
Abstract
Leveraging deep learning (DL) approaches in genomics data has led to significant advances in cancer prediction. The continuous availability of gene expression datasets over the preceding years has made them one of the most accessible sources of genome-wide data, advancing cancer bioinformatics research and advanced prediction of cancer genomic data. To contribute to this topic, the proposed work is based on DL prediction in both convolutional neural network (CNN) and recurrent neural network (RNN) for five classes in brain cancer using gene expression data obtained from Curated Microarray Database (CuMiDa). This database is used for cancer classification and is publicly accessible on the official CuMiDa website. This paper implemented DL approaches using a One Dimensional-Convolutional Neural Network (1D-CNN) followed by an RNN classifier with and without Bayesian hyperparameter optimization (BO). The accuracy of this hybrid model combination of (BO + 1D-CNN + RNN) produced the highest classification accuracy of 100% instead of the 95% for the ML model in prior work and 90% for the (1D-CNN + RNN) algorithm considered in the paper. Therefore, the classification of brain cancer gene expression according to the hybrid model (BO + 1D-CNN + RNN) provides more accurate and useful assessments for patients with different types of brain cancers. Thus, gene expression data are used to create a DL classification-based- hybrid model that will hold senior promise in the treatment of brain cancer.
Similar content being viewed by others


Explore related subjects
Find the latest articles, discoveries, and news in related topics.Avoid common mistakes on your manuscript.
Introduction
Understanding cancer gene expression data on patient survival is important to boost treatment decisions but also to improve cancer classification [1]. Thus, handling cancer gene expression data remains a major challenge and scientific efforts are needed to support the clinician in cancer diagnosis, cancer classification, and the outcome prognosis [2]. Gene expression in digital matrix format built from a DNA microarray sample obtained from transcribed DNA sequences in a high-density array on the side of a glass microscope. Also, microarray gene expression data consists of samples and features that employ classification efficiency to find the genomic biomarker [3]. The limitations of gene expression depend on a high level of noise due to a few dozen samples containing thousands of gene expressions that require the deletion of the irrelevant genes. Thus, many factors affected the classification accuracy of cancer gene expression such as memory requirements, generalization, and control for complexity.
Brain cancer is the most common fatal malignancy in children and adults, resulting in an incidence of 24,810 cases in the United States in 2023 [4]. Nevertheless, the presence of cancer in the brain leads to the initial stages of diagnosis being much more challenging [5]. Based on the WHO classification [6], the most prevalent form of primary brain cancer among these is glioma. According to their morphological appearance, gliomas are divided into several groups, including astrocytomas, oligodendrogliomas, ependymomas, and glioblastomas [7].
There are many frameworks [8] for brain cancer classification that are based on different types of databases such as magnetic resonance (MR) brain images, clinical data, and gene expression. The literature on brain cancer images is not accurate in classification research [9]. On the other hand, clinical data for brain cancer collected from the Surveillance, Epidemiology, and End Results (SEER) has a high accuracy for brain cancer survivability only, but may not be suitable for classification tasks [10]. In view of gene expression, Li et al. [11] suggested a classification model for different types of gliomas and confirmed that gene expression data is not suitable for brain cancer survivability.
The complex characteristics underlie brain cancer that can result from oncogenes and tumor-suppressor genes that have undergone genetic alteration, and mutation in gene expression [12]. Naqvi et al. [13] confirmed that the expression variations of certain genes may be linked to a higher risk of developing brain cancer. Therefore, it is important to continue to promote the design of innovative surrogate classifications and criteria for bioinformatics approaches to early diagnosis in brain cancer research.
Recently, publicly available cancer genomics data has given a more detailed genetic understanding of brain cancer and those significant classes. Zhao et al. [14] proposed a public resource for the brain cancer gene database (BCGene) which comprises 40 categories of brain cancer. Another research [15] used gene expression of glioblastoma as a predictive biomarker.
Based on gene expression data, machine learning (ML) has become a routine method for cancer prediction. DL is still relatively new for this purpose, therefore opinions on its effectiveness and usefulness are still divided [16]. Compared with ML, DL is an end-to-end approach that automatically extracts features from raw data without the need for preprocessing.
Although DL approaches have attained cutting-edge outcomes in cancer research and their implementation has become commonplace in the past years, few consider the use of cancer gene expression data in DL approaches because there is a lack of structure in gene expression data that makes it different from image or text [17]. Recently, CNN models have been applied for precise cancer-type prediction using gene expression data from The Cancer Genome Atlas (TCGA) [18].
Moreover, there are many attempts to create gene expression datasets such as inSilicodb [19], datamicroarray [20], BioLab [21], and AROMA [22]. In one of the modern attempts, Feltes et al. [23] constructed the Curated Microarray Database (CuMiDa) which consists of 78 gene expression datasets including 13 different human cancer types, and applied it to ML techniques. Noteworthy, the CuMiDa is the best one because it provides more file download options and cancer classification results using ML different approaches. This database is the first benchmark for cancer gene expression that serves as an important tool in the oncology community. Based on the analysis of CuMiDa, overexpression of the uncontrolled underlying mechanism is more closely associated with the tumorigenic process than underexpression of a specific gene cluster [24]. Also, re-analysis of gene expression data confirmed that there are unrecognized commonalities of gene expression among several cancer types. On the other hand, the relevance aggregation process [25] can be used to address the biased and poor algorithms on tabular data using breast cancer gene expression extracted from CuMiDa. It can be used to ensure that the performance of the different classifiers may be regularly improved by the higher-rated features.
In this paper, the proposed model exploited DL approaches including 1D-CNN and RNN classifiers on CuMiDa brain cancer gene expression data. It is clear that none of the previous DL studies evaluated different CNN /RNN model constructions and their effect on the CuMiDa brain cancer gene expression data. Indeed, we trained five classes in CuMiDa brain cancer gene expression data to perform the best classification accuracy using two DL approaches and compared their results with those of ML approaches as in [23]. The major contributions of this work are based on the development of RNN [26] and BO [27] hyperparameter with ID-CNN structure [28] for the classification of five classes of brain cancer genes.
The novelty of the proposed model is the first challenge using the combination of two DL architectures which are CNN and RNN on CuMiDa brain cancer gene expression data. Then, the evaluation criteria were compared on the test database for both (BO + 1D-CNN + RNN) and (1D-CNN + RNN) models to classify brain cancer categories. Also, the overall performance of the proposed model outperformed the previous studies based on ML [23].
Related works
Microarray cancer data is now widely used in computational and biological research using ML approaches that can assist with the diagnosis and predict various diseases [23]. Although the best ML classifier for Microarray cancer data is still up for debate [29], some research indicates that support vector machine (SVM) and random forest (RF) are the two most compelling options [30]. The clustering techniques are another application of ML on microarray data by aiding in the identification of new information about a certain set of genes by autonomously classifying samples based on the expression of those genes based on certain similarity criteria [31].
Even though ML approaches are still being utilized in cancer research, it is becoming more difficult to locate fresh databases that offer a suitable standard of cancer-focused microarray databases to be used for comparing or evaluating ML algorithms. It was recently argued that the development of bioinformatics depends on the appropriate use and construction of benchmarks for comparing the output of new tools [32]. However, the hybrid DL algorithm achieved a higher level of recognition accuracy compared with the existing techniques [33, 34].
As can be observed, all of the earlier gene expression datasets [19,20,21,22] were rather old and low-quality samples without any update while the latest one is CuMiDa which was released in 2019 with high-quality control and updates in different formats. Since all of the earlier datasets provided data that has already been carefully chosen and processed by the bioinformatics community, they do not provide a standard procedure for selecting and working with data sets [23]. Also, CuMiDa provided a public repository of cancer datasets only using most ML algorithms, making the CuMiDa database a useful addition to the existing databases.
Based on the recent microarray database [23], CuMiDa contains 13 cancer types and is applied to six ML strategies such as SVM, RF, k-nearest neighbors (k-NN), decision tree (DT), Naive Bayes (NB), and multilayer perceptron (MLP). We focused on dataset GSE50161 for brain cancer gene expression from CuMiDa with five classes, 54,676 genes, and 130 samples.
The comparison to existing ML methods on the CuMiDa brain cancer gene expression is presented to classify the five classes according to evaluation performance in terms of accuracy values [23]. The accuracy percentages in the testing process of six ML models were 95% for SVM, 82% for MLP, 81% for RF, 87% for KNN, and 85% for both DT and NB.
The weaknesses of previous studies were based on low classification accuracy using ML models, and each model required extensive preprocessing steps before being introduced into the classification process.
On the other hand, the strength of our study relied on the use of DL algorithms on a state-of-the-art microarray database to improve the accuracy of the classifier without any preprocessing steps. One advantage of our proposed model is to use the BO optimizer [27] to reduce the value of the model loss function by finding the optimal way to obtain the correct model parameters. The effectiveness of the proposed model was evaluated not only on the basis of accuracy but also on the basis of other performance standards.
Materials and methods
Proposed hybrid model
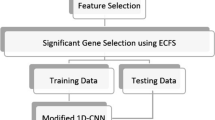
As shown in the graphical illustration in Fig. 1, we used a hybrid approach (1D-CNN + RNN) to classify the CuMiDa brain cancer-implicated genes [23]. The CuMiDa brain cancer dataset includes 4 distinct forms of brain cancer in addition to healthy tissue. Regarding this database, Ependymoma and glioblastoma are represented by 35%, and 26% of the whole database, respectively. All medulloblastoma, pilocytic astrocytoma, and normal are represented by 39% of the whole database.
The major steps of the proposed model include:
-
i.
The input gene expression dataset is divided into training, validation, and testing by 80%, 5%, and 15% respectively.
-
ii.
Bayesian model-based optimization is used to reduce the execution times of the objective function by using the optimal collection of hyperparameters to select the best classifier.
-
iii.
The proposed hybrid model (1D-CNN + RNN) is applied through training and validation sets.
-
iv.
The evaluation process was performed by using the test set that was classified using this best classifier.
-
v.
The feedback process was adopted to run the results back to the BO for the best results.

