
Abstract
Introduction
Etanercept, a tumor necrosis factor inhibitor, is used to treat rheumatoid arthritis (RA) and psoriatic arthritis (PsA), and is administered via subcutaneous injection. Injection site pain (ISP) associated with subcutaneous administration may affect compliance or hinder initiation of prescribed medications. To improve the patient experience, a new phosphate-free formulation of etanercept was evaluated for reduced ISP associated with administration.
Methods
This phase 3b, multicenter, randomized, double-blind, cross-over study compared the prior formulation of etanercept to a phosphate-free formulation. Etanercept-naïve adults with RA or PsA indicated for treatment with etanercept were eligible. Patients were randomized (1:1) to receive both etanercept formulations (50 mg) in one of two crossover sequences: prior formulation followed by phosphate-free formulation (sequence AB) or phosphate-free formulation followed by prior formulation (sequence BA) at visits 1 week apart. Patients self-reported ISP using a fit-for-purpose 100-mm visual analog scale within 30 s after injection. Safety outcomes included incidence of treatment-emergent adverse events. Mixed-effects analysis of variance model was used to assess ISP, with treatment, study period, sequence, and disease indication as fixed-effect covariates and patient-within-sequence as random effect.
Results
A total of 111 patients enrolled (56 sequence AB; 55 sequence BA). Mean ISP score for prior formulation was 23.1 mm and for phosphate-free formulation was 19.1 mm (mean difference − 4 mm; 95% confidence interval: − 8.0, 0.0; P = 0.048). Patients with the highest ISP scores from the prior formulation (by quartile cut points) had the largest reduction in pain with phosphate-free formulation. Injection site reactions were few in number and similar between formulations; no new safety signals were observed.
Conclusions
The new phosphate-free formulation of etanercept had statistically significantly lower mean pain scores than the prior formulation, with largest pain reductions observed among patients who reported highest pain with the prior formulation.
Trial Registration
ClinicalTrials.gov: NCT02986139.
Funding
Amgen Inc, Thousand Oaks, CA USA.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a systemic, inflammatory, autoimmune disease that causes joint damage and synovitis, and affects approximately 1% of the global population [1]. Psoriatic arthritis (PsA) is a form of inflammatory arthritis with heterogeneous presentation, including peripheral arthritis, axial disease, enthesitis, dactylitis, psoriasis, and nail disease, which affects approximately 0.25% of the global population with regional variations [2]. Disease-modifying antirheumatic drugs (DMARDs) are used to treat both RA [3] and PsA (after failure of nonsteroidal anti-inflammatory drugs [NSAIDs] and intra-articular glucocorticoid injections in patients with PsA) [4]. Tumor necrosis factor inhibitors (TNFis) are a class of biologic DMARDs that have been shown to be effective for the treatment of RA and PsA [5]. Etanercept is a dimeric fusion protein comprising the extracellular ligand-binding portion of the human 75 kD (p75) TNF receptor linked to the Fc portion of human immunoglobulin G1 [6]. Etanercept has been shown to be efficacious for the treatment of moderate-to-severe RA and active PsA and to inhibit long-term radiographic progression of both diseases [7,8,9]. Etanercept is approved in the US for the treatment of moderate-to-severe RA and active PsA [6].
Protein-based medications are not compatible with oral administration [10], and etanercept is therefore administered via subcutaneous (SC) injection [6]. SC administration is an effective method for delivering medications slowly and at a sustained rate, as fatty SC tissue allows uptake of large proteins via the lymphatic system [10, 11]. Injection of medications into the SC space can be associated with local pain and irritation from the needle puncture, the composition of the medication excipients and formulation, volume of medication administered, and location of the injection site [10, 12, 13]. Patients may have individual reasons for discomfort with SC-administered medications, such as needle phobia [14]. Injection site reactions have been reported at various rates in clinical trials of all current biologic medications [15].
Injection-related pain has been shown to contribute to poor compliance with drug administration. For example, 41% of patients with RA reported the injection experience as the primary or secondary reason for discontinuing biologic DMARD therapy in an Internet-based survey [16]. Minimizing the pain and discomfort associated with injections is therefore important to improve medication compliance and the patient experience.
In the interest of improving the patient experience with etanercept, a new phosphate-free etanercept formulation was engineered. This double-blind, crossover study was designed to investigate the injection site pain (ISP) experience with the new formulation vs. the prior formulation of etanercept in patients with RA or PsA. ISP associated with the formulations was assessed and described by disease indication. In addition, the safety of both formulations of etanercept was evaluated.
Methods
Study Design and Outcomes
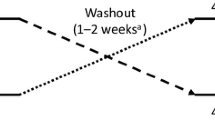
This was a phase 3b, multicenter, randomized, double-blind, crossover study. The study included a screening period up to 14 days, a treatment period of 2 weeks, and a 30-day safety follow-up period (Fig. 1). Blood samples were obtained during the screening period to determine eligibility. Treatment A comprised a single SC 50-mg dose of the prior formulation of etanercept and Treatment B comprised a single SC 50-mg dose of the new phosphate-free formulation; both formulations were administered in a prefilled SureClick® autoinjector. In sequence AB, patients received Treatment A during the first period, followed by Treatment B during the second period. In sequence BA, patients received Treatment B during the first period and Treatment A during the second period. Prior to administration, patients were required to demonstrate competency to correctly self-administer the SC dose. At each study visit, patients administered the blinded treatment in the presence of the investigator. Injections were performed in the thigh or abdomen; if the first injection was given in the thigh, the second injection was performed on the contralateral limb.

Study schema. ISP injection site pain, SC subcutaneous, sequence AB treatment A followed by sequence B, sequence BA sequence B followed by sequence A, VAS visual analog scale
The outcome of ISP severity was measured using a fit-for-purpose single-item visual analog scale (VAS) administered immediately (within 30 s) following the patient injection. The item was developed for content validity through qualitative interviews and a cognitive debriefing process. Existing general pain items were used to develop the ISP item. Fourteen adult patients participated in individual interviews, with a mean time of 1 week since receiving an injection. Four sequential groups reviewed a draft ISP item that was refined and then retested in subsequent interviews. Revisions included item wording, instructions, highest and lowest scale descriptors, and some of the graphic depictions. The final item was a 100-mm VAS specific to severity of ISP from a recent injection. Patients indicated the severity of their ISP on the VAS with 0 mm indicating “No Pain At All” and 100 mm indicating “Worst Pain Imaginable.” Content validity was demonstrated based on qualitative studies.
Safety was assessed throughout the study. A safety follow-up call was performed 30 days after the last dose of etanercept. After study completion, patients were allowed to continue with the prior formulation of etanercept. Safety outcomes included the incidence of all treatment-emergent adverse events (AEs). AEs were coded using the Medical Dictionary for Regulatory Activities (MedDRA) version 20.0, and severity of AEs was graded using the Common Terminology Criteria for Adverse Events (CTCAE) version 4.0.
This study was approved by Schulman Associates IRB (Cincinnati, OH). All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study. This study was registered under ClinicalTrials.gov identifier NCT02986139.
Patients
Men or women ≥ 18 years of age with a diagnosis of RA or PsA who were indicated for treatment with etanercept according to the US prescribing information but were naïve to etanercept were eligible to participate in this study. Patients were excluded if they had used a biologic DMARD ≤ 3 months prior to screening; had skin conditions that would interfere with evaluations, active infections, history of tuberculosis infection, recent history of malignancy, or concomitant medical condition. Patients were also excluded if they were receiving continuous treatment with acetaminophen, NSAIDs, or tramadol, hydrocodone, oxycodone, codeine, and/or propoxyphene and received doses within 4 h before study visit and the dose was not stable for ≥ 2 weeks before first dose of etanercept. At least 20 patients with PsA were planned to be enrolled in the study.
