
Abstract
Mechanistic evidence suggests that arsenic exposure from drinking water increases the production of reactive oxygen species and influences inflammatory responses and endothelial nitric oxide homeostasis. These arsenic-induced events may lead to endothelial dysfunction that increases the risk of atherosclerosis and cardiovascular disease. We reviewed accumulating epidemiologic evidence that evaluated the association between arsenic exposure and intermediate markers and subclinical measures that predict future cardiovascular risk. Cross-sectional studies have indicated positive associations between high or low-to-moderate levels of arsenic exposure with indices of subclinical atherosclerosis, QT interval prolongation, and circulating markers of endothelial dysfunction. The evidence is limited for other intermediate endpoints such as markers of oxidative stress and inflammation, QT dispersion, and lipid profiles. Prospective studies are needed to enhance the causal inferences of the effects of arsenic on subclinical endpoints of cardiovascular disease, especially at lower arsenic exposure levels.
Similar content being viewed by others

Introduction
Arsenic is an abundant element of the Earth’s crust and can enter drinking water supplies from natural deposits. Areas affected by arsenic exposure via contaminated drinking water span the globe, and significant exposures have been identified in Bangladesh, India, Taiwan, China, México, Argentina, Chile, and the US. Recent data also raise concerns for arsenic exposure via foods including appreciable levels of inorganic arsenic (InAs) in rice and rice products, fruit juices, and chicken [1–5]. These data suggest that arsenic exposure may be more widespread than previously considered and be of massive public health significance. As a group 1 human carcinogen, arsenic exposure from drinking water causes cancers of the skin, lung, and bladder [6–8]. In addition, arsenic exposure has been related to elevated risks of non-cancerous outcomes such as skin lesions [9, 10], lung disease [11, 12], and cardiovascular disease (CVD) [13].
High levels of arsenic exposure (>500 μg/L) have been related to an array of CVD in southwestern Taiwan, Bangladesh, and Chile [14–18]. Cross-sectional studies from northeastern Taiwan and prospective studies from Bangladesh reported that moderate-to-high levels of arsenic exposure (<300 μg/L) were associated with CVD prevalence, incidence, and mortality [19–22]. Positive associations of arsenic exposure at low levels (<100 μg/L) have also been found with CVD mortality in a series of ecological studies from the US and Spain [23–28], and recently, with the risk of overall and subtypes of CVD in a prospective cohort study in the US [29•]. Despite the clear epidemiologic links between arsenic exposure and CVD risk, the underlying mechanisms have not been well established.
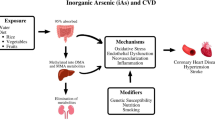
Experimental studies have suggested that arsenic can increase the production of reactive oxygen species [30, 31], interfere with the activity of key antioxidant enzymes [32–34], and induce lipid peroxidation [33, 34]. The induction of oxidative stress may in turn mediate abnormal gene expression, inflammatory responses, or impaired nitric oxide homeostasis [35–37]. Arsenic up-regulates the expression of various inflammatory molecules [36, 38–40] and inhibits key regulators of lipid homeostasis [41], two key components in the initiation of atherosclerosis. These events may ultimately lead to endothelial dysfunction, which disrupts the balance in vasomotor tone between relaxation and contraction and increases CVD risk [42]. Arsenic may also enhance platelet aggregation and reduce fibrinolysis leading to arterial thrombosis and atherosclerosis [43]. Many mechanistic studies have suggested the possible pathways underlying the cardiovascular effects of arsenic exposure. In addition, epidemiologic studies that evaluate the associations between arsenic exposure and intermediate phenotypes along these pathways are accumulating. Reviews of epidemiologic studies on arsenic exposure and CVD [13, 44] as well as on the effects of arsenic on CVD risk factors such as diabetes mellitus and hypertension [45, 46] have been published. Here, we review the epidemiologic evidence on arsenic exposure and subclinical endpoints of CVD, including subclinical atherosclerosis (Table 1), electrocardiographic abnormalities (Table 2), markers of oxidative stress, inflammation, and endothelial dysfunction, as well as lipid profiles (Table 3). We also discuss studies on the individual susceptibility to some of these subclinical endpoints due to arsenic methylation capacity and genetic polymorphisms. The schematic depiction of epidemiologic evidence on possible mechanisms underlying the cardiovascular effects of arsenic exposure is presented in Fig. 1.

Schematic depiction of epidemiologic evidence on possible mechanisms underlying the cardiovascular effects of arsenic exposure. Epidemiologic studies have demonstrated that arsenic may lead to cardiovascular diseases via its effects on subclinical endpoints which may be modified by individual susceptibility. Ox-LDL oxidized low-density lipoproteins, CRP C-reactive protein, PAI-1 plasminogen activator inhibitor-1, sICAM-1 soluble intercellular adhesion molecule 1, sVCAM-1 soluble vascular adhesion molecule 1, cIMT carotid artery intima-media thickness, QTc heart-rate corrected QT, QTD QT dispersion, TDR transmural dispersion of repolarization, CVD cardiovascular disease
Subclinical Atherosclerosis
Carotid artery intima-media thickness (cIMT) and plaque, which can be detected non-invasively with reasonable precision using high-resolution ultrasound techniques, reflect a thickening of the carotid artery wall or the presence of large irregular arterial wall deposits, respectively. cIMT and plaque are established measures of subclinical atherosclerosis. Multiple independent studies have established consistent association of carotid phenotypes with coronary events and stroke in prospective studies of young, middle-aged, and older adults [47, 48], and recent consensus prevention guidelines cite cIMT as a potentially useful measure for prediction of these events [49]. In a cross-sectional study of 463 subjects conducted in southwestern Taiwan with high levels of arsenic exposure (median in villages ranges from 700 to 930 μg/L), there was a dose–response relationship between well-water arsenic and carotid atherosclerosis [50•]. Carotid atherosclerosis in this study was defined using three indices including presence of plaque or cIMT ≥1.0 mm, cIMT ≥1.0 mm, and presence of plaque. Measures of long-term arsenic exposure including the duration of consuming well water, the average arsenic concentration in well water, and the cumulative arsenic exposure were all significantly associated with the prevalence of carotid atherosclerosis in a dose–response manner [50•]. In a study from northeastern Taiwan with low-to-moderate arsenic exposure (median in villages from undetectable to 140 μg/L), the prevalence of carotid atherosclerosis was significantly increased with an adjusted odds ratio (OR) of 2.6 (95 % confidence interval [CI] 1.3–5.0) among the highest exposure group (>100 μg/L) compared with the lowest group (≤50 μg/L) [51].
Some recent studies conducted in Bangladesh with more precise exposure measurements at the individual level also evaluated the effect of low-to-moderate levels of arsenic exposure on subclinical atherosclerosis. In a pilot study of 66 healthy, normotensive, relatively young (mean age 35 years) participants selected from the Health Effects of Arsenic Longitudinal Study (HEALS) [52], a prospective cohort study of more than 20,000 participants in Araihazar, Bangladesh, the adjusted ORs for cIