182, miR-183, miR-210, and miR-126 levels in patients with early-stage non-small cell lung cancer. PLoS One. 2016;11(4):e0153046. https://doi.org/10.1371/journal.pone.0153046.
Powrozek T, Kuznar-Kaminska B, Dziedzic M, Mlak R, Batura-Gabryel H, Sagan D, et al. The diagnostic role of plasma circulating precursors of miRNA-944 and miRNA-3662 for non-small cell lung cancer detection. Pathol Res Pract. 2017;213(11):1384–7. https://doi.org/10.1016/j.prp.2017.09.011.
Shin VY, Siu JM, Cheuk I, Ng EKO, Kwong A. Circulating cell-free miRNAs as biomarker for triple-negative breast cancer. Br J Cancer. 2015;112(11):1751–9. https://doi.org/10.1038/bjc.2015.143.
Pang PC, Shi XY, Huang WL, Sun K. miR-497 as a potential serum biomarker for the diagnosis and prognosis of osteosarcoma. Eur Rev Med Pharmacol Sci. 2016;20(18):3765–9.
Fang ZQ, Dai W, Wang XW, Chen W, Shen CX, Ye G, et al. Circulating miR-205: a promising biomarker for the detection and prognosis evaluation of bladder cancer. Tumor Biol. 2016;37(6):8075–82. https://doi.org/10.1007/s13277-015-4698-y.
Gong L, Wang CJ, Gao Y, Wang J. Decreased expression of microRNA-148a predicts poor prognosis in ovarian cancer and associates with tumor growth and metastasis. Biomed Pharmacother. 2016;83:58–63. https://doi.org/10.1016/j.biopha.2016.05.049.
Shen J, Kong WW, Wu YN, Ren HZ, Wei J, Yang Y, et al. Plasma mRNA as liquid biopsy predicts chemo-sensitivity in advanced gastric cancer patients. J Cancer. 2017;8(3):434–42. https://doi.org/10.7150/jca.17369.
Shen J, Wang H, Wei J, Yu LX, Xie L, Qian XP, et al. Thymidylate synthase mRNA levels in plasma and tumor as potential predictive biomarkers for raltitrexed sensitivity in gastric cancer. Int J Cancer. 2012;131(6):E938–E45. https://doi.org/10.1002/ijc.27530.
Shen J, Wei J, Guan WX, Wang H, Ding YT, Qian XP, et al. Plasma mRNA expression levels of BRCA1 and TS as potential predictive biomarkers for chemotherapy in gastric cancer. J Transl Med. 2014;12:355. https://doi.org/10.1186/s12967-014-0355-2.
Kang Y, Zhang JC, Sun PC, Shang J. Circulating cell-free human telomerase reverse transcriptase mRNA in plasma and its potential diagnostic and prognostic value for gastric cancer. Int J Clin Oncol. 2013;18(3):478–86. https://doi.org/10.1007/s10147-012-0405-9.
March-Villalba JA, Martinez-Jabaloyas JM, Herrero MJ, Santamaria J, Alino SF, Dasi F. Cell-free circulating plasma hTERT mRNA is a useful marker for prostate cancer diagnosis and is associated with poor prognosis tumor characteristics. PLoS One. 2012;7(8):e43470. https://doi.org/10.1371/journal.pone.0043470.
Miura N, Maeda Y, Kanbe T, Yazama H, Takeda Y, Sato R, et al. Serum human telomerase reverse transcriptase messenger RNA as a novel tumor marker for hepatocellular carcinoma. Clin Cancer Res. 2005;11(9):3205–9. https://doi.org/10.1158/1078-0432.Ccr-04-1487.
Terrin L, Rampazzo E, Pucciarelli S, Agostini M, Bertorelle R, Esposito G, et al. Relationship between tumor and plasma levels of hTERT mRNA in patients with colorectal cancer: implications for monitoring of neoplastic disease. Clin Cancer Res. 2008;14(22):7444–51. https://doi.org/10.1158/1078-0432.Ccr-08-0478.
Chen XQ, Bonnefoi H, Pelte MF, Lyautey J, Lederrey C, Movarekhi S, et al. Telomerase RNA as a detection marker in the serum of breast cancer patients. Clin Cancer Res. 2000;6(10):3823–6.
Pritchard CC, Kroh E, Wood B, Arroyo JD, Dougherty KJ, Miyaji MM, et al. Blood cell origin of circulating MicroRNAs: a cautionary note for cancer biomarker studies. Cancer Prev Res. 2012;5(3):492–7. https://doi.org/10.1158/1940-6207.Capr-11-0370.
Kirschner MB, Kao SC, Edelman JJ, Armstrong NJ, Vallely MP, van Zandwijk N, et al. Haemolysis during sample preparation alters microRNA content of plasma. PLoS One. 2011;6(9):e24145. https://doi.org/10.1371/journal.pone.0024145.
Jarry J, Schadendorf D, Greenwood C, Spatz A, van Kempen LC. The validity of circulating microRNAs in oncology: five years of challenges and contradictions. Mol Oncol. 2014;8(4):819–29. https://doi.org/10.1016/j.molonc.2014.02.009.
Montani F, Marzi MJ, Dezi F, Dama E, Carletti RM, Bonizzi G, et al. miR-Test: a blood test for lung cancer early detection. Jnci-J Natl Cancer I. 2015;107(6):djv063. https://doi.org/10.1093/jnci/djv063.
Veronesi G, Bellomi M, Mulshine JL, Pelosi G, Scanagatta P, Paganelli G, et al. Lung cancer screening with low-dose computed tomography: a non-invasive diagnostic protocol for baseline lung nodules. Lung Cancer. 2008;61(3):340–9. https://doi.org/10.1016/j.lungcan.2008.01.001.
Sozzi G, Boeri M, Rossi M, Verri C, Suatoni P, Bravi F, et al. Clinical utility of a plasma-based miRNA signature classifier within computed tomography lung cancer screening: a correlative MILD trial study. J Clin Oncol. 2014;32(8):768–73. https://doi.org/10.1200/Jco.2013.50.4357.
Gittelman MC, Hertzman B, Bailen J, Williams T, Koziol I, Henderson RJ, et al. PCA3 molecular urine test as a predictor of repeat prostate biopsy outcome in men with previous negative biopsies: a prospective multicenter clinical study. J Urol. 2013;190(1):64–9. https://doi.org/10.1016/j.juro.2013.02.018.
Lee GL, Dobi A, Srivastava S. Prostate cancer: diagnostic performance of the PCA3 urine test. Nat Rev Urol. 2011;8(3):123–4. https://doi.org/10.1038/nrurol.2011.10.
Groskopf J, Aubin SMJ, Deras IL, Blase A, Bodrug S, Clark C, et al. APTIMA PCA3 molecular urine test: development of a method to aid in the diagnosis of prostate cancer. Clin Chem. 2006;52(6):1089–95. https://doi.org/10.1373/clinchem.2005.063289.
Sokoll LJ, Ellis W, Lange P, Noteboom J, Elliott DJ, Deras IL, et al. A multicenter evaluation of the PCA3 molecular urine test: pre-analytical effects, analytical performance, and diagnostic accuracy. Clin Chim Acta. 2008;389(1-2):1–6. https://doi.org/10.1016/j.cca.2007.11.003.
Van Neste L, Hendriks RJ, Dijkstra S, Trooskens G, Cornel EB, Jannink SA, et al. Detection of high-grade prostate cancer using a urinary molecular biomarker-based risk score. Eur Urol. 2016;70(5):740–8. https://doi.org/10.1016/j.eururo.2016.04.012.
Govers TM, Caba L, Resnick MJ. Cost-effectiveness of urinary biomarker panel in prostate cancer risk assessment. J Urol. 2018;200(6):1221–6. https://doi.org/10.1016/j.juro.2018.07.034.
Hessels D, Govers T, Van Criekinge W, Vlaeminck-Guillem V, Schmitz-drager B, Stief C, et al. Cost-effectiveness of selectmdx for prostate cancer in four European countries: a modelling study. J Urol. 2018;199(4):E614–E5.
Holyoake A, O'Sullivan P, Pollock R, Best T, Watanabe J, Kajita Y, et al. Development of a multiplex RNA urine test for the detection and stratification of transitional cell carcinoma of the bladder. Clin Cancer Res. 2008;14(3):742–9. https://doi.org/10.1158/1078-0432.CCR-07-1672.
Darling D, Luxmanan C, O’Sullivan P, Lough T, Suttie J. Clinical utility of Cxbladder for the diagnosis of urothelial carcinoma. Adv Ther. 2017;34(5):1087–96. https://doi.org/10.1007/s12325-017-0518-7.
O'Sullivan P, Sharples K, Dalphin M, Davidson P, Gilling P, Cambridge L, et al. A multigene urine test for the detection and stratification of bladder cancer in patients presenting with hematuria. J Urol. 2012;188(3):741–7. https://doi.org/10.1016/j.juro.2012.05.003.
Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R, et al. A draft map of the human proteome. Nature. 2014;509(7502):575–81. https://doi.org/10.1038/nature13302.
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Tissue-based map of the human proteome. Science. 2015;347(6220):1260419. https://doi.org/10.1126/science.1260419.
Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Blal HA, et al. A subcellular map of the human proteome. Science. 2017;356(6340):eaal3321. https://doi.org/10.1126/science.aal3321.
Wilhelm M, Schlegl J, Hahne H, Gholami AM, Lieberenz M, Savitski MM, et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509(7502):582–7. https://doi.org/10.1038/nature13319.
Jimenez CR, Zhang H, Kinsinger CR, Nice EC. The cancer proteomic landscape and the HUPO Cancer Proteome Project. Clin Proteomics. 2018;15:4. https://doi.org/10.1186/s12014-018-9180-6.
Fuzery AK, Levin J, Chan MM, Chan DW. Translation of proteomic biomarkers into FDA approved cancer diagnostics: issues and challenges. Clin Proteomics. 2013;10:13. https://doi.org/10.1186/1559-0275-10-13.
Zhang Z, Chan DW. The road from discovery to clinical diagnostics: lessons learned from the first FDA-cleared in vitro diagnostic multivariate index assay of proteomic biomarkers. Cancer Epidemiol Biomark Prev. 2010;19(12):2995–9. https://doi.org/10.1158/1055-9965.Epi-10-0580.
Sawyers CL. The cancer biomarker problem. Nature. 2008;452(7187):548–52. https://doi.org/10.1038/nature06913.
Drucker E, Krapfenbauer K. Pitfalls and limitations in translation from biomarker discovery to clinical utility in predictive and personalised medicine. EPMA Journal. 2013;4(1):7. https://doi.org/10.1186/1878-5085-4-7.
Gold P, Freedman SO. Specific carcinoembryonic antigens of the human digestive system. J Exp Med. 1965;122(3):467–81. https://doi.org/10.1084/jem.122.3.467.
Wang MC, Valenzuela LA, Murphy GP, Chu TM. Purification of a human prostate specific antigen. Investig Urol. 1979;17(2):159–63.
Stenman UH, Tiitinen A, Alfthan H, Valmu L. The classification, functions and clinical use of different isoforms of HCG. Hum Reprod Update. 2006;12(6):769–84. https://doi.org/10.1093/humupd/dml029.
Tatarinov IS. Content of the embryo-specific alpha-globulin in the serum of the fetus, newborn infant and adult man with primary liver cancer. Vopr Med Khim. 1965;11(2):20–4.
Bourreille J, Metayer P, Sauger F, Matray F, Fondimare A. Existence of alpha feto protein during gastric-origin secondary cancer of the liver. Presse Med. 1970;78(28):1277–8.
Rucker P, Antonio SM, Braden B. Elevated fibrinogen-fibrin degradation products (FDP) in serum of colorectal cancer patients. Anal Lett. 2004;37(14):2965–76. https://doi.org/10.1081/Al-200035849.
Lee S, Huh SJ, Oh SY, Koh MS, Kim SH, Lee JH, et al. Clinical significance of coagulation factors in operable colorectal cancer. Oncol Lett. 2017;13(6):4669–74. https://doi.org/10.3892/ol.2017.6058.
Bingle L, Singleton V, Bingle CD. The putative ovarian tumour marker gene HE4 (WFDC2), is expressed in normal tissues and undergoes complex alternative splicing to yield multiple protein isoforms. Oncogene. 2002;21(17):2768–73. https://doi.org/10.1038/sj/onc/1205363.
McGill MR. The past and present of serum aminotransferases and the future of liver injury biomarkers. EXCLI J. 2016;15:817–28. https://doi.org/10.17179/excli2016-800.
Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol. 2009;27(36):6199–206. https://doi.org/10.1200/Jco.2009.23.4799.
Agarwala SS, Keilholz U, Gilles E, Bedikian AY, Wu J, Kay R, et al. LDH correlation with survival in advanced melanoma from two large, randomised trials (Oblimersen GM301 and EORTC 18951). Eur J Cancer. 2009;45(10):1807–14. https://doi.org/10.1016/j.ejca.2009.04.016.
Bast RC, Feeney M, Lazarus H, Nadler LM, Colvin RB, Knapp RC. Reactivity of a monoclonal-antibody with human ovarian-carcinoma. J Clin Investig. 1981;68(5):1331–7. https://doi.org/10.1172/Jci110380.
Kabawat SE, Bast RC, Welch WR, Knapp RC, Colvin RB. Immunopathologic characterization of a monoclonal-antibody that recognizes common surface-antigens of human ovarian-tumors of serous, endometrioid, and clear cell-types. Am J Clin Pathol. 1983;79(1):98–104. https://doi.org/10.1093/ajcp/79.1.98.
Frenette PS, Thirlwell MP, Trudeau M, Thomson DMP, Joseph L, Shuster JS. The diagnostic-value of Ca-27-29, Ca-15-3, Mucin-like carcinoma antigen, carcinoembryonic antigen and Ca-19-9 in breast and gastrointestinal malignancies. Tumor Biol. 1994;15(5):247–54. https://doi.org/10.1159/000217898.
Magnani JL, Brockhaus M, Smith DF, Ginsburg V, Blaszczyk M, Mitchell KF, et al. A monosialoganglioside is a monoclonal antibody-defined antigen of colon-carcinoma. Science. 1981;212(4490):55–6. https://doi.org/10.1126/science.7209516.
Pavlou MP, Diamandis EP, Blasutig IM. The long journey of cancer biomarkers from the bench to the clinic. Clin Chem. 2013;59(1):147–57. https://doi.org/10.1373/clinchem.2012.184614.
Borrebaeck CAK. Precision diagnostics: moving towards protein biomarker signatures of clinical utility in cancer. Nat Rev Cancer. 2017;17(3):199–204. https://doi.org/10.1038/nrc.2016.153.
Duffy MJ. Tumor markers in clinical practice: a review focusing on common solid cancers. Med Princ Pract. 2013;22(1):4–11. https://doi.org/10.1159/000338393.
Mordente A, Meucci E, Martorana GE, Silvestrini A. Cancer biomarkers discovery and validation: state of the art, problems and future perspectives. In: Scatena R, editor. Advances in cancer biomarkers: from biochemistry to clinic for a critical revision. Advances in Experimental Medicine and Biology, vol. 867. Dordrecht: Springer Netherlands; 2015. p. 9–26.
Anderson NL. The clinical plasma proteome: a survey of clinical assays for proteins in plasma and serum. Clin Chem. 2010;56(2):177–85. https://doi.org/10.1373/clinchem.2009.126706.
Scherl A. Clinical protein mass spectrometry. Methods. 2015;81:3–14. https://doi.org/10.1016/j.ymeth.2015.02.015.
Smith LM, Kelleher NL. Consortium top down proteomics. Proteoform: a single term describing protein complexity. Nat Methods. 2013;10(3):186–7. https://doi.org/10.1038/nmeth.2369.
Hoofnagle AN, Wener MH. The fundamental flaws of immunoassays and potential solutions using tandem mass spectrometry. J Immunol Methods. 2009;347(1-2):3–11. https://doi.org/10.1016/j.jim.2009.06.003.
Morgan BR, Tarter TH. Serum heterophile antibodies interfere with prostate specific antigen test and result in over treatment in a patient with prostate cancer. J Urol. 2001;166(6):2311–2. https://doi.org/10.1016/S0022-5347(05)65565-6.
de la Mora JF, Van Berkel GJ, Enke CG, Cole RB, Martinez-Sanchez M, Fenn JB. Electrochemical processes in electrospray ionization mass spectrometry - Discussion. J Mass Spectrom. 2000;35(8):939–52.
Hillenkamp F, Karas M, Beavis RC, Chait BT. Matrix-assisted laser desorption ionization mass-spectrometry of biopolymers. Anal Chem. 1991;63(24):A1193–A202.
Tang N, Tornatore P, Weinberger SR. Current developments in SELDI affinity technology. Mass Spectrom Rev. 2004;23(1):34–44. https://doi.org/10.1002/mas.10066.
MacLean B, Tomazela DM, Abbatiello SE, Zhang SC, Whiteaker JR, Paulovich AG, et al. Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Anal Chem. 2010;82(24):10116–24. https://doi.org/10.1021/ac102179j.
Wolf-Yadlin A, Hautaniemi S, Lauffenburger DA, White FM. Multiple reaction monitoring for robust quantitative proteomic analysis of cellular signaling networks. P Natl Acad Sci USA. 2007;104(14):5860–5. https://doi.org/10.1073/pnas.0608638104.
Vidova V, Spacil Z. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition. Anal Chim Acta. 2017;964:7–23. https://doi.org/10.1016/j.aca.2017.01.059.
Kiyonami R, Schoen A, Prakash A, Peterman S, Zabrouskov V, Picotti P, et al. Increased selectivity, analytical precision, and throughput in targeted proteomics. Mol Cell Proteomics. 2011;10(2):M110.002931. https://doi.org/10.1074/mcp.M110.002931.
Aebersold R, Mann M. Mass-spectrometric exploration of proteome structure and function. Nature. 2016;537(7620):347–55. https://doi.org/10.1038/nature19949.
Huang Y, Zhu H. Protein array-based approaches for biomarker discovery in cancer. Genom Proteom Bioinf. 2017;15(2):73–81. https://doi.org/10.1016/j.gpb.2017.03.001.
