
Opinion statement
Cutaneous fibrosing disorders encompass a diverse array of diseases united by the presence of varying degrees of dermal sclerosis. The quality and distribution of skin involvement, presence or absence of systemic complications, and unique associated laboratory abnormalities often help to distinguish between these diseases. It is imperative that an effort is made to accurately differentiate between scleroderma and its mimics, in order to guide long-term management and facilitate implementation of the appropriate treatment modality where indicated.
Similar content being viewed by others


Introduction
Scleroderma is a rare autoimmune disorder, with a national annual incidence of 20 cases per million in the USA and an estimated prevalence of 150–300 cases per million [1, 2]. This complex connective tissue disease is characterized by diverse clinical manifestations and multisystem involvement, with fibrosis of the skin and internal organs [3]. In his dissertation On Epidemics, Hippocrates describes treating a patient from Athens whose skin was so hard that “it was not possible to raise it in folds” [4]. Indeed, the nomenclature scleroderma is derived from the Greek words skleros (hard) and derma (skin) alluding to the clinical hallmark, taught tethering of the skin. In addition to the cutaneous manifestations, other clinical features include Raynaud’s phenomenon and abnormal nail-fold capillaries as well as systemic features contingent on the areas of extra-cutaneous involvement.
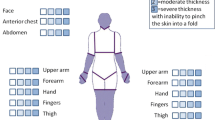
Despite the seemingly unique traits of scleroderma, several conditions have been similarly characterized by varying degrees of cutaneous fibrosis. Table 1 summarizes a list of scleroderma mimics, categorized as immune-mediated, metabolic, hereditary, deposition, and toxin-induced disorders. A broad differential diagnosis is therefore required when evaluating a patient with increased thickness of the skin. Nevertheless, a proper review of the constellation of signs and symptoms, with which the patient presents, will assist the astute clinician in differentiating between scleroderma and some of its more common mimics. Particular attention should be paid to the quality and distribution of cutaneous involvement, the presence or absence of Raynaud’s phenomenon, findings on nail-fold capillaroscopy, and any association with coexisting conditions or abnormal laboratory parameters, which may aid in refining the diagnosis.
Table 2 outlines the key features of some of the more common mimickers of scleroderma, while comparing and contrasting their similarities and differences, respectively. Beyond academic interest, it is imperative that one endeavors to distinguish between these disease entities, as the definitive diagnosis may portend a favorable or poor prognosis, guide further screening, and direct appropriate therapy as deemed necessary. This article aims to provide a comprehensive review of the more common cutaneous fibrosing disorders which mimic scleroderma. Specifically, the disorders of scleredema, scleromyxedema, eosinophilic fasciitis, pansclerotic morphea, nephrogenic systemic fibrosis, and diabetic chieroarthropathy will be discussed. Particular emphasis will be placed on the etiopathogensis, clinical features, diagnosis, and treatment and prognosis of these diseases, as well as an update on the most recent data published in the last 3 years.
Scleredema
Epidemiology and etiopathogenesis
Scleredema is a sclerotic skin disease which was first described by German dermatologist Abraham Buschke in 1902 [5]. Three subtypes of this disease exist. Type 1 often termed Scleredema adultorum of Buschke is the most prevalent type and primarily affects individuals less than 20 years, occasionally occurring in children. It demonstrates a predilection for the female gender. Both viral and bacterial infectious etiologies have been implicated in this subtype, the onset of which is often heralded by a febrile illness. Pathogenic organisms associated with type 1 scleredema include streptococcus, measles, mumps, influenza, and varicella. The onset is usually rapid but gives way to a frequently self-limited benign clinical course, with resolution of symptoms within months to a few years. It is the exception that periodic exacerbations may occur [6]. Type 2 scleredema bears a strong association with the occurrence of paraproteinemias, in particular monoclonal gammopathy of the IgG kappa type. Its clinical course is overall chronic and progressive. The sequela of overt multiple myeloma has been noted in 25 to 45 % of patients [7, 8]. Type 3 scleredema, aptly termed scleredema diabeticorum, is more commonly observed in patients with long-standing uncontrolled diabetes mellitus. The patients are phenotypically male, more than 40 years of age, and with concomitant microvascular complications from their diabetes [9]. Overall, data is lacking as it pertains to the prevalence of this disease.
Clinical features
Affected individuals may experience signs and symptoms of urticaria and erythema with dermographism [10]. Irrespective of the subtype, the cutaneous manifestation of scleredema is characteristically a woody, non-pitting induration with symmetric distribution throughout the upper and mid back, chest, neck, face, and arms. The distal extremities (hands and feet) are notably spared, a distinguishing feature when compared to scleroderma in which sclerodactyly is almost universal. Cases of systemic involvement albeit rare have been reported, resulting in cardiac dysfunction, ocular palsy, and esophageal involvement with dysphagia [6, 11–14].
Diagnostic evaluation
Biopsy of the affected skin reveals swollen collagen fibers underlying a thickened dermis, with intervening spaces of amorphous material composed of hyaluronic acid [15]. This supports but is not routinely required to make a diagnosis of scleredema. The diagnosis is primarily a clinical one, based on the nature of skin involvement and presence of an associated condition [16].
Treatment and prognosis
A variety of reports have advocated the use of diverse therapies, including ultraviolet light, radiation therapy, immunosuppressants, corticosteroids, and intravenous immunoglobulins (IVIg) [17–21]. Varying degrees of therapeutic benefit have been observed. In some instances, type 3 scleredema may be effectively treated by management of the underlying diabetes mellitus [22]. Overall, the scleredema subtype influences the prognosis; type 1 is relatively self-limited while types 2 and 3 demonstrate a more indolent course.