Randomization and Blinding
Randomization was performed using an interactive voice/web response system (IVRS). An unblinded vendor created the patient list, created blocks with strata using a stratified fixed permuted block randomization, and assigned treatment based on the treatment ratio within blocks. Treatment was assigned by the IVRS vendor in sequence to maintain the random allocation and treatment ratio within blocks. Randomization was stratified by indication (RA, PsA) with a block size of 6. Patients and investigators were blinded to treatment.
Statistical Considerations
A planned sample size of 112 patients (56 per sequence) was estimated to provide > 90% power to detect a difference in means of 8 mm in ISP score between the two formulations with a corresponding standard deviation (SD) of 25 using a t test (crossover analysis of variance) with a two-sided significance level of 0.05. Estimated ISP scores among patients with PsA were assumed to be similar to those with RA.
The primary analysis set comprised all patients who received both formulations and completed the ISP assessment during each study period. The safety analysis set included all patients who received at least one dose of etanercept. Subgroup analyses were conducted based on baseline physician global assessment (PGA) score, age category, and sex. Subgroup analyses stratified by PGA score at baseline were based on median PGA score for the full analysis set (N = 111).
A mixed-effects analysis of variance model was used to assess ISP with the phosphate-free formulation as the test treatment and the prior formulation as the reference treatment. Treatment, study period, sequence, and disease indication were evaluated as fixed-effect covariates, and patient-within-sequence was included as a random effect. The least square mean ISP score and the difference between formulations were estimated, and the corresponding two-sided 95% confidence intervals (CI) and P value were provided. A post hoc analysis was conducted for subgroups defined by quartile cut points of prior formulation ISP scores. No adjustment for multiplicity was performed and data were analyzed as observed without imputation for missing data. Analyses were conducted using SAS version 9.4 (SAS Institute, Cary, NC, USA).
Results
Patients
A total of 111 patients were enrolled from 23 centers in the US between November 29, 2016 (first patient enrolled) and October 9, 2017 (last patient follow-up). Of these, 56 patients were randomized to sequence AB and 55 were randomized to sequence BA. The mean (SD) age was 55.3 (13.7) years, 69% of the population was female, 78% had a diagnosis of RA, and 22% had PsA (Table 1). Overall, 109 (98.2%) patients received at least one dose of etanercept (safety analysis set: 84 patients with RA, 25 patients with PsA) and 104 (93.7%) patients received both doses of etanercept and recorded ISP (primary analysis set: 81 patients with RA, 23 patients with PsA). Five (4.5%) patients discontinued the study: one patient in sequence AB and two patients in sequence BA discontinued based on sponsor decision; one patient in sequence BA withdrew consent; and one patient in sequence BA was lost to follow-up. For the prior formulation injections, 56 (52.8%) patients administered study drug in the abdomen and 50 (47.2%) administered in the thigh. For the phosphate-free formulation injections, 57 (53.3%) patients administered the study drug in the abdomen and 50 (46.7%) administered in the thigh.
ISP Outcomes
The mean ISP VAS score for the prior formulation was 23.1 mm and for the phosphate-free formulation was 19.1 mm (Table 2) for a mean difference (95% CI) of − 4 mm (− 8.0, 0.0; P = 0.048). When analyzed by arithmetic mean based on the prior formulation (23.6 mm), patients above the mean (n = 39) had a reduction of 12.2 mm (95% CI 3.1, 21.3) and those below the arithmetic mean (n = 65) had a reduction of − 0.9 mm (95% CI − 3.9, 2.1) (Table 2). For patients with RA, the mean ISP score for the prior formulation was 23.7 mm and for the phosphate-free formulation was 20.5 mm. For patients with PsA, the mean ISP score for the prior formulation was 23.3 mm and for the phosphate-free formulation was 17.2 mm. In covariate analyses, indication (RA vs. PsA; P = 0.68) and treatment sequence (AB vs. BA; P = 0.74) were not statistically significant. However, a period effect was observed across indications: the mean ISP score for Period 1 was 18.8 mm and for Period 2 was 23.4 mm, for a mean difference of 4.6 mm (95% CI 0.6, 8.5; P = 0.024).