Zhu H, Bilgin M, Bangham R, Hall D, Casamayor A, Bertone P, et al. Global analysis of protein activities using proteome chips. Science. 2001;293(5537):2101–5. https://doi.org/10.1126/science.1062191.
Haab BB, Dunham MJ, Brown PO. Protein microarrays for highly parallel detection and quantitation of specific proteins and antibodies in complex solutions. Genome Biol. 2001;2(2):RESEARCH0004.
Hudson ME, Pozdnyakova I, Haines K, Mor G, Snyder M. Identification of differentially expressed proteins in ovarian cancer using high-density protein microarrays. P Natl Acad Sci USA. 2007;104(44):17494–9. https://doi.org/10.1073/pnas.0708572104.
Paweletz CP, Charboneau L, Bichsel VE, Simone NL, Chen T, Gillespie JW, et al. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene. 2001;20(16):1981–9. https://doi.org/10.1038/sj.onc.1204265.
Moore CD, Ajala OZ, Zhu H. Applications in high-content functional protein microarrays. Curr Opin Chem Biol. 2016;30:21–7. https://doi.org/10.1016/j.cbpa.2015.10.013.
Spurrier B, Ramalingam S, Nishizuka S. Reverse-phase protein lysate microarrays for cell signaling analysis. Nat Protoc. 2008;3(11):1796–808. https://doi.org/10.1038/nprot.2008.179.
Rapkiewicz A, Espina V, Zujewski JA, Lebowitz PF, Filie A, Wulfkuhle J, et al. The needle in the haystack: application of breast fine-needle aspirate samples to quantitative protein microarray technology. Cancer Cytopathol. 2007;111(3):173–84. https://doi.org/10.1002/cncr.22686.
Kim CH, Tworoger SS, Stampfer MJ, Dillon ST, Gu XS, Sawyer SJ, et al. Stability and reproducibility of proteomic profiles measured with an aptamer-based platform. Sci Rep. 2018;8:8382. https://doi.org/10.1038/s41598-018-26640-w.
Thiagarajan M CPTAC phase II final report. Cancer Res. 2017;77. https://doi.org/10.1158/1538-7445.Am2017-399.
Hannick LI. NCI's CPTAC Phase III, proteogenomic analysis of additonal cancer types. Cancer Res. 2017;77. https://doi.org/10.1158/1538-7445.Am2017-400.
Mertins P, Mani DR, Ruggles KV, Gillette MA, Clauser KR, Wang P, et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534(7605):55–62. https://doi.org/10.1038/nature18003.
Fiore LD, Rodriguez H, Shriver CD. Collaboration to accelerate proteogenomics cancer care: The Department of Veterans Affairs, Department of Defense, and the National Cancer Institute's Applied Proteogenomics OrganizationaL Learning and Outcomes (APOLLO) Network. Clin Pharmacol Ther. 2017;101(5):619–21. https://doi.org/10.1002/cpt.658.
Rodriguez H, Pennington SR. Revolutionizing precision oncology through collaborative proteogenomics and data sharing. Cell. 2018;173(3):533–7. https://doi.org/10.1016/j.cell.2018.04.008.
Djulbegovic M, Beyth RJ, Neuberger MM, Stoffs TL, Vieweg J, Djulbegovic B, et al. Screening for prostate cancer: systematic review and meta-analysis of randomised controlled trials. Bmj-Brit Med J. 2010;341. https://doi.org/10.1136/bmj.c4543.
Henderson JT, Webber EM, Sawaya GF. Screening for ovarian cancer updated evidence report and systematic review for the US Preventive Services Task Force. Jama-J Am Med Assoc. 2018;319(6):595–606. https://doi.org/10.1001/jama.2017.21421.
Ilic D, Djulbegovic M, Jung JH, Hwang EC, Zhou Q, Cleves A, et al. Prostate cancer screening with prostate-specific antigen (PSA) test: a systematic review and meta-analysis. BMJ. 2018;362:k3519. https://doi.org/10.1136/bmj.k3519.
Sorensen CG, Karlsson WK, Pommergaard HC, Burcharth J, Rosenberg J. The diagnostic accuracy of carcinoembryonic antigen to detect colorectal cancer recurrence - a systematic review. Int J Surg. 2016;25:134–44. https://doi.org/10.1016/j.ijsu.2015.11.065.
Yilmaz A, Ece F, Bayramgurler B, Akkaya E, Baran R. The value of Ca 125 in the evaluation of tuberculosis activity. Respir Med. 2001;95(8):666–9. https://doi.org/10.1053/rmed.2001.1121.
Chao M, Gibbs P. Caution is required before recommending routine carcinoembryonic antigen and imaging follow-up for patients with early-stage colon cancer. J Clin Oncol. 2009;27(36):e279–e80. https://doi.org/10.1200/Jco.2009.25.6156.
Tan E, Gouvas N, Nicholls RJ, Ziprin P, Xynos E, Tekkis PP. Diagnostic precision of carcinoembryonic antigen in the detection of recurrence of colorectal cancer. Surg Oncol. 2009;18(1):15–24. https://doi.org/10.1016/j.suronc.2008.05.008.
Park IJ, Choi GS, Lim KH, Kang BM, Jun SH. Serum carcinoembryonic antigen monitoring after curative resection for colorectal cancer: clinical significance of the preoperative level. Ann Surg Oncol. 2009;16(11):3087–93. https://doi.org/10.1245/s10434-009-0625-z.
Fenton JJ, Weyrich MS, Durbin S, Liu Y, Bang H, Melnikow J. Prostate-specific antigen–based screening for prostate cancer. Jama. 2018;319(18):1914–31. https://doi.org/10.1001/jama.2018.3712.
Stamey TA. Preoperative serum prostate-specific antigen (PSA) below 10 mu g/L predicts neither the presence of prostate cancer nor the rate of postoperative PSA failure. Clin Chem. 2001;47(4):631–4.
Adhyam M, Gupta AK. A review on the clinical utility of PSA in cancer prostate. Indian J Surg Oncol. 2012;3(2):120–9. https://doi.org/10.1007/s13193-012-0142-6.
Palmer SR, Erickson LA, Ichetovkin I, Knauer DJ, Markovic SN. Circulating serologic and molecular biomarkers in malignant melanoma. Mayo Clin Proc. 2011;86(10):981–90. https://doi.org/10.4065/mcp.2011.0287.
Bell AW, Deutsch EW, Au CE, Kearney RE, Beavis R, Sechi S, et al. A HUPO test sample study reveals common problems in mass spectrometry-based proteomics. Nat Methods. 2009;6(6):423–30. https://doi.org/10.1038/nmeth.1333.
Addona TA, Abbatiello SE, Schilling B, Skates SJ, Mani DR, Bunk DM, et al. Multi-site assessment of the precision and reproducibility of multiple reaction monitoring-based measurements of proteins in plasma. Nat Biotechnol. 2009;27(7):633–U85. https://doi.org/10.1038/nbt.1546.
Carr SA, Abbatiello SE, Ackermann BL, Borchers C, Domon B, Deutsch EW, et al. Targeted peptide measurements in biology and medicine: best practices for mass spectrometry- based assay development using a fit- for- purpose approach. Mol Cell Proteomics. 2014;13(3):907–17. https://doi.org/10.1074/mcp.M113.036095.
Villanueva J, Shaffer DR, Philip J, Chaparro CA, Erdjument-Bromage H, Olshen AB, et al. Differential exoprotease activities confer tumor-specific serum peptidome patterns. J Clin Investig. 2006;116(1):271–84. https://doi.org/10.1172/Jci26022.
Timms JF, Cramer R, Camuzeaux S, Tiss A, Smith C, Burford B, et al. Peptides generated ex vivo from serum proteins by tumor-specific exopeptidases are not useful biomarkers in ovarian cancer. Clin Chem. 2010;56(2):262–71. https://doi.org/10.1373/clinchem.2009.133363.
Mor G, Visintin I, Lai Y, Zhao H, Schwartz P, Rutherford T, et al. Serum protein markers for early detection of ovarian cancer. P Natl Acad Sci USA. 2005;102(21):7677–82. https://doi.org/10.1073/pnas.0502178102.
Visintin I, Feng Z, Longton G, Ward DC, Alvero AB, Lai YL, et al. Diagnostic markers for early detection of ovarian cancer. Clin Cancer Res. 2008;14(4):1065–72. https://doi.org/10.1158/1078-0432.Ccr-07-1569.
Cramer DW, Bast RC, Berg CD, Diamandis EP, Godwin AK, Hartge P, et al. Ovarian cancer biomarker performance in prostate, lung, colorectal, and ovarian cancer screening trial specimens. Cancer Prev Res. 2011;4(3):365–74. https://doi.org/10.1158/1940-6207.Capr-10-0195.
McIntosh M, Anderson G, Drescher C, Hanash S, Urban N, Brown P, et al. Ovarian cancer early detection claims are biased. Clin Cancer Res. 2008;14(22):7574. https://doi.org/10.1158/1078-0432.Ccr-08-0623.
Petricoin EF, Ardekani AM, Hitt BA, Levine PJ, Fusaro VA, Steinberg SM, et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet. 2002;359(9306):572–7. https://doi.org/10.1016/S0140-6736(02)07746-2.