Block diagram for classification of CuMiDa brain cancer gene expression data using proposed hybrid model (BO + 1D-CNN + RNN)
CuMiDa brain cancer gene expression data
The CuMiDa [23] gene expression is an open resource that includes large-scale cancer microarray datasets curated from 30,000 Gene Expression Omnibus (GEO) studies.
As indicated in the CuMiDa, many classes had to be eliminated because they did not meet the minimal standard for ML approaches [24]. The CuMiDa dataset has an unbalance of samples and ML algorithms performed much worse with fewer samples. All datasets were carefully adjusted to remove incorrect probes before being manually corrected for background, normalized, and sample quality analysis.
In this paper, we used the brain cancer gene expression data obtained from CuMiDa which consists of five classes including ependymoma, glioblastoma, medulloblastoma, pilocytic astrocytoma, and normal. The CuMiDa brain cancer is represented in a CSV file using 130 cells (samples) in rows and 54,678 features (genes) in columns. This CuMiDa brain cancer is divided into 46 cells from ependymoma, 34 cells from glioblastoma, 22 cells from medulloblastoma, 15 cells from pilocytic astrocytoma, and 13 cells from normal. In this paper, this database was split into training, validation, and test sets as in Table 1.
According to previous ML results on CuMiDa brain cancer gene expression data [23], the highest accuracy was 95% using the SVM classifier.
Bayesian optimization (BO)
The hyperparameters of each class in the database are optimized using BO [27] that based on a probability model. It is utilized for cancer classification on gene expression data [35] to identify the optimal solution in less number of iterations than random algorithms. By selecting hyperparameters, the loss function is minimized by altering the network’s parameters during the training process. According to Koul et al. [35], using the BO approach to cancer gene expression gets closer to the optimal solution with each hyperparameter choice. The BO approach is a useful tool for cancer gene expression rather than the previous optimizer techniques. However, it needs significant computational time to provide a better group of hyperparameters.
Using objective function evaluations as training data, the BO maintains an internal Gaussian process model of the objective function as shown in Fig. 2. The acquisition function assesses the “quality” of each point based on the posterior distribution of the objective function [27].
For the hyperparameters tuning task, we used four parameters including Gated Recurrent Unit (GRU), fully connected layers (FC1, FC2), and bidirectional Long Short-Term Memory (BiLSTM). The values of four hyperparameters in the BO are shown in Table 2. We chose these four parameters because of the characteristics of our dataset which includes a time series with up to 50 iterations. Table 3 lists four hyperparameters for 1–5 iterations during the tuning task. Table 4 shows the best values for 15 iterations to optimize the learning process.

Layout of BO structure
The function of GRU on gene expression [36] is based on the association between data at the beginning and finish of the spectral series. GRU has used the update gate and reset gate to selectively filter and memorize historical data. In this work, GRU offers a simpler network with equivalent performance to solve the issue of vanishing gradients and preserves the impact of long-term dependencies as shown in Eqs. 1–4.
Where zt is a vector used for update gate with weight matrices Wz and Uz. While rt is the vector used for reset gate with weight matrices Wz and Uz. ⊙ stands for the element-wise multiplication and σ for the sigmoid function. The input at time step t is given by xt, the preceding hidden state by ht1, and trainable bias vectors by bz, br, and bh. The update gate determines if the hidden state is updated with a new hidden state\(\:\:{\stackrel{\sim}{\text{h}}}_{\text{t}}\), whereas the reset gate determines whether the previous hidden state ht1, is disregarded in this design.
The BiLSTM [37] is a sequential processing approach constructed out of two LSTMs, one of which accepts input in one direction and the other in the opposite. It is used to control the long-term dependency issues faced by RNNs. It successfully increases the amount of data that the network has access to, giving the algorithm greater context by using some gates. Instead of addressing each point in the sequence individually, LSTM can process whole sequences and store relevant information about earlier data points to aid in the processing of later data points.
The FC1 and FC2 have the same function which produces a temporal sequence of predicted parameters. The Eq. 5 of the FC layer [38] is depicted as follows.
Where w, σ, and \(\:{\:h}_{i}^{l-i}\)are expressed as the weight of the ith node for layer l − 1 and the jth node for layer, nonlinear activation function, and bias, respectively.
This pilot study lists finding high-performance hyperparameter choices by using the BO method, along with its performance comparison without BO. Therefore, we take advantage of the selected four hyperparameters to refine the brain cancer classification model on CuMiDa gene expression data.
Hybrid model
In this paper, the hybrid model consists of 1D-CNN and RNN architecture with 18 layers. According to Fig. 1, the sequence layer, two 1D-CNN layers, two normalization layers, two Rectified Linear Unit (ReLU) layers, two BiLSTM layers, two dropout layers, average global, GRU layer, FC layers, softmax layer, and classification layer are utilized to classify brain cancer types extracted from CuMiDa gene expression data.
Initially, the output of the sequence layer is passed to the 1D-CNN layer and the output of the 1D-CNN layer is passed to the ReLU layer. The two ReLU layers help to stop the occurrence of the vanishing gradient” problem that frequently occurs when using sigmoidal functions. Also, the ReLU layer pooled as much data as possible because the FC layer can erase spatial characteristics and have an unfavorable impact on anticipated results.
