
Abstract
Patients with chronic kidney disease (CKD) have a greatly enhanced risk of cardiovascular morbidity and mortality. Over the past decade it has come clear that a disturbed calcium-phosphate metabolism, with Fibroblast Growth Factor-23 as a key hormone, is partly accountable for this enhanced risk. Numerous studies have been performed unravelling FGF23s actions and its association with clinical conditions. As FGF23 is strongly associated with adverse outcome it may be a promising biomarker for risk prediction or, even more important, targeting FGF23 may be a strategy to improve patient outcome. This review elaborates on the clinical usefulness of FGF23 measurement. Firstly it discusses the reliability of the FGF23 measurement. Secondly, it evaluates whether FGF23 measurement may lead to improved patient risk classification. Finally, and possibly most importantly, this review evaluates if lowering of FGF23 should be a target for therapy. For this, the review discusses the current evidence indicating that FGF23 may be in the causal pathway to cardiovascular pathology, provides an overview of strategies to lower FGF23 levels and discusses the current evidence concerning the benefit of lowering FGF23.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
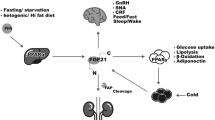
Chronic kidney disease (CKD) is a major health concern, given its high prevalence and associated cardiovascular morbidity and mortality, leading to a high rate of health care consumption [1]. This high burden of cardiovascular disease (CVD) is seen in CKD stage 3 and beyond. Although traditional risk factors, such as hypertension, diabetes and smoking contribute to the development of CVD in CKD, they cannot fully explain the high incidence of cardiovascular mortality in these patients [2, 3]. Disturbances in calcium-phosphate homeostasis are probably contributing to this high mortality risk [4]. A key hormone, in the regulation of calcium-phosphate homeostasis is the 32-kDa peptide Fibroblast Growth Factor-23 (FGF23). This hormone was discovered in the early 2000s in patients with autosomal dominant hypophosphataemic rickets (ADHR) [5]. This disease is characterized by hypophosphatemia and hyperphosphaturia resulting in growth retardation, bone deformities and rickets [6, 7]. FGF23 appeared to be the humoral factor to induce this excess renal phosphate loss. FGF23 is secreted by osteocytes in bone and is one of the three regulators of phosphate homeostasis, together with PTH and 1,25 dihydroxycholecalciferol (1,25(OH)2D3). The first two hormones both have phosphate lowering effects by decreasing tubular phosphate reabsorption by downregulation of the sodium dependent phosphate transporters (NaPi2a and NaPi2c) [8], but have opposite effects on vitamin D regulation, which is activated by PTH, but catabolized by FGF23 [9,10,11,12] (see Fig. 1). FGF23 acts on its main target organs, the kidney and parathyroid, by binding to the FGF23 receptors with α-Klotho as a co-receptor. This co-receptor is generally considered to be necessary to induce intracellular signal transduction, at least so for FGFR1 [13,14,15].

Figure with permission adapted from Vervloet et al. Nature Reviews Nephrology 2017 (155) FGF23; Fibroblast growth factor 23, factor-23; Na-Pi-2a; Sodium-Phosphate co=transporter 2a
FGF23 physiology.
Nowadays FGF23 has gained wide attention in chronic kidney disease associated mineral bone disease (CKD-MBD) and appears to be a candidate as missing link between chronic kidney disease and cardiovascular morbidity and mortality. FGF23 levels increase during progression of CKD [16, 17]. Although this, initially physiological, adaptation is crucial for maintaining phosphate balance in early CKD, prolonged exposure and extreme concentrations in advanced CKD may have deleterious effects, in particular on the cardiovascular system [18,19,20]. Several observational studies showed an independent association between FGF23 levels and adverse outcome through all stages of CKD.
Over the last decade a legion of studies has been published on FGF23, unravelling its biology, physiological actions and its association with clinical conditions. Some epidemiological data and experimental studies suggest that FGF23 not only acts as regulator of parathyroid hormone (PTH), vitamin D, or phosphorus, but may actually be in the causal pathway to cardiovascular pathology. However, definite proof of causality is lacking, since many questions still remain. This review will focus on the clinical usefulness of FGF23 as a biomarker and its potential use as a target for therapy.
Reliability of FGF23 measurement
Currently, FGF23 is rarely measured in routine clinical practice. There are four immunoassays commercially available for measurement of FGF23: Immutopics (1st and 2nd generation, San Clemente, USA), Kainos (Tokyo, Japan), Millipore (Billerica, USA) and DiaSorin (Saluggia, Italy). Most assays measure the intact 251 amino-acid protein (iFGF23) by simultaneous recognition of epitopes on the N- and C-terminal domains close to the proteolytic cleavage site. Additionally, Immutopics has an assay which measures both iFGF23 and the C-terminal fragment of FGF23 (cFGF23) by two antibodies against two epitopes within the C-terminal portion.
The four assays differ substantially as they are using different antibodies targeting different epitopes on the FGF23 protein. Besides different reported units [iFGF23 in picograms per milliliter (pg/ml) and cFGF23 in relative units (RU) per milliliter], absolute values between the assays vary substantially due to different calibration, and no harmonization has ever been conducted [21, 22].
If FGF23 would be used as a new biomarker certain issues need to be assessed. An ideal biomarker would be stable (no degradation ex vivo), show minimal diurnal variability and the analysis should be accurate, reproducible and affordable [23].
Stability of FGF23
There are several studies performed to assess the stability of FGF23, since intact FGF23 may be degraded by proteases or modified after blood withdrawal. First of all, iFGF23 is significantly more stable in plasma (EDTA) than in serum [24]. Even if samples after venepuncture are directly centrifuged and processed, there is the possibility of direct post-venepuncture instability of FGF23. The latter was investigated by Dirks et al. who found no differences between FGF23 concentrations in normal EDTA collecting tubes compared to tubes pre-coated with a protease-inhibitor, suggesting that no immediate protein proteolysis occurs after normal blood withdrawal [25].
However, when centrifugation is delayed a significant decrease of intact FGF23 concentrations was observed in several studies, both in healthy volunteers as well as in patients on dialysis, of 12% with the Immutopics assay, 7% with the Millipore assay and 5% with the Kainos assay (all p < 0.05) [25]. This confirmed two earlier studies that showed 23% reduction of FGF23 measured after a 8 h delay of centrifugation (compared with prompt centrifugation) using the second generation Immutopics assay [21]. Comparable reductions were found with the Millipore and Kainos assays [21, 26]. With the DiaSorin assay no such decrease in FGF23 concentrations was found [27].
Also, post-centrifugal stability of intact FGR 23 has been tested. No decrease of intact FGF23 concentrations are observed when in directly centrifuged samples delayed measurement of intact FGF23 was performed (after 8 h or more) with all the four currently used assays [25, 28, 29]. Besides, there are no indications of degradation of intact FGF23 after storage of processed samples at − 80 °C [25].
Biological variability
In healthy subjects, iFGF23 is subject to significant diurnal variation as iFGF23 concentrations peak in the early morning and fall during the day (mean decrease of 25%) [30, 31]. In contrast, cFGF23 concentrations show only a modest non-significant increase during the day [31]. The inequality of diurnal variation between iFGF23 and cFGF23 concentrations likely reflects the difference in clearance of the intact protein or its C-terminal fragment [32]. iFGF23 concentrations increase after phosphate intake with a delay of at least 12 h [31, 33]. Therefore it is preferable when measuring iFGF23 to use fasting samples or early morning samples as it is shown that iFGF23 concentrations in early morning samples are comparable to fasting samples [30]. In contrast, cFGF23 shows no significant postprandial changes in healthy individuals or patients with early CKD [31, 34, 35]. It is unlikely that a circadian rhythm is of clinical relevance in situations like advanced CKD, where FGF23 levels are extremely elevated.
Accuracy and reproducibility of methods for FGF23 measurement of different assays
In healthy adults, the 95% reference limits for plasma iFGF23 is 11.7–48.6 pg/ml and for cFGF23 21.6–91.0 RU/ml [30]. The intra- and inter-assay coefficient of variation are respectively, < 2.4% and < 4.7% for the second generation Immutopics cFGF23, < 9.7% and < 14% for the Kainos assay [36], < 2.9% and < 6.3% for the DiaSorin assay [28], and lastly, < 10% and < 8% for the Millipore assay. These latter variations are values provided by the manufacturer. The Millipore assay also reports to have a wider functional analytical range, this comes at the expense of poor sensitivity at low concentrations [21]. The first generation intact assay of Immutopics had an unacceptably high inter-assay coefficient of variation [36], that was substantially improved in the second generation assay to < 5% [37].
In patients on dialysis FGF23 concentrations become very high (frequently exceeding 100,000 RU/ml in prevalent patients). As the functional analytical range of the available assays is limited, large dilutions may be necessary to bring the concentration within this range. Another problem is that in patients on haemodialysis substantial intra-individual (week-to-week) variation in cFGF23 concentration has been reported [38, 39]. For all commercially available assays applies that they have not been validated for clinical use. Age- and renal function adapted reference ranges have not been established yet.
Using intact or c-terminal FGF23 assay and when?
The measurement of cFGF23 has, as earlier stated, the advantage of little diurnal variation and has more desirable variance characteristics with higher inter-individual than intra-individual variation. Besides, cFGF23 is more consistently associated with outcome as shown in the meta-analysis of Xiao et al. in which c-term FGF23 was associated with mortality in HD patients whereas iFGF23 measurement did not correlate to mortality [40]. Furthermore cFGF23 is a better predictor for identifying patients with declining renal function [41], atherosclerosis associated cardiovascular disease and heart failure [42]. Therefore, the cFGF23 assay may outperform the iFGF23 assay for clinical use, especially for the purpose of patients individuals risk assessment.
However, iFGF23 may better represent the biological effect of FGF23 [43], especially since it is reported that the c-terminal fragments might have counter-regulator effects to the biologically active full-length hormone [32]. A recent study on the effect of dietary phosphate restriction on FGF23 levels found a more pronounced effect on iFGF23, than on cFGF23 [44].
FGF23 as a risk predictor
FGF23 is associated with progression of kidney failure and initiation of dialysis [41, 45,46,47,48,49]. This raises the question if FGF23 measurement might be a useful tool for risk prediction for progression of CKD and for other adverse outcome. Tangri et al. developed a model to predict progression of CKD that was validated in thirty-one cohorts, including 721,357 participants with CKD stages 3 to 5 (including age, gender, eGFR, albuminuria, serum calcium, serum phosphate, serum bicarbonate, and serum albumin) with a good performance of overall C-statistics (0.90; 95% CI 0.89–0.92). Several studies evaluated if FGF23 was able to improve this predictive value using specific statistics such as the area under the ROC curve and net reclassification index (NRI) statistics [50]. However, only one small study found that adding FGF23 to a base model improved the agreement of predicted and observed probability of renal function decline [51], all other studies found no significant improvement of risk prediction for decline in renal function or ESRD with the addition of FGF23 [49, 52,53,54].