Sorace JM, Zhan M. A data review and re-assessment of ovarian cancer serum proteomic profiling. Bmc Bioinformatics. 2003;4:24. https://doi.org/10.1186/1471-2105-4-24.
Baggerly KA, Morris JS, Coombes KR. Reproducibility of SELDI-TOF protein patterns in serum: comparing datasets from different experiments. Bioinformatics. 2004;20(5):777–U10. https://doi.org/10.1093/bioinformatics/btg484.
Committee on the Review of Omics-Based Tests for Predicting Patient Outcomes in Clinical Trials. Evolution of translational omics: lessons learned and the path forward. Washington (DC): Institute of Medicine of the National Academies; 2012.
Pepe MS, Feng ZD, Janes H, Bossuyt PM, Potter JD. Pivotal evaluation of the accuracy of a biomarker used for classification or prediction: standards for study design. J Natl Cancer I. 2008;100(20):1432–8. https://doi.org/10.1093/jnci/djn326.
Kim J, Bamlet WR, Oberg AL, Chaffee KG, Donahue G, Cao XJ, et al. Detection of early pancreatic ductal adenocarcinoma with thrombospondin-2 and CA19-9 blood markers. Sci Transl Med. 2017;9(398):eaah5583. https://doi.org/10.1126/scitranslmed.aah5583.
Pepe MS, Etzioni R, Feng Z, Potter JD, Thompson ML, Thornquist M, et al. Phases of biomarker development for early detection of cancer. J Natl Cancer Inst. 2001;93(14):1054–61.
Kim J, Hoffman JP, Alpaugh RK, Rhim AD, Reichert M, Stanger B, et al. An iPSC line from human pancreatic ductal adenocarcinoma undergoes early to invasive stages of pancreatic cancer progression. Cell Rep. 2013;3(6):2088–99. https://doi.org/10.1016/j.celrep.2013.05.036.
Nanjappa V, Thomas JK, Marimuthu A, Muthusamy B, Radhakrishnan A, Sharma R, et al. Plasma Proteome Database as a resource for proteomics research: 2014 update. Nucleic Acids Res. 2014;42(D1):D959–D65. https://doi.org/10.1093/nar/gkt1251.
Fung ET. A recipe for proteomics diagnostic test development: the OVA1 test, from Biomarker Discovery to FDA Clearance. Clin Chem. 2010;56(2):327–9. https://doi.org/10.1373/clinchem.2009.140855.
Ueland FR, Desimone CP, Seamon LG, Miller RA, Goodrich S, Podzielinski I, et al. Effectiveness of a multivariate index assay in the preoperative assessment of ovarian tumors. Obstet Gynecol. 2011;117(6):1289–97. https://doi.org/10.1097/AOG.0b013e31821b5118.
Bristow RE, Smith A, Zhang Z, Chan DW, Crutcher G, Fung ET, et al. Ovarian malignancy risk stratification of the adnexal mass using a multivariate index assay. Gynecol Oncol. 2013;128(2):252–9. https://doi.org/10.1016/j.ygyno.2012.11.022.
Coleman RL, Herzog TJ, Chan DW, Munroe DG, Pappas TC, Smith A, et al. Validation of a second-generation multivariate index assay for malignancy risk of adnexal masses. Am J Obstet Gynecol. 2016;215(1):82.e1–82.e11. https://doi.org/10.1016/j.ajog.2016.03.003.
Fidler MJ, Fhied CL, Roder J, Basu S, Sayidine S, Fughhi I, et al. The serum-based VeriStrat (R) test is associated with proinflammatory reactants and clinical outcome in non-small cell lung cancer patients. BMC Cancer. 2018;18:310. https://doi.org/10.1186/s12885-018-4193-0.
Taguchi F, Solomon B, Gregorc V, Roder H, Gray R, Kasahara K, et al. Mass spectrometry to classify non-small-cell lung cancer patients for clinical outcome after treatment with epidermal growth factor receptor tyrosine kinase inhibitors: a multicohort cross-institutional study. J Natl Cancer I. 2007;99(11):838–46. https://doi.org/10.1093/jnci/djk195.
Gregorc V, Novello S, Lazzari C, Barni S, Aieta M, Mencoboni M, et al. Predictive value of a proteomic signature in patients with non-small-cell lung cancer treated with second-line erlotinib or chemotherapy (PROSE): a biomarker-stratified, randomised phase 3 trial. Lancet Oncol. 2014;15(7):713–21. https://doi.org/10.1016/S1470-2045(14)70162-7.
Silvestri GA, Tanner NT, Kearney P, Vachani A, Massion PP, Porter A, et al. Assessment of plasma proteomics biomarker’s ability to distinguish benign from malignant lung nodules results of the PANOPTIC (Pulmonary Nodule Plasma Proteomic Classifier) Trial. Chest. 2018;154(3):491–500. https://doi.org/10.1016/j.chest.2018.02.012.
Ingvarsson J, Wingren C, Carlsson A, Ellmark P, Wahren B, Engstrom G, et al. Detection of pancreatic cancer using antibody microarray-based serum protein profiling. Proteomics. 2008;8(11):2211–9. https://doi.org/10.1002/pmic.200701167.
Wingren C, Sandstrom A, Segersvard R, Carlsson A, Andersson R, Lohr M, et al. Identification of serum biomarker signatures associated with pancreatic cancer. Cancer Res. 2012;72(10):2481–90. https://doi.org/10.1158/0008-5472.Can-11-2883.
Gerdtsson AS, Malats N, Sall A, Real FX, Porta M, Skoog P, et al. A multicenter trial defining a serum protein signature associated with pancreatic ductal adenocarcinoma. Int J Proteomics. 2015;2015:587250. https://doi.org/10.1155/2015/587250.
Gerdtsson AS, Wingren C, Persson H, Delfani P, Nordstrom M, Ren H, et al. Plasma protein profiling in a stage defined pancreatic cancer cohort - implications for early diagnosis. Mol Oncol. 2016;10(8):1305–16. https://doi.org/10.1016/j.molonc.2016.07.001.
Mellby LD, Nyberg AP, Johansen JS, Wingren C, Nordestgaard BG, Bojesen SE, et al. Serum biomarker signature-based liquid biopsy for diagnosis of early-stage pancreatic cancer. J Clin Oncol. 2018;36(28):2887–94. https://doi.org/10.1200/Jco.2017.77.6658.
Carlsson A, Wingren C, Kristensson M, Rose C, Ferno M, Olsson H, et al. Molecular serum portraits in patients with primary breast cancer predict the development of distant metastases. P Natl Acad Sci USA. 2011;108(34):14252–7. https://doi.org/10.1073/pnas.1103125108.
Nordstrom M, Wingren C, Rose C, Bjartell A, Becker C, Lilja H, et al. Identification of plasma protein profiles associated with risk groups of prostate cancer patients. Proteomics Clin Appl. 2014;8(11-12):951–62. https://doi.org/10.1002/prca.201300059.
Mehan MR, Williams SA, Siegfried JM, Bigbee WL, Weissfeld JL, Wilson DO, et al. Validation of a blood protein signature for non-small cell lung cancer. Clin Proteomics. 2014;11(1):32. https://doi.org/10.1186/1559-0275-11-32.
Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21(3):297–308. https://doi.org/10.1016/j.ccr.2012.02.014.
Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8(6):519–30. https://doi.org/10.1085/jgp.8.6.519.
DeBerardinis RJ, Chandel NS. Fundamentals of cancer metabolism. Sci Adv. 2016;2(5):e1600200. https://doi.org/10.1126/sciadv.1600200.
Boroughs LK, DeBerardinis RJ. Metabolic pathways promoting cancer cell survival and growth. Nat Cell Biol. 2015;17(4):351–9. https://doi.org/10.1038/ncb3124.
Yan H, Parsons DW, Jin GL, McLendon R, Rasheed BA, Yuan WS, et al. IDH1 and IDH2 mutations in gliomas. New Engl J Med. 2009;360(8):765–73. https://doi.org/10.1056/NEJMoa0808710.
Kang MR, Kim MS, Oh JE, Kim YR, Song SY, Seo SI, et al. Mutational analysis of IDH1 codon 132 in glioblastomas and other common cancers. Int J Cancer. 2009;125(2):353–5. https://doi.org/10.1002/ijc.24379.
Oivares O, Dabritz JHM, King A, Gottlieb E, Halsey C. Research into cancer metabolomics: towards a clinical metamorphosis. Semin Cell Dev Biol. 2015;43:52–64. https://doi.org/10.1016/j.semcdb.2015.09.008.
Antoniewicz MR. A guide to (13)C metabolic flux analysis for the cancer biologist. Exp Mol Med. 2018;50(4):19. https://doi.org/10.1038/s12276-018-0060-y.
Ranjan R, Sinha N. Nuclear magnetic resonance (NMR)-based metabolomics for cancer research. NMR Biomed. 2018;32:e3916. https://doi.org/10.1002/nbm.3916.
Dai C, Arceo J, Arnold J, Sreekumar A, Dovichi NJ, Li J, et al. Metabolomics of oncogene-specific metabolic reprogramming during breast cancer. Cancer Metab. 2018;6:5. https://doi.org/10.1186/s40170-018-0175-6.