Then, the output of the ReLU layer is passed to the normalization layer. The normalization layer can restrict the processed data to a certain range, thus eliminating the dimensional effect between each data point and facilitating the training of the model.
In the training process, the two dropout layers are used for preventing overfitting while two BiLSTM layers are used for learning representations. The dropout value was set to 0.25 as widely used in previous work [39] to solve the overfitting problem. The inclusion of the dropout layer in the 1D-CNN model successfully increases the model’s capacity for generalization and enhances the accuracy of cross-load learning. In this model, the output of the GRU layer is passed to the BiLSTM layer. Next, the dropout layer receives the output of the BiLSTM layer as in Fig. 1. The dropout layer is a mask that preserves the functionality of all neurons while removing some neurons’ contributions to the subsequent layer.
The FC layer is used to transform the sequential output into a fixed-length representation suitable for the classification process. The FC layer is carried out by the classification of the training sets and test sets. Then, the outputs of FC are fed into the softmax layer and classification layer. Finally, the retrieved results from the classification process are subsequently passed into the BO structure to improve the model performance.
Following the hybrid model, 1D-CNN and RNN architectures are employed, where manual feature selection that caused some information loss is not performed. It has been observed that the hybrid model yielded better results on a larger dataset than methods in the previous studies. Therefore, the hybrid model is appropriate for the classification of CuMiDa brain cancer gene expression data. This is also demonstrated by the improved performance after BO optimization in the hybrid model.
One dimensional- convolutional neural network (1D-CNN)
CNN is a DL model built primarily using convolutional filters. It consists of convolutional, pooling layers and additional regularization layers such as batch normalization or dropout layers. A CNN-based model for gene expression analysis was described by Xiao et al. [40] to classify cancer types with a precision of 98% when applied to three publicly available datasets. Elbashir et al. [41] suggested CNN architecture for breast cancer classification employing gene expression data with a precision of 98.76%. 1D-CNN model [28] is one type of CNN architecture that works well with time-series data and has shown a lot of promise for numerical classification tasks. The 1D-CNN is substantially simpler because it only has one convolutional layer and does not demand a specific order for the inputs. 1D-CNN consists of a vector input layer, convolutional layer, and FC layer.
In this model, the input for 1D-CNN is a sequence layer as shown in Fig. 1. The RNN is preprocessed using 1D-CNN to create shorter sequences of higher-level features from an input sequence.
Recurrent neural network (RNN)
RNN [26] is based on consecutive activities which provides a memory function that allows it to use previous observations to comprehend the present observation or forecast upcoming observations in an input sequence. RNN forms three layers including input, hidden, and output. Using a recurrent connection, the hidden layer receives both information about its current inputs at time step t and details about its previous hidden state at time step t-1 as follows: h(t) = θ(xt, ht−1). Where θ is a non-linearity function such as tanh or sigmoid.
The RNN, in contrast to other kinds of neural networks, shares the same parameters throughout all phases, which reduces the number of parameters the networks require to learn. It is simple to train the RNN to produce the cancer classification accurately in sequential data [42]. RNN model [26] on gene expression data used to determine the transcriptional target factor. In gene expression analysis, the RNN model provides some benefits because it increases model effectiveness by allowing the model to recognize and remember sequential feature information [43]. On the other hand, there are some drawbacks of RNN architecture related to gene expression analysis, including longer processing times than CNNs and other comparable techniques. It leads to slower, more complicated training procedures and a worse capacity to capture relationships in larger genomic sequences than other neural networks.
Evaluation process
The effectiveness of the DL models’ design for various data sets is an essential problem. This study evaluates the effectiveness of the created methodology and other comparison methods by using five metrics including accuracy, sensitivity, specificity, precision, and F1-score as shown in Eqs. 6–10.
According to the literature [44, 45], the confusion matrix and these five performance metrics are popular in multiclass classification tasks.
Where C is five classes. TP stands for true positive for correctly diagnosed cancer cells, TN stands for true negative for correctly diagnosed normal cells, FN stands for false negative for incorrectly diagnosed normal cells and FP stands for false positive for erroneously diagnosed cancer cells.
Results
This study is representative of a hybrid model to aid in the classification of disease-related genomic data. Overall, there are certain restrictions on the gene expression database, but these can be circumvented by the DL model with proper hyperparameter adjustment using optimization.
This proposed model is the first combination of (BO + 1D-CNN + RNN) on five classes of CuMiDa brain cancer gene expression [23]. In this paper, CuMiDa brain cancer is divided into 80% of training sets, 15% of test sets, and 5% of validation sets. A matrix of gene expression values from the CuMiDa database, with each column denoting a feature and each row denoting a cell, serves as the model’s input. We applied the two hybrid architectures to the CuMiDa brain cancer database to classify the brain cancer types. For implementation, the suggested model was executed on 5 GB RAM GPU and MATLAB 2022b software.
In the initial step, we used a BO [27] structure to thoroughly examine four hyperparameter combinations of the number of GRU-FC1-BiLSTM-FC2 layers. When dealing with sequence data, it is common to use GRU and BiLSTM to capture temporal dependencies and improve sequence processing with an RNN model [38]. We adjusted each hyperparameter separately while maintaining the values of the other hyperparameters among the tuning of hyperparameters. We assessed the performance change concerning the change in each hyperparameter by recurring the training process. The optimization result for the best values of hyperparameters at 15 iterations is shown in Fig. 3.