Scleromyxedema
Epidemiology and etiopathogenesis
In 1954, Gottron coined the term scleromyxedema, while describing his encounter with a female patient affected by a peculiar chronic dermatosis [23]. Scleromyxedema, also known as Arndt-Gottron disease, papular mucinosis, and lichen myxedematosus, is an extremely rare primary cutaneous mucinosis. The exact incidence and prevalence of this disease is unknown. It demonstrates an equal gender distribution, with the mean age of onset in the sixth decade of life [24]. Occurrence of this disease has not been reported in the pediatric population. The pathogenesis of this condition remains elusive. A prominent paraproteinemia has been noted in approximately 80 % of patients, raising questions about its etiological role [25]. Results from studies examining the relationship between the paraproteinemia and cutaneous mucinosis have however yielded conflicting evidence [26–28].
Clinical features
The list of primary cutaneous mucinoses is quite extensive. Owing to somewhat inconsistent terminology, a classification schema has been proposed which makes a distinction between scleromyxedema, a systemic disease, and other limited cutaneous mucinoses [29]. The distinguishing feature of scleromyxedema is the presence of cutaneous mucin deposition giving rise to multiple flesh-colored lichenoid papules measuring 2–3 mm in diameter. The skin accordingly has a characteristic “cobblestone” texture. These papules are particularly prominent on the glabella and neck and in the posterior auricular area. Cutaneous involvement also extends to the extremities, as well as the trunk in a linear symmetric distribution. The dorsum of the hands and fingers are commonly involved resulting in sclerodactyly, Fig. 1a; palms are notably spared. Patients with progressive long-standing disease may demonstrate leonine “mask-like” facies due to the transition to confluent plaque formation, with the ensuing thickening of skin folds [10]. Systemic involvement has been reported with the occurrence of dysphagia, myopathies, and cardiopulmonary compromise [30]. In 10 to 15 % of patients, milder neurological complications occur including carpal tunnel syndrome, peripheral neuropathies, and headache [24, 31]. Dermato-neuro syndrome, a potentially fatal complication of this disease, is characterized by fever, convulsions, and coma and may occur in up to 10 % of untreated patients [32–34].

a Characteristic subcutaneous nodules, depicted in the hand of a patient with scleromyxedema. b Pansclerotic morphea involving the feet.
Diagnostic evaluation
Dermal infiltration with an amorphous material that separates the collagen fibers is apparent on skin biopsy histopathology. In addition, a mild to moderate superficial perivascular dermal inflammatory infiltrate may be noted, which is composed of lymphocytes and plasma cells [10, 15]. Scleromyxedema is frequently associated with a benign gammopathy, usually of the IgG type (both kappa and lambda) [35]. Diagnostic guidelines have been proposed which entail the four criteria detailed below:
-
1.
Papular cutaneous eruption (in a characteristic scleroderma-like distribution)
-
2.
Skin biopsy, which demonstrates the cardinal features of scleromyxedema (dermal mucin deposition, proliferation of spindle-like fibroblasts, increase in collagen)
-
3.
Presence of monoclonal gammopathy in peripheral blood
-
4.
Absence of thyroid dysfunction
All four criteria should be present to comfortably make a definitive diagnosis [36]. Clinically, scleromyxedema may be distinguished from scleroderma by the characteristic papular quality of the skin, with involvement of the ears and mid back, areas generally spared in scleroderma.
Treatment and prognosis
The majority of the proposed treatment modalities for scleromyxedema have paralleled regimens used for other plasma cell dyscrasias, in particular multiple myeloma. These agents include corticosteroids, thalidomide, melphalan, and bortezomib [36, 37]. Autologous stem cell transplant has also been attempted [38]. Promising results have been noted with the use of IVIg therapy in two larger cohorts [24, 25]. Patients receiving IVIg may experience partial or even complete therapeutic response [39, 40]. Ultimately, in a small subset of patients, systemic complications may be associated with a poor outcome.
Eosinophilic fasciitis
Epidemiology and etiopathogenesis
Shulman, in 1975, described two cases of men with a scleroderma-like disease affecting the extremities, associated with profound peripheral eosinophilia, hypergammaglobulinemia, and elevated erythrocyte sedimentation rate. It was thought to be a distinct clinical entity from scleroderma, due to its predilection for the forearm while sparing the fingers, absence of Raynaud’s phenomenon, and favorable response to corticosteroid therapy [41]. Rodnan et al. later proposed that this condition be termed eosinophilic fasciitis, owing to the marked thickening of the fascia and an intense lymphoplasmocytic inflammatory infiltrate noted on skin biopsy [42]. The incidence and prevalence of this very rare condition is unknown. It has a male predominance and is more common in persons of Caucasian ethnicity. Eosinophilic fasciitis has been reported to primarily occur in the third to sixth decades of life [43]. The underlying etiopathogenesis is unknown. An array of cases has been reported, suggesting associations with potential etiological agents including infectious (Borrelia burgdorferi), post-radiotherapy, insect bite, and drug induced [44–50]. Many cases occur in the context of trauma or after vigorous exercise [51]. A predominantly humoral immune process has been suggested, in which eosinophils releasing transforming growth factor beta mediate the activation of fibroblasts and rapid fibrosis ensues within days to weeks [52].
