
Abstract
The investigation of the fractionation of S compounds in forest soils is a powerful tool for interpreting S dynamics and S biogeochemistry in forest ecosystems. Beech stands on high pH (nutrient-rich) sites on Flysch and on low pH (nutrient-poor) sites on Molasse were selected for testing the influence of stemflow, which represents a high input of water and dissolved elements to the soil, on spatial patterns of sulfur (S) fractions. Soil cores were taken at six distances from a beech stem per site at 55 cm uphill and at 27, 55, 100, 150 and 300 cm downhill from the stem. The cores were divided into the mineral soil horizons 0–3, 3–10, 10–20, 20–30 and 30–50 cm. Soil samples were characterized for pH, Corg, pedogenic Al and Fe oxides and S fractions. Sequential extraction by NH4Cl, NH4H2PO4 and HCl yielded readily available sulfate-S (RAS), adsorbed sulfate-S (AS) and HCl-soluble sulfate-S (HCS). Organic sulfur (OS) was estimated as the difference between total sulfur (ToS) and inorganic sulfur (RAS + AS + HCS). Organic sulfur was further divided into ester sulfate-S (ES, HI-reduction) and carbon bonded sulfur (CS). On Flysch, RAS represented 3–6%, AS 2–12%, HCS 0–8% and OS 81–95% of ToS. On Molasse, RAS amounted 1–6%, AS 1–60%, HCS 0–8% and OS 37–95% of ToS. Spatial S distribution patterns with respect to the distance from the tree stem base could be clearly observed at all investigated sites. The presented data is a contribution to current reports on negative input–output S budgets of forest watersheds, suggesting that mineralization of OS on nutrient rich soils and desorption of historic AS on nutrient-poor soils are the dominant S sources, which have to be considered in future modeling of sulfur.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Due to the combustion of fossil fuels, European sulfur (S) emissions and deposition increased steadily since the industrial revolution in the middle of the nineteenth century. As a consequence, sulfur deposition in forested ecosystems of north and central Europe peaked in the early 1980s, reaching, in certain cases, loads of more than 100 kg S ha−1 year−1 (Prechtel et al. 2001). Legislation to reduce acidifying emissions has taken place at an international level, and, e.g., in Austria, SO2 emissions declined by 77% from 1990 (75,000 t year−1) to 2013 (17,000 t year−1; Umweltbundesamt 2015). Throughfall (plus stemflow) fluxes in beech (Fagus sylvatica) forests of eastern Austria declined from 23 to 6 kg S ha−1 year−1 from 1984 to 2013 (Berger and Muras 2016).
Release of previously stored S delays the recovery of pH of soils and surface waters, because sulfate is the only S output in the leachate. Leaching of sulfate is associated with loss of base cations and soil acidification. Reuss and Johnson (1986) called the time prior the peak of S deposition, when soils adsorbed S, a grace period, since base cations remained available for plant uptake and sulfate adsorption neutralized deposited acids resulting from the replacement of –OH− groups on the soil exchanger surface by SO4 2−. Sulfate adsorption/desorption is a concentration-dependent process, meaning that the amount of adsorbed sulfate increased with increasing sulfate soil solution concentrations in the past but desorption is expected in the future due to declining S deposition and associated decreased sulfate soil solution concentrations. However, a time lag may be expected until this new equilibrium is reached, causing higher sulfate outputs than inputs of forest ecosystems.
In fact, in many regions the chemistry of soils and surface waters has not recovered as expected (Berger et al. 2016; Lawrence et al. 2015). Several calibrated watersheds are currently monitored throughout eastern North America and Europe, reporting net losses of base cations in recent decades as reviewed by Watmough et al. (2005). It is striking that the same authors reported that SO4 2− export exceeded input at 18 out of 21 studied catchments by 6–76 kg SO4 2− ha−1 year−1. This discrepancy may be explained by net desorption of inorganic S and net mineralization of organic S (Likens et al. 2002).
Inorganic S, determined as the sum of readily available (also termed water-extractable; Morche 2008), adsorbed (also phosphate-extractable; Likens et al. 2002) and carbonate occluded SO4 2− (also HCl-extractable S, Chen et al. 1997) represent in general less than 10% of the total S (Likens et al. 2002; Tisdale et al. 1993; Vannier and Guillet 1994). Organic S compounds represent in general over 90% of the total S in soils (Likens et al. 2002; Tisdale et al. 1993; Vannier and Guillet 1994). The organic S is often divided in two groups: ester sulfates (ES; a variety of alkyl-, aryl-, phenol sulfates, etc.) and carbon bonded S (CS; amino acids, sulfoxides, etc.; Tabatabai 1996). The mineralization of organic S compounds to SO4 2− depends on the microbial activity, which is affected by many parameters (soil temperature, humidity, pH, use of soil; Morche 2008). Aside the soil microflora, the type of vegetation growing on the soil determines whether ES or CS will be mineralized (Morche 2008).
Many biological and chemical processes in the soil are pH-dependent. Mineralization of organic S, as a result of high S plant uptake rates in the past, coupled with increasing soil pHs after the end of the acid rain period, provides additional current sulfate sources. The density of net positive surface charges in the soil (humus, clay minerals, Al and Fe oxides and hydroxides) decreases with increasing soil pH (Likens et al. 2002), expecting a further increased desorption rate of sulfate after acid rain had ceased. Precipitation/dissolution processes of aluminum hydroxy sulfate are expected to release sulfate at higher soil pH as well (Prietzel and Hirsch 1998).
Stemflow of beech represents a high input of water and dissolved elements to the forest soil. Due to the particular canopy structure and bark composition stemflow effects are confined to beech trees only and is negligible for other tree species (Berger et al. 2008). Therefore, deposition of acidifying substances (NO3 −, SO4 2−) has been shown to be significantly higher close to a beech stem than in areas affected by throughfall only (Kazda 1983; Kazda and Glatzel 1984; Koch and Matzner 1993; Sonderegger 1981). Comparison of soil chemistry between the infiltration zone (near the base of the stem) and the “between trees area” in old beech stands by Lindebner (1990) and Rampazzo and Blum (1992) in the “Vienna Woods” proved a significant impact of deposition of atmospheric pollutants: soil acidification, heavy metal accumulation, increased total sulfur contents and loss of the base cations, especially in the infiltration zone. Berger and Muras (2016) estimated that routing 50% of measured stemflow fluxes in 1984 through the soil profile at 27 cm downhill of a beech stem in the “Vienna woods” resulted in a deposition load (throughfall plus stemflow) of 215 kg S ha−1 year−1 while the input at 300 cm distance from the stem (throughfall only) amounted only 15 kg S ha−1 year−1. Meanwhile (2013), these loads declined to 30 kg S ha−1 year−1 (stem base at 27 cm) and 5 kg S ha−1 year−1 (between trees area at 300 cm distance), respectively. Thus, focusing on the spatial heterogeneity of soil chemistry related to the distance from beech stems enables the study of recovery of differently polluted soil within the same stand. Assuming that increasing soil solution fluxes with decreasing distance from the stem cause a quicker steady state of soil SO4 2− pools in response to currently decreased inputs, studying soil gradients from the base of the stem to “bulk soil” from the between trees area should enable to better characterize soil acidification recovery dynamics (similar to a false chronosequence), as suggested by Berger and Muras (2016).
Sulfur content and the distribution of S into different fractions were investigated in S-deficient podzolic and chernozemic forest soil profiles (Lowe 1965), in spruce forest soils (Vannier and Guillet 1994) and, to a smaller extent, in hardwood forest soils (Likens et al. 2002). All listed S fractions proved to be of importance for S dynamics and S biogeochemistry (e.g., Chen et al. 1997). As pointed out input–output budgets of sulfate are critical for prediction of soil recovery from acid rain. For that reason the relation between inorganic and organic soil S fractions is crucial which may depend on the applied analytical methods. It is suggested that analyzing the micro-spatial heterogeneity of soil columns downhill of a beech stem enables predictions of soil recovery as a function of historic acid loads and time (Berger and Muras 2016). Hence, we analyzed S fractions at different distances to the stem base of beech at nutrient-rich, high pH (Flysch) and nutrient-poor, low pH (Molasse) sites and hypothesized that high historic S deposition coupled with high water inputs via stemflow result in spatial soil patterns of S fractions. The hypothesis was developed into three specific research questions to address above issues:
-
1.
How are S fractions distributed and related to each other in low pH (nutrient-poor) and high pH (nutrient-rich) forest soils?
-
2.
Are stemflow induced soil changes reflected in spatial patterns of S fractions?
-
3.
Do these patterns match the reported negative S-budgets or recovery from acid rain?
Materials and methods
Study sites
Three study sites on two different bedrocks (Flysch and Molasse) each were selected in pure old-growth beech stands. The sites J, E and W on Flysch (Table 1) are located in the Vienna Woods along a distance gradient from the urban area from south–east to north–west (prevailing wind direction during high-pressure episodes). The impact of atmospheric pollutants in the early 1980s on soil characteristics of these sites was documented by Sonderegger (1981) and Kazda (1983). The beech stands B1, B2 and F on Molasse are located in the “Kobernaußerwald”, Upper Austria, and have been extensively studied before (Berger et al. 2006, 2009a, b). The study sites on Molasse are characterized by higher mean annual precipitation (1050 mm) and lower mean annual temperature (7.8 °C) than the sites on Flysch (742 mm, 9.2 °C), as indicated by long term means (1971–2000) of nearby weather stations.
Flysch consists mainly of old tertiary and mesozoic sandstones and clayey marls. Nutrient release from this bedrock is high and, consequently, the prevalent humus forms are mull, indicating quick turnover of the forest litter layer (usually less than 2 cm thickness). All soils of these study sites were classified as stagnic cambisol.
Molasse is composed of tertiary sediments (“Hausruck-Kobernausserwald” gravel), which consist mainly of quartz and other siliceous material (granite, gneiss, hornblende schist, pseudotachylite and colored sandstone). Because of this acidic bedrock with low rates of nutrient release, the dominant soil types are dystric cambisols to podzols. Humus form is acidic moder and the thickness of the forest litter layer varies between 5 and 10 cm, indicating slow turnover and accumulation of nutrients.
Soil sampling and preparation
Soil samples in the vicinity of one beech stem per site were collected in June 2010 (Flysch) and in October 2011 (Molasse). Soil cores were taken with a core sampler of 70 mm diameter to a depth of 50 cm. Two soil cores were sampled at each of the following six distances from one beech stem per site: at 55 cm uphill (further labeled −55), and at 27, 55, 100, 150 and 300 cm downhill from the stem. The cores were divided into the mineral soil horizons 0–3, 3–10, 10–20, 20–30 and 30–50 cm depth and both replicates were mixed. The samples were sieved (mesh size 2 mm), homogenized and oven dried (105 °C, 24 h). Masses of roots (only one diameter class) in the individual soil horizons did not show clear patterns in relation to the distance to the stem base. Hence, uniform S plant uptake rates are assumed within 3 m distance to the stem. Soil pH was measured in 0.01 mol L−1 CaCl2, according to ISO 10390:2005. Basic soil parameters, relevant for this study, are listed in Table 2. More details on sampling and soil characterization are given by Muras (2012) and Kaliwoda (2015). Organic C was calculated total C (Wösthoff Carmhomat ADG 8, Germany, ÖNORM L1080) minus CCaCO3 (Scheibler method: reaction of carbonates with HCl and volumetric determination of emerging CO2 according to ÖNORM L1084). In general, soils on Molasse contained more organic carbon, were characterized by wider Corg/Ntot (mineral soil) and Corg/Stot ratios (total soil profile), were more acidic, more sandy and less supplied with nutrients than soils on Flysch. Total soil S stocks (S m−2 per horizon) were within the same range for all study sites (due to higher bulk densities of loamy to clayey soils on Flysch than of sandy soils on Molasse).