The relation between minimum objective and number of function evaluations for 15 iterations during the optimization process
In the second step, we performed two hybrid models on the CuMiDa brain cancer database by using the training and validation sets. The first hybrid model consists of (BO + 1D-CNN + RNN) while the second hybrid model consists of (1D-CNN + RNN) without BO structure.
Performance of CuMiDa brain cancer database in a hybrid model with/without BO
By using the (1D-CNN + RNN) model, the number of hidden layers is fixed in hyperparameters, where GRU, BiLSTM, FC1, and FC2 are 1000, 1000,100, and 50 respectively.
By using the (BO + 1D-CNN + RNN) model, the number of hidden layers changes in hyperparameters according to the BO structure to select the best values for hyperparameters. Afterward, we test the output of the classification layer by using five metrics.
Based on the hyperparameters chosen, the classification results of the hybrid model (BO + 1D-CNN + RNN) are better than the existing ML model on the CuMiDa brain cancer database. Thus, we report the BO method substantially influences classification success on the CuMiDa brain cancer database. This (BO + 1D-CNN + RNN) architecture outperforms the (1D-CNN + RNN) architecture during the training process as demonstrated in Figs. 4 and 5.
On the other hand, it was noted that the hybrid model (1D-CNN + RNN) performed poorly in the CuMiDa brain cancer database. Meanwhile, the hyperparameter optimization values at various time steps were suitably changed to make sure that the final output model is more similar to the ideal model.
Comparison of CuMiDa performance with other models
In this work, we compare the performance of CuMiDa brain cancer gene expression against two hybrid models (BO + 1D-CNN + RNN) and (1D-CNN + RNN) in terms of accuracy, sensitivity, specificity, precision, and F1-score.
Table 5 lists the performance comparison of two hybrid architectures on test sets from the CuMiDa brain cancer database by using five metrics. Compared with them, the accuracy of the (BO + 1D-CNN + RNN) and (1D-CNN + RNN) are improved by 100% and 90% respectively. As depicted from the confusion matrix in Fig. 6, the accuracy of (BO + 1D-CNN + RNN) architecture on ependymoma, glioblastoma, medulloblastoma, pilocytic astrocytoma, and normal cells extracted from the CuMiDa brain cancer database is achieved 100%.
On the other hand, we applied the hybrid model to another cancer type e.g. prostate cancer gene expression extracted from the CuMiDa database. The prostate cancer gene expression results were observed to be 83.3% accuracy, 90% sensitivity, 81.8% precision, and 85.7% F1-score. This means that the proposed model on prostate cancer gene expression achieved the best accuracy instead of ML results as computed in [23]. This indicates that there is generalization of this hybrid model to any other cancer data.
To the best of our information, there is no DL model applied to the CuMiDa brain cancer gene expression database. As aforementioned, we leveraged the CuMiDa brain cancer database (features, cells) learned from the previous ML models [23] to classify brain cancer genes based on a hybrid model that consists of RNN coupled with 1D-CNN and BO. The empirical results reveal the importance of the BO method in accurately classifying the CuMiDa brain cancer gene expression database.

Layout of training progress using (BO + 1D-CNN + RNN) architecture on CuMiDa brain cancer gene expression database

Layout of training progress using (1D-CNN + RNN) architecture on CuMiDa brain cancer gene expression database

Confusion matrix of (BO + 1D-CNN + RNN) architecture on test data extracted from CuMiDa brain cancer gene expression
Discussion
The patients’ molecular profiles are extensively generated using gene expression technology. One of the major difficulties is creating new tools for gene expression interpretation, particularly in oncology. There is proof that genetic factors are associated with childhood and family-related cancers [46]. Only a small number of experimental studies have assessed DL approaches in gene expression analysis and contrasted them with cutting-edge ML [47]. Although the DL approach has been successful in other fields, it hasn’t been fully investigated in gene expression cancer [48]. The problem of gene expression is based on the small number of samples but a large number of features [49]. Recently, the CuMiDa gene expression database has been enriched in diseases related to different types of cancer such as breast, brain, prostate, ovary, liver, bladder, lung, throat, renal, and leukemia.
The main concern with using a gene expression database is high-dimensional data, repetitive features, and weekly irrelevant/uninformative features that lead to overfitting during the training process. This paper discusses a solution to this problem without selecting genes, which takes a lot of processing time to detect important feature sets in a gene expression database. Also, another problem is that the gene expression database is applied to non-open access and incorrect data.
In this paper, the proposed model used the CuMiDa brain cancer gene expression database to distinguish four types of classes (ependymoma, glioblastoma, medulloblastoma, pilocytic astrocytoma) against the normal class. The number of samples in the CuMiDa brain cancer dataset is comparatively low for a DL-based model to train correctly without any overfitting problems. Also, this data has class imbalance problems. To handle these issues, we employed a hybrid model (BO + 1D-CNN + RNN) to achieve higher accuracy rather than the previous study based on an ML model on the same dataset [23].