Clinical features
Eosinophilic fasciitis usually presents with the subacute onset of tender, painful symmetric induration of the subcutaneous tissues, occurring over a course of weeks to months. This may be accompanied by constitutional symptoms of fever, weight loss, and generalized malaise. The skin is initially edematous bearing a “peau d’orange” appearance. Progression of the disease gives rise to a more woody induration of the subcutaneous tissues with evidence of puckering of the skin and the characteristic “venous groove sign” evident on elevation of the affected limb. Of note, the integrity of the superficial dermis is maintained, yielding folds when the examining physician attempts to pinch the skin. The distribution of skin involvement demonstrates preferential involvement of the upper extremities, sparing the hands. The feet and lower extremities, trunk, and neck are also commonly involved. A symmetric non-erosive arthritis is frequently seen, complicated by disfiguring joint contractures. Periarticular fibrosis may occur, and, in this context, carpal tunnel syndrome and other peripheral compressive neuropathies may be noted. Raynaud’s phenomenon may be infrequently present, but, in contrast to scleroderma, nail-fold capillaroscopy is normal. Systemic complications are rare. In a significant percentage of patients, synchronous morphea-like cutaneous lesions have occurred [51, 53]. Associations with autoimmune conditions including thyroiditis, autoimmune cytopenias, diabetes mellitus, and connective tissue diseases have been reported [54–56]. In less than 10 % of patients, myeloproliferative disorders and leukemias including chronic lymphocytic and myelomonocytic leukemias may occur [51, 57, 58].
Diagnostic evaluation
The hallmark histopathologic feature of eosinophilic fasciitis is the presence of dermal-hypodermal sclerosis associated with fibrotic thickening of the subcutaneous adipose lobular septae, superficial fascia, and perimysium [51, 57]. The epidermis is spared. Full-thickness skin biopsy is considered by some to be the gold standard for diagnosis. In lieu of this invasive procedure, the diagnosis is often made on the basis of clinical and laboratory findings with magnetic resonance imaging (MRI) when deemed necessary. In as much as 63 to 93 % of patients, peripheral eosinophilia may be present at the onset, but this is often transient and disappears rapidly with steroid exposure [51, 57, 59]. In addition, the magnitude of eosinophilia does not correlate with disease severity. Inflammatory markers are often elevated, and an associated hypergammaglobulinemia is sometimes present. The aldolase may be elevated even in the wake of a normal creatinine phosphokinase [16, 60]. It has been proposed that this may be due to the associated perimysial inflammation [16]. MRI has proven to be a useful, non-invasive, diagnostic tool that may also help to monitor treatment. In the acute phases, fascial thickening may be evident on T1, T2, and STIR imaging. Fascial enhancement is prominent on administration of gadolinium [61].
Treatment and prognosis
Corticosteroids are considered to be the first-line therapeutic agent, with demonstrated efficacy in more than 70 % of patients [51, 62]. A high-dose prednisone regimen is initially implemented, with doses of 0.5 to 1 mg/kg daily [63]. This dose is usually maintained for a few weeks until a clinical response is evident. Thereafter, a slow taper may be attempted, with judicious monitoring for regression, particularly when doses less than 20 mg are attained. A complete response to steroid therapy may not be evident until after 12 to 18 months. Patients who present with an initially aggressive clinical course as evidenced by extensive body surface area involvement and constitutional symptoms often require additional immunosuppressive therapy. Agents such as mycophenolate mofetil and methotrexate have been used for their steroid-sparing effects while allowing the physician to curb the disease progression in a more timely manner [64]. Conservative management with adjunctive physiotherapy is also recommended to minimize the development of flexion contractures and long-term disability. Ultimately, the goal of treatment is to achieve complete remission; this is contingent on the early implementation of appropriate immunosuppressive therapy. Overall, the prognosis of eosinophilic fasciitis is good, with the majority of cases achieving full remission. In the absence of a definitive intervention, spontaneous resolution has been reported in some cases.
Localized scleroderma
Epidemiology and etiopathogenesis
Localized scleroderma is a group of heterogeneous inflammatory disorders characterized by increased collagen deposition with sclerosis of the skin and subcutaneous tissues. It is rare, with estimates of the incidence ranging between 0.34 and 2.7 cases per 100,000 population per annum [65, 66]. Various subtypes of this disorder exist differentiated by the distribution of skin involvement and extent of subcutaneous fibrosis. A classification system was proposed by Peterson et al. in 1995, describing categories of plaque, generalized, bullous, linear, and deep morphea [67]. This has since been revised to include five main subtypes of circumscribed, linear, generalized, pansclerotic, and mixed morphea [68]. Pansclerotic morphea is an extremely aggressive and very rare variant. It occurs primarily in children, but cases of adult-onset pansclerotic morphea have been reported [69]. The underlying pathogenesis is yet to be completely elucidated. There however appears to be trauma, hypoxia, infection, or post-radiation-mediated endothelial cell damage with subsequent lymphocyte recruitment, proinflammatory cytokine secretion, and fibroblast activation, resulting in remodeling with tissue and vascular fibrosis [70–73].
Clinical features
The onset is rapid, occurring within months. Two main presentations of pansclerotic morphea exist, a more commonly generalized sclerosis and the rare asymmetric localized plaque. The cutaneous lesions are circumscribed with an ill-defined active erythematous or violaceous border and a hypopigmented fibrotic center [10]. The solitary plaque of morphea profunda typically occurs in a paraspinal location on the upper trunk [74]. In the more generalized form, there is symmetric homogenous distribution over large surface areas, with involvement of the trunk and proximal extremities. The feet are often involved while the hands are characteristically spared, Fig. 1b. In both forms, there is extensive subcutaneous fibrosis of adipose tissue, fascia, and muscle. The ensuing severe contractures, skin necrosis, and large poorly healing ulcers are often quite disabling [75]. Raynaud’s phenomenon is typically absent, and nail-fold capillaroscopy is normal. Arthralgias and myalgias are common. Patients may develop dysphagia secondary to external compression [76]. In the setting of normal CT scans, restrictive patterns have been observed on pulmonary function tests, attributed to external limitations of rib cage mobility [77]. Other extra-dermal manifestations are rare.