Determination of sulfur fractions
Inorganic S is determined as the sum of readily available (also termed water-extractable; Morche 2008), adsorbed (also phosphate-extractable; Likens et al. 2002) and carbonate occluded SO4 2− (also HCl-extractable S, Chen et al. 1997). A sequential extraction is applied to obtain these three inorganic S fractions (Chen et al. 1997; Kulhanek et al. 2011; Lowe 1965; Morche 2008; Shan et al. 1992). Although the extraction processes are not sulfate-specific (it cannot be excluded that hydrophilic organic S compounds are co-extracted), it is not likely that the amount of readily available or adsorbed SO4 2− is overestimated significantly, as demonstrated by Shan et al. (1992). In the case of carbonate occluded SO4 2−, our preliminary measurements have shown that a sulfate-specific measurement, i.e., ion chromatography (IC), is required to not overestimate this fraction, and, consequently, the total inorganic S amount in accordance to Shan et al. (1992).
Organic S can be calculated as the difference between total S and inorganic S. To divide it into ES and CS, the soil was reduced by HI as described by Johnson and Nishita (1952), modified by Shan et al. (1992) and applied routinely since then (Chen et al. 1997; Kulhanek et al. 2011; Likens et al. 2002; Morche 2008). All reagents, salts and acids applied were of pro analysis grade.
Total sulfur (ToS)
The ToS was determined according to the ÖNORM L 1096 using LECO SC 444 (LECO Corp., St. Joseph, MI, USA). LECO 302–508 was used as a standard.
Soil sulfur extraction
The extraction method as described by Tabatabai (1996) and Kulhanek et al. (2011) was slightly modified (NH4 + instead of Na+ salt solutions were used in this work at the same anion concentrations as used by Kulhanek et al. 2011) and applied for extraction of soil S fractions. Soil was extracted separately for each fraction (not sequentially). Each extraction was repeated three times. Each extract was filtered through a syringe filter (0.45 µm). Double sub-boiled HNO3 was used for acidification of extracts to 2% (w/w) HNO3 prior to analysis. RTS-1 (CCRMP, Ottawa, ON, CA) certified soil reference material was used for method validation. The reference material was certified for both total S content and adsorbed sulfate. The following S fractions were extracted:
Readily available sulfate-S (RAS) 1.0 g soil sample was shaken with 6 mL 0.02 mol L−1 NH4Cl solution for 3 h,
Phosphate extractable sulfate-S (PES) 1.0 g soil sample was shaken in 6 mL 0.02 mol L−1 NH4H2PO4 solution for 3 h,
HCl-extractable sulfate-S (HCE) 1.0 g soil sample was shaken in 20 mL cold 1 mol L−1 HCl for 1 h.
HI-reduction of soil sulfur
Samples from one site per bedrock (W on Flysch and B1 on Molasse), representing the lowest and highest soil Stot stocks according to Table 2, were investigated in more details: W and B1 soil samples were reduced by HI as described by Johnson and Nishita (1952), modified by Shan et al. (1992) and applied routinely since then (Chen et al. 1997; Kulhanek et al. 2011; Likens et al. 2002; Morche 2008).
HI-reducible sulfur (HIS) 0.5 g soil sample was reduced using a HI-reducing agent (Shan et al. 1992). Each sample was processed twice.
Sulfur content measurement
Sulfur contents in all extracts were determined (detection limit 0.5 µg L−1) by ICP-MS (Element XR, Thermo Scientific, Waltham, MA, USA) operated in medium mass resolution, applying external calibration and internal normalization (In and Sb, both single element ICP standards). However, given amounts of HCE (sulfate-S) throughout the paper were determined (detection limit 0.1 mg L−1) by means of ion-exchange chromatography (IC; ICS 1600, Dionex, USA) with suppressed conductivity, applying the IonPac AS11-HC (Dionex, USA) guard and analytical columns, and the ASRS 300-4 mm suppressor (Dionex, USA). 30 mmol L−1 KOH was used as the effluent (flow rate 1 mL min−1).
Calculation of soil S species
Adsorbed sulfate-S (AS) mass fraction: AS = PES − RAS
HCl-soluble sulfate-S (HCS) mass fraction: HCS = HCE − PES
Organic sulfur (OS) mass fraction: OS = ToS − HCE
For soil profile imaging, OS was further divided into
Ester sulfate-S (ES) mass fraction: ES = HIS − HCE
Carbon bonded sulfur (CS) mass fraction: CS = ToS − HIS
Determination of pedogenic Al and Fe oxides
Pedogenic Al and Fe oxides (comprising oxides, oxyhydroxides, and hydrated oxides) were analyzed for the sites W (Flysch) and B1 (Molasse). The dithionite-soluble Fe (Fed) and Al (Ald) fractions were determined according to Holmgren (1967): 2.0 g soil sample were mixed with 2.0 g Na2S2O4 and shaken for 16 h in 100 mL 0.3 mol L−1 sodium citrate − 1.0 mol L−1 NaHCO3 (4:1) solution. The contents of the oxalate-soluble Fe (Feo) and Al (Alo) were determined according to ÖNORM L 1201: 1.0 g soil sample was shaken in 50 mL 0.2 mol L−1 ammonium oxalate − 0.2 mol L−1 oxalic acid solution (4:3) for 4 h in the dark. The first 10 mL of the filtered extract were not used for measurement. Contents of Fe and Al in these extracts were analyzed by ICP-OES (ICP-OES, Optima 3000 XL, Perkin Elmer, USA), using external calibration.
Applied statistics
One-way analysis of variance (ANOVA) and Duncan post hoc analyses were applied using IBM SPSS Statistics 21 software (IBM Corporation, Armonk, NY, USA). Mean values of pH, RAS, AS, HCS, OS and ToS were evaluated with respect to soil depth and distance from a tree stem. ANOVA and the associated multiple range test was used as the data were normally distributed in most cases. In a few cases (AS on Molasse at 100 and 150 cm distance, and pH and ToS on Flysch at 30–50 and 0–3 cm, respectively), this precondition for ANOVA was fulfilled after a simple 1/square root transformation of the data. All statements on significance throughout the text are based on applied statistics. Bivariate linear correlation matrices were carried out for all combinations of pH, Corg, Fed, Feo, Ald, Alo and individual S fractions for the sites W (Flysch) and B1 (Molasse) using IBM SPSS Statistics 21 software.
Results
Tables and figures
Mean soil pH and sulfur mass fractions in soil profiles at −55 and 27, 55, 100, 150 and 300 cm distance to the base of one beech stem per site are presented for individual horizons on (a) Flysch and (b) Molasse in Table 3. The study sites W (Flysch) and B1 (Molasse), for which HIS was measured (enabling the calculation of ES and CS), were selected for graphical illustration of all possible S fractions in the soil (Fig. 1). The relative S distribution at W (Fig. 1a) and B1 (Fig. 1b) matches well to the mean values of the corresponding S fractions of all three sites on Flysch and Molasse, respectively (Table 3). Bivariate coefficients of linear correlations (R, Pearson) among pH, Corg, Fed, Feo, Ald, Alo and individual S fractions in the soil are given for the sites W and B1 in Table 4. Significant (p < 0.01) linear correlations are debated in the “Discussion” section.

Distribution of readily available sulfate-S (RAS), adsorbed sulfate-S (AS), HCl-soluble sulfate-S (HCS), ester sulfate-S (ES), carbon bonded sulfur (CS) and total sulfur (ToS) on a Flysch (study site W) and b Molasse (study site B1). Values were measured in the soil horizons 0–3 (a), 3–10 (b), 10–20 (c), 20–30 (d) and 30–50 (e) cm in −55, 27, 55, 100, 150 and 300 cm distance from a tree trunk. Values between these points were calculated as moving average. Maximum and minimum values are given in the legend of each sub-plot. All data are given in µg S g−1
Soil pH and Corg
The pH values of mean soil profiles were significantly lower on Molasse than on Flysch, except for the profiles at 27 and 55 cm distance (Table 3).
Organic C contents (Corg) ranged from 5/21 (30–50 cm depth) to 106/243 (0–3 cm; W/B1, data in mg g1) and declined significantly with increasing depth (mean contents of all distances per depth were compared by posthoc Duncan multiple range test; p < 0.05). The mean (N = 6 horizons × 6 soil depth = 30) Corg content was significantly (p < 0.001) lower at W (20 mg g−1) than at B1 (76 mg g−1). Though there was a clear trend of enriched Corg contents in the soil profile at 27 cm on Flysch (W), no significant impact of distance on Corg patterns was recorded (data not shown).
Sulfur fractions
On average (mean geometric horizon data of three study sites), the RAS fraction represented 3–6%, AS 2–12%, HCS 0–8% and OS 81–95% of ToS on Flysch. On Molasse, RAS amounted 1–6%, AS 1–60%, HCS 0–8% and OS 37–95% of ToS.
The contents of all S fractions were significantly higher on Molasse than on Flysch (see statistics for each average soil profile in Table 3), with the exception of not significant differences for RAS (27, 55), AS (27) and HCS (all distances). On both bedrocks, distance from the stem base had no significant effect on ToS and OS fractions. However, these fractions declined with increasing soil depth (Table 3).
Significantly higher RAS values were found in the topsoil on both bedrocks. The RAS maxima were reached at distances around 100 cm from the stem base. The AS fraction increased with depth on Molasse, while AS tended to decline (not significantly) with depth on Flysch. Average AS of the soil profile next to the stem base (27 cm) was significantly reduced on Flysch. A similar behavior was observed on Molasse (not significant differences, see Table 3). The HCS fraction was significantly enriched next to the stem base (27 cm) on Molasse. Since 1 mol L−1 HCl represents the strongest extractant whithin this study, it is suggested that the generally very low HCS contents, measured by IC (0–8% of ToS; Table 3) represented the remaining sulfate adsorbed on soil particles that was not exchanged by phosphate.
Pedogenic Al and Fe oxides
Dithionite-soluble Fe (Fed) amounted 3.8–6.6 and 5.5–14.4 mg g−1 at W and B1, respectively. The mean Fed content was significantly (p < 0.001) lower at W (4.6 mg g−1) than at B1 (10.8 mg g−1). Oxalate-soluble Fe (Feo) amounted 2.3–3.3 and 2.5–14.0 mg g−1 at W and B1, respectively. The mean Feo content was significantly (p < 0.001) lower at W (2.7 mg g−1) than at B1 (9.9 mg g− 1). The higher Feo/Fed ratio at B1 (0.9) than at W (0.6) indicated higher crystallinity and lower amounts of more active forms of Fe (Rampazzo et al. 1999). Duncan multiple range tests showed that the content of the mean soil profile at 27 cm was higher than at 55 cm for Fed and higher than at all other distances for Feo on Flysch (W), but there was no effect by distance on Molasse (B1; data not shown).
Dithionite-soluble Al (Ald) amounted 1.1–1.9 and 2.2–9.7 mg g−1 at W and B1, respectively. The mean Ald content was significantly (p < 0.001) lower at W (1.4 mg g−1) than at B1 (5.7 mg g−1). Oxalate-soluble Al (Alo) amounted 0.8–1.2 and 1.6–7.6 mg g−1 at W and B1, respectively. The mean Alo content was significantly (p < 0.001) lower at W (1.0 mg g−1) than at B1 (4.8 mg g−1; data not shown). It must be pointed out that Alo and Ald contents did not differ between mean soil depths at W but increased significantly with increasing depth at B1 (Ald: 0–3, 3–10 < 10–20 < 20–30, 30–50; Alo: 0–3, 3–10 ≤ 20–30, 30–50; soil horizon ranges in cm).
Discussion
How are S fractions distributed and related to each other in low pH (nutrient-poor) and high pH (nutrient-rich) forest soils?
Inorganic SO4 2− accounts generally for less than 10% of the total S (Tisdale et al. 1993). In the presented study, however, these theoretical limits were exceeded on both investigated bedrocks for mean horizon values (Table 3): the sum of RAS, AS and HCS reached up to 19 and 63% in some points on Flysch and Molasse, respectively. The AS fraction contributed mainly to these high values. According to a simplified S flow diagram of an upland forest ecosystem by Reuss and Johnson (1986), SO4 2− is the only S output in the leachate. In turn, the SO4 2− soil solution pool is connected to two S pools: the adsorbed SO4 2− pool and the organic S pool. In accordance with Reuss and Johnson (1986), we suggest that the observed high inorganic SO4 2− pools exert a strong control on leaching of SO4 2− and associated loss of base cations and soil acidification.
Sulfur contained in soil organic matter (OS) represented large pools of up to 95% of total S (ToS) on both bedrocks which is in accordance with data reported by Vannier et al. (1993) and Erkenberg et al. (1996). The ES fraction amounted 38 and 54% of OS at W and B1 (mean of average soil profiles), respectively. This is in contradiction to Havlin et al. (2005), suggesting that ES represents the majority of the organic S. However, data of ES (33 and 39% of ToS on W and B1, respectively) are higher than reported contributions of 16% by David et al. (1982) in forest soils, but close to the 38% contribution as found at the Hubbard Brook Experimental Forest (Likens et al. 2002). The total S pool of a haplic podzol in the control watershed Schluchsee consisted of >90% OS, 64% of ToS was CS, and 28% ES. However, in a dystric cambisol in the control watershed Villingen, inorganic S bound in the mineral soil accounted for 67% of the ToS pool (Prietzel et al. 2001).
Bivariate Pearson correlation coefficients (R) are given in Table 4. Caution must be exercised when interpreting correlations because they do not give information about “controlling factors or processes”. Conclusions may be drawn from cases where the coefficients for W and B1 were very contrary. e.g., high positive correlation between OS and AS at W (0.75; p < 0.001) may be explained by high mineralization rates on Flysch, while the lack of this relation (even negative sign) at B1 may indicate that immobilization (microbial uptake) is an important process on Molasse. Ester sulfate-S, that is considered to be easily mineralized after hydrolysis (Morche 2008), was positively correlated with AS at W (indicating mineralization on Flysch) but vice versa at B1 (indicating immobilization on Molasse).
Bivariate relations among S fractions, which were similar for both sites, can be summarized as follows (Table 4): Organic sulfur increased with decreasing pH since acidic conditions promote the accumulation of organic matter and vice versa (i.e., organic matter accumulation produces organic acids). Organic sulfur was positively correlated with all other S fractions at both sites, except with AS at B1 (see assumption on preferential S immobilization on Molasse above), HCS (probably due to the fact that most data were zero) and with ES at B1 (which was distributed more patchy than at W; see Fig. 1). Extremely high coefficients were recorded for both sites between OS and ToS (W: 1.00; B1: 0.98), which is not surprising as OS represents in total the major S fraction in the investigated soils.
Organic C was positively related to RAS, OS and ToS (p < 0.001) stating the general link between C and S in terrestrial ecosystems (Table 4). The facts that Corg was (a) positively related to AS at W but negatively at B1, and (b) positively to ES at W but not significant at B1 is striking. Possible interpretations are suggested below.
Plant debris (Corg) provided CS at both substrates (R, Corg/CS: 0.94–0.96). Carbon bonded S seemed to be rather stable at B1 (R, CS/ES: ns; suggesting immobilization) but contributed to labile ES at W1 (R, CS/OS: 0.86; suggesting mineralization). According to Ghani et al. (1991), carbon-bonded S is converted, probably oxidatively, to ES and is then mineralized to SO4 2−. Labile ES probably contributed to RAS at W (0.62; note: R, ES/RAS at B1: ns) which, finally, showed up as adsorbed sulfate-S at W (R, RAS/AS: 0.67 vs ns at B1). This underlines the suggestion that AS originates mainly from decomposition (mineralization) of organic material at Flysch (see high accumulation of AS in the top soil of Fig. 1) while AS is accumulated in the deep soil at Molasse (Bs horizons with high contents of Al and Fe oxides) as visible in Fig. 1. The fact that a considerable proportion of atmospherically deposited SO4 2− may be cycled through the organic S pool before being released to soil solution, as suggested for the Flysch sites, was indicated by 34S/32S ratios (Alewell et al. 1999; Zhang et al. 1998). Selected water samples of rainwater (precipitation and throughfall) and soil solution at the study site W were measured for δ34SVCDT by Hanousek et al. (2016) and ranged between 4 and 6‰, the ratio in soil solution being slightly lower. The lower ratio indicated that a considerable portion of the atmospherically deposited sulfate was cycled through the organic S pool before being released to the soil solution and supports our conclusions for the site W above, deduced from relations between the different S-fractions.
Interpreting bi-variate correlation coefficients of Table 4 is difficult due to many inter-correlated variables. e.g., we would expect a positive relation between Ald and Corg at B1 (which was negative: R = −0.82; p < 0.001) as was recorded at W (R: 0.47; p < 0.01), since Corg was coupled with negative soil pH (R: −0.42 and −0.83 at W and B1, respectively) promoting the formation of Al oxides via silicate weathering (Rampazzo et al. 1999). Because Fe and Al oxides increased significantly with increasing depth at B1 (Bs horizons of podzols on Molasse) but Corg contents decreased (similar to depth profiles of OS, Table 3) this relation Ald (Alo) to Corg was negative. As a consequence, the relations of Ald (Alo) to RS (readily available S is mainly provided from the humus layer), CS, OS and ToS were all negative as well. However, Al oxides played a major role for adsorption of AS, as indicated by positive correlations at B1 (R, Ald/AS 0.48, p < 0.05; Alo/AS 0.55, p < 0.01). At W (Flysch), there was a declining trend of Ald with increasing depth, causing significant positive relations to Corg and all other S-fractions (except HCS). This is why, AS was correlated negatively with soil pH (R = −0.33) and positively with Corg (R = 0.73) at W (due to increasing positive charges of humus particles and Al oxides for adsorbing sulfate) but positively with soil pH (R = 0.65) and negatively with Corg (R = −0.40) at B1 (due to increasing Al oxides with increasing depth at increasing soil pH but decreasing Corg contents).
Dithionite-soluble Fed at B1 showed similar coefficients as Ald, though relations with Corg and AS were not significant (Table 4). At high pH-soils on Flysch (W), the formation of Fe oxides was lower and its role to adsorb sulfate was negligible.
Are stemflow induced soil changes reflected in spatial patterns of S fractions?
The average pH values of the six total soil profiles per site indicated clearly soil acidification at near downhill distances on Flysch but not on Molasse (Table 3). As soil pHs were lower on Molasse, stemflow induced acidification was not significant as a consequence of the logarithmic pH-scale, of lower base contents (compare Table 2), which do not allow redistribution of large amounts of base cations, and of buffering by the aluminum system at soil pH below 4.2.
Wide soil pH ranges (3.2–7.0) on Flysch (pH: W < E < J; Table 3) are the reason that pH did neither differ with depth within individual soil profiles nor with distance from the stem within one soil depth. On Molasse, at relatively narrow soil pH ranges (2.8–4.4), top soil acidification was recorded for the average soil profiles at all six distances as related to podzolization (Table 3).
Changes within an individual horizon over distance and vice versa (Table 3) were compared to disentangle the soil depth effect on S fractions from stemflow induced spatial patterns. There was no significant effect of stemflow (distance) on distribution of ToS and OS within the mineral soil, however, total S and OS (both for ES and CS at W) tended to be higher at 27 cm distance in the top soil of Flysch (Table 3a; Fig. 1a). Total S and OS showed a general decline with increasing depth. This decline was steeper on Molasse (Table 3b) due to surface accumulation of organic matter on podzolic soils, characterized by slower decomposition and mineralization rates than on Flysch.
Mean contents of total S of 97 beech sites in the Vienna Woods decreased markedly in the stemflow area (0–5 cm soil depth; 20 cm downhill from the base of the stem) but slightly increased in the between trees area (0–5 cm soil depth; at least 3 m away from beech stems) from 1984 to 2012 (Berger et al. 2016), corresponding to a S loss (0–5 cm horizon) of 18.2 g m−2 and a S gain of 8.2 g m−2, respectively (unpublished data by the same authors). Therefore, high amounts of atmospherically deposited SO4 2−, partly cycled through the organic S pool, must have been mineralized on Flysch since the 1980s. Berger et al. (2009a, b) concluded for comparable beech sites on Flysch that net mineralization of S in the top soil is the major reason for negative SO4 2− input - output budgets. Preferential mineralization over immobilization on Flysch was suggested from the correlation analysis (Table 4). Narrow mineral soil Ctot/Stot ratios on Flysch (Table 2) support this conclusion.
Net immobilization of S on Molasse may be another reason why no acidifying effects of stemflow were visible (Table 3b). In fact, SO4 2− stemflow fluxes caused an accumulation of ES along the tree stem (Fig. 1b). Wide mineral soil Ctot/Stot ratios on Molasse (Table 2) support the theory of high microbial S immobilization rates. High amounts of precipitated Al3+ and SO4 2− as Al hydroxy sulfates in the stemflow area (27 cm) were likely reflected via HCl extractions (as indicated as HCS in Table 3b and Fig. 1b), since, according to Prietzel and Hirsch (1998, 2000), phosphate extraction (AS) may underestimate inorganic S in acidic soils. A high rate of precipitation/dissolution of Al hydroxy sulfates in the stemflow area would be another reason for buffering acidic input via stemflow.
Desorption of SO4 2− in response to input of high amounts of water with low SO4 2− concentrations at the stem after the end of the 1980s is put forward to explain significantly reduced AS at the stem base (27 cm) on Flysch (Table 3a). A similar (not significant) trend was visible on Molasse (Table 3b). This trend can be explained by “natural water extractions” (i.e., desorption) over historic time-periods.
Do these patterns match the reported negative S-budgets or recovery from acid rain?
Contents of AS were much lower on the high pH soils on Flysch than on the low pH soils on Molasse, since the density of net positive surface charges decreases with increasing soil pH. Hence, the conclusion by Berger et al. (2009a, b) that top soil mineralization of organic S is the major source of net SO4 2− output on Flysch agrees with our data. The mean 2-year (2005–2007) net SO4-S balance on comparable beech stands (between trees area) on Flysch was estimated −2.0 kg S ha−1 year−1 (Berger et al. 2009a).
The fact that AS increased with depth on Molasse can be explained by the vertical wash-out direction (desorption starts at the top soil) and increasing amounts of Fe- and Al oxides or hydroxides in deeper soil layers (adsorption in the deeper soil; Table 3), as stated in the results section. Reuss and Johnson (1986) pointed out that a long time may be required for passage of the SO4 2− front to any particular soil depth. It is striking that the soil profile next to the stem (27 cm) is the only one, which did not show a significant vertical increase of adsorbed SO4 2− (AS). It can be suggested that this passage from top to deeper soil horizons has ceased due to accelerated recovery after the reduction of S deposition. Solute SO4 2− flux profiles (between trees area) for the same beech sites on Molasse by Berger et al. (2009a) showed a steady increase with depth. Hence, desorption of historically deposited S below 10 cm can be assumed as the major source of net S loss for these acidic soils. The mean 2-year (2005–2007) net SO4-S balance on the same beech stands on Molasse (between trees area) of this study was estimated −1.4 kg S ha−1 year−1 (Berger et al. 2009a).
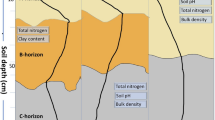
In accordance to Reuss and Johnson (1986), it can be concluded that increasing soil solution fluxes with decreasing distance from the stem cause a quicker steady state of soil SO4 2− pools in response to currently decreased inputs. The presented data on AS support this hypothesis. Comparison of old and recent top soil pH values indicates a higher increase in the infiltration zone of beech stemflow than in the “between trees area” (Berger et al. 2016). The data (Table 3) did not show any horizontal pH depression close to the stem, as reported for Flysch sites in the Vienna Woods in the 1980s (Berger and Muras 2016; Kazda 1983; Sonderegger 1981). In contrast to the reported negative S budgets for whole forested watersheds, the stem area of beech seems to have recovered from acid rain - if recovery is defined as the state where input–output S budgets turn from negative to zero. It is concluded that reduced atmospheric sulfate inputs affected soil conditions. Our results match nicely with predictions by Berger and Muras (2016) that the top soil will recover from acid deposition, as already recorded in the infiltration zone of stemflow near the base of the stem. However, in the between trees areas and especially in deeper soil horizons recovery may be highly delayed (Fig. 2).