In subgroup analyses, patients with baseline PGA score above the median (score ≥ 50; vs. those with PGA below the median), patients ≥ 65 years of age (vs. those < 65 years of age), and males (vs. females) showed greater numerical differences in reporting less ISP with the phosphate-free formulation vs. the prior formulation of etanercept (Table 3). A post hoc analysis using patient ISP scores from the prior formulation administration analyzed by quartile cut points of ≥ 6 mm, ≥ 17.5 mm, and ≥ 35 mm showed mean reductions between the prior and phosphate-free formulations of 6.2 mm (nominal P = 0.017), 9.5 mm (P = 0.011), and 17.2 mm (P = 0.003), respectively (Fig. 2).

Improvement in ISP score with phosphate-free formulation by subgroups defined by prior formulation pain scores. Mean change in ISP for quartiles based on prior formulation ISP are shown. Error bars represent 95% CI. CI confidence interval, ISP injection site pain
Safety Outcomes
Of 109 patients in the safety analysis set, 17 (15.6%) had at least one treatment-emergent AE. Ten patients had an AE after receiving prior formulation and eight patients had an AE after receiving phosphate-free formulation. The only serious AE was a case of viral bronchitis (with the phosphate-free formulation). No grade 4 or greater event or fatal event occurred during the study. Injection site reactions were few in number and similar between formulations (Table 4). The only non-injection site AEs that occurred in more than one patient were hypoesthesia, which occurred in one patient following prior formulation and two patients following phosphate-free formulation, and extremity pain, which occurred in one patient following prior formulation and two patients following phosphate-free formulation.
Discussion
Route of administration, physical and chemical attributes of injected drug products, and physical attributes of needles used to administer biologic drugs play a role in injection pain associated with drug administration [17,18,19,20]. Reduction of ISP can occur through modification of the solution composition. A new phosphate-free etanercept formulation has been shown to reduce the ISP related to its administration. In this study, we showed that the new phosphate-free formulation of etanercept had statistically significantly lower mean ISP scores than the prior formulation. This observation may be useful in patient–physician discussions of treatment selection.
The subset analysis based on disease activity (PGA) suggested that patients with higher disease activity (i.e., those with PGA ≥ median) may also realize greater improvements in ISP compared to patients with less severe disease. Although not statistically significant, older patients (≥ 65 years) had greater reductions in ISP score than patients < 65 years of age, and males had greater reductions in mean ISP score than women. Disease was not included in subgroup analyses; however, patients with RA and PsA had numerically similar improvements in ISP with the phosphate-free formulation.
Pain is an individual experience, and can only be measured by the patient. For this study, we used a content-valid, fit-for-purpose VAS to assess ISP. Although many studies have attempted to establish a minimal clinically important difference (MCID) or minimal clinically important change (MCIC) in pain VAS scores, the estimates vary widely based on the pain source, chronicity, and disease. In this study, the pain source is specific to ISP and the timing is acute. Where there are reported MCIDs in patients with RA for joint pain, these may not translate to ISP assessments. In the setting of acute joint pain, an MCID range of 10–16 mm has been suggested, and in chronic joint pain, a range of 5–11 mm has been suggested [21,22,23]. Baseline pain scores in studies of joint pain were much higher than baseline scores observed with ISP in our study (mean 23.1 mm), and may not be comparable to the current study.
The trial enrolled patients without prespecifying a minimum ISP score at screening or baseline. Our results are broadly generalizable although the overall magnitude of the reduction in the mean pain scores for all patients was relatively modest. A post hoc analysis found that patients with higher pain scores with the prior formulation had correspondingly greater reductions in ISP with the phosphate-free formulation, an observation of practical significance. A limitation of the study was the small sample size, which precluded our ability to perform more rigorous statistical analyses on patient subgroups. In addition, an MCID or MCIC has not been established for ISP, making interpretation of the effects difficult.
Conclusions
In conclusion, the phosphate-free etanercept formulation reduced the mean ISP score compared to the prior formulation. Patients with higher ISP scores on the prior formulation experienced greater reductions in ISP with the phosphate-free formulation. The safety results from this study were consistent with the known safety profile of etanercept. These results may be helpful for patients and physicians when selecting a TNFi therapy.
References
Gibofsky A. Epidemiology, pathophysiology, and diagnosis of rheumatoid arthritis: a synopsis. Am J Manag Care. 2014;20(7 Suppl):S128–35.
McArdle A, Pennington S, FitzGerald O. Clinical features of psoriatic arthritis: a comprehensive review of unmet clinical needs. Clin Rev Allergy Immunol. 2017. https://doi.org/10.1007/s12016-017-8630-7.
Singh JA, Saag KG, Bridges SL Jr, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. 2016;68:1–26.
Gossec L, Smolen JS, Ramiro S, et al. European League Against Rheumatism (EULAR) recommendations for the management of psoriatic arthritis with pharmacological therapies: 2015 update. Ann Rheum Dis. 2016;75:499–510.
Nam JL, Takase-Minegishi K, Ramiro S, et al. Efficacy of biological disease-modifying antirheumatic drugs: a systematic literature review informing the 2016 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann Rheum Dis. 2017;76:1113–36.
Enbrel® (etanercept) prescribing information. Immunex Corporation, Thousand Oaks, CA. 2017. https://www.pi.amgen.com/~/media/amgen/repositorysites/pi-amgen-com/enbrel/enbrel_pi.pdf.
Lethaby A, Lopez-Olivo MA, Maxwell L, Burls A, Tugwell P, Wells GA. Etanercept for the treatment of rheumatoid arthritis. Cochrane Datab Syst Rev. 2013;2013:CD004525.
Gladman DD, Bombardier C, Thorne C, et al. Effectiveness and safety of etanercept in patients with psoriatic arthritis in a Canadian clinical practice setting: the REPArE trial. J Rheumatol. 2011;38:1355–62.
Mease PJ, Kivitz AJ, Burch FX, et al. Continued inhibition of radiographic progression in patients with psoriatic arthritis following 2 years of treatment with etanercept. J Rheumatol. 2006;33:712–21.
Jones GB, Collins DS, Harrison MW, Thyagarajapuram NR, Wright JM. Subcutaneous drug delivery: an evolving enterprise. Sci Transl Med. 2017. https://doi.org/10.1126/scitranslmed.aaf9166.
Kim H, Park H, Lee SJ. Effective method for drug injection into subcutaneous tissue. Sci Rep. 2017;7:9613.
Zijlstra E, Jahnke J, Fischer A, Kapitza C, Forst T. Impact of injection speed, volume, and site on pain sensation. J Diabetes Sci Technol. 2018;12:163–8.
Hamuro L, Kijanka G, Kinderman F, et al. Perspectives on subcutaneous route of administration as an immunogenicity risk factor for therapeutic proteins. J Pharm Sci. 2017;106:2946–54.
Hamilton JG. Needle phobia: a neglected diagnosis. J Fam Pract. 1995;41:169–75.
Henderson Berg MH, Carrasco D. Injection site reactions to biologic agents used in psoriasis and psoriatic arthritis. J Drugs Dermatol. 2017;16:695–8.
Bolge SC, Goren A, Tandon N. Reasons for discontinuation of subcutaneous biologic therapy in the treatment of rheumatoid arthritis: a patient perspective. Patient Prefer Adherence. 2015;9:121–31.
Palmon SC, Lloyd AT, Kirsch JR. The effect of needle gauge and lidocaine pH on pain during intradermal injection. Anesth Analg. 1998;86:379–81.
Robb DM, Kanji Z. Comparison of two needle sizes for subcutaneous administration of enoxaparin: effects on size of hematomas and pain on injection. Pharmacotherapy. 2002;22:1105–9.
Laursen T, Hansen B, Fisker S. Pain perception after subcutaneous injections of media containing different buffers. Basic Clin Pharmacol Toxicol. 2006;98:218–21.
Flanagan T, Wahl MJ, Schmitt MM, Wahl JA. Size doesn’t matter: needle gauge and injection pain. Gen Dent. 2007;55:216–7.
Kelly AM. The minimum clinically significant difference in visual analogue scale pain score does not differ with severity of pain. Emerg Med J. 2001;18:205–7.
Wolfe F, Michaud K. Assessment of pain in rheumatoid arthritis: minimal clinically significant difference, predictors, and the effect of anti-tumor necrosis factor therapy. J Rheumatol. 2007;34:1674–83.
Gallagher EJ, Bijur PE, Latimer C, Silver W. Reliability and validity of a visual analog scale for acute abdominal pain in the ED. Am J Emerg Med. 2002;20:287–90.
Acknowledgements
We thank the patients who generously shared their time to participate in this study.
Funding
This study was sponsored by Amgen Inc, Thousand Oaks, CA, USA. All authors had full access to the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis. Amgen Inc. funded the article processing charges.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Medical writing assistance
Linda Rice (Amgen Inc.) and Julia R. Gage (on behalf of Amgen Inc.) provided assistance with the preparation of this article. This medical writing support was funded by Amgen Inc.
Disclosures
S Cohen is a consultant and investigator for Amgen Inc. A. Samad is an employee and shareholder of Amgen Inc. E. Karis is an employee and shareholder of Amgen Inc. B.S. Stolshek is an employee and shareholder of Amgen Inc. H. Zhang is an employee and shareholder of Amgen Inc. G. Kricorian is an employee and shareholder of Amgen Inc. J.B. Chung is an employee and shareholder of Amgen Inc. M. Trivedi is a current employee of Gilead Sciences, Inc. but was an employee and shareholder of Amgen Inc. at the time of the study. G.A. Aras is a current employee of Esperion Therapeutics but was an employee and shareholder of Amgen Inc. at the time of the study.
Compliance with Ethics Guidelines
This study was approved by Schulman Associates IRB, Cincinnati, OH. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants included in the study.
Data Availability
There is a plan to share data. This may include de-identified individual patient data for variables necessary to address the specific research question in an approved data-sharing request; also related data dictionaries, study protocol, statistical analysis plan, informed consent form, and/or clinical study report. Data sharing requests relating to data in this manuscript will be considered after the publication date and (1) this product and indication (or other new use) have been granted marketing authorization in both the US and Europe, or (2) clinical development discontinues and the data will not be submitted to regulatory authorities. There is no end date for eligibility to submit a data sharing request for these data. Qualified researchers may submit a request containing the research objectives, the Amgen product(s) and Amgen study/studies in scope, endpoints/outcomes of interest, statistical analysis plan, data requirements, publication plan, and qualifications of the researcher(s). In general, Amgen does not grant external requests for individual patient data for the purpose of re-evaluating safety and efficacy issues already addressed in the product labeling. A committee of internal advisors reviews requests. If not approved, a Data Sharing Independent Review Panel will arbitrate and make the final decision. Upon approval, information necessary to address the research question will be provided under the terms of a data sharing agreement. This may include anonymized individual patient data and/or available supporting documents, containing fragments of analysis code where provided in analysis specifications. Further details are available at the following: http://www.amgen.com/datasharing.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to: https://doi.org/10.6084/m9.figshare.7874471.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Cohen, S., Samad, A., Karis, E. et al. Decreased Injection Site Pain Associated with Phosphate-Free Etanercept Formulation in Rheumatoid Arthritis or Psoriatic Arthritis Patients: A Randomized Controlled Trial. Rheumatol Ther 6, 245–254 (2019). https://doi.org/10.1007/s40744-019-0152-8
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40744-019-0152-8