Diagnostic evaluation
The histopathologic features of localized and systemic sclerosis are similar, making it difficult to distinguish between the two solely on the basis of skin biopsy results. An early inflammatory and a late fibrotic phase have been described [75]. Initially, thick collagen bundles develop within the reticular dermis surrounded by dense inflammatory primarily lymphocytic infiltrates. The epidermis may be spared or become thin and atrophic. In the late phase, fibrotic changes are evident in the deep dermis with potential extension to the muscles and tendons. Antinuclear antibody positivity may be noted but is non-specific [16].
Treatment and prognosis
Treatment has proven to be challenging, primarily due to lack of validated outcome measures to assess efficacy. Various treatment modalities have been utilized, including topical and systemic agents. Several retrospective studies support the use of an immunosuppressive regimen consisting of methotrexate, alone or in combination with systemic corticosteroids and phototherapy [73, 78, 79, 80••, 81, 82•]. In the absence of definitive therapy, there is only a limited tendency to regression of the associated fibrotic changes [75].
Nephrogenic systemic fibrosis
Epidemiology and etiopathogenesis
In 2000, Cowper et al. reported a case series of 14 patients with end-stage renal disease who developed a scleromyxedema-like cutaneous disease [83]. This disorder was initially referred to as “nephrogenic fibrosing dermopathy” [84]. By 2003, in recognition of its systemic involvement, the terminology was subsequently changed to nephrogenic systemic fibrosis (NSF) [85, 86]. In 2006, reports were made on the consequential association between exposure to gadolinium-based contrast agents (GBCAs) and the development of NSF in patients with renal failure [87, 88]. It has been proposed that, in patients with impaired renal function, the ionized form of gadolinium (Gd3+) dissociates from its stable ligand complex to form insoluble molecules, which ultimately precipitate in the stroma and stimulate the production of profibrotic cytokines [89]. There is no gender or racial predilection for NSF. Patients with NSF are usually middle-aged, but it has been reported to affect patients at both extremes of age. Reports of this disease have primarily arisen from investigators in the USA and Europe [90]. The vast majority of cases of NSF have been observed in patients with underlying renal impairment, usually end-stage renal disease requiring replacement therapy, acute kidney injury, and chronic kidney disease stage IV or V [91, 92]. The risk of NSF increases with the degree of renal impairment [93]. The US Food and Drug Administration recognizing the putative association between GBCAs and NSF issued a black box warning in 2007 which cautioned against the use of GBCAs in patients with acute or chronic severe renal insufficiency (http://www.fda.gov/Drugs/DrugSafety/ucm223966.htm). To date, 380 cases of NSF have been recorded in the US NSF registry [94]. The incidence of NSF has declined significantly since the black box warning was issued and with the implementation of guidelines regarding the use of GBCAs in patients with renal impairment [95].
Clinical features
NSF commonly presents with the acute onset of edema and progressively enlarging nodules and papules, which coalesce into brawny plaques. These cutaneous lesions may be pruritic and develop within days to weeks after gadolinium exposure, although some late cases have been reported. The distal extremities are initially affected, with proximal extension to the elbows and knees. Occasionally, the trunk is involved, and the face is however spared. The fibrotic process affects the skin, muscle, and fascia. The skin may ultimately become attached to the underlying fascia, and severe joint contractures develop. Cardiac, pulmonary, and gastrointestinal complications may occur due to rapidly progressive fibrosis with internal organ involvement. Patients may experience a burning sensation and significant pain with evidence of a peripheral neuropathy on nerve conduction studies. Raynaud’s phenomenon is not usually present, and nail-fold capillaroscopy is normal.
Diagnostic evaluation
A full-thickness skin biopsy reveals fibrosis of the dermis with extension to the subcutaneous fat septae. An inflammatory infiltrate is notably absent. The identification of CD34-positive, procollagen I-positive fibrocytes, cutaneous mucin, and collagen deposition in the absence of a monoclonal IgG paraprotein is considered to be distinctive features of NSF [96].
Treatment and prognosis
There is no uniformly efficacious therapy for NSF. Immunosuppressive therapeutic regimens have been proposed, consisting of prednisone, cyclophosphamide, and methotrexate, but evidence supporting their use is at best anecdotal [90, 97]. Promising results have been demonstrated with imatinib mesylate, a tyrosine kinase inhibitor [98]. The use of this agent has resulted in a reduction in skin thickening and tethering as well as an improvement in joint contractures in patients with NSF. Unfortunately however, these effects were short lived as relapses occurred in all patients on discontinuation of the medication [99]. Varying outcomes have been noted status post renal transplant [100, 101]. The efficacy of this intervention appears to be influenced by the timing of transplantation and severity of the underlying disease. Conservative management is key and should consist of pain management and physiotherapy, to minimize disability and improve function [102•]. Overall, it is imperative that primary emphasis be placed on prevention, through avoidance of GBCAs or judicious use of these agents in at-risk patients with renal impairment.
Diabetic chieroarthropathy
Epidemiology and etiopathogenesis
Diabetic chieroarthropathy or limited joint mobility syndrome is a recognized complication of uncontrolled diabetes mellitus, which may occur in both type 1 and type 2 diabetic patients. The risk of developing this disorder increases with increased glycated hemoglobin A1c levels in patients with long-standing diabetes mellitus [103]. Diabetic chieroarthropathy is believed to occur due to the non-enzymatic glycosylation of collagen with subsequent stimulation of cutaneous fibroblasts to produce excess amounts of matrix proteins [104].