Tolstikov VV, Lommen A, Nakanishi K, Tanaka N, Fiehn O. Monolithic silica-based capillary reversed-phase liquid chromatography/electrospray mass spectrometry for plant metabolomics. Anal Chem. 2003;75(23):6737–40. https://doi.org/10.1021/ac034716z.
Soga T, Ohashi Y, Ueno Y, Naraoka H, Tomita M, Nishioka T. Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J Proteome Res. 2003;2(5):488–94. https://doi.org/10.1021/pr034020m.
Armitage EG, Barbas C. Metabolomics in cancer biomarker discovery: current trends and future perspectives. J Pharmaceut Biomed. 2014;87:1–11. https://doi.org/10.1016/j.jpba.2013.08.041.
Dias DA, Koal T. Progress in metabolomics standardisation and its significance in future clinical laboratory medicine. EJIFCC. 2016;27(4):331–43.
Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462(7274):739–U52. https://doi.org/10.1038/nature08617.
Lombardi G, Corona G, Bellu L, Della Puppa A, Pambuku A, Fiduccia P, et al. Diagnostic value of plasma and urinary 2-hydroxyglutarate to identify patients with isocitrate dehydrogenase-mutated glioma. Oncologist. 2015;20(5):562–7. https://doi.org/10.1634/theoncologist.2014-0266.
Wang D, DuBois RN. Prostaglandins and cancer. Gut. 2006;55(1):115–22. https://doi.org/10.1136/gut.2004.047100.
Cui Y, Shu XO, Li HL, Yang G, Wen WQ, Gao YT, et al. Prospective study of urinary prostaglandin E2 metabolite and pancreatic cancer risk. Int J Cancer. 2017;141(12):2423–9. https://doi.org/10.1002/ijc.31007.
Kim S, Taylor JA, Milne GL, Sandler DP. Association between urinary prostaglandin E-2 metabolite and breast cancer risk: a prospective, case-cohort study of postmenopausal women. Cancer Prev Res. 2013;6(6):511–8. https://doi.org/10.1158/1940-6207.Capr-13-0040.
Morris PG, Zhou XK, Milne GL, Goldstein D, Hawks LC, Dang CT, et al. Increased levels of urinary PGE-M, a biomarker of inflammation, occur in association with obesity, aging, and lung metastases in patients with breast cancer. Cancer Prev Res. 2013;6(5):428–36. https://doi.org/10.1158/1940-6207.Capr-12-0431.
Casero RA, Stewart TM, Pegg AE. Polyamine metabolism and cancer: treatments, challenges and opportunities. Nat Rev Cancer. 2018;18(11):681–95. https://doi.org/10.1038/s41568-018-0050-3.
Nakajima T, Katsumata K, Kuwabara H, Soya R, Enomoto M, Ishizaki T, et al. Urinary polyamine biomarker panels with machine-learning differentiated colorectal cancers, benign disease, and healthy controls. Int J Mol Sci. 2018;19(3):E756. https://doi.org/10.3390/ijms19030756.
Venalainen MK, Roine AN, Hakkinen MR, Vepsalainen JJ, Kumpulainen PS, Kiviniemi MS, et al. Altered polyamine profiles in colorectal cancer. Anticancer Res. 2018;38(6):3601–7. https://doi.org/10.21873/anticanres.12634.
Niemi RJ, Roine AN, Hakkinen MR, Kumpulainen PS, Keinanen TA, Vepsalainen JJ, et al. Urinary polyamines as biomarkers for ovarian cancer. Int J Gynecol Cancer. 2017;27(7):1360–6. https://doi.org/10.1097/Igc.0000000000001031.
Tsoi TH, Chan CF, Chan WL, Chiu KF, Wong WT, Ng CF, et al. Urinary polyamines: a pilot study on their roles as prostate cancer detection biomarkers. PLoS One. 2016;11(9):e0162217. https://doi.org/10.1371/journal.pone.0162217.
Falegan OS, Ball MW, Shaykhutdinov RA, Pieroraio PM, Farshidfar F, Vogel HJ, et al. Urine and serum metabolomics analyses may distinguish between stages of renal cell carcinoma. Metabolites. 2017;7(1):6. https://doi.org/10.3390/metabo7010006.
Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, Mayers JR, et al. Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nat Med. 2017;23(2):235–41. https://doi.org/10.1038/nm.4256.
Mayers JR, Wu C, Clish CB, Kraft P, Torrence ME, Fiske BP, et al. Elevation of circulating branched-chain amino acids is an early event in human pancreatic adenocarcinoma development. Nat Med. 2014;20(10):1193–8. https://doi.org/10.1038/nm.3686.
Handa H, Usuba A, Maddula S, Baumbach JI, Mineshita M, Miyazawa T. Exhaled breath analysis for lung cancer detection using ion mobility spectrometry. PLoS One. 2014;9(12):e114555. https://doi.org/10.1371/journal.pone.0114555.
Westhoff M, Litterst P, Freitag L, Urfer W, Bader S, Baumbach JI. Ion mobility spectrometry for the detection of volatile organic compounds in exhaled breath of patients with lung cancer: results of a pilot study. Thorax. 2009;64(9):744–8. https://doi.org/10.1136/thx.2008.099465.
Saalberg Y, Wolff M. VOC breath biomarkers in lung cancer. Clin Chim Acta. 2016;459:5–9. https://doi.org/10.1016/j.cca.2016.05.013.
Cala MP, Aldana J, Medina J, Sanchez J, Guio J, Wist J, et al. Multiplatform plasma metabolic and lipid fingerprinting of breast cancer: a pilot control-case study in Colombian Hispanic women. PLoS One. 2018;13(2):e0190958. https://doi.org/10.1371/journal.pone.0190958.
Ritchie SA, Ahiahonu PWK, Jayasinghe D, Heath D, Liu J, Lu YS, et al. Reduced levels of hydroxylated, polyunsaturated ultra long-chain fatty acids in the serum of colorectal cancer patients: implications for early screening and detection. BMC Med. 2010;8:13. https://doi.org/10.1186/1741-7015-8-13.
Gatto F, Nookaew I, Nielsen J. Chromosome 3p loss of heterozygosity is associated with a unique metabolic network in clear cell renal carcinoma. P Natl Acad Sci USA. 2014;111(9):E866–E75. https://doi.org/10.1073/pnas.1319196111.
Gatto F, Volpi N, Nilsson H, Nookaew I, Maruzzo M, Roma A, et al. Glycosaminoglycan profiling in patients’ plasma and urine predicts the occurrence of metastatic clear cell renal cell carcinoma. Cell Rep. 2016;15(8):1822–36. https://doi.org/10.1016/j.celrep.2016.04.056.
Gatto F, Maruzzo M, Magro C, Basso U, Nielsen J. Prognostic value of plasma and urine glycosaminoglycan scores in clear cell renal cell carcinoma. Front Oncol. 2016;6:253. https://doi.org/10.3389/fonc.2016.00253.
Gatto F, Blum KA, Hosseini SS, Ghanaat M, Kashan M, Maccari F, et al. Plasma glycosaminoglycans as diagnostic and prognostic biomarkers in surgically treated renal cell carcinoma. Eur Urol Oncol. 2018;1(5):364–77. https://doi.org/10.1016/j.euo.2018.04.015.
Johnson CH, Patterson AD, Idle JR, Gonzalez FJ. Xenobiotic metabolomics: major impact on the metabolome. Annu Rev Pharmacol. 2012;52:37–56. https://doi.org/10.1146/annurev-pharmtox-010611-134748.
Wishart DS, Feunang YD, Marcu A, Guo AC, Liang K, Vazquez-Fresno R, et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018;46(D1):D608–D17. https://doi.org/10.1093/nar/gkx1089.
Sreekumar A, Poisson LM, Rajendiran TM, Khan AP, Cao Q, Yu JD, et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature. 2009;457(7231):910–4. https://doi.org/10.1038/nature07762.
Jentzmik F, Stephan C, Miller K, Schrader M, Erbersdobler A, Kristiansen G, et al. Sarcosine in urine after digital rectal examination fails as a marker in prostate cancer detection and identification of aggressive tumours. Eur Urol. 2010;58(1):12–8. https://doi.org/10.1016/j.eururo.2010.01.035.
Struys EA, Heijboer AC, van Moorselaar J, Jakobs C, Blankenstein MA. Serum sarcosine is not a marker for prostate cancer. Ann Clin Biochem. 2010;47(Pt 3):282. https://doi.org/10.1258/acb.2010.009270.
Wu H, Liu TT, Ma CG, Xue RY, Deng CH, Zeng HZ, et al. GC/MS-based metabolomic approach to validate the role of urinary sarcosine and target biomarkers for human prostate cancer by microwave-assisted derivatization. Anal Bioanal Chem. 2011;401(2):635–46. https://doi.org/10.1007/s00216-011-5098-9.
Wang M, Zou LH, Liang J, Wang X, Zhang DL, Fang Y, et al. The urinary sarcosine/creatinine ratio is a potential diagnostic and prognostic marker in prostate cancer. Med Sci Monit. 2018;24:3034–41. https://doi.org/10.12659/Msm.909949.
Noguchi Y, Zhang QW, Sugimoto T, Furuhata Y, Sakai Y, Mori M, et al. Network analysis of plasma and tissue amino acids and the generation of an amino index for potential diagnostic use. Am J Clin Nutr. 2006;83(2):513 s–9 s.
Kimura T, Noguchi Y, Shikata N, Takahashi M. Plasma amino acid analysis for diagnosis and amino acid-based metabolic networks. Curr Opin Clin Nutr. 2009;12(1):49–53. https://doi.org/10.1097/MCO.0b013e3283169242.
Anayama T, Higashiyama M, Yamamoto H, Kikuchi S, Ikeda A, Okami J, et al. Post-operative AICS status in completely resected lung cancer patients with pre-operative AICS abnormalities: predictive significance of disease recurrence. Sci Rep. 2018;8:12378. https://doi.org/10.1038/s41598-018-30685-2.
Miyagi Y, Higashiyama M, Gochi A, Akaike M, Ishikawa T, Miura T, et al. Plasma free amino acid profiling of five types of cancer patients and its application for early detection. PLoS One. 2011;6(9):e24143. https://doi.org/10.1371/journal.pone.0024143.
Katayama K, Higuchi A, Yamamoto H, Ikeda A, Kikuchi S, Shiozawa M. Perioperative dynamics and significance of plasma-free amino acid profiles in colorectal cancer. BMC Surg. 2018;18:11. https://doi.org/10.1186/s12893-018-0344-0.
Okamoto N. Use of AminoIndex technology for cancer screening. Ningen Dock. 2012;26(6):911–22.
Amal H, Leja M, Funka K, Lasina I, Skapars R, Sivins A, et al. Breath testing as potential colorectal cancer screening tool. Int J Cancer. 2016;138(1):229–36. https://doi.org/10.1002/ijc.29701.
Adam ME, Fehervari M, Boshier PR, Chin S-T, Lin G-P, Romano A, et al. Mass-spectrometry analysis of mixed-breath, isolated-bronchial-breath, and gastric-endoluminal-air volatile fatty acids in esophagogastric cancer. Anal Chem. 2019;91(5):3740–6. https://doi.org/10.1021/acs.analchem.9b00148.
Markar SR, Wiggins T, Antonowicz S, Chin ST, Romano A, Nikolic K, et al. Assessment of a noninvasive exhaled breath test for the diagnosis of oesophagogastric cancer. Jama Oncol. 2018;4(7):970–6. https://doi.org/10.1001/jamaoncol.2018.0991.
Markar SR, Brodie B, Chin ST, Romano A, Spalding D, Hanna GB. Profile of exhaled-breath volatile organic compounds to diagnose pancreatic cancer. Brit J Surg. 2018;105(11):1493–500. https://doi.org/10.1002/bjs.10909.
Tai YL, Chen KC, Hsieh JT, Shen TL. Exosomes in cancer development and clinical applications. Cancer Sci. 2018;109(8):2364–74. https://doi.org/10.1111/cas.13697.
Harding C, Heuser J, Stahl P. Receptor-mediated endocytosis of transferrin and recycling of the transferrin receptor in rat reticulocytes. J Cell Biol. 1983;97(2):329–39. https://doi.org/10.1083/jcb.97.2.329.
Pan BT, Johnstone RM. Fate of the transferrin receptor during maturation of sheep reticulocytes in vitro: selective externalization of the receptor. Cell. 1983;33(3):967–78. https://doi.org/10.1016/0092-8674(83)90040-5.
Rajagopal C, Harikumar KB. The origin and functions of exosomes in cancer. Front Oncol. 2018;8:66. https://doi.org/10.3389/fonc.2018.00066.
Kalluri R. The biology and function of exosomes in cancer. J Clin Investig. 2016;126(4):1208–15. https://doi.org/10.1172/Jci81135.
Sun W, Luo JD, Jiang H, Duan DD. Tumor exosomes: a double-edged sword in cancer therapy. Acta Pharmacol Sin. 2018;39(4):534–41. https://doi.org/10.1038/aps.2018.17.
Gao D, Jiang LL. Exosomes in cancer therapy: a novel experimental strategy. Am J Cancer Res. 2018;8(11):2165–75.
Kibria G, Ramos EK, Wan Y, Gius DR, Liu HP. Exosomes as a drug delivery system in cancer therapy: potential and challenges. Mol Pharm. 2018;15(9):3625–33. https://doi.org/10.1021/acs.molpharmaceut.8b00277.
Yang JL, Hagen J, Guntur KV, Allette K, Schuyler S, Ranjan J, et al. A next generation sequencing based approach to identify extracellular vesicle mediated mRNA transfers between cells. BMC Genomics. 2017;18:987. https://doi.org/10.1186/s12864-017-4359-1.
Sun Y, Huo CH, Qao Z, Shang Z, Uzzaman A, Liu S, et al. Comparative proteomic analysis of exosomes and microvesicles in human saliva for lung cancer. J Proteome Res. 2018;17(3):1101–7. https://doi.org/10.1021/acs.jproteome.7b00770.
Puhka M, Takatalo M, Nordberg ME, Valkonen S, Nandania J, Aatonen M, et al. Metabolomic profiling of extracellular vesicles and alternative normalization methods reveal enriched metabolites and strategies to study prostate cancer-related changes. Theranostics. 2017;7(16):3824–41. https://doi.org/10.7150/thno.19890.
Tauro BJ, Greening DW, Mathias RA, Ji H, Mathivanan S, Scott AM, et al. Comparison of ultracentrifugation, density gradient separation, and immunoaffinity capture methods for isolating human colon cancer cell line LIM1863-derived exosomes. Methods. 2012;56(2):293–304. https://doi.org/10.1016/j.ymeth.2012.01.002.
Thery C, Amigorena S, Raposo G, Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol. 2006;Chapter 3:3.22.1–3.9. https://doi.org/10.1002/0471143030.cb0322s30.
Ludwig AK, De Miroschedji K, Doeppner TR, Borger V, Ruesing J, Rebmann V, et al. Precipitation with polyethylene glycol followed by washing and pelleting by ultracentrifugation enriches extracellular vesicles from tissue culture supernatants in small and large scales. J Extracell Vesicles. 2018;7(1):1528109. https://doi.org/10.1080/20013078.2018.1528109.
Martins TS, Catita J, Rosa IM, Silva OABDE, Henriques AG. Exosome isolation from distinct biofluids using precipitation and column-based approaches. PLoS One. 2018;13(6):e0198820. https://doi.org/10.1371/journal.pone.0198820.
Clayton A, Court J, Navabi H, Adams M, Mason MD, Hobot JA, et al. Analysis of antigen presenting cell derived exosomes, based on immuno-magnetic isolation and flow cytometry. J Immunol Methods. 2001;247(1-2):163–74. https://doi.org/10.1016/S0022-1759(00)00321-5.
Merchant ML, Powell DW, Wilkey DW, Cummins TD, Deegens JK, Rood IM, et al. Microfiltration isolation of human urinary exosomes for characterization by MS. Proteomics Clin Appl. 2010;4(1):84–96. https://doi.org/10.1002/prca.200800093.
Lane RE, Korbie D, Hill MM, Trau M. Extracellular vesicles as circulating cancer biomarkers: opportunities and challenges. Clin Transl Med. 2018;7. https://doi.org/10.1186/s40169-018-0192-7.
Gamez-Valero A, Monguio-Tortajada M, Carreras-Planella L, Franquesa M, Beyer K, Borras FE. Size-exclusion chromatography-based isolation minimally alters extracellular vesicles’ characteristics compared to precipitating agents. Sci Rep. 2016;6:33641. https://doi.org/10.1038/srep33641.
Van Deun J, Mestdagh P, Sormunen R, Cocquyt V, Vermaelen K, Vandesompele J, et al. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J Extracell Vesicles. 2014;3. https://doi.org/10.3402/jev.v3.24858.
Rekker K, Saare M, Roost AM, Kubo AL, Zarovni N, Chiesi A, et al. Comparison of serum exosome isolation methods for microRNA profiling. Clin Biochem. 2014;47(1-2):135–8. https://doi.org/10.1016/j.clinbiochem.2013.10.020.
Van Deun J, Mestdagh P, Agostinis P, Akay O, Anand S, Anckaert J, et al. EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat Methods. 2017;14(3):228–32. https://doi.org/10.1038/nmeth.4185.
Arraud N, Linares R, Tan S, Gounou C, Pasquet JM, Mornet S, et al. Extracellular vesicles from blood plasma: determination of their morphology, size, phenotype and concentration. J Thromb Haemost. 2014;12(5):614–27. https://doi.org/10.1111/jth.12554.
Rikkert LG, Nieuwland R, Terstappen LWMM, Coumans FAW. Quality of extracellular vesicle images by transmission electron microscopy is operator and protocol dependent. Journal of Extracellular Vesicles. 2019;8(1):1555419. https://doi.org/10.1080/20013078.2018.1555419.
van der Pol E, Hoekstra AG, Sturk A, Otto C, van Leeuwen TG, Nieuwland R. Optical and non-optical methods for detection and characterization of microparticles and exosomes. J Thromb Haemost. 2010;8(12):2596–607. https://doi.org/10.1111/j.1538-7836.2010.04074.x.
Caby MP, Lankar D, Vincendeau-Scherrer C, Raposo G, Bonnerot C. Exosomal-like vesicles are present in human blood plasma. Int Immunol. 2005;17(7):879–87. https://doi.org/10.1093/intimm/dxh267.
Thakur BK, Zhang H, Becker A, Matei I, Huang Y, Costa-Silva B, et al. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 2014;24(6):766–9. https://doi.org/10.1038/cr.2014.44.
Allenson K, Castillo J, San Lucas FA, Scelo G, Kim DU, Bernard V, et al. High prevalence of mutant KRAS in circulating exosome-derived DNA from early-stage pancreatic cancer patients. Ann Oncol. 2017;28(4):741–7. https://doi.org/10.1093/annonc/mdx004.
Yang SJ, Che SPY, Kurywchak P, Tavormina JL, Gansmo LB, de Sampaio PC, et al. Detection of mutant KRAS and TP53 DNA in circulating exosomes from healthy individuals and patients with pancreatic cancer. Cancer Biol Ther. 2017;18(3):158–65. https://doi.org/10.1080/15384047.2017.1281499.
Kim KM, Abdelmohsen K, Mustapic M, Kapogiannis D, Gorospe M. RNA in extracellular vesicles. Wiley Interdiscip Rev RNA. 2017;8(4). https://doi.org/10.1002/wrna.1413.
Bayraktar R, Van Roosbroeck K, Calin GA. Cell-to-cell communication: microRNAs as hormones. Mol Oncol. 2017;11(12):1673–86. https://doi.org/10.1002/1878-0261.12144.
Srivastava AK, Singh PK, Rath SK, Dalela D, Goel MM, Bhatt MLB. Appraisal of diagnostic ability of UCA1 as a biomarker of carcinoma of the urinary bladder. Tumor Biol. 2014;35(11):11435–42. https://doi.org/10.1007/s13277-014-2474-z.
Goldvaser H, Gutkin A, Beery E, Edel Y, Nordenberg J, Wolach O, et al. Characterisation of blood-derived exosomal hTERT mRNA secretion in cancer patients: a potential pan-cancer marker. Br J Cancer. 2017;117(3):353–7. https://doi.org/10.1038/bjc.2017.166.
Ferrari E, De Palma A, Mauri P. Emerging MS-based platforms for the characterization of tumor-derived exosomes isolated from human biofluids: challenges and promises of MudPIT. Expert Rev Proteomic. 2017;14(9):757–67. https://doi.org/10.1080/14789450.2017.1364629.
Ogawa Y, Miura Y, Harazono A, Kanai-Azuma M, Akimoto Y, Kawakami H, et al. Proteomic analysis of two types of exosomes in human whole saliva. Biol Pharm Bull. 2011;34(1):13–23. https://doi.org/10.1248/bpb.34.13.
Webber J, Stone TC, Katilius E, Smith BC, Gordon B, Mason MD, et al. Proteomics analysis of cancer exosomes using a novel modified aptamer-based array (SOMAscan(TM)) platform*. Mol Cell Proteomics. 2014;13(4):1050–64. https://doi.org/10.1074/mcp.M113.032136.
Melo SA, Luecke LB, Kahlert C, Fernandez AF, Gammon ST, Kaye J, et al. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature. 2015;523(7559):177–U82. https://doi.org/10.1038/nature14581.
Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17(6):816–26. https://doi.org/10.1038/ncb3169.
Alegre E, Zubiri L, Perez-Gracia JL, Gonzalez-Cao M, Soria L, Martin-Algarra S, et al. Circulating melanoma exosomes as diagnostic and prognosis biomarkers. Clin Chim Acta. 2016;454:28–32. https://doi.org/10.1016/j.cca.2015.12.031.
Im H, Shao HL, Park YI, Peterson VM, Castro CM, Weissleder R, et al. Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nat Biotechnol. 2014;32(5):490–U219. https://doi.org/10.1038/nbt.2886.
San Lucas FA, Allenson K, Bernard V, Castillo J, Kim DU, Ellis K, et al. Minimally invasive genomic and transcriptomic profiling of visceral cancers by next-generation sequencing of circulating exosomes. Ann Oncol. 2016;27(4):635–41. https://doi.org/10.1093/annonc/mdv604.
Madhavan B, Yue SJ, Galli U, Rana S, Gross W, Muller M, et al. Combined evaluation of a panel of protein and miRNA serum-exosome biomarkers for pancreatic cancer diagnosis increases sensitivity and specificity. Int J Cancer. 2015;136(11):2616–27. https://doi.org/10.1002/ijc.29324.
Yoshioka Y, Kosaka N, Konishi Y, Ohta H, Okamoto H, Sonoda H, et al. Ultra-sensitive liquid biopsy of circulating extracellular vesicles using ExoScreen. Nat Commun. 2014;5:3591. https://doi.org/10.1038/ncomms4591.
Jorgensen M, Baek R, Pedersen S, Sondergaard EK, Kristensen SR, Varming K. Extracellular Vesicle (EV) Array: microarray capturing of exosomes and other extracellular vesicles for multiplexed phenotyping. J Extracell Vesicles. 2013;2. https://doi.org/10.3402/jev.v2i0.20920.
Kanwar SS, Dunlay CJ, Simeone DM, Nagrath S. Microfluidic device (ExoChip) for on-chip isolation, quantification and characterization of circulating exosomes. Lab Chip. 2014;14(11):1891–900. https://doi.org/10.1039/c4lc00136b.
Liga A, Vliegenthart ADB, Oosthuyzen W, Dear JW, Kersaudy-Kerhoas M. Exosome isolation: a microfluidic road-map. Lab Chip. 2015;15(11):2388–94. https://doi.org/10.1039/c5lc00240k.
Zhao Z, Yang Y, Zeng Y, He M. A microfluidic ExoSearch chip for multiplexed exosome detection towards blood-based ovarian cancer diagnosis. Lab Chip. 2016;16(3):489–96. https://doi.org/10.1039/c5lc01117e.
Escola JM, Kleijmeer MJ, Stoorvogel W, Griffith JM, Yoshie O, Geuze HJ. Selective enrichment of tetraspan proteins on the internal vesicles of multivesicular endosomes and on exosomes secreted by human B-lymphocytes. J Biol Chem. 1998;273(32):20121–7. https://doi.org/10.1074/jbc.273.32.20121.
Woo HK, Sunkara V, Park J, Kim TH, Han JR, Kim CJ, et al. Exodisc for rapid, size-selective, and efficient isolation and analysis of nanoscale extracellular vesicles from biological samples. ACS Nano. 2017;11(2):1360–70. https://doi.org/10.1021/acsnano.6b06131.
Ko J, Carpenter E, Issadore D. Detection and isolation of circulating exosomes and microvesicles for cancer monitoring and diagnostics using micro-/nano-based devices. Analyst. 2016;141(2):450–60. https://doi.org/10.1039/c5an01610j.
Li P, Kaslan M, Lee SH, Yao J, Gao ZQ. Progress in exosome isolation techniques. Theranostics. 2017;7(3):789–804. https://doi.org/10.7150/thno.18133.
McKiernan J, Donovan MJ, O'Neill V, Bentink S, Noerholm M, Belzer S, et al. A novel urine exosome gene expression assay to predict high-grade prostate cancer at initial biopsy. Jama Oncology. 2016;2(7):882–9. https://doi.org/10.1001/jamaoncol.2016.0097.
Brinkmann K, Enderle D, Flinspach C, Meyer L, Skog J, Noerholm M. Exosome liquid biopsies of NSCLC patients for longitudinal monitoring of ALK fusions and resistance mutations. J Clin Oncol. 2018;36(15):e24090. https://doi.org/10.1200/JCO.2018.36.15_suppl.e24090.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. https://doi.org/10.1016/j.cell.2011.02.013.
Vafaee F, Diakos C, Kirschner MB, Reid G, Michael MZ, Horvath LG, et al. A data-driven, knowledge-based approach to biomarker discovery: application to circulating microRNA markers of colorectal cancer prognosis. NPJ Syst Biol Appl. 2018;4:20. https://doi.org/10.1038/s41540-018-0056-1.
Kourou K, Exarchos TP, Exarchos KP, Karamouzis MV, Fotiadis DI. Machine learning applications in cancer prognosis and prediction. Comput Struct Biotec. 2015;13:8–17. https://doi.org/10.1016/j.csbj.2014.11.005.
Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173(2):400–16 e11. https://doi.org/10.1016/j.cell.2018.02.052.
Hudson TJ, Anderson W, Aretz A, Barker AD, Bell C, Bernabe RR, et al. International network of cancer genome projects. Nature. 2010;464(7291):993–8. https://doi.org/10.1038/nature08987.
Li Y, Chen L. Big biological data: challenges and opportunities. Genomics Proteomics Bioinformatics. 2014;12(5):187–9. https://doi.org/10.1016/j.gpb.2014.10.001.
Ristevski B, Chen M. Big data analytics in medicine and healthcare. J Integr Bioinform. 2018;15(3). https://doi.org/10.1515/jib-2017-0030.