For the optimization task, The BO [50] is implemented to find the best values of four selected hyperparameters including GRU, FC1, BiLSTM, and FC2 layers. GRU performs admirably while processing sequences [51]. In short, BiLSTM indicates that the input sequence flows backward in the additional LSTM layer. Following that, the outputs from the two LSTM layers are merged using a variety of techniques to convey information from both the past and the present [52].
For the hybrid model, the 1D-CNN network learns regional patterns sequentially. During sequence processing, a 1D- CNN layer can compete with RNN at a significantly lower computing cost by detecting local patterns in sequences. The RNN classifier can theoretically store information in any long sequences. However, it was restricted to a few steps of glancing back.
The results of applying the two hybrid models with and without BO on the CuMiDa brain cancer database for the classification task are discretized in Table 5. The test results confirmed that the accuracy, sensitivity, specificity, precision, and F1-score were achieved 100% using the proposed (BO + 1D-CNN + RNN) model on the CuMiDa brain cancer database. On the other hand, the accuracy, sensitivity, specificity, precision, and F1-score were achieved as 90%, 91.52%, 97.70%, 86%, and 86.16% respectively using the proposed (1D-CNN + RNN) model on CuMiDa brain cancer database as shown in Table 5.
In two experiments, the performance of two hybrid models was compared with 15% of test sets by running 100 epochs with 15 batch sizes and 600 iterations using the Adam optimizer. It should be mentioned that the learning rate is set with a value of 0.001 during the training process to improve model performance. For comparison with the previous model [23], the classification accuracy was 95% using the SVM classifier on the CuMiDa brain cancer database as outlined in Table 5. Therefore, the proposed (BO + 1D-CNN + RNN) model is superior to the previous model [23] for classifying the CuMiDa brain cancer database.
It was noted that as the number of iterations increases, the training loss of the (BO + 1D-CNN + RNN) model decreases faster than that of the other (1D-CNN + RNN) model. Thus, the execution time is 4 min for the (BO + 1D-CNN + RNN) model and 12 min for the (1D-CNN + RNN) model as shown in Figs. 4 and 5. Although the RNN model has a long execution time which leads to a slower and more complex training process, the RNN model with BO achieved faster time instead of just RNN.
It is observed that the work of hybrid models on gene expression databases is much less when compared to ML and DL models. Apparently, it needs more developments from researchers and biologists to collect and analyze more databases related to gene expression.
In general, it has been pointed out that the performance of classification models depends on specific model-specific databases. Moreover, this particular model cannot be deduced as the best model for all types of databases.
For Gene expression-based CuMiDa brain cancer classification, the ML model is presented in [23] with 95% accuracy. However, our proposed hybrid model focuses on DL and BO with 100% accuracy which opens a new avenue for cancer classification. The BO is used in this case to close the gap between the difficulty of hyperparameters adjusting and a computational optimization approach. Finally, the hybrid model has a good performance in terms of classification accuracy and execution time for handling the gene expression database. Our model’s strengths are as follows:
-
(1)
High accuracy: The hybrid model (BO + 1D-CNN + RNN) showed consistently high accuracy on CuMiDa brain cancer gene expression. The accuracy exceeded previous models’ by a significant margin, confirming the effectiveness of BO in fine-tuning the models’ parameters to achieve more accurate predictions.
-
(2)
Performance metrics: Using the five metrics including accuracy, sensitivity, specificity, precision, and F1-score directly affects a hybrid model’s effectiveness.
-
(3)
Optimized hyperparameters: Using BO to optimize hyperparameters is crucial to the performance of our model. This improves the discriminative capacity of our model by enabling it to fine-tune its configurations to extract the most prominent hyperparameters from the data.
-
(4)
Sturdy generalization: The DL model showed excellent generalization across the CuMiDa brain cancer dataset, indicating that it can efficiently manage different patterns and adjust to a variety of network conditions. Models’ performance may be not influenced by the particular features of the dataset during the training process.
Furthermore, the following list represents our model’s limitations:
-
(1)
Computational difficulty: When using BO, there is a certain amount of computational complexity involved, particularly in the hyperparameter optimization process.
-
(2)
Data imbalance: Class imbalance occurs when there are substantially fewer samples for a disease than other samples for another disease in a dataset. Although our models demonstrate resilience to this obstacle.
Conclusions
The applications of DL have multiplied recently in many areas of biological research. However, the use of the DL models on gene expression data remains a technological difficulty due to their extensive layers, functional components, and operations. To overcome this challenge, we developed an RNN model by fusing it with BO + 1D-CNN architecture which aims to successfully classify the CuMiDa brain cancer database. We showed how the optimization step can be used to improve the hybrid model performance. Finally, we anticipate that the hybrid model of (BO + 1D-CNN + RNN) will provide insight into the classification of other cancer types obtained from the gene expression database among DL models.
References
Tao Z, Shi A, Li R et al (2017) Microarray bioinformatics in cancer—A review. J BUON 22:838–843
Gupta S, Gupta MK, Shabaz M, Sharma A (2022) Deep learning techniques for cancer classification using microarray gene expression data. Front Physiol 13:952709
Netzer M, Baumgartner C, Baumgarten D (2022) Predicting prediction: a systematic workflow to analyze factors affecting the classification performance in genomic biomarker discovery. PLoS ONE 17(11):e0276607
Siegel RL, Miller KD, Wagle NS, Jemal A (2023) Cancer statistics, 2023. CA Cancer J Clin 73(1):17–48
Müller Bark J, Kulasinghe A, Chua B, Day BW, Punyadeera C (2020) Circulating biomarkers in patients with Glioblastoma. Br J Cancer 122(3):295–305
Rodriguez FJ (2022) The WHO classification of tumors of the central nervous system-finally here, and welcome. Brain Pathol 32:e13077
Gladson CL, Prayson RA, Liu WM (2010) The pathobiology of glioma tumors. Annu Rev Pathol Mech Dis 5:33–50
Gopal Nath A, Coursey Y, Li S, Prabhu H, Garg SC, Halder (2023) Saptarshi Sengupta, an interactive web-based tool for predicting and exploring brain cancer survivability. Healthc Analytics 3:100132
Kickingereder P, Bonekamp D, Nowosielski M, Kratz A, Sill M, Burth S, Wick A, Eidel O, Schlemmer H-P, Radbruch A, Debus J, Herold-Mende C, Unterberg A, Jones D, Pfister S, Wick W, von Deimling A, Bendszus M, Capper D (2016) Radiogenomics of glioblastoma: machine learning–based classification of molecular characteristics by using multiparametric and multiregional MR imaging features. Radiology 281(3):907–918
Liu H, Qin X, Zhao L, Zhao G, Wang Y (2021) Epidemiology and survival of patients with brainstem gliomas: a population-based study using the SEER database. Front Oncol 11
Li A, Walling J, Ahn S, Kotliarov Y, Su Q, Quezado M, Oberholtzer JC, Park J, Zenklusen JC, Fine HA (2009) Unsupervised analysis of transcriptomic profiles reveals six glioma subtypes. Cancer Res 69(5):2091–2099
Silantyev AS, Falzone L, Libra M, Gurina OI, Kardashova KS, Nikolouzakis TK et al (2019) Current and future trends on diagnosis and prognosis of Glioblastoma: from Molecular Biology to Proteomics. Cells 8:8
Naqvi AZ, Mahjabeen I, Ameen S, Ahmed MW, Khan AU, Akram Z, Kayani MA (2020) Genetic and expression variations of cell cycle pathway genes in brain tumor patients. Biosci Rep 40(5):BSR20190629
Zhao M, Liu Y, Ding G et al (2021) Online database for brain cancer-implicated genes: exploring the subtype-specific mechanisms of brain cancer. BMC Genomics 22:458
Li Q, Aishwarya S, Li J-P, Pan D-X, Shi J-P (2022) Gene expression profiling of Glioblastoma to recognize potential biomarker candidates. Front Genet 13:832742
Hanczar B, Bourgeais V, Zehraoui F (2022) Assessment of deep learning and transfer learning for cancer prediction based on gene expression data. BMC Bioinformatics 23:262
Danaee P, Ghaeini R, Hendrix DA (2017) A deep learning approach for cancer detection and relevant gene identification. In: Pacific symposium on biocomputing 2017. World Scientific; pp. 219–29
Mostavi M, Chiu Y-C, Huang Y, Chen Y (2020) Convolutional neural network models for cancer type prediction based on gene expression. BMC Med Genomics 13(5):1–13
Taminau J, Steenhoff D, Coletta A et al (2011) Insilicodb: an r/bioconductor package for accessing human affymetrix expert-curated datasets from geo. Bioinformatics 27:3204–3205
https://www.rdocumentation.org/packages/datamicroarray/versions/0.2.3
Mramor M, Leban G, Demsˇar J et al (2007) Visualization-based cancer Microarray data Classif Anal Bioinf 23:2147–2154
An open-source r framework for your microarray analysis (2018) http://www.aroma-project.org/
Feltes BC, Chandelier EB, Grisci BI, Dorn M (2019) Cumida: an extensively curated microarray database for benchmarking and testing of machine learning approaches in cancer research. J Comput Biol 26:376–386
Feltes BC, Poloni JF, Nunes IJG, Faria SS, Dorn M (2020) Multi-approach Bioinformatics Analysis of Curated Omics Data provides a gene expression Panorama for multiple Cancer types. Front Genet 11:586602
Grisci BI, Krause MJ, Dorn M (2021) Relevance aggregation for neural networks interpretability and knowledge discovery on tabular data, Information sciences. 559:111–129
Zhao Y, Joshi P, Shin D-G, Recurrent Neural Network for Gene Regulation Network Construction on Time Series Expression Data, 2019 IEEE International Conference on Bioinformatics and, Biomedicine (2019) (BIBM), San Diego, CA, USA, pp. 610–615, https://doi.org/10.1109/BIBM47256.2019.8983068
Snoek J, Larochelle H, Adams RP (2012) Practical Bayesian optimization of machine learning algorithms. In: NIPS’12: Proceedings of the 25th International Conference on Neural Information Processing Systems. 2:2951–2959
Shang H, Shang L, Wu J, Xu Z, Zhou S, Wang Z, Wang H, Yin J (2023) NIR spectroscopy combined with 1D-convolutional neural network for breast cancerization analysis and diagnosis. Spectrochim Acta Mol Biomol Spectrosc 287(Pt 1):121990
Allison DB, Cui X, Page GP et al (2006) Microarray data analysis: from disarray to consolidation and consensus. Nat Rev Genet 7:55
Statnikov A, Wang L, Aliferis CF (2008) A comprehensive comparison of random forests and support vector machines for microarray-based cancer classification. BMC Bioinformatics 9:319
Whitworth GB (2010) An introduction to microarray data analysis and visualization, 19–50. Methods in Enzymology, vol 470. Elsevier, San Francisco, CA
Peters B, Brenner S, Wang E et al (2018) Putting benchmarks in their rightful place: the heart of computational biology. PLoS Comput Biol 14, e1006494
Chakravarthi B, Ng SC, Ezilarasan MR, Leung MF (2022) EEG-based emotion recognition using hybrid CNN and LSTM classification. Front Comput Neurosci 16:1019776
Song X, Wu N, Song S, Zhang Y, Stojanovic V Bipartite synchronization for cooperative-competitive neural networks with reaction–diffusion terms via dual event-triggered mechanism, Neurocomputing,550,2023,126498.