Clinical features
Patients may present with “pseudoscleroderma,” the bilateral symmetric induration and thickening of the skin involving the fingers and dorsum of the hands. The “prayer sign” may be demonstrated, due to an inability to fully appose the fingers as a result of permanent joint contractures. Other joints of the upper and lower extremities may be affected, with the development of joint contractures leading to significant loss of function. In contrast to scleroderma, Raynaud’s phenomenon is absent and nail-fold capillaroscopy is normal.
Diagnostic evaluation
Dermal fibrosis and sclerosis of the tendon sheath may be evident on histology. Sonographic findings often reveal thickening of the subcutaneous tissues and flexor tendon sheath [105]. Thickening and enhancement of the flexor tendon sheath may also be evident on MRI [106].
Treatment and prognosis
Treatment is primarily aimed at appropriate glycemic control to halt disease progression [107]. Adjunctive physiotherapy has been advocated to minimize functional disability.
Conclusions
A variety of conditions may mimic scleroderma. While similarities exist between scleroderma and its aforementioned mimics, knowledge of their key distinguishing features will prove helpful in determining the etiology of a patient’s cutaneous fibrosis and ultimately facilitate the implementation of appropriate therapy where indicated.
References and Recommended Reading
Papers of particular interest, published recently have been highlighted as: • Of importance •• Of major importance
Mayes MD, Lacey Jr JV, Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003;48(8):2246–55.
Mayes MD. Scleroderma epidemiology. Rheum Dis Clin N Am. 2003;29(2):239–54.
Wollheim FA. Classification of systemic sclerosis. Visions and reality. Rheumatology (Oxford). 2005;44(10):1212–6.
Pasero G, Marson P. Hippocrates and rheumatology. Clin Exp Rheumatol. 2004;22(6):687–9.
A. B. Ueber Skleroedem Berl Klin Wochenschr. 1902 (39):955.
Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35(6):355–9.
Dziadzio M, Anastassiades CP, Hawkins PN, Potter M, Gabrielli A, Brough GM, et al. From scleredema to AL amyloidosis: disease progression or coincidence? Review of the literature. Clin Rheumatol. 2006;25(1):3–15.
Ratip S, Akin H, Ozdemirli M, Yucelten D, Kus S, Ekenel M, et al. Scleredema of Buschke associated with Waldenstrom’s macroglobulinaemia. Br J Dermatol. 2000;143(2):450–2.
Tate BJ, Kelly JW, Rotstein H. Scleredema of Buschke: a report of seven cases. Australas J Dermatol. 1996;37(3):139–42.
Fernandez-Flores A, Gatica-Torres M, Ruelas-Villavicencio AL, Saeb-Lima M. Morphological clues in the diagnosis of sclerodermiform dermatitis. Am J Dermatopathol. 2014;36(6):449–64.
Wright RA, Bernie H. Scleredema adultorum of Buschke with upper esophageal involvement. Am J Gastroenterol. 1982;77(1):9–11.
Ulmer A, Schaumburg-Lever G, Bauer J, Kotter I, Fierlbeck G. Scleredema adultorum Buschke. Case report and review of the literature. Der Hautarzt Zeitschrift fur Dermatologie, Venerologie, und verwandte Gebiete. 1998;49(1):48–54.
Ioannidou DI, Krasagakis K, Stefanidou MP, Karampekios S, Panayiotidis J, Tosca AD. Scleredema adultorum of Buschke presenting as periorbital edema: a diagnostic challenge. J Am Acad Dermatol. 2005;52(2 Suppl 1):41–4.
Paz RA, Badra RE, Marti HM, Maxit MJ. Systemic Buschke’s scleredema with cardiomyopathy, monoclonal IgG kappa gammopathy and amyloidosis. Case report with autopsy. Medicina. 1998;58(5 Pt 1):501–3.
Rongioletti F, Rebora A. Cutaneous mucinoses: microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23(3):257–67.
Nashel J, Steen V. Scleroderma mimics. Curr Rheumatol Rep. 2012;14(1):39–46.
Eastham AB, Femia AN, Velez NF, Smith HP, Vleugels RA. Paraproteinemia-associated scleredema treated successfully with intravenous immunoglobulin. JAMA Dermatol. 2014;150(7):788–9.
Breuckmann F, Appelhans C, Harati A, Rotterdam S, Altmeyer P, Kreuter A. Failure of low-dose methotrexate in the treatment of scleredema diabeticorum in seven cases. Dermatology. 2005;211(3):299–301.
Nakajima K, Iwagaki M, Ikeda M, Kodama H. Two cases of diabetic scleredema that responded to PUVA therapy. J Dermatol. 2006;33(11):820–2.
Tuchinda C, Kerr HA, Taylor CR, Jacobe H, Bergamo BM, Elmets C, et al. UVA1 phototherapy for cutaneous diseases: an experience of 92 cases in the United States. Photodermatol Photoimmunol Photomed. 2006;22(5):247–53.
Konemann S, Hesselmann S, Bolling T, Grabbe S, Schuck A, Moustakis C, et al. Radiotherapy of benign diseases-scleredema adultorum Buschke. Strahlentherapie und Onkologie Organ der Deutschen Rontgengesellschaft. 2004;180(12):811–4.
Rho YW, Suhr KB, Lee JH, Park JK. A clinical observation of scleredema adultorum and its relationship to diabetes. J Dermatol. 1998;25(2):103–7.
Gottron HA. Scleromyxedema; a strange manifestation of myxothesaurodermy. Arch Klin Exp Dermatol. 1954;199(1):71–91.
Rongioletti F, Merlo G, Cinotti E, Fausti V, Cozzani E, Cribier B, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69(1):66–72.
Blum M, Wigley FM, Hummers LK. Scleromyxedema: a case series highlighting long-term outcomes of treatment with intravenous immunoglobulin (IVIG). Medicine (Baltimore). 2008;87(1):10–20.
Harper RA, Rispler J. Lichen myxedematosus serum stimulates human skin fibroblast proliferation. Science (New York). 1978;199(4328):545–7.
Yaron M, Yaron I, Yust I, Brenner S. Lichen myxedematosus (scleromyxedema) serum stimulates hyaluronic acid and prostaglandin E production by human fibroblasts. J Rheumatol. 1985;12(1):171–5.
Ferrarini M, Helfrich DJ, Walker ER, Medsger Jr TA, Whiteside TL. Scleromyxedema serum increases proliferation but not the glycosaminoglycan synthesis of dermal fibroblasts. J Rheumatol. 1989;16(6):837–41.
Rongioletti F. Lichen myxedematosus (papular mucinosis): new concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25(2):100–4.
Pomann JJ, Rudner EJ. Scleromyxedema revisited. Int J Dermatol. 2003;42(1):31–5.
Jackson EM, English 3rd JC. Diffuse cutaneous mucinoses. Dermatol Clin. 2002;20(3):493–501.
Fleming KE, Virmani D, Sutton E, Langley R, Corbin J, Pasternak S, et al. Scleromyxedema and the dermato-neuro syndrome: case report and review of the literature. J Cutan Pathol. 2012;39(5):508–17.
Landais AF, Duchemin CM, Bourhis VM. Scleromyxedema (papular mucinosis) with dermato-neuro syndrome: a rare, potentially fatal complication. Presse Med. 2015;44(7–8):850–1.
Gonzalez J, Palangio M, Schwartz J, Klainer AS, Bisaccia E. Scleromyxedema with dermato-neuro syndrome. J Am Acad Dermatol. 2000;42(5 Pt 2):927–8.
Jablonska S, Blaszczyk M. Scleroderma-like disorders. Semin Cutan Med Surg. 1998;17(1):65–76.
Hummers LK. Scleromyxedema. Curr Opin Rheumatol. 2014;26(6):658–62.
Canueto J, Labrador J, Roman C, Santos-Briz A, Contreras T, Gutierrez NC, et al. The combination of bortezomib and dexamethasone is an efficient therapy for relapsed/refractory scleromyxedema: a rare disease with new clinical insights. Eur J Haematol. 2012;88(5):450–4.
Lacy MQ, Hogan WJ, Gertz MA, Dispenzieri A, Rajkumar SV, Hayman S, et al. Successful treatment of scleromyxedema with autologous peripheral blood stem cell transplantation. Arch Dermatol. 2005;141(10):1277–82.
Caudill L, Howell E. Scleromyxedema: a case clinically and histologically responsive to intravenous immunoglobulin. J Clin Aesthet Dermatol. 2014;7(5):45–7.
Devos T, Thiessen S, Cuyle PJ, Meersseman W, Delforge M. Long-term follow-up in a patient with the dermato-neuro syndrome treated with high-dose melphalan, thalidomide, and intravenous immunoglobulins for more than 7 years. Ann Hematol. 2014;93(11):1927–8.
Shulman LE. Diffuse fasciitis with eosinophilia: a new syndrome? Trans Assoc Am Phys. 1975;88:70–86.
Rodnan GP, DiBartolomeo A, Medsger Jr TA. Proceedings: eosinophilic fasciitis. Report of six cases of a newly recognized scleroderma-like syndrome. Arthritis Rheum. 1975;18(5):525.
Doyle JA, Ginsburg WW. Eosinophilic fasciitis. Med Clin N Am. 1989;73(5):1157–66.
Mosconi S, Streit M, Bronimann M, Braathen L. Eosinophilic fasciitis (Shulman syndrome). Dermatology. 2002;205(2):204–6.
Granter SR, Barnhill RL, Duray PH. Borrelial fasciitis: diffuse fasciitis and peripheral eosinophilia associated with Borrelia infection. Am J Dermatopathol. 1996;18(5):465–73.
Choquet-Kastylevsky G, Kanitakis J, Dumas V, Descotes J, Faure M, Claudy A. Eosinophilic fasciitis and simvastatin. Arch Intern Med. 2001;161(11):1456–7.
DeGiovanni C, Chard M, Woollons A. Eosinophilic fasciitis secondary to treatment with atorvastatin. Clin Exp Dermatol. 2006;31(1):131–2.
Sherber NS, Wigley FM, Paget SA. Diffuse fasciitis with eosinophilia developing after local irradiation for breast cancer. Clin Rheumatol. 2009;28(6):729–32.
Mallepalli JR, Quinet RJ, Sus R. Eosinophilic fasciitis induced by fire ant bites. Ochsner J. 2008;8(3):114–8.
Chazerain P, Vigneron AM, Grossin M, Meyer O, Kahn MF. Posttraumatic diffuse eosinophilic fasciitis accepted for workers’ compensation. Revue du rhumatisme. 1997;64(6):433–4.
Lakhanpal S, Ginsburg WW, Michet CJ, Doyle JA, Moore SB. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum. 1988;17(4):221–31.
Kahari VM, Heino J, Niskanen L, Fraki J, Uitto J. Eosinophilic fasciitis. Increased collagen production and type I procollagen messenger RNA levels in fibroblasts cultured from involved skin. Arch Dermatol. 1990;126(5):613–7.
Miller 3rd JJ. The fasciitis-morphea complex in children. Am J Dis Child. 1992;146(6):733–6.
Bachmeyer C, Monge M, Dhote R, Sanguina M, Aractingi S, Mougeot-Martin M. Eosinophilic fasciitis following idiopathic thrombocytopenic purpura, autoimmune hemolytic anemia and Hashimoto’s disease. Dermatology. 1999;199(3):282.
Hur JW, Lee HS, Uhm WS, Jun JB, Bae SC, Park CK, et al. Eosinophilic fasciitis associated with autoimmune thyroiditis. Korean J Intern Med. 2005;20(2):180–2.
Baffoni L, Frisoni M, Maccaferri M, Ferri S. Systemic lupus erythematosus and eosinophilic fasciitis: an unusual association. Clin Rheumatol. 1995;14(5):591–2.
Lebeaux D, Frances C, Barete S, Wechsler B, Dubourg O, Renoux J, et al. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology (Oxford). 2012;51(3):557–61.
Bischoff L, Derk CT. Eosinophilic fasciitis: demographics, disease pattern and response to treatment: report of 12 cases and review of the literature. Int J Dermatol. 2008;47(1):29–35.
Endo Y, Tamura A, Matsushima Y, Iwasaki T, Hasegawa M, Nagai Y, et al. Eosinophilic fasciitis: report of two cases and a systematic review of the literature dealing with clinical variables that predict outcome. Clin Rheumatol. 2007;26(9):1445–51.
Fujimoto M, Sato S, Ihn H, Kikuchi K, Yamada N, Takehara K. Serum aldolase level is a useful indicator of disease activity in eosinophilic fasciitis. J Rheumatol. 1995;22(3):563–5.
Baumann F, Bruhlmann P, Andreisek G, Michel BA, Marincek B, Weishaupt D. MRI for diagnosis and monitoring of patients with eosinophilic fasciitis. AJR Am J Roentgenol. 2005;184(1):169–74.
Antic M, Lautenschlager S, Itin PH. Eosinophilic fasciitis 30 years after - what do we really know? Report of 11 patients and review of the literature. Dermatology. 2006;213(2):93–101.
Boin F, Hummers LK. Scleroderma-like fibrosing disorders. Rheum Dis Clin N Am. 2008;34(1):199–220. ix.
Tyndall A, Fistarol S. The differential diagnosis of systemic sclerosis. Curr Opin Rheumatol. 2013;25(6):692–9.
Fett N, Werth VP. Update on morphea: part I. Epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol. 2011;64(2):217–28. quiz 29–30.
Peterson LS, Nelson AM, Su WP, Mason T, O'Fallon WM, Gabriel SE. The epidemiology of morphea (localized scleroderma) in Olmsted County 1960–1993. J Rheumatol. 1997;24(1):73–80.
Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995;70(11):1068–76.
Laxer RM, Zulian F. Localized scleroderma. Curr Opin Rheumatol. 2006;18(6):606–13.
Maragh SH, Davis MD, Bruce AJ, Nelson AM. Disabling pansclerotic morphea: clinical presentation in two adults. J Am Acad Dermatol. 2005;53(2 Suppl 1):S115–9.
Leitenberger JJ, Cayce RL, Haley RW, Adams-Huet B, Bergstresser PR, Jacobe HT. Distinct autoimmune syndromes in morphea: a review of 245 adult and pediatric cases. Arch Dermatol. 2009;145(5):545–50.
Tomimura S, Ogawa F, Iwata Y, Komura K, Hara T, Muroi E, et al. Autoantibodies against matrix metalloproteinase-1 in patients with localized scleroderma. J Dermatol Sci. 2008;52(1):47–54.
Kurzinski K, Torok KS. Cytokine profiles in localized scleroderma and relationship to clinical features. Cytokine. 2011;55(2):157–64.
Sartori-Valinotti JC, Tollefson MM, Reed AM. Updates on morphea: role of vascular injury and advances in treatment. Autoimmune Dis. 2013;2013:467808.
Azad J, Dawn G, Shaffrali FC, Holmes SC, Barnetson RJ, Forsyth A. Does solitary morphoea profunda progress? Clin Exp Dermatol. 2004;29(1):25–7.
Kreuter A. Localized scleroderma. Dermatol Ther. 2012;25(2):135–47.
Arif T, Masood Q, Singh J, Hassan I. Assessment of esophageal involvement in systemic sclerosis and morphea (localized scleroderma) by clinical, endoscopic, manometric and pH metric features: a prospective comparative hospital based study. BMC Gastroenterol. 2015;15:24.
Kim A, Marinkovich N, Vasquez R, Jacobe HT. Clinical features of patients with morphea and the pansclerotic subtype: a cross-sectional study from the morphea in adults and children cohort. J Rheumatol. 2014;41(1):106–12.
Zulian F, Vallongo C, Patrizi A, Belloni-Fortina A, Cutrone M, Alessio M, et al. A long-term follow-up study of methotrexate in juvenile localized scleroderma (morphea). J Am Acad Dermatol. 2012;67(6):1151–6.
Torok KS, Arkachaisri T. Methotrexate and corticosteroids in the treatment of localized scleroderma: a standardized prospective longitudinal single-center study. J Rheumatol. 2012;39(2):286–94.
Li SC, Torok KS, Pope E, Dedeoglu F, Hong S, Jacobe HT, et al. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res. 2012;64(8):1175–85. Consensus guidelines for the treatment of juvenile localized scleroderma.
Marsol B. Update on the classification and treatment of localized scleroderma. Actas Dermosifiliogr. 2013;104:654–66.