However, for the prediction of all-cause mortality it has been reported that FGF23 might have added value in patients with CKD. [49, 53, 54] In a large study from the CRIC cohort by Edmonston et al. an improved prediction for all-cause mortality and hospital admission for heart failure was found when FGF23 concentration was added to the model, but the NRI did not reach statistical significance. For cardiovascular mortality no improved risk prediction was found [54]. Furthermore, in patients on hemodialysis, addition ofFGF23 did not improve risk prediction for mortality [55, 56].
Concerning cardiovascular events, FGF23 does not consistently improve prediction of novel events in hemodialysis patients [57]. Although one study showed that FGF23 improved prediction of fatal and non-fatal cardiovascular events in predialysis patients [49], another study showed small improvement of prediction only, yet without improved (NRI) classification. However, other studies showed no improvement at all [42, 54]. Interestingly the study by Emrich et al. found that when NT proBNP was added to the model the predictive value of FGF23 was largely eliminated and NT proBNP had a much stronger discriminating ability than FGF23 [42]. Overall, FGF23 only marginally improved the prediction for outcome. An overview of several risk predicting studies is provided in Table 1.
Future studies should evaluate whether multiple measurements of FGF23 may be advantageous compared to a single measurement for individual patients risk assessment in those with CKD, as it was shown that especially increasing FGF23 concentrations over time are associated with increased mortality [58, 59].
Apart from being a risk predictor, FGF23 might serve as an useful tool to identify patients to benefit from certain therapy. Udell et al. showed in their study among patients with stable ischemic heart disease that FGF23 was able to identify patients profiting from angiotensin-converting enzyme inhibitor therapy resulting in reduced cardiovascular death or incident heart failure [60].
Indications of FGF23 toxicity from epidemiological studies
The question however arises if FGF23, besides being a potential risk predictor for adverse outcomes, might also have an instrumental role in the pathogenesis of complications. A great number of epidemiological studies sought an answer to this question.
Mortality
One of the first studies to report an association between FGF23 and mortality was the study by Gutierrez et al. [61] In this nested case control study among incident haemodialysis patients, a concentration dependent effect of FGF23 levels on mortality was observed. Even more interesting, this association became stronger after multiple adjustments, including adjustment for serum phosphate. This observation was confirmed in subsequent studies that followed, mainly in incident HD patients [62,63,64,65]. However, this finding is not consistent, as other studies found no association between FGF23 and mortality in patients on haemodialysis [66,67,68,69,70]. Overall, when 8 studies in patients on haemodialysis were pooled, a relative risk for the highest third of FGF23 versus the lowest third of FGF23 of 1.5 (95% CI 1.29–1.73) for all-cause mortality and of 1.42 (95% CI 0.96–2.39) for cardiovascular mortality was found the meta-analysis by Marthi et al. [71] Remarkably, the association of FGF23 with mortality is stronger in CKD patients not on dialysis despite much lower absolute levels of FGF23 [45, 46, 49, 53, 72,73,74,75]. Concerning the general population, although there are a few studies that found no association of FGF23 with all-cause mortality ([76, 77] most epidemiological studies (some consisting of great number of participants), report modest associations, even when adjusted for eGFR [75, 77,78,79,80].
Cardiovascular disease; myocardial infarction and stroke
In a post hoc analysis of the EVOLVE trial (vide infra) by Moe et al. among nearly three thousand patients on dialysis, FGF23 was statistically significantly associated with the incidence of myocardial infarction [81], an association also found in CKD [45, 75] and in the general population [82, 83]. However, for ischaemic stroke, no consistent association with FGF23 was found in in patient on dialysis [81], nor in the general population [75, 84]. Although some reports do suggest an association may exist with haemorrhagic stroke or thromboembolic stroke [82, 85, 86]. Concerning patients with pre-dialysis CKD, one cohort consisting of nearly four thousand patients found an association between FGF23 and a composite endpoint including myocardial infarction, stroke and peripheral vascular disease [87], an observation confirmed in other CKD cohorts and in the meta-analysis by Marthi et al. [45, 71, 75].
Left ventricular hypertrophy
There are epidemiological data linking FGF23 and left ventricular hypertrophy (LVH). The relatively small studies by Hsu et al. in 2009 and by Kirkpantur et al. in 2011 found an positive association between FGF23 and left ventricular mass in haemodialysis patients [66, 88]. However, in a sub analysis of the Evolve trial, among nearly three thousand haemodialysis patients, there was no association of FGF23 with heart failure [71, 81]. In CKD patients not on dialysis, the association with heart failure is more consistent. Although the study by Bouma -de Krijger et al. in in the Masterplan cohort found no association between FGF23 and congestive heart failure [73], other studies did report such an association [18, 75, 87, 89, 90]. Most epidemiological studies in the general population, one consisting of eleven thousand participants [79], also found an association between FGF23 and heart failure [79, 83, 91,92,93]. Combining several population studies [79, 83, 84, 91], the meta-analysis by Marthi et al. calculated a relative risk of FGF23 on heart failure of 1.24 (95% CI 1.29–1.69) for the highest versus the lowest tertile of FGF23 [71].
Is there evidence that FGF23 can directly induce tissue pathology leading to organ damage?
Apart from its associations with clinical events in etiological driven epidemiological analyses, numerous experimental studies investigated potential mechanisms by which FGF23 might induce cardiovascular pathology.
FGF23 as a cause for left ventricular hypertrophy
Left ventricular hypertrophy (LVH) is an important contributor to cardiovascular morbidity in patients with CKD and LVH is associated with high FGF23 concentration [18]. Several studies explored the potential mechanisms by which FGF23 might induce LVH. For this, Faul et al. administered recombinant FGF23 (rFGF23) to isolated cardiomyocytes and to wild type and klotho deficient mice, where subsequently hypertrophic growth of the myocytes and cardiac hypertrophy in mice was observed, even so in the klotho knock-out animals [18]. FGF23 activated the FGF Receptor leading to calcineurin and nuclear factor of activated T cells (NFAT) signalling in cardiomyocytes. When a pan-FGF Receptor blocker was added to the CKD mice model, LVH was attenuated [18, 94]. Subsequent studies identified FGFR4 as the klotho-independent receptor for FGF23 on cardiomyocytes [95]. Specific blockade of FGFR4 by an antibody inhibited hypertrophy in the isolated cardiac myocytes and mice lacking the FGFR4 did not develop LVH in response to FGF23 [96], establishing FGFR4 as the receptor involved in FGF23-induced LVH [97]. However, these findings are not consistently reported, as in a transgenic mouse model of CKD, with high serum phosphate and FGF23, no signs of pathological cardiac remodelling were found [98].
Interestingly, LVH itself causes cardiac expression of FGF23. Matsui et al. developed two mice models of LVH. In both the transgenic and the pressure overload models, increased expression of FGF23 in the cardiomyocyte (inducing NFAT signaling) followed the development of LVH, while bone expression of FGF23 remained normal [99]. These findings were confirmed in an experimental animal models and in humans where, after myocardial infarction, expression of FGF23 in the heart is described [100, 101]. The upregulation of FGFR4 receptor, the culprit receptor for FGF23-induced cardiotoxicity, in the myocardium might further contribute to hypertrophy, which would imply a feedforward loop [102].
However, contradictory to the above findings, are the results of different mouse models of x-linked hypophosphatemia (XLH). Mice models of XLH have excess FGF23 production, yet those mice do not develop cardiac hypertrophy [103, 104]. This finding is confirmed in XLH patients, where no cardiac hypertrophy is observed [105]. There are several possible explanations for these contractionary findings. Unlike the situation in CKD, XLH is accompanied with low serum phosphate, normal blood pressure, serum calcium and renal function, and the absence of vascular calcification, and this very different phenotype may explain the discordancy. Also, in end stage CKD, FGF23 concentrations can reach values that are more than 1000 fold above normal, and as such much higher than in patients with XLH. Also, the different animal models of LVH had a variety of systemic alterations, such as high phosphate, uraemia or hypertension. It is possible that the deleterious effect of high FGF23 levels might result from a synergy between those abnormalities.
FGF23 and vascular calcification
Another mechanism contributing to the high burden of cardiovascular disease and mortality in CKD is arterial stiffness [106]. This can be worsened, among other causes, by vascular calcification or endothelial dysfunction. Arterial stiffness, regardless of its cause, increases pulse wave velocity, promotes the development of left ventricular hypertrophy, and can result in heart failure. An unresolved question is whether FGF23 can directly act on vascular cells to promote or inhibit matrix calcification. Two studies, by Scialla et al. and Lindberg et al., performed with vascular smooth muscle cells in vitro, showed no calcification when FGF23 was added [107, 108]. Also aortic rings or ex vivo mesenteric arteries of mice showed no calcification or changed vasoreactivity in response to FGF23 [107, 108]. On the other hand, Zhu et al. reported that addition of recombinant FGF23 had a protective effect on calcification in cultured murine SMCs [109]. In contrast, Jimbo et al. showed that FGF23 amplified Pi-induced calcification in cultured human vascular SMCs overexpressing α-Klotho [110]. Other studies focussed on the presence of α-klotho expression, necessary for FGFR1-mediated FGF23 action, in the vasculature. However, these results are also conflicting. Although some studies reported α-klotho expression and FGF23 signalling through FGFR1s receptor activation in the arterial wall [109,110,111], more compelling evidence refutes its presence [107, 112, 113].
FGF23 and endothelial dysfunction
As outlined, besides medial layer calcification, endothelial dysfunction can also contribute to arterial stiffness. Yilmaz et al. found a negative association between FGF23 and flow-mediated vasodilation (FMD) accompanied by increased concentrations of asymmetrical dimethyl arginine (ADMA), an endogenous competitive inhibitor of the vasodilator nitric oxide (NO) [114]. Since FGF23 and ADMA are both associated with progression of CKD, ADMA was added to a statistical model and was found to attenuate the effect of FGF23 on FMD. In two cohorts of CKD patients Tripepi et al. found a strong competitive interaction between FGF23 and ADMA suggesting that FGF23 is a modifier for ADMA levels, leading to dysregulation of the nitric oxide system associated with CKD progression [115].
Different experimental models have been used to explore this aspect of potential FGF23 toxicity. In an ex vivo model of isolated mice aortic rings, addition of recombinant FGF23 increased superoxide levels and reduced the bioavailability of nitric oxide in endothelial cells resulting in impaired relaxation [116]. When a pan FGF23 blocker was administered this effect was eliminated. Another experimental study suggested that the effect of FGF23 on endothelial cells is mediated by reactive oxygen species (ROS) that negatively influence arterial vasodilator capacity [117]. In that study mouse and human aortic rings (the latter obtained after aortic valve bypass surgery) and umbilical cord subjected to high concentrations of recombinant FGF23, recombinant soluble Klotho or phosphate in parallel showed increased ROS production. Addition of sKlotho attenuated the effect of FGF23 and Pi through increasing NO production, thereby protecting the vessel to some extend against the potentially noxious effects of high phosphate or FGF23 concentrations. In the study by Verkaik et al. resistance arteries from mice with renal failure and healthy mouse were studied ex vivo. They showed that pre-treatment with recombinant FGF23 impaired acetylcholine (Ach)-induced vasodilatation, which was restored after administration of FGF23 blocking antibodies regardless of the presence of renal failure.
Collectively, there is evidence that FGF23 induces arterial stiffness. This can be attributed to a large extent to impaired endothelial function, but unlikely to arterial calcification. This effect on endothelial layer mediated arterial stiffness would provide a rationale to target FGF23 as treatment goal. Obviously, before implementing such an approach, this needs clinical proof from prospective trials.
Is FGF23 modifiable?
There are several potential strategies for reducing excess FGF23 levels or bioactivity, principally through dietary phosphate restriction or use of oral phosphate binders, by inhibiting FGFR signalling, by FGF23 blocking agents, and by the use of calcimimetics. In addition, in patients on dialysis, hemodiafiltration is able to reduce of FGF23 levels.
Dietary phosphate restriction to lower FGF23
Several studies evaluated if FGF23 could be reduced by the use of dietary phosphate restriction. Most studies in healthy individuals with normal kidney function reported a decline of intact FGF23 with dietary phosphate restriction and an increased iFGF23 after a phosphate-enriched diet. Studies in CKD found also a reduction of intact FGF23 with a phosphate restricted diet and increased FGF23 with dietary phosphate loading [118,119,120]. Interestingly, studies that measured C-terminal FGF23 did not report modification of cFGF23 with either phosphate loading or restriction, in healthy participants [31, 34, 35], as well as in patients with CKD [121, 122]. A possible explanation for this different findings for iFGF23 and cFGF23 was postulated by Smith et al. in their review on the different FGF23 assays [23]. They hypothesized that dietary phosphate loading might lead to enhanced FGF23 stability, thus a greater proportion of biologically active intact compared to the C-terminal peptide, in order to restore phosphate homeostasis. Of importance is that not only the absolute phosphate content in food counts, but also the source of phosphate matters, since bioavailability of phosphate is different between organic and inorganic phosphate. Phosphate as a food additive (for taste or conservation) is inorganic phosphate and is easily absorbed (bioavailability above 90%) compared to organic phosphate derived from vegetables such as in peas, nuts and cereals (absorption between 40 and 60%). The study by Moe et al. in CKD patients, elegantly showed that a vegetarian diet compared to a meat diet, despite both diets containing comparable amounts of phosphate, induced a decrease in iFGF23, whereas iFGF23 increased in the meat diet [123].
Lowering FGF23 by phosphate binders
Table 2 provides an overview of several different intervention trials using phosphate binders that also reported effects on FGF23 levels. When calcium-containing phosphate binders were used, all of six studies reported no decrease of FGF23 [124,125,126,127,128,129]. Block et al. even found in their placebo controlled trial among 148 patients with moderate CKD (eGFR 20–45 ml/min/1.73 m2), an increase of intact FGF23 with the use of calcium acetate as a phosphate binder [127]. In that study, in which patients were randomized to either placebo, lanthanum carbonate, calcium acetate or sevelamer carbonate, only the group of patients receiving sevelamer carbonate had a significant decline of intact FGF23 compared to placebo. Interestingly, these findings were assay-dependent as these results were not found when the c-terminal assay was used. Other prospective intervention studies that evaluated non-calcium based oral phosphate binders showed a similar pattern. Most studies in CKD patients measuring the intact FGF23 assay found a decrease of FGF23 with the use of either sevelamer [124,125,126, 128], or lanthanum carbonate [129,130,131] in CKD stage 2–5 patients. Ketteler and colleagues did report a 64% reduction of intact FGF23 using sucroferric oxyhydroxide (Velphoro) in patients on dialysis [132]. However, several other studies in predialysis CKD did not find a reduction of FGF23 levels with the use of sevelamer carbonate measuring intact FGF23 [133,134,135,136]. In addition, in several studies measuring c-terminal FGF23, no reduction was found after treatment with either lanthanum carbonate or sevelamer carbonate, sometimes even not so when combined with a low phosphate diet [121, 135, 137,138,139,140]. Only one of studies measuring c-terminal FGF23 found a reduction of FGF23 with dietary phosphate restriction [122]. However, the group with FGF23 reduction in this study had a higher baseline FGF23 concentration than the other groups making interpretation difficult. Another factor that might influence whether or not FGF23 reduction was achieved in the different studies, is the duration of phosphate binder use. Duration of therapy possibly should be 3-6 months or more since it is demonstrated that in kidney transplant patients high FGF23 levels may sustain for this period even when overt hypophosphatemia exists [141, 142], suggesting autonomous FGF23 production, which vanishes only after time. As outlined in Table 2, most studies were of relatively short duration.
FGF23 antibodies and FGF23 receptor blockers
Burosumab is an FDA approved monoclonal antibody targeting FGF23 and was developed for the treatment of XLH. In FGF23-mediated hypophosphataemic disorders it improves hypophosphatemia and bone abnormalities in children [143]. However, there are no data on its use in CKD and its use might be even induce harm by disturbing the adaptive response of FGF23, as was shown in an experimental model of CKD [144]. In that animal study, FGF23 antibodies resulted in decrease of FGF23 and ameliorated uremic hyperparathyroidism. However, urinary phosphate excretion decreased and hyperphosphatemia developed, promoting vascular calcification and increased mortality. The same observations were found in an experimental study with a pan-FGFR inhibitor [145], showing that FGF23 remains necessary to maintain serum Pi levels within range, at least in non-dialysis dependent CKD. A more promising strategy might be to specifically inhibit FGFR4 signalling to prevent the development of LVH associated with high FGF23 levels [96]. However, this was only tested in an animal model and there are currently no data available to suggest a clinical benefit for patients with CKD.
Calcimimetics
Several studies showed that FGF23 concentrations can be reduced by the use of cinacalcet or etelcalcetide. In a randomized trial by Wetmore et al. it was demonstrated that patients assigned to cinacalcet compared to low dose calcitriol had a decrease of serum FGF23 concentrations [146]. However, it was not clear if this was caused by the relatively low dose of active vitamin D in the cinacalcet treatment arm, considering that active vitamin D being a strong stimulator of FGF23. The study by Koizumi et al. found that the decrease of FGF23 during cinacalcet treatment was independent of active vitamin D [147]. Also data from the Evolve trial showed that cinacalcet is a potent suppressor of FGF23 [81]. The newer intravenously used calcimimetic etelcalcetide appears to be an even more potent suppressor of FGF23 compared to cinacalcet [148]. However, the mechanism by which cinacalcet or etelcalcetide suppresses FGF23 is unclear. Both a direct effect on the calcium sensing receptor on osteocytes or a decline of the calcium-phosphate product may have accomplished this effect.
Haemodiafiltration (HDF)
Previous studies have shown that FGF23 with its 32 kDa middle molecular size can be cleared by HDF and not by low-flux HD [149]. Several studies have shown a percent reduction of serum FGF23 within a single HDF session of around 50% (± 25%), with FGF23 detected in spent dialysate samples [150,151,152]. The study by Bouma et al. showed a sustained decline FGF23 over time, of greater size with higher convection volume [70].
Is it useful to measure serial FGF23 concentrations?
Whether change of FGF23 better reflects risks and possibly impacts on outcomes is an open question. There are some studies that do shed some light in this issue. Probably the best of these is the study by Isakova on the CRIC cohort, consisting of 1135 patients with a mean eGFR of 46.3 (± 14.7) ml/min per 1.73 m2. Although, the majority of patients from this cohort had stable FGF23 concentrations during the 5 years of follow-up, patients with rapidly increasing FGF23 concentrations had an exceptionally high mortality risk [58]. Also, in a sub analysis of the CONTRAST study in prevalent haemodialysis patients, increasing levels of FGF23 were associated with increased mortality [70]. Both studies found, in different populations, that increasing FGF23 is disadvantageous. However, the study by Jovanovich in over 900 dialysis patients showed that, although over 24 month stable low FGF23 concentration was associated with a favourable outcome compared to stable high FGF23 concentrations, the group with high FGF23 in which FGF23 further increased over time, had no further increased risk for all-cause mortality [59]. More importantly, this study also showed that in patient with high baseline, and subsequently decreasing FGF23 concentrations had no improved risk for mortality. Likewise, in the sub analysis of the CONTRAST study, a decrease in FGF23 was not associated with improved mortality compared to a stable FGF23 concentration. A secondary analysis of the EVOLVE trial [153] analysed the impact of cinacalcet-induced reductions in FGF23. This analysis, different from the studies discussed above, did suggest that decreasing FGF23 might be beneficial [81]. Here, a more than 30% reduction of FGF23 in 20 weeks among participants allocated to cinacalcet, was associated with a reduced risk on the composite outcome of cardiovascular mortality, sudden cardiac death and heart failure. Remarkably, in the placebo treated group there were also (yet fewer) patients with more than 30% FGF23 reduction, but in those patients no association with reduced risk on outcome was found. This suggests that not the decline of FGF23 itself, but the way it was achieved, determined the more favourable outcome. Since the main outcome of the EVOLVE trial demonstrated no improvement of the primary combined outcome in cinacalcet treated patients, FGF23 reduction probably just identified patients who might benefit more from cinacalcet treatment [153]. Since FGF23 has been implicated with the development of LVH, Seifert et al. studied CKD patients with an increase of LVH over 12 months. This worsening of LVH was not associated with an increase of FGF23 [154]. Moreover, Chue et al. identified in their study, among CKD patients treated with sevelamer, a subgroup with a decrease of FGF23, which was however not accompanied with a change in arterial stiffness, left ventricular mass or cardiac function [133]. Therefore, there is currently no compelling evidence that FGF23 reduction leads to improved outcome and this leaves the role of FGF23 in the pathway to adverse outcome still under debate.
FGF23 are we ready to use it in clinical practice? (conclusion)
In conclusion, FGF23 is a promising biomarker in CKD. Although the different FGF23 assays should be harmonized and assay specific reference intervals should be established, FGF23 measurement has shown to be consistently associated for adverse outcome in different populations. Furthermore, FGF23 measurement was shown, and validated, to predict mortality in CKD. However, FGF23 has limited value in predicting progression of renal failure in CKD or in risk prediction in patients on dialysis. Future studies should evaluate the predictive validity of repeated FGF23 testing. Increasing FGF23 concentrations over time, both in CKD and dialysis patients are associated with dismal outcomes.
Considering the fact that there is currently no, or very limited, evidence that FGF23 reduction leads to improved outcome, it is preliminary to use FGF23 concentrations as target for therapy in everyday clinical practice, despite the ability of dietary phosphate restriction and phosphate binders to lower FGF23 concentrations in CKD. For patients on haemodialysis both calcimimetics, non-calcium containing phosphate binders and HDF are effective modes to lower FGF23. The availability of interventions that lower FGF23 sets the stage for clinical trials that target FGF23 (and not phosphate concentrations) and have clinical events as primary endpoint.
References
Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY (2004) Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 351(13):1296–1305
Kestenbaum B, Sampson JN, Rudser KD, Patterson DJ, Seliger SL, Young B, Sherrard DJ, Andress DL (2005) Serum phosphate levels and mortality risk among people with chronic kidney disease. J Am Soc Nephrol 16(2):520–528
Hecking M, Moissl U, Genser B, Rayner H, Dasgupta I, Stuard S, Stopper A, Chazot C, Maddux FW, Canaud B, Port FK, Zoccali C, Wabel P (2018) Greater fluid overload and lower interdialytic weight gain are independently associated with mortality in a large international hemodialysis population. Nephrol Dial Transplant 33(10):1832–1842. https://doi.org/10.1093/ndt/gfy083
Covic A, Kothawala P, Bernal M, Robbins S, Chalian A, Goldsmith D (2009) Systematic review of the evidence underlying the association between mineral metabolism disturbances and risk of all-cause mortality, cardiovascular mortality and cardiovascular events in chronic kidney disease. Nephrol Dial Transplant 24(5):1506–1523
Consortium A (2000) Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet 26(3):345–348. https://doi.org/10.1038/81664
Bastepe M, Juppner H (2008) Inherited hypophosphatemic disorders in children and the evolving mechanisms of phosphate regulation. Rev Endocr Metab Disord 9(2):171–180. https://doi.org/10.1007/s11154-008-9075-3
Goldsweig BK, Carpenter TO (2015) Hypophosphatemic rickets: lessons from disrupted FGF23 control of phosphorus homeostasis. Curr Osteoporos Rep 13(2):88–97. https://doi.org/10.1007/s11914-015-0259-y
Farrow EG, Davis SI, Summers LJ, White KE (2009) Initial FGF23-mediated signaling occurs in the distal convoluted tubule. J Am Soc Nephrol 20(5):955–960. https://doi.org/10.1681/ASN.2008070783
Saito H, Kusano K, Kinosaki M, Ito H, Hirata M, Segawa H, Miyamoto K, Fukushima N (2003) Human fibroblast growth factor-23 mutants suppress Na+ -dependent phosphate co-transport activity and 1 alpha, 25-dihydroxyvitamin D3 production. J Biol Chem 278(4):2206–2211
Shimada T, Hasegawa H, Yamazaki Y, Muto T, Hino R, Takeuchi Y, Fujita T, Nakahara K, Fukumoto S, Yamashita T (2004) FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J Bone Miner Res 19(3):429–435
Baum M, Schiavi S, Dwarakanath V, Quigley R (2005) Effect of fibroblast growth factor-23 on phosphate transport in proximal tubules. Kidney Int 68(3):1148–1153
Ben-Dov IZ, Galitzer H, Lavi-Moshayoff V, Goetz R, Mohammadi M, Sirkis R, Naveh-Many T, Silver J (2007) The parathyroid is a target organ for FGF23 in rats. J Clin Invest 117(12):4003–4008. https://doi.org/10.1172/jci32409
Urakawa I, Yamazaki Y, Shimada T, Iijima K, Hasegawa H, Okawa K, Fujita T, Fukumoto S, Yamashita T (2006) Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444(7120):770–774
Kurosu H, Ogawa Y, Miyoshi M, Yamamoto M, Nandi A, Rosenblatt KP, Baum MG, Schiavi S, Hu MC, Moe OW, K-o M (2006) Regulation of fibroblast growth factor-23 signaling by klotho. J Biol Chem 281(10):6120–6123
Nakatani T, Sarraj B, Ohnishi M, Densmore MJ, Taguchi T, Goetz R, Mohammadi M, Lanske B, Razzaque MS (2009) In vivo genetic evidence for klotho-dependent, fibroblast growth factor 23 (FGF23) -mediated regulation of systemic phosphate homeostasis. FASEB J 23(2):433–441
Ix JH, Shlipak MG, Wassel CL, Whooley MA (2010) Fibroblast growth factor-23 and early decrements in kidney function: the Heart and Soul Study. Nephrol Dial Transplant 25(3):993–997
Isakova T, Wahl P, Vargas GS, Gutierrez OM, Scialla J, Xie H, Appleby D, Nessel L, Bellovich K, Chen J, Hamm L, Gadegbeku C, Horwitz E, Townsend RR, Anderson CA, Lash JP, Hsu CY, Leonard MB, Wolf M (2011) Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int 79(12):1370–1378
Faul C, Amaral AP, Oskouei B, Hu MC, Sloan A, Isakova T, Gutierrez OM, Guillon-Prada R, Lincoln J, Hare JM, Mundel P, Morales A, Scialla J, Fischer M, Soliman EZ, Chen J, Go AS, Rosas SE, Nessel L, Townsend RR, Feldman HI, St John SM, Ojo A, Gadegbeku C, Di Marco GS, Reuter S, Kentrup D, Tiemann K, Brand M, Hill JA, Moe OW, Kuro O, Kusek JW, Keane MG, Wolf M (2011) FGF23 induces left ventricular hypertrophy. J Clin Invest 121(11):4393–4408
Mirza MA, Larsson A, Lind L, Larsson TE (2009) Circulating fibroblast growth factor-23 is associated with vascular dysfunction in the community. Atherosclerosis 205(2):385–390
Yilmaz MI, Sonmez A, Saglam M, Yaman H, Kilic S, Demirkaya E, Eyileten T, Caglar K, Oguz Y, Vural A, Yenicesu M, Zoccali C (2010) FGF-23 and vascular dysfunction in patients with stage 3 and 4 chronic kidney disease. Kidney Int 78(7):679–685
Smith ER, McMahon LP, Holt SG (2013) Method-specific differences in plasma fibroblast growth factor 23 measurement using four commercial ELISAs. Clin Chem Lab Med 51(10):1971–1981. https://doi.org/10.1515/cclm-2013-0208
Sinha MD, Turner C, Goldsmith DJ (2013) FGF23 concentrations measured using “intact” assays similar but not interchangeable. Int Urol Nephrol 45(6):1821–1823. https://doi.org/10.1007/s11255-013-0451-x
Smith ER, McMahon LP, Holt SG (2014) Fibroblast growth factor 23. Ann Clin Biochem 51(Pt 2):203–227. https://doi.org/10.1177/0004563213510708
Fassbender WJ, Brandenburg V, Schmitz S, Sandig D, Simon SA, Windolf J, Stumpf UC (2009) Evaluation of human fibroblast growth factor 23 (FGF-23) C-terminal and intact enzyme-linked immunosorbent-assays in end-stage renal disease patients. Clin Lab 55(3–4):144–152
Dirks NF, Smith ER, van Schoor NM, Vervloet MG, Ackermans MT, de Jonge R, Heijboer AC (2019) Pre-analytical stability of FGF23 with the contemporary immunoassays. Clin Chim Acta 493:104–106. https://doi.org/10.1016/j.cca.2019.02.032
Cui S, Vaingankar SM, Stenger A, Waikar SS, Leaf DE (2017) Stability of fibroblast growth factor 23 in human plasma. J Appl Lab Med 6:729–734
van Helden J, Weiskirchen R (2018) Technical and diagnostic performance of a new fully automated immunoassay for the determination of intact fibroblast growth factor 23 (FGF23). Scand J Clin Lab Invest 78(7–8):584–590. https://doi.org/10.1080/00365513.2018.1526411
Souberbielle JC, Prie D, Piketty ML, Rothenbuhler A, Delanaye P, Chanson P, Cavalier E (2017) Evaluation of a new fully automated assay for plasma intact FGF23. Calcif Tissue Int 101(5):510–518. https://doi.org/10.1007/s00223-017-0307-y
El-Maouche D, Dumitrescu CE, Andreopoulou P, Gafni RI, Brillante BA, Bhattacharyya N, Fedarko NS, Collins MT (2016) Stability and degradation of fibroblast growth factor 23 (FGF23): the effect of time and temperature and assay type. Osteoporos Int 27(7):2345–2353. https://doi.org/10.1007/s00198-016-3543-5
Smith ER, Cai MM, McMahon LP, Holt SG (2012) Biological variability of plasma intact and C-terminal FGF23 measurements. J Clin Endocrinol Metab 97(9):3357–3365. https://doi.org/10.1210/jc.2012-1811
Vervloet MG, van Ittersum FJ, Buttler RM, Heijboer AC, Blankenstein MA, ter Wee PM (2011) Effects of dietary phosphate and calcium intake on fibroblast growth factor-23. Clin J Am Soc Nephrol 6(2):383–389
Goetz R, Nakada Y, Hu MC, Kurosu H, Wang L, Nakatani T, Shi M, Eliseenkova AV, Razzaque MS, Moe OW, Kuro-o M, Mohammadi M (2010) Isolated C-terminal tail of FGF23 alleviates hypophosphatemia by inhibiting FGF23–FGFR–klotho complex formation. Proc Natl Acad Sci USA 107(1):407–412. https://doi.org/10.1073/pnas.0902006107
Nishida Y, Taketani Y, Yamanaka-Okumura H, Imamura F, Taniguchi A, Sato T, Shuto E, Nashiki K, Arai H, Yamamoto H, Takeda E (2006) Acute effect of oral phosphate loading on serum fibroblast growth factor 23 levels in healthy men. Kidney Int 70(12):2141–2147. https://doi.org/10.1038/sj.ki.5002000
Larsson T, Nisbeth U, Ljunggren O, Juppner H, Jonsson KB (2003) Circulating concentration of FGF-23 increases as renal function declines in patients with chronic kidney disease, but does not change in response to variation in phosphate intake in healthy volunteers. Kidney Int 64(6):2272–2279. https://doi.org/10.1046/j.1523-1755.2003.00328.x
Burnett SM, Gunawardene SC, Bringhurst FR, Juppner H, Lee H, Finkelstein JS (2006) Regulation of C-terminal and intact FGF-23 by dietary phosphate in men and women. J Bone Miner Res 21(8):1187–1196
Heijboer AC, Levitus M, Vervloet MG, Lips P, ter Wee PM, Dijstelbloem HM, Blankenstein MA (2009) Determination of fibroblast growth factor 23. Ann Clin Biochem 46(Pt 4):338–340
Smith ER (2014) The use of fibroblast growth factor 23 testing in patients with kidney disease. Clin J Am Soc Nephrol 9(7):1283–1303. https://doi.org/10.2215/CJN.10941013
Seiler S, Lucisano G, Ege P, Fell LH, Rogacev KS, Lerner-Graber A, Klingele M, Ziegler M, Fliser D, Heine GH (2013) Single FGF-23 measurement and time-averaged plasma phosphate levels in hemodialysis patients. Clin J Am Soc Nephrol 8(10):1764–1772. https://doi.org/10.2215/cjn.13021212
Cavalier E, Delanaye P, Moranne O (2013) Variability of new bone mineral metabolism markers in patients treated with maintenance hemodialysis: implications for clinical decision making. Am J Kidney Dis 61(5):847–848. https://doi.org/10.1053/j.ajkd.2012.12.013
Xiao Y, Luo X, Huang W, Zhang J, Peng C (2014) Fibroblast growth factor 23 and risk of all-cause mortality and cardiovascular events: a meta-analysis of prospective cohort studies. Int J Cardiol 174(3):824–828. https://doi.org/10.1016/j.ijcard.2014.04.138
Fliser D, Kollerits B, Neyer U, Ankerst DP, Lhotta K, Lingenhel A, Ritz E, Kronenberg F, Kuen E, Konig P, Kraatz G, Mann JF, Muller GA, Kohler H, Riegler P (2007) Fibroblast growth factor 23 (FGF23) predicts progression of chronic kidney disease: the Mild to Moderate Kidney Disease (MMKD) Study. J Am Soc Nephrol 18(9):2600–2608
Emrich IE, Brandenburg V, Sellier AB, Schauerte J, Wiedenroth J, Untersteller K, Lennartz CS, Seiler-Mussler S, Wagenpfeil S, Fliser D, Heine GH (2019) Strength of fibroblast growth factor 23 as a cardiovascular risk predictor in chronic kidney disease weaken by ProBNP adjustment. Am J Nephrol 49(3):203–211. https://doi.org/10.1159/000497125
Shimada T, Muto T, Urakawa I, Yoneya T, Yamazaki Y, Okawa K, Takeuchi Y, Fujita T, Fukumoto S, Yamashita T (2002) Mutant FGF-23 responsible for autosomal dominant hypophosphatemic rickets is resistant to proteolytic cleavage and causes hypophosphatemia in vivo. Endocrinology 143(8):3179–3182. https://doi.org/10.1210/endo.143.8.8795
Tsai WC, Wu HY, Peng YS, Hsu SP, Chiu YL, Chen HY, Yang JY, Ko MJ, Pai MF, Tu YK, Hung KY, Chien KL (2018) Effects of lower versus higher phosphate diets on fibroblast growth factor-23 levels in patients with chronic kidney disease: a systematic review and meta-analysis. Nephrol Dial Transplant. https://doi.org/10.1093/ndt/gfy005
Kendrick J, Cheung AK, Kaufman JS, Greene T, Roberts WL, Smits G, Chonchol M (2011) FGF-23 associates with death, cardiovascular events, and initiation of chronic dialysis. J Am Soc Nephrol 22(10):1913–1922
Isakova T, Xie H, Yang W, Xie D, Anderson AH, Scialla J, Wahl P, Gutierrez OM, Steigerwalt S, He J, Schwartz S, Lo J, Ojo A, Sondheimer J, Hsu CY, Lash J, Leonard M, Kusek JW, Feldman HI, Wolf M (2011) Fibroblast growth factor 23 and risks of mortality and end-stage renal disease in patients with chronic kidney disease. JAMA 305(23):2432–2439
Titan SM, Zatz R, Graciolli FG, dos Reis LM, Barros RT, Jorgetti V, Moyses RM (2011) FGF-23 as a predictor of renal outcome in diabetic nephropathy. Clin J Am Soc Nephrol 6(2):241–247
Scialla JJ, Astor BC, Isakova T, Xie H, Appel LJ, Wolf M (2013) Mineral metabolites and CKD progression in African Americans. J Am Soc Nephrol 24(1):125–135. https://doi.org/10.1681/asn.2012070713
Alderson HV, Ritchie JP, Middleton R, Larsson A, Larsson TE, Kalra PA (2016) FGF-23 and osteoprotegerin but not fetuin-A are associated with death and enhance risk prediction in non-dialysis chronic kidney disease stages 3-5. Nephrology (Carlton) 21(7):566–573. https://doi.org/10.1111/nep.12664
Pencina MJ, D’Agostino RB Sr, D’Agostino RB Jr, Vasan RS (2008) Evaluating the added predictive ability of a new marker: from area under the ROC curve to reclassification and beyond. Stat Med 27(2):157–172. https://doi.org/10.1002/sim.2929(discussion 207–112)
Smith ER, Ford ML, Tomlinson LA, McMahon LP, Rajkumar C, Holt SG (2012) FGF23 adds value to risk prediction in patients with chronic kidney disease. Bone 51(4):830–831. https://doi.org/10.1016/j.bone.2012.05.017(author reply 832–833)
Nowak N, Skupien J, Smiles AM, Yamanouchi M, Niewczas MA, Galecki AT, Duffin KL, Breyer MD, Pullen N, Bonventre JV, Krolewski AS (2018) Markers of early progressive renal decline in type 2 diabetes suggest different implications for etiological studies and prognostic tests development. Kidney Int 93(5):1198–1206. https://doi.org/10.1016/j.kint.2017.11.024
Levin A, Rigatto C, Barrett B, Madore F, Muirhead N, Holmes D, Clase CM, Tang M, Djurdjev O, Can PI (2014) Biomarkers of inflammation, fibrosis, cardiac stretch and injury predict death but not renal replacement therapy at 1 year in a Canadian chronic kidney disease cohort. Nephrol Dial Transplant 29(5):1037–1047. https://doi.org/10.1093/ndt/gft479
Edmonston D, Wojdyla D, Mehta R, Cai X, Lora C, Cohen D, Townsend RR, He J, Go AS, Kusek J, Weir MR, Isakova T, Pencina M, Wolf M, Investigators CS (2019) Single measurements of carboxy-terminal fibroblast growth factor 23 and clinical risk prediction of adverse outcomes in CKD. Am J Kidney Dis. https://doi.org/10.1053/j.ajkd.2019.05.026
Artunc F, Nowak A, Muller C, Peter A, Heyne N, Haring HU, Friedrich B (2014) Mortality prediction using modern peptide biomarkers in hemodialysis patients–a comparative analysis. Kidney Blood Press Res 39(6):563–572. https://doi.org/10.1159/000368468
Yamashita K, Mizuiri S, Nishizawa Y, Shigemoto K, Doi S, Masaki T (2018) Addition of novel biomarkers for predicting all-cause and cardiovascular mortality in prevalent hemodialysis patients. Ther Apher Dial 22(1):31–39. https://doi.org/10.1111/1744-9987.12593
Nakano C, Hamano T, Fujii N, Obi Y, Matsui I, Tomida K, Mikami S, Inoue K, Shimomura A, Nagasawa Y, Okada N, Tsubakihara Y, Rakugi H, Isaka Y (2012) Intact fibroblast growth factor 23 levels predict incident cardiovascular event before but not after the start of dialysis. Bone 50(6):1266–1274. https://doi.org/10.1016/j.bone.2012.02.634
Isakova T, Cai X, Lee J, Xie D, Wang X, Mehta R, Allen NB, Scialla JJ, Pencina MJ, Anderson AH, Talierco J, Chen J, Fischer MJ, Steigerwalt SP, Leonard MB, Hsu CY, de Boer IH, Kusek JW, Feldman HI, Wolf M, Chronic Renal Insufficiency Cohort Study I (2018) Longitudinal FGF23 trajectories and mortality in patients with CKD. J Am Soc Nephrol 29(2):579–590. https://doi.org/10.1681/ASN.2017070772
Jovanovich A, You Z, Isakova T, Nowak K, Cheung A, Wolf M, Chonchol M, Kendrick J (2019) Fibroblast growth factor 23 trajectories in chronic hemodialysis patients: lessons from the HEMO Study. Am J Nephrol 49(4):263–270. https://doi.org/10.1159/000497445
Udell JA, Morrow DA, Jarolim P, Sloan S, Hoffman EB, O’Donnell TF, Vora AN, Omland T, Solomon SD, Pfeffer MA, Braunwald E, Sabatine MS (2014) Fibroblast growth factor-23, cardiovascular prognosis, and benefit of angiotensin-converting enzyme inhibition in stable ischemic heart disease. J Am Coll Cardiol 63(22):2421–2428. https://doi.org/10.1016/j.jacc.2014.03.026
Gutierrez OM, Mannstadt M, Isakova T, Rauh-Hain JA, Tamez H, Shah A, Smith K, Lee H, Thadhani R, Juppner H, Wolf M (2008) Fibroblast growth factor 23 and mortality among patients undergoing hemodialysis. N Engl J Med 359(6):584–592
Scialla JJ, Parekh RS, Eustace JA, Astor BC, Plantinga L, Jaar BG, Shafi T, Coresh J, Powe NR, Melamed ML (2015) Race, mineral homeostasis and mortality in patients with end-stage renal disease on dialysis. Am J Nephrol 42(1):25–34. https://doi.org/10.1159/000438999
Nowak A, Friedrich B, Artunc F, Serra AL, Breidthardt T, Twerenbold R, Peter M, Mueller C (2014) Prognostic value and link to atrial fibrillation of soluble Klotho and FGF23 in hemodialysis patients. PLoS ONE 9(7):e100688. https://doi.org/10.1371/journal.pone.0100688
Jean G, Terrat JC, Vanel T, Hurot JM, Lorriaux C, Mayor B, Chazot C (2009) High levels of serum fibroblast growth factor (FGF)-23 are associated with increased mortality in long haemodialysis patients. Nephrol Dial Transplant 24(9):2792–2796
Chonchol M, Greene T, Zhang Y, Hoofnagle AN, Cheung AK (2016) Low vitamin D and high fibroblast growth factor 23 serum levels associate with infectious and cardiac deaths in the HEMO Study. J Am Soc Nephrol 27(1):227–237. https://doi.org/10.1681/ASN.2014101009
Hsu HJ, Wu MS (2009) Fibroblast growth factor 23: a possible cause of left ventricular hypertrophy in hemodialysis patients. Am J Med Sci 337(2):116–122
Olauson H, Qureshi AR, Miyamoto T, Barany P, Heimburger O, Lindholm B, Stenvinkel P, Larsson TE (2010) Relation between serum fibroblast growth factor-23 level and mortality in incident dialysis patients: are gender and cardiovascular disease confounding the relationship? Nephrol Dial Transplant 25(9):3033–3038. https://doi.org/10.1093/ndt/gfq191
Sugimoto H, Ogawa T, Iwabuchi Y, Otsuka K, Nitta K (2014) Relationship between serum fibroblast growth factor-23 level and mortality in chronic hemodialysis patients. Int Urol Nephrol 46(1):99–106. https://doi.org/10.1007/s11255-013-0386-2
Komaba H, Fuller DS, Taniguchi M, Yamamoto S, Nomura T, Bieber BA, Robinson BM, Pisoni RL, Fukagawa M (2017) FGF23 and mortality in a large cohort of prevalent hemodialysis patients: results from the J-DOPPS. ASN Kidney Week, abstract #: TH-PO787
Bouma-de Krijger A, De Roij van Zuijdewijn CLM, Nubé MJ, Grooteman MPC, Vervloet MG (2020) Change of FGF23 concentrations over time and its association with all-cause mortality in patients treated with hemodialysis or hemodiafiltration. Clin Kidney J (publication accepted, follows in 2020)
Marthi A, Donovan K, Haynes R, Wheeler DC, Baigent C, Rooney CM, Landray MJ, Moe SM, Yang J, Holland L, di Giuseppe R, Bouma-de Krijger A, Mihaylova B, Herrington WG (2018) Fibroblast growth factor-23 and risks of cardiovascular and noncardiovascular diseases: a meta-analysis. J Am Soc Nephrol 29(7):2015–2027. https://doi.org/10.1681/ASN.2017121334
Baia LC, Humalda JK, Vervloet MG, Navis G, Bakker SJ, de Borst MH, Consortium N (2013) Fibroblast growth factor 23 and cardiovascular mortality after kidney transplantation. Clin J Am Soc Nephrol 8(11):1968–1978. https://doi.org/10.2215/CJN.01880213
Bouma-de KA, Bots ML, Vervloet MG, Blankestijn PJ, Ter Wee PW, van Zuilen AD, Wetzels JF (2014) Time-averaged level of fibroblast growth factor-23 and clinical events in chronic kidney disease. Nephrol Dial Transplant 29(1):88–97. https://doi.org/10.1093/ndt/gft456
Munoz Mendoza J, Isakova T, Cai X, Bayes LY, Faul C, Scialla JJ, Lash JP, Chen J, He J, Navaneethan S, Negrea L, Rosas SE, Kretzler M, Nessel L, Xie D, Anderson AH, Raj DS, Wolf M, Investigators CS (2017) Inflammation and elevated levels of fibroblast growth factor 23 are independent risk factors for death in chronic kidney disease. Kidney Int 91(3):711–719. https://doi.org/10.1016/j.kint.2016.10.021
Ix JH, Katz R, Kestenbaum BR, Chonchol M, Mukamal KJ, Rifkin D, Siscovick DS, Sarnak MJ, Shlipak MG (2012) Fibroblast growth factor-23 and death, heart failure, and cardiovascular events in community-living individuals: CHS (Cardiovascular Health Study). J Am Coll Cardiol 60(3):200–207
Arnlov J, Carlsson AC, Sundstrom J, Ingelsson E, Larsson A, Lind L, Larsson TE (2013) Higher fibroblast growth factor-23 increases the risk of all-cause and cardiovascular mortality in the community. Kidney Int 83(1):160–166. https://doi.org/10.1038/ki.2012.327
Westerberg PA, Tivesten A, Karlsson MK, Mellstrom D, Orwoll E, Ohlsson C, Larsson TE, Linde T, Ljunggren O (2013) Fibroblast growth factor 23, mineral metabolism and mortality among elderly men (Swedish MrOs). BMC Nephrol 14:85. https://doi.org/10.1186/1471-2369-14-85
Brandenburg VM, Kleber ME, Vervloet MG, Tomaschitz A, Pilz S, Stojakovic T, Delgado G, Grammer TB, Marx N, Marz W, Scharnagl H (2014) Fibroblast growth factor 23 (FGF23) and mortality: the Ludwigshafen Risk and Cardiovascular Health Study. Atherosclerosis 237(1):53–59. https://doi.org/10.1016/j.atherosclerosis.2014.08.037
Lutsey PL, Alonso A, Selvin E, Pankow JS, Michos ED, Agarwal SK, Loehr LR, Eckfeldt JH, Coresh J (2014) Fibroblast growth factor-23 and incident coronary heart disease, heart failure, and cardiovascular mortality: the Atherosclerosis Risk in Communities study. J Am Heart Assoc 3(3):e000936. https://doi.org/10.1161/JAHA.114.000936
Souma N, Isakova T, Lipiszko D, Sacco RL, Elkind MS, DeRosa JT, Silverberg SJ, Mendez AJ, Dong C, Wright CB, Wolf M (2016) Fibroblast growth factor 23 and cause-specific mortality in the general population: the Northern Manhattan Study. J Clin Endocrinol Metab 101(10):3779–3786. https://doi.org/10.1210/jc.2016-2215
Moe SM, Chertow GM, Parfrey PS, Kubo Y, Block GA, Correa-Rotter R, Drueke TB, Herzog CA, London GM, Mahaffey KW, Wheeler DC, Stolina M, Dehmel B, Goodman WG, Floege J (2015) Evaluation of cinacalcet HTtLCETI, cinacalcet, fibroblast growth factor-23, and cardiovascular disease in hemodialysis: the evaluation of cinacalcet HCl therapy to lower cardiovascular events (EVOLVE) trial. Circulation 132(1):27–39. https://doi.org/10.1161/CIRCULATIONAHA.114.013876
di Giuseppe R, Kuhn T, Hirche F, Buijsse B, Dierkes J, Fritsche A, Kaaks R, Boeing H, Stangl GI, Weikert C (2015) Plasma fibroblast growth factor 23 and risk of cardiovascular disease: results from the EPIC-Germany case-cohort study. Eur J Epidemiol 30(2):131–141. https://doi.org/10.1007/s10654-014-9982-4
Parker BD, Schurgers LJ, Brandenburg VM, Christenson RH, Vermeer C, Ketteler M, Shlipak MG, Whooley MA, Ix JH (2010) The associations of fibroblast growth factor 23 and uncarboxylated matrix Gla protein with mortality in coronary artery disease: the Heart and Soul Study. Ann Intern Med 152(10):640–648. https://doi.org/10.7326/0003-4819-152-10-201005180-00004
Kestenbaum B, Sachs MC, Hoofnagle AN, Siscovick DS, Ix JH, Robinson-Cohen C, Lima JA, Polak JF, Blondon M, Ruzinski J, Rock D, de Boer IH (2014) Fibroblast growth factor-23 and cardiovascular disease in the general population: the Multi-Ethnic Study of Atherosclerosis. Circ Heart Fail 7(3):409–417. https://doi.org/10.1161/CIRCHEARTFAILURE.113.000952
Wright CB, Dong C, Stark M, Silverberg S, Rundek T, Elkind MS, Sacco RL, Mendez A, Wolf M (2014) Plasma FGF23 and the risk of stroke: the Northern Manhattan Study (NOMAS). Neurology 82(19):1700–1706. https://doi.org/10.1212/WNL.0000000000000410
Panwar B, Jenny NS, Howard VJ, Wadley VG, Muntner P, Kissela BM, Judd SE, Gutierrez OM (2015) Fibroblast growth factor 23 and risk of incident stroke in community-living adults. Stroke 46(2):322–328. https://doi.org/10.1161/STROKEAHA.114.007489
Scialla JJ, Xie H, Rahman M, Anderson AH, Isakova T, Ojo A, Zhang X, Nessel L, Hamano T, Grunwald JE, Raj DS, Yang W, He J, Lash JP, Go AS, Kusek JW, Feldman H, Wolf M (2014) Fibroblast Growth Factor-23 and Cardiovascular Events in CKD. J Am Soc Nephrol 25(2):349–360. https://doi.org/10.1681/asn.2013050465
Kirkpantur A, Balci M, Gurbuz OA, Afsar B, Canbakan B, Akdemir R, Ayli MD (2011) Serum fibroblast growth factor-23 (FGF-23) levels are independently associated with left ventricular mass and myocardial performance index in maintenance haemodialysis patients. Nephrol Dial Transplant 26(4):1346–1354
Seiler S, Rogacev KS, Roth HJ, Shafein P, Emrich I, Neuhaus S, Floege J, Fliser D, Heine GH (2014) Associations of FGF-23 and sKlotho with cardiovascular outcomes among patients with CKD stages 2-4. Clin J Am Soc Nephrol 9(6):1049–1058. https://doi.org/10.2215/CJN.07870713
Gutierrez OM, Januzzi JL, Isakova T, Laliberte K, Smith K, Collerone G, Sarwar A, Hoffmann U, Coglianese E, Christenson R, Wang TJ, deFilippi C, Wolf M (2009) Fibroblast growth factor 23 and left ventricular hypertrophy in chronic kidney disease. Circulation 119(19):2545–2552. https://doi.org/10.1161/CIRCULATIONAHA.108.844506
di Giuseppe R, Buijsse B, Hirche F, Wirth J, Arregui M, Westphal S, Isermann B, Hense HW, Dierkes J, Boeing H, Stangl GI, Weikert C (2014) Plasma fibroblast growth factor 23, parathyroid hormone, 25-hydroxyvitamin D3, and risk of heart failure: a prospective, case-cohort study. J Clin Endocrinol Metab 99(3):947–955. https://doi.org/10.1210/jc.2013-2963
Mirza MA, Larsson A, Melhus H, Lind L, Larsson TE (2009) Serum intact FGF23 associate with left ventricular mass, hypertrophy and geometry in an elderly population. Atherosclerosis 207(2):546–551
Jovanovich A, Ix JH, Gottdiener J, McFann K, Katz R, Kestenbaum B, de Boer IH, Sarnak M, Shlipak MG, Mukamal KJ, Siscovick D, Chonchol M (2013) Fibroblast growth factor 23, left ventricular mass, and left ventricular hypertrophy in community-dwelling older adults. Atherosclerosis 231(1):114–119. https://doi.org/10.1016/j.atherosclerosis.2013.09.002
Di Marco GS, Reuter S, Kentrup D, Grabner A, Amaral AP, Fobker M, Stypmann J, Pavenstadt H, Wolf M, Faul C, Brand M (2014) Treatment of established left ventricular hypertrophy with fibroblast growth factor receptor blockade in an animal model of CKD. Nephrol Dial Transplant 29(11):2028–2035. https://doi.org/10.1093/ndt/gfu190
Grabner A, Amaral AP, Schramm K, Singh S, Sloan A, Yanucil C, Li J, Shehadeh LA, Hare JM, David V, Martin A, Fornoni A, Di Marco GS, Kentrup D, Reuter S, Mayer AB, Pavenstadt H, Stypmann J, Kuhn C, Hille S, Frey N, Leifheit-Nestler M, Richter B, Haffner D, Abraham R, Bange J, Sperl B, Ullrich A, Brand M, Wolf M, Faul C (2015) Activation of cardiac fibroblast growth factor receptor 4 causes left ventricular hypertrophy. Cell Metab 22(6):1020–1032. https://doi.org/10.1016/j.cmet.2015.09.002
Grabner A, Schramm K, Silswal N, Hendrix M, Yanucil C, Czaya B, Singh S, Wolf M, Hermann S, Stypmann J, Di Marco GS, Brand M, Wacker MJ, Faul C (2017) FGF23/FGFR4-mediated left ventricular hypertrophy is reversible. Sci Rep 7(1):1993. https://doi.org/10.1038/s41598-017-02068-6
Verkaik M, Oranje M, Abdurrachim D, Goebel M, Gam Z, Prompers JJ, Helmes M, Ter Wee PM, van der Velden J, Kuster DW, Vervloet MG, Eringa EC, Consortium N (2018) High fibroblast growth factor 23 concentrations in experimental renal failure impair calcium handling in cardiomyocytes. Physiol Rep 6(7):e13591. https://doi.org/10.14814/phy2.13591
Christov M, Clark AR, Corbin B, Hakroush S, Rhee EP, Saito H, Brooks D, Hesse E, Bouxsein M, Galjart N, Jung JY, Mundel P, Juppner H, Weins A, Greka A (2018) Inducible podocyte-specific deletion of CTCF drives progressive kidney disease and bone abnormalities. JCI Insight. https://doi.org/10.1172/jci.insight.95091
Matsui I, Oka T, Kusunoki Y, Mori D, Hashimoto N, Matsumoto A, Shimada K, Yamaguchi S, Kubota K, Yonemoto S, Higo T, Sakaguchi Y, Takabatake Y, Hamano T, Isaka Y (2018) Cardiac hypertrophy elevates serum levels of fibroblast growth factor 23. Kidney Int 94(1):60–71. https://doi.org/10.1016/j.kint.2018.02.018
Andrukhova O, Slavic S, Odorfer KI, Erben RG (2015) Experimental myocardial infarction upregulates circulating fibroblast growth factor-23. J Bone Miner Res 30(10):1831–1839. https://doi.org/10.1002/jbmr.2527
Richter M, Lautze HJ, Walther T, Braun T, Kostin S, Kubin T (2015) The failing heart is a major source of circulating FGF23 via oncostatin M receptor activation. J Heart Lung Transplant 34(9):1211–1214. https://doi.org/10.1016/j.healun.2015.06.007
Leifheit-Nestler M, Grabner A, Hermann L, Richter B, Schmitz K, Fischer DC, Yanucil C, Faul C, Haffner D (2017) Vitamin D treatment attenuates cardiac FGF23/FGFR4 signaling and hypertrophy in uremic rats. Nephrol Dial Transplant 32(9):1493–1503. https://doi.org/10.1093/ndt/gfw454
Pastor-Arroyo EM, Gehring N, Krudewig C, Costantino S, Bettoni C, Knopfel T, Sabrautzki S, Lorenz-Depiereux B, Pastor J, Strom TM, Hrabe de Angelis M, Camici GG, Paneni F, Wagner CA, Rubio-Aliaga I (2018) The elevation of circulating fibroblast growth factor 23 without kidney disease does not increase cardiovascular disease risk. Kidney Int 94(1):49–59. https://doi.org/10.1016/j.kint.2018.02.017
Liu ES, Thoonen R, Petit E, Yu B, Buys ES, Scherrer-Crosbie M, Demay MB (2018) Increased circulating FGF23 does not lead to cardiac hypertrophy in the male hyp mouse model of XLH. Endocrinology 159(5):2165–2172. https://doi.org/10.1210/en.2018-00174
Takashi Y, Kinoshita Y, Hori M, Ito N, Taguchi M, Fukumoto S (2016) Patients with FGF23-related hypophosphatemic rickets/osteomalacia do not present with left ventricular hypertrophy. Endocr Res. https://doi.org/10.1080/07435800.2016.1242604
Moe S, Drueke T, Cunningham J, Goodman W, Martin K, Olgaard K, Ott S, Sprague S, Lameire N, Eknoyan G, Kidney Disease: Improving Global O (2006) Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney Int 69(11):1945–1953. https://doi.org/10.1038/sj.ki.5000414
Lindberg K, Olauson H, Amin R, Ponnusamy A, Goetz R, Taylor RF, Mohammadi M, Canfield A, Kublickiene K, Larsson TE (2013) Arterial klotho expression and FGF23 effects on vascular calcification and function. PLoS One 8(4):e60658. https://doi.org/10.1371/journal.pone.0060658
Scialla JJ, Lau WL, Reilly MP, Isakova T, Yang HY, Crouthamel MH, Chavkin NW, Rahman M, Wahl P, Amaral AP, Hamano T, Master SR, Nessel L, Chai B, Xie D, Kallem RR, Chen J, Lash JP, Kusek JW, Budoff MJ, Giachelli CM, Wolf M, Chronic Renal Insufficiency Cohort Study I (2013) Fibroblast growth factor 23 is not associated with and does not induce arterial calcification. Kidney Int 83(6):1159–1168. https://doi.org/10.1038/ki.2013.3
Zhu D, Mackenzie NC, Millan JL, Farquharson C, MacRae VE (2013) A protective role for FGF-23 in local defence against disrupted arterial wall integrity? Mol Cell Endocrinol 372(1–2):1–11. https://doi.org/10.1016/j.mce.2013.03.008
Jimbo R, Kawakami-Mori F, Mu S, Hirohama D, Majtan B, Shimizu Y, Yatomi Y, Fukumoto S, Fujita T, Shimosawa T (2014) Fibroblast growth factor 23 accelerates phosphate-induced vascular calcification in the absence of klotho deficiency. Kidney Int 85(5):1103–1111. https://doi.org/10.1038/ki.2013.332
Lim K, Lu TS, Molostvov G, Lee C, Lam FT, Zehnder D, Hsiao LL (2012) Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation 125(18):2243–2255. https://doi.org/10.1161/CIRCULATIONAHA.111.053405
Mencke R, Harms G, Mirkovic K, Struik J, Van Ark J, Van Loon E, Verkaik M, De Borst MH, Zeebregts CJ, Hoenderop JG, Vervloet MG, Hillebrands JL, Consortium N (2015) Membrane-bound Klotho is not expressed endogenously in healthy or uraemic human vascular tissue. Cardiovasc Res 108(2):220–231. https://doi.org/10.1093/cvr/cvv187
Tripepi G, Kollerits B, Leonardis D, Yilmaz MI, Postorino M, Fliser D, Mallamaci F, Kronenberg F, Zoccali C (2015) Competitive interaction between fibroblast growth factor 23 and asymmetric dimethylarginine in patients with CKD. J Am Soc Nephrol 26 (4):935-944. doi: https://doi.org/10.1681/asn.2013121355
Tripepi G, Kollerits B, Leonardis D, Yilmaz MI, Postorino M, Fliser D, Mallamaci F, Kronenberg F, Zoccali C (2015) Competitive interaction between fibroblast growth factor 23 and asymmetric dimethylarginine in patients with CKD. J Am Soc Nephrol 26(4):935–944. https://doi.org/10.1681/asn.2013121355
Silswal N, Touchberry CD, Daniel DR, McCarthy DL, Zhang S, Andresen J, Stubbs JR, Wacker MJ (2014) FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am J Physiol Endocrinol Metab 307 (5):E426-E436. doi:ajpendo.00264.2014 [pii];https://doi.org/10.1152/ajpendo.00264.2014 [doi]
Di Iorio B, Di Micco L, Torraca S, Sirico ML, Russo L, Pota A, Mirenghi F, Russo D (2012) Acute effects of very-low-protein diet on FGF23 levels: a randomized study. Clin J Am Soc Nephrol 7(4):581–587. https://doi.org/10.2215/CJN.07640711
Sigrist M, Tang M, Beaulieu M, Espino-Hernandez G, Er L, Djurdjev O, Levin A (2013) Responsiveness of FGF-23 and mineral metabolism to altered dietary phosphate intake in chronic kidney disease (CKD): results of a randomized trial. Nephrol Dial Transplant 28(1):161–169. https://doi.org/10.1093/ndt/gfs405
Goto S, Nakai K, Kono K, Yonekura Y, Ito J, Fujii H, Nishi S (2014) Dietary phosphorus restriction by a standard low-protein diet decreased serum fibroblast growth factor 23 levels in patients with early and advanced stage chronic kidney disease. Clin Exp Nephrol 18(6):925–931. https://doi.org/10.1007/s10157-014-0947-4
Isakova T, Gutierrez OM, Smith K, Epstein M, Keating LK, Juppner H, Wolf M (2011) Pilot study of dietary phosphorus restriction and phosphorus binders to target fibroblast growth factor 23 in patients with chronic kidney disease. Nephrol Dial Transplant 26(2):584–591
Isakova T, Barchi-Chung A, Enfield G, Smith K, Vargas G, Houston J, Xie H, Wahl P, Schiavenato E, Dosch A, Gutierrez OM, Diego J, Lenz O, Contreras G, Mendez A, Weiner RB, Wolf M (2013) Effects of dietary phosphate restriction and phosphate binders on FGF23 levels in CKD. Clin J Am Soc Nephrol 8(6):1009–1018. https://doi.org/10.2215/cjn.09250912
Moe SM, Zidehsarai MP, Chambers MA, Jackman LA, Radcliffe JS, Trevino LL, Donahue SE, Asplin JR (2011) Vegetarian compared with meat dietary protein source and phosphorus homeostasis in chronic kidney disease. Clin J Am Soc Nephrol 6(2):257–264. https://doi.org/10.2215/CJN.05040610
Oliveira RB, Cancela AL, Graciolli FG, dos Reis LM, Draibe SA, Cuppari L, Carvalho AB, Jorgetti V, Canziani ME, Moyses RM (2010) Early control of PTH and FGF23 in normophosphatemic CKD patients: a new target in CKD-MBD therapy? Clin J Am Soc Nephrol 5(2):286–291
Yilmaz MI, Sonmez A, Saglam M, Yaman H, Kilic S, Eyileten T, Caglar K, Oguz Y, Vural A, Yenicesu M, Mallamaci F, Zoccali C (2012) Comparison of calcium acetate and sevelamer on vascular function and fibroblast growth factor 23 in CKD patients: a randomized clinical trial. Am J Kidney Dis 59(2):177–185
Vlassara H, Uribarri J, Cai W, Goodman S, Pyzik R, Post J, Grosjean F, Woodward M, Striker GE (2012) Effects of sevelamer on HbA1c, inflammation, and advanced glycation end products in diabetic kidney disease. Clin J Am Soc Nephrol 7(6):934–942. https://doi.org/10.2215/cjn.12891211
Block GA, Wheeler DC, Persky MS, Kestenbaum B, Ketteler M, Spiegel DM, Allison MA, Asplin J, Smits G, Hoofnagle AN, Kooienga L, Thadhani R, Mannstadt M, Wolf M, Chertow GM (2012) Effects of phosphate binders in moderate CKD. J Am Soc Nephrol 23(8):1407–1415
Koiwa F, Kazama JJ, Tokumoto A, Onoda N, Kato H, Okada T, Nii-Kono T, Fukagawa M, Shigematsu T (2005) Sevelamer hydrochloride and calcium bicarbonate reduce serum fibroblast growth factor 23 levels in dialysis patients. Ther Apher Dial 9(4):336–339
Chang YM, Tsai SC, Shiao CC, Liou HH, Yang CL, Tung NY, Hsu KS, Chen IL, Liu MC, Kao JL, Jhen RN, Huang YT (2017) Effects of lanthanum carbonate and calcium carbonate on fibroblast growth factor 23 and hepcidin levels in chronic hemodialysis patients. Clin Exp Nephrol 21(5):908–916. https://doi.org/10.1007/s10157-016-1362-9
Gonzalez-Parra E, Gonzalez-Casaus ML, Galan A, Martinez-Calero A, Navas V, Rodriguez M, Ortiz A (2011) Lanthanum carbonate reduces FGF23 in chronic kidney disease stage 3 patients. Nephrol Dial Transplant 26(8):2567–2571
Zhang C, Wang S, Zhao S, Zhang X (2017) Effect of lanthanum carbonate on coronary artery calcification and bone mineral density in maintenance hemodialysis patients with diabetes complicated with adynamic bone disease: a prospective pilot study. Medicine (Baltimore) 96(45):e8664. https://doi.org/10.1097/MD.0000000000008664
Ketteler M, Sprague SM, Covic AC, Rastogi A, Spinowitz B, Rakov V, Walpen S, Floege J (2018) Effects of sucroferric oxyhydroxide and sevelamer carbonate on chronic kidney disease-mineral bone disorder parameters in dialysis patients. Nephrol Dial Transplant. https://doi.org/10.1093/ndt/gfy127
Chue CD, Townend JN, Moody WE, Zehnder D, Wall NA, Harper L, Edwards NC, Steeds RP, Ferro CJ (2013) Cardiovascular effects of sevelamer in stage 3 CKD. J Am Soc Nephrol 24(5):842–852. https://doi.org/10.1681/asn.2012070719
Liabeuf S, Ryckelynck JP, El Esper N, Urena P, Combe C, Dussol B, Fouque D, Vanhille P, Frimat L, Thervet E, Mentaverri R, Prie D, Choukroun G, Collaborators FS (2017) Randomized clinical trial of sevelamer carbonate on serum klotho and fibroblast growth factor 23 in CKD. Clin J Am Soc Nephrol 12(12):1930–1940. https://doi.org/10.2215/CJN.03030317
Seifert ME, de las FL, Rothstein M, Dietzen DJ, Bierhals AJ, Cheng SC, Ross W, Windus D, Davila-Roman VG, Hruska KA (2013) Effects of phosphate binder therapy on vascular stiffness in early-stage chronic kidney disease. Am J Nephrol 38(2):158–167. https://doi.org/10.1159/000353569
Ix JH, Isakova T, Larive B, Raphael KL, Raj DS, Cheung AK, Sprague SM, Fried LF, Gassman JJ, Middleton JP, Flessner MF, Block GA, Wolf M (2019) Effects of nicotinamide and lanthanum carbonate on serum phosphate and fibroblast growth factor-23 in CKD: the COMBINE trial. J Am Soc Nephrol 30(6):1096–1108. https://doi.org/10.1681/ASN.2018101058
Bleskestad IH, Bergrem H, Hartmann A, Godang K, Goransson LG (2012) Fibroblast growth factor 23 and parathyroid hormone after treatment with active vitamin D and sevelamer carbonate in patients with chronic kidney disease stage 3b, a randomized crossover trial. BMC Nephrol 13:49. https://doi.org/10.1186/1471-2369-13-49
Spatz C, Roe K, Lehman E, Verma N (2013) Effect of a non-calcium-based phosphate binder on fibroblast growth factor 23 in chronic kidney disease. Nephron Clin Pract 123(1–2):61–66. https://doi.org/10.1159/000351811
Brandenburg VM, Schlieper G, Heussen N, Holzmann S, Busch B, Evenepoel P, Vanholder R, Meijers B, Meert N, Fassbender WJ, Floege J, Jahnen-Dechent W, Ketteler M (2010) Serological cardiovascular and mortality risk predictors in dialysis patients receiving sevelamer: a prospective study. Nephrol Dial Transplant 25(8):2672–2679. https://doi.org/10.1093/ndt/gfq053
Bouma-de Krijger A, van Ittersum FJ, Hoekstra T, Ter Wee PM, Vervloet MG (2019) Short-term effects of sevelamer-carbonate on fibroblast growth factor 23 and pulse wave velocity in patients with normophosphataemic chronic kidney disease stage 3. Clin Kidney J 12(5):678–685. https://doi.org/10.1093/ckj/sfz027
Seeherunvong W, Wolf M (2011) Tertiary excess of fibroblast growth factor 23 and hypophosphatemia following kidney transplantation. Pediatr Transplant 15(1):37–46. https://doi.org/10.1111/j.1399-3046.2010.01405.x
Prasad N, Jaiswal A, Agarwal V, Kumar S, Chaturvedi S, Yadav S, Gupta A, Sharma RK, Bhadauria D, Kaul A (2016) FGF23 is associated with early post-transplant hypophosphataemia and normalizes faster than iPTH in living donor renal transplant recipients: a longitudinal follow-up study. Clin Kidney J 9(5):669–676. https://doi.org/10.1093/ckj/sfw065
Carpenter TO, Whyte MP, Imel EA, Boot AM, Hogler W, Linglart A, Padidela R, Van’t Hoff W, Mao M, Chen CY, Skrinar A, Kakkis E, San Martin J, Portale AA (2018) Burosumab therapy in children with x-linked hypophosphatemia. N Engl J Med 378(21):1987–1998. https://doi.org/10.1056/NEJMoa1714641
Shalhoub V, Shatzen EM, Ward SC, Davis J, Stevens J, Bi V, Renshaw L, Hawkins N, Wang W, Chen C, Tsai MM, Cattley RC, Wronski TJ, Xia X, Li X, Henley C, Eschenberg M, Richards WG (2012) FGF23 neutralization improves chronic kidney disease-associated hyperparathyroidism yet increases mortality. J Clin Invest 122(7):2543–2553
Yanochko GM, Vitsky A, Heyen JR, Hirakawa B, Lam JL, May J, Nichols T, Sace F, Trajkovic D, Blasi E (2013) Pan-FGFR inhibition leads to blockade of FGF23 signaling, soft tissue mineralization, and cardiovascular dysfunction. Toxicol Sci 135(2):451–464. https://doi.org/10.1093/toxsci/kft161
Wetmore JB, Liu S, Krebill R, Menard R, Quarles LD (2010) Effects of cinacalcet and concurrent low-dose vitamin D on FGF23 levels in ESRD. Clin J Am Soc Nephrol 5(1):110–116. https://doi.org/10.2215/CJN.03630509
Koizumi M, Komaba H, Nakanishi S, Fujimori A, Fukagawa M (2012) Cinacalcet treatment and serum FGF23 levels in haemodialysis patients with secondary hyperparathyroidism. Nephrol Dial Transplant 27(2):784–790. https://doi.org/10.1093/ndt/gfr384
Wolf M, Block GA, Chertow GM, Cooper K, Fouqueray B, Moe S, Sun Y, Tomlin H, Vervloet M, Oberbauer R (2019) Effects of etelcalcetide on fibroblast growth factor 23 in patients with secondary hyperparathyroidism receiving hemodialysis. Clin Kidney J 1:1–10
Humalda JK, Riphagen IJ, Assa S, Hummel YM, Westerhuis R, Vervloet MG, Voors AA, Navis G, Franssen CF, de Borst MH, Consortium N (2016) Fibroblast growth factor 23 correlates with volume status in haemodialysis patients and is not reduced by haemodialysis. Nephrol Dial Transplant 31(9):1494–1501. https://doi.org/10.1093/ndt/gfv393
Patrier L, Dupuy AM, Granger Vallee A, Chalabi L, Morena M, Canaud B, Cristol JP (2013) FGF-23 removal is improved by on-line high-efficiency hemodiafiltration compared to conventional high flux hemodialysis. J Nephrol 26(2):342–349. https://doi.org/10.5301/jn.5000150
Cernaro V, Lucisano S, Canale V, Bruzzese A, Caccamo D, Costantino G, Buemi M, Santoro D (2018) Acetate-free biofiltration to remove fibroblast growth factor 23 in hemodialysis patients: a pilot study. J Nephrol 31(3):429–433. https://doi.org/10.1007/s40620-017-0393-y
Choo SZ, Polkinghorne KR, Kerr PG (2018) Biochemical comparison of 8-hour haemodialysis and 4-hour haemodiafiltration, and two dialysis membranes, in a randomised cross-over trial. Nephrology (Carlton). https://doi.org/10.1111/nep.13397
Investigators ET, Chertow GM, Block GA, Correa-Rotter R, Drueke TB, Floege J, Goodman WG, Herzog CA, Kubo Y, London GM, Mahaffey KW, Mix TC, Moe SM, Trotman ML, Wheeler DC, Parfrey PS (2012) Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med 367(26):2482–2494. https://doi.org/10.1056/NEJMoa1205624
Seifert ME, de Las Fuentes L, Ginsberg C, Rothstein M, Dietzen DJ, Cheng SC, Ross W, Windus D, Davila-Roman VG, Hruska KA (2014) Left ventricular mass progression despite stable blood pressure and kidney function in stage 3 chronic kidney disease. Am J Nephrol 39(5):392–399. https://doi.org/10.1159/000362251
Author information
Authors and Affiliations
Contributions
This review was written on invitation from Antonio Bellasi, who invented the title of the manuscript. The content was an idea from MGV and AB-dK, the literature search was performed by AB-dK, the manuscript was drafted by AB-dK. MGV critically revised the work.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
Not applicable.
Informed consent
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bouma-de Krijger, A., Vervloet, M.G. Fibroblast growth factor 23: are we ready to use it in clinical practice?. J Nephrol 33, 509–527 (2020). https://doi.org/10.1007/s40620-020-00715-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40620-020-00715-2