(redrawn from Berger and Muras 2016)
Generalizing spatial soil recovery of the top soil, expressed as pH change
Conclusions
Fractionation of S compounds in forest soils is a powerful tool for interpreting S dynamics and S biogeochemistry in forest ecosystems. Stemflow resulted in spatial soil patterns of S fractions (overall hypothesis). On Flysch, RAS represented 3–6%, AS 2–12%, HCS 0–8% and OS 81–95% of ToS. On Molasse, RAS amounted 1–6%, AS 1–60%, HCS 0–8% and OS 37–95% of ToS. It was possible to discuss relations between S fractions in regard to important soil processes, e.g., mineralization/immobilization and adsorption/desorption (question 1). Stemflow clearly caused spatial soil patterns of the inorganic S fractions (question 2). Desorption of SO4 2− in response to input of high amounts of water with low SO4 2− concentrations at the stem after the end of the 1980s is put forward to explain reduced AS at the stem base. Our data contribute to current reports on negative input–output S budgets of forest watersheds, suggesting that mineralization of OS on nutrient rich soils and desorption of historic AS on nutrient-poor soils are the dominant S sources (question 3), which have to be considered in future modeling of sulfur. Finally, we conclude that the impact of heavy S deposition loads via stemflow and recovery from these inputs can be fingerprinted from the S fraction distribution in the soil. We suggest that combining the soil S fractionation method with analyses of stable isotopes of S and in situ soil solution studies will complete our knowledge on S biogeochemistry.
Abbreviations
- RAS :
-
Readily available sulfate-S
- AS :
-
Adsorbed sulfate-S
- HCS :
-
HCl-soluble sulfate-S
- OS :
-
Organic sulfur
- ToS :
-
Total sulfur
- ES :
-
Ester sulfate-S
- CS :
-
Carbon bonded sulfur
- HIS :
-
HI-reducible sulfur
- PES :
-
Phosphate-extractable sulfate-S
- HCE :
-
HCl-extractable sulfate-S
References
Alewell C, Mitchell MJ, Likens GE, Krouse HR (1999) Sources of stream sulfate at the Hubbard Brook Experimental Forest: long-term analyses using stable isotopes. Biogeochemistry 44:281–299
Berger TW, Muras A (2016) Predicting recovery from acid rain using the micro-spatial heterogeneity of soil columns downhill the infiltration zone of beech stemflow—introduction of a hypothesis. Model Earth Syst Environ 2:154
Berger TW, Swoboda S, Prohaska T, Glatzel G (2006) The role of calcium uptake from deep soils for spruce (Picea abies) and beech (Fagus sylvatica). For Ecol Manag 229:234–246
Berger TW, Untersteiner H, Schume H, Jost G (2008) Throughfall fluxes in a secondary spruce (Picea abies), a beech (Fagus sylvatica) and a mixed spruce-beech stand. For Ecol Manag 255:605–618
Berger TW, Inselsbacher E, Mutsch F, Pfeffer M (2009a) Nutrient cycling and soil leaching in eighteen pure and mixed stands of beech (Fagus sylvatica) and spruce (Picea abies). For Ecol Manag 258:2578–2592
Berger TW, Untersteiner H, Toplitzer M, Neubauer C (2009b) Nutrient fluxes in pure and mixed stands of spruce (Picea abies) and beech (Fagus sylvatica). Plant Soil 322:317–342
Berger TW, Türtscher S, Berger P, Lindebner L (2016) A slight recovery of soils from Acid Rain over the last three decades is not reflected in the macro nutrition of beech (Fagus sylvatica) at 97 forest stands of the Vienna Woods. Environ Pollut 216:624–635
Chen B, Shan X, Shen D (1997) Nature of the HCl-soluble sulfate in the sequential extraction for sulfur speciation in soils. Fresenius J Anal Chem 357:941–945
David MB, Mitchell MJ, Nakas JP (1982) Organic and inorganic sulfur constituents of a forest soil and their relationship to microbial activity. Soil Sci Soc Am J 46:847–852
Erkenberg A, Prietzel J, Rehfuess K-E (1996) Schwefelausstattung ausgewählter europäischer Waldböden in Abhängigkeit vom atmogenen S-Eintrag. Z Pflanz Bodenk 159:101–109
Ghani A, McLaren RG, Swift RS (1991) Sulphur mineralization in some New Zealand soils. Biol Fertil Soils 11:68–74
Hanousek O, Berger TW, Prohaska T (2016) MC ICP-MS δ34SVCDT measurement of dissolved sulfate in environmental aqueous samples after matrix separation by means of an anion exchange membrane. Anal Bioanal Chem 408:399–407
Havlin JL, Beaton JD, Tisdale SL, Nelson WL (2005) Soil fertility and fertilizers. Prentice Hall, Upper Saddle River
Holmgren GGS (1967) A rapid citrate–dithionite extractable iron procedure. Soil Sci Soc Am Proc 31:210–211
Johnson CM, Nishita H (1952) Microestimation of sulfur in plant materials, soils, and irrigation waters. Anal Chem 24:736–742
Kaliwoda M (2015) Soil acidification and nutrient status in the stemflow area of beech in the Kobernaußer Wald (Upper Austria). Master thesis, BOKU-University of Natural Resources and Life Sciences Vienna, Austria (in German)
Kazda M (1983) Schwermetalleintrag in das Buchenwaldökosystem des Wienerwaldes. Master thesis, BOKU-University of Natural Resources and Life Sciences Vienna, Austria
Kazda M, Glatzel G (1984) Schwermetallanreicherung und Schwermetallverfügbarkeit im Einsickerungsbereich von Stammablaufwasser in Buchenwäldern (Fagus sylvatica) des Wienerwaldes. Z Pflanz Bodenk 147:743–752
Koch AS, Matzner E (1993) Heterogeneity of soil and soil solution chemistry under Norway spruce (Picea abies Karst.) and European beech (Fagus sylvatica L.) as influenced by distance from the stem basis. Plant Soil 151:227–237
Kulhanek M, Cerny J, Balik J, Vanek V, Sedlar O (2011) Influence of the nitrogen–sulfur fertilizing on the content of different sulfur fractions in soil. Plant Soil Environ 57:553–558
Lawrence GP, Hazlett PW, Fernandez IJ, Ouimet R, Bailey SW, Shortle WC, Smith KT, Antidormi MR (2015) Declining acidic deposition begins reversal of forest-soil acidification in the northeastern US and eastern Canada. Environ Sci Technol 49:13103–13111
Likens GE, Driscoll CT, Buso DC, Mitchell MJ, Lovett GM, Bailey SW, Siccama TG, Reiners WA, Alewell C (2002) The biogeochemistry of sulfur at Hubbard Brook. Biogeochemistry 60:235–316
Lindebner L (1990) The status of forest soils in beech (Fagus sylvatica, L.) stands of the Vienna Woods, with special reference to changes due to the deposition of air pollutants. PhD thesis, BOKU-University of Natural Resources and Life Sciences Vienna, Vienna (in German)
Lowe LE (1965) Sulphur fractions of selected Alberta soil profiles of the chernozemic and podzolic orders. Can. J Soil Sci 45:297–303
Morche L (2008) S-fluxes and spatial alterations of inorganic and organic sulphur fractions in soil as well as their accumulation and depletion in the rhizosphere of agricultural crops by partial use of the radioisotope 35S. PhD thesis, Rheinische Friedrich-Wilhelms-Universität Bonn, Germany (in German)
Muras A (2012) Impact of declining emissions over the last 25 years on the soil status of beech stands in the Vienna Woods. Master thesis, BOKU-University of Natural Resources and Life Sciences Vienna, Austria (in German)
Prechtel A, Alewell C, Armbruster M, Bittersohl J, Cullen JM, Evans CD, Helliwell R, Kopáček J, Marchetto A, Matzner E, Meesenburg H, Moldan F, Moritz K, Veselý J, Wright RF (2001) Response of sulphur dynamics in European catchments to decreasing sulphate deposition. Hydrol Earth Syst Sci 5:311–325
Prietzel J, Hirsch C (1998) Extractability and dissolution kinetics of pure and soil-added synthesized aluminium hydroxy sulphate minerals. Eur J Soil Sci 49:669–681
Prietzel J, Hirsch C (2000) Ammonium fluoride extraction for determining inorganic sulphur in acid forest soils. Eur J Soil Sci 51:323–333
Prietzel J, Weick C, Korintenberg J, Seybold G, Thumerer T, Treml B (2001) Effects of repeated (NH4)2SO4 application on sulfur pool in soil, soil microbial biomass, and ground vegetation of two watersheds in the Black Forest/Germany. Plant Soil 230:287–305
Rampazzo N, Blum WEH (1992) Changes in chemstry and mineralogy of forest soils by acid rain. Water Air Soil Pollut 61:209–220
Rampazzo N, Schwertmann U, Blum WEH, Mentler A (1999) Effect of soil acidification on the formation of Fe-, Al-, and Mn-oxides and the stability of soil aggregates. Int Agrophys 13:283–291
Reuss JO, Johnson DW (1986) Acid deposition and the acidification of soils and water. Springer, New York
Shan X, Chen B, Jin L, Zheng Y, Hou X, Mou S (1992) Determination of sulfur fractions in soils by sequential extraction, inductively coupled plasma-optical emission spectroscopy and ion chromatography. Chem Spec Bioavailab 4:97–103
Sonderegger E (1981) Bodenschädigung durch sauren Stammablauf in Buchenbeständen der Flyschzone. Master thesis, BOKU-University of Natural Resources and Life Sciences Vienna, Austria
Tabatabai MA (1996) Sulfur. In: Sparks DL et al (eds) Methods of soil analysis. Part 3. Chemical methods. Book series number 5. Soil Science Society of America and American Society of Agronomy, Madison, pp 921–960
Tisdale SL, Nelson WL, Beaton JD, Havlin JL (1993) Soil fertility and fertilizers. Macmillan Publishing Company, New York
Umweltbundesamt (2015) Emissions trends 1990–2013: Ein Überblick über die Verursacher von Luftschadstoffen in Österreich (Datenstand 2015). Report REP-0543, ISBN 978-3-99004-354-7. Umweltbundesamt GmbH, Vienna
Vannier C, Guillet B (1994) Sulphur forms in the organic fractions of an upland forest soil (Mont Lozère, France). Soil Biol Biochem 26:149–151
Vannier C, Didon-Lescot JF, Lelong F, Guillet B (1993) Distribution of sulphur forms in soils from beech and spruce forests of Mont Lozère (France). Plant Soil 154:197–209
Watmough SA, Aherne J, Alewell C, Arp P, Bailey S, Clair T, Dillon P, Duchesne L, Eimers C, Fernandez I, Foster N, Larssen T, Miller E, Mitchell M, Page S (2005) Sulphate, nitrogen and base cation budgets at 21 forested catchments in Canada, The United States and Europe. Environ Monit Assess 109:1–36
Zhang Y, Mitchell MJ, Christ M, Likens GE, Krouse HR (1998) Stable sulfur isotopic biogeochemistry of the Hubbard Brook Experimental Forest, New Hampshire. Biogeochemistry 41:259–275
Acknowledgements
Open access funding provided by Austrian Science Fund (FWF). This study was funded by the Austrian Science Fund (FWF, Project Number P23861-B16) and the Commission for Interdisciplinary Ecological Studies (KIÖS) at the Austrian Academy of Sciences (Project Number 2010-05), both granted to T. W. Berger, and the FWF Project P23647, granted to T. Prohaska. The analyses of HI-reducible sulfur (HIS) were supported by the Ministry of Agriculture of the Czech Republic, Project Number QJ1530171. The determination of HCE (sulfate-S) by means of ion-exchange chromatography was supported by the Czech University of Life Sciences Prague (Internal Project Number CIGA 20152011). We thank Walter Wenzel for arranging the scientific research in Prague and Alexander Muras and Michael Kaliwoda for physically processing the soil samples. Franz Zehetner kindly supervised the dithionite- and oxalate extractions at the Institute of Soil Science (BOKU). Marcel Hirsch, Frauke Neumann, Sylvie Bonnet and Melanie Diesner thankfully supported performing the chemical analyses. We thank the forest owners (Austrian Federal Forests, City of Vienna) for the possibility to perform this research on their properties. Finally, we thank two excellent reviewers for their critical comments for the improvement of this paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Hanousek, O., Prohaska, T., Kulhanek, M. et al. Fractionation of sulfur (S) in beech (Fagus sylvatica) forest soils in relation to distance from the stem base as useful tool for modeling S biogeochemistry. Model. Earth Syst. Environ. 3, 1065–1079 (2017). https://doi.org/10.1007/s40808-017-0353-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40808-017-0353-5