Koul N, Manvi SS (2021) Framework for classification of cancer gene expression data using bayesian hyper-parameter optimization. Med Biol Eng Comput 59:2353–2371
Yang B, Liu F, Ren C, Ouyang Z, Xie Z, Bo X et al (2017) BiRen: predicting enhancers with a deep-learning-based model using the DNA sequence alone. Bioinformatics 33(13):1930–1936
Li Y, Wang L (2022) Human Activity Recognition Based on Residual Network and BiLSTM. Sensors 22:635
Fazlipour Z, Mashhour E, Joorabian M (2022) A deep model for short-term load forecasting applying a stacked autoencoder based on LSTM supported by a multi-stage attention mechanism. Appl Energy 327:120063
Srivastava N, Hinton G, Krizhevsky A, Dropout (2014) A simple way to prevent neural networks from overfitting. J Mach Learn Res 15:1929–1958
Xiao Y, Wu J, Lin Z, Zhao XA (2018) Deep learning-based Multi-model Ensemble Method for Cancer Prediction. Comput Methods Programs Biomed 153:1–9
Elbashir MK, Ezz M, Mohammed M, Saloum SS (2019) Lightweight convolutional neural network for breast Cancer classification using RNA-Seq gene expression data. IEEE Access 7:185338–185348
Chen L, Pan X, Zhang YH, Liu M, Huang T, Cai YD (2018) Classification of widely and rarely expressed genes with recurrent neural network. Comput Struct Biotechnol J 17:49–60
Chowdhury S, Dong X, Li X (2019) Recurrent Neural Network Based Feature Selection for High Dimensional and Low Sample Size Micro-Array Data. In Proceedings of the 2019 IEEE International Conference on Big Data (Big Data), Los Angeles, CA, USA, 9–12 December ; pp. 4823–4828
Hossin M, Sulaiman M (2015) A review on evaluation metrics for data classification evaluations. Int J Data Min Knowl Manag Process 5(2):1
Chen D, Lu Y, Hsu C-Y (2022) Measurement Invariance Investigation for Performance of Deep Learning Architectures, in IEEE Access, vol. 10, pp. 78070–78087, https://doi.org/10.1109/ACCESS.2022.3192468
Garber JE, Offit K (2005) Hereditary Cancer Predisposition syndromes. J Clin Oncol 23(2):276–292 pmid:15637391
Mazlan AU, Sahabudin NA, Remli MA, Ismail NSN, Mohamad MS, Nies HW, Abd Warif NB (2021) A review on recent progress in machine learning and deep learning methods for cancer classification on gene expression data. Processes 9(8):1466
Oyewola DO, Dada EG, Omotehinwa TO, Emebo O, Oluwagbemi OO (2022) Application of deep learning techniques and bayesian optimization with Tree Parzen Estimator in the classification of Supply Chain pricing datasets of Health medications. Appl Sci 12:10166
Ravindran U, Gunavathi C (2023) A survey on gene expression data analysis using deep learning methods for cancer diagnosis. Prog Biophys Mol Biol 177:1–13
Wu J, Chen X, Zhang H, Xiong L, Lei H, Deng S (2019) Hyperparameter optimization for machine learning models based on bayesian optimization. J Electron Sci Technol 17(1):26–40
Le NQK, Yapp EKY, Yeh H-Y (2019b) Et-gru: using multi-layer gated recurrent units to identify electron transport proteins. BMC Bioinform 20:377
Xu G, Meng Y, Qiu X, Ziheng Yu, Xu, Wu (2019) Sentiment Analysis of Comment Text Based on BiLSTM, IEEE Access, vol. 7, Mar 31. pp. 51522–51532
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Afify, H., Mohammed, K. & Hassanien, A. Leveraging hybrid 1D-CNN and RNN approach for classification of brain cancer gene expression. Complex Intell. Syst. (2024). https://doi.org/10.1007/s40747-024-01555-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40747-024-01555-4