Mohammed A, Biegert G, Adamec J, Helikar T. CancerDiscover: an integrative pipeline for cancer biomarker and cancer class prediction from high-throughput sequencing data. Oncotarget. 2018;9(2):2565–73. https://doi.org/10.18632/oncotarget.23511.
Labuzzetta CJ, Antonio ML, Watson PM, Wilson RC, Laboissonniere LA, Trimarchi JM, et al. Complementary feature selection from alternative splicing events and gene expression for phenotype prediction. Bioinformatics. 2016;32(17):421–9. https://doi.org/10.1093/bioinformatics/btw430.
Azuaje F. Artificial intelligence for precision oncology: beyond patient stratification. Npj Precis Oncol. 2019;3:6. https://doi.org/10.1038/s41698-019-0078-1.
Esteva A, Robicquet A, Ramsundar B, Kuleshov V, DePristo M, Chou K, et al. A guide to deep learning in healthcare. Nat Med. 2019;25(1):24–9. https://doi.org/10.1038/s41591-018-0316-z.
Esteva A, Kuprel B, Novoa RA, Ko J, Swetter SM, Blau HM, et al. Dermatologist-level classification of skin cancer with deep neural networks (vol 542, pg 115, 2017). Nature. 2017;546(7660):686. https://doi.org/10.1038/nature22985.
Coudray N, Ocampo PS, Sakellaropoulos T, Narula N, Snuderl M, Fenyo D, et al. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat Med. 2018;24(10):1559–67. https://doi.org/10.1038/s41591-018-0177-5.
Deulofeu M, Kolarova L, Salvado V, Pena-Mendez EM, Almasi M, Stork M, et al. Rapid discrimination of multiple myeloma patients by artificial neural networks coupled with mass spectrometry of peripheral blood plasma. Sci Rep. 2019;9:7975. https://doi.org/10.1038/s41598-019-44215-1.
Elias KM, Fendler W, Stawiski K, Fiascone SJ, Vitonis AF, Berkowitz RS, et al. Diagnostic potential for a serum miRNA neural network for detection of ovarian cancer. Elife. 2017;6:e28932. https://doi.org/10.7554/eLife.28932.001.
Towards trustable machine learning. Nat Biomed Eng. 2018;2(10):709-10. https://doi.org/10.1038/s41551-018-0315-x.
Yu KH, Beam AL, Kohane IS. Artificial intelligence in healthcare. Nat Biomed Eng. 2018;2(10):719–31. https://doi.org/10.1038/s41551-018-0305-z.
Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018;359(6378):926–30. https://doi.org/10.1126/science.aar3247.
Bartsch DK, Gercke N, Strauch K, Wieboldt R, Matthai E, Wagner V, et al. The combination of MiRNA-196b, LCN2, and TIMP1 is a potential set of circulating biomarkers for screening individuals at risk for familial pancreatic cancer. J Clin Med. 2018;7(10):E295. https://doi.org/10.3390/jcm7100295.
Moller A, Olsson H, Gronberg H, Eklund M, Aly M, Nordstrom T. The Stockholm3 blood-test predicts clinically-significant cancer on biopsy: independent validation in a multi-center community cohort. Prostate Cancer Prostatic Dis. 2019;22(1):137–42. https://doi.org/10.1038/s41391-018-0082-5.
Ulz P, Thallinger GG, Auer M, Graf R, Kashofer K, Jahn SW, et al. Inferring expressed genes by whole-genome sequencing of plasma DNA. Nat Genet. 2016;48(10):1273–8. https://doi.org/10.1038/ng.3648.
Wan N, Weinberg D, Liu T-Y, Niehaus K, Delubac D, Kannan A et al. Machine learning enables detection of early-stage colorectal cancer by whole-genome sequencing of plasma cell-free DNA. BMC Cancer 2018;19:832. 478065. https://doi.org/10.1101/478065.
Frantzi M, Latosinska A, Kontostathi G, Mischak H. Clinical proteomics: closing the gap from discovery to implementation. Proteomics. 2018;18(14):1700463. https://doi.org/10.1002/pmic.201700463.
Poste G. Biospecimens, biomarkers, and burgeoning data: the imperative for more rigorous research standards. Trends Mol Med. 2012;18(12):717–22. https://doi.org/10.1016/j.molmed.2012.09.003.
Diamandis EP. The failure of protein cancer biomarkers to reach the clinic: why, and what can be done to address the problem? BMC Med. 2012;10:87. https://doi.org/10.1186/1741-7015-10-87.
Moore HM, Kelly A, Jewell SD, McShane LM, Clark DP, Greenspan R, et al. Biospecimen reporting for improved study quality. Biopreserv Biobank. 2011;9(1):57–70. https://doi.org/10.1089/bio.2010.0036.
Perez-Gracia JL, Sanmamed MF, Bosch A, Patino-Garcia A, Schalper KA, Segura V, et al. Strategies to design clinical studies to identify predictive biomarkers in cancer research. Cancer Treat Rev. 2017;53:79–97. https://doi.org/10.1016/j.ctrv.2016.12.005.
Sauerbrei W, Taube SE, McShane LM, Cavenagh MM, Altman DG. Reporting recommendations for tumor marker prognostic studies (REMARK): an abridged explanation and elaboration. Jnci-J Natl Cancer I. 2018;110(8):803–11. djy088. https://doi.org/10.1093/jnci/djy088.
Turner L, Shamseer L, Altman DG, Weeks L, Peters J, Kober T, et al. Consolidated standards of reporting trials (CONSORT) and the completeness of reporting of randomised controlled trials (RCTs) published in medical journals. Cochrane Db Syst Rev. 2012;11:MR000030. https://doi.org/10.1002/14651858.MR000030.pub2.
Bossuyt PM, Reitsma JB, Bruns DE, Gatsonis CA, Glasziou PP, Irwig LM, et al. The STARD statement for reporting studies of diagnostic accuracy: explanation and elaboration. Clin Chem. 2003;49(1):7–18. https://doi.org/10.1373/49.1.7.
Poste G, Compton CC, Barker AD. The national biomarker development alliance: confronting the poor productivity of biomarker research and development. Expert Rev Mol Diagn. 2015;15(2):211–8. https://doi.org/10.1586/14737159.2015.974561.
Ioannidis JPA, Bossuyt PMM. Waste, leaks, and failures in the biomarker pipeline. Clin Chem. 2017;63(5):963–72. https://doi.org/10.1373/clinchem.2016.254649.
Food and Drug Administration HHS. Good laboratory practice for nonclinical laboratory studies (81 FR 58341). 2016. https://www.federalregister.gov/documents/2016/08/24/2016-19875/good-laboratory-practice-for-nonclinical-laboratory-studies.
CDC. Clinical Laboratory Improvement Amendments (CLIA). https://www.cdc.gov/clia/law-regulations.html.
De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol. 2008;19(3):508–15. https://doi.org/10.1093/annonc/mdm496.
Karapetis CS, Khambata-Ford S, Jonker DJ, O'Callaghan CJ, Tu D, Tebbutt NC, et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. New Engl J Med. 2008;359(17):1757–65. https://doi.org/10.1056/NEJMoa0804385.
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G, et al. Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol. 2014;15(8):852–61. https://doi.org/10.1016/S1470-2045(14)70228-1.
Okajima W, Komatsu S, Ichikawa D, Miyamae M, Ohashi T, Imamura T, et al. Liquid biopsy in patients with hepatocellular carcinoma: Circulating tumor cells and cell-free nucleic acids. World J Gastroenterol. 2017;23(31):5650–68. https://doi.org/10.3748/wjg.v23.i31.5650.
Yadav DK, Bai X, Yadav RK, Singh A, Li G, Ma T, et al. Liquid biopsy in pancreatic cancer: the beginning of a new era. Oncotarget. 2018;9(42):26900–33. https://doi.org/10.18632/oncotarget.24809.
Muinelo-Romay L, Casas-Arozamena C, Abal M. Liquid biopsy in endometrial cancer: new opportunities for personalized oncology. Int J Mol Sci. 2018;19(8):2311. https://doi.org/10.3390/ijms19082311.
Shankar GM, Balaj L, Stott SL, Nahed B, Carter BS. Liquid biopsy for brain tumors. Expert Rev Mol Diagn. 2017;17(10):943–7. https://doi.org/10.1080/14737159.2017.1374854.
Acknowledgments
The authors wish to acknowledge the following grant support: Knut and Alice Wallenbergs Foundation (Dnr 2018.0266). The authors would like to give acknowledgements to Professor Robert Langer for inspiring our work in the field of cancer biology. His many contributions in the field of science and engineering, and in particular his demonstrations on how science can be translated for use in the society, have served as a great inspiration for us in our work. His mentoring and support for our work related to cancer biomarkers is deeply appreciated.
Funding
Open access funding provided by Chalmers University of Technology.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
FG and JN are shareholders in Elypta AB. Additionally, FG is employed at Elypta AB.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Bratulic, S., Gatto, F. & Nielsen, J. The Translational Status of Cancer Liquid Biopsies. Regen. Eng. Transl. Med. 7, 312–352 (2021). https://doi.org/10.1007/s40883-019-00141-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40883-019-00141-2