Strickland N, Patel G, Strickland A, Jacobe H. Attitudes and trends in the treatment of morphea: a national survey. J Am Acad Dermatol. 2015;72(4):727–8. An update on the treatment of morphea.
Cowper SE, Robin HS, Steinberg SM, Su LD, Gupta S, LeBoit PE. Scleromyxoedema-like cutaneous diseases in renal-dialysis patients. Lancet. 2000;356(9234):1000–1.
Cowper SE, Su LD, Bhawan J, Robin HS, LeBoit PE. Nephrogenic fibrosing dermopathy. Am J Dermatopathol. 2001;23(5):383–93.
Ting WW, Stone MS, Madison KC, Kurtz K. Nephrogenic fibrosing dermopathy with systemic involvement. Arch Dermatol. 2003;139(7):903–6.
Cowper SE. Nephrogenic systemic fibrosis: the nosological and conceptual evolution of nephrogenic fibrosing dermopathy. Am J Kidney Dis. 2005;46(4):763–5.
Marckmann P, Skov L, Rossen K, Dupont A, Damholt MB, Heaf JG, et al. Nephrogenic systemic fibrosis: suspected causative role of gadodiamide used for contrast-enhanced magnetic resonance imaging. J Am Soc Nephrol. 2006;17(9):2359–62.
Boyd AS, Zic JA, Abraham JL. Gadolinium deposition in nephrogenic fibrosing dermopathy. J Am Acad Dermatol. 2007;56(1):27–30.
Baranyai Z, Palinkas Z, Uggeri F, Maiocchi A, Aime S, Brucher E. Dissociation kinetics of open-chain and macrocyclic gadolinium(III)-aminopolycarboxylate complexes related to magnetic resonance imaging: catalytic effect of endogenous ligands. Chem Weinheim Bergstr Ger. 2012;18(51):16426–35.
Bernstein EJ, Schmidt-Lauber C, Kay J. Nephrogenic systemic fibrosis: a systemic fibrosing disease resulting from gadolinium exposure. Best Pract Res Clin Rheumatol. 2012;26(4):489–503.
Braverman IM, Cowper S. Nephrogenic systemic fibrosis. F1000 medicine reports. 2010;2:84.
Abu-Alfa A. The impact of NSF on the care of patients with kidney disease. J Am Coll Radiol. 2008;5(1):45–52.
Kuo PH, Kanal E, Abu-Alfa AK, Cowper SE. Gadolinium-based MR contrast agents and nephrogenic systemic fibrosis. Radiology. 2007;242(3):647–9.
Cowper S. Nephrogenic systemic fibrosis [ICNSFR Website]. 2001-2013 (http://www.icnsfr.org Accessed 19 Sep 2015).
Morcos SK, Haylor J. Pathophysiology of nephrogenic systemic fibrosis: a review of experimental data. World J Radiol. 2010;2(11):427–33.
Galan A, Cowper SE, Bucala R. Nephrogenic systemic fibrosis (nephrogenic fibrosing dermopathy). Curr Opin Rheumatol. 2006;18(6):614–7.
Marckmann P, Skov L. Nephrogenic systemic fibrosis: clinical picture and treatment. Radiol Clin N Am. 2009;47(5):833–40. vi.
Kay J, High WA. Imatinib mesylate treatment of nephrogenic systemic fibrosis. Arthritis Rheum. 2008;58(8):2543–8.
Kaewlai R, Abujudeh H. Nephrogenic systemic fibrosis. AJR Am J Roentgenol. 2012;199(1):W17–23.
Cuffy MC, Singh M, Formica R, Simmons E, Abu Alfa AK, Carlson K, et al. Renal transplantation for nephrogenic systemic fibrosis: a case report and review of the literature. Nephrol Dial Transplant. 2011;26(3):1099–101.
Panesar M, Yacoub R. What is the role of renal transplantation in a patient with nephrogenic systemic fibrosis? Semin Dial. 2011;24(4):373–4.
Daftari Besheli L, Aran S, Shaqdan K, Kay J, Abujudeh H. Current status of nephrogenic systemic fibrosis. Clin Radiol. 2014;69(7):661–8. Highlights the latest advances in the treatment of nephrogenic systemic fibrosis and provides insight into the underlying pathogenesis of this disease.
Silverstein JH, Gordon G, Pollock BH, Rosenbloom AL. Long-term glycemic control influences the onset of limited joint mobility in type 1 diabetes. J Pediatr. 1998;132(6):944–7.
Haustein UF. Scleroderma-like lesions in insulin-dependent diabetes mellitus. J Eur Acad Dermatol Venereol. 1999;13(1):50–3.
Ismail AA, Dasgupta B, Tanqueray AB, Hamblin JJ. Ultrasonographic features of diabetic cheiroarthropathy. Br J Rheumatol. 1996;35(7):676–9.
Khanna G, Ferguson P. MRI of diabetic cheiroarthropathy. AJR Am J Roentgenol. 2007;188(1):W94–5.
Abate M, Schiavone C, Salini V, Andia I. Management of limited joint mobility in diabetic patients. Diab Metab Syndr Obes Targets Ther. 2013;6:197–207.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Nadia D. Morgan declares that she has no conflict of interest. Laura K. Hummers declares that she has no conflict of interest. Morgan reports that research reported in this publication was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases under T32 Award Number AR048522-13. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Scleroderma
Rights and permissions
About this article

Cite this article
Morgan, N.D., Hummers, L.K. Scleroderma Mimickers. Curr Treat Options in Rheum 2, 69–84 (2016). https://doi.org/10.1007/s40674-016-0038-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40674-016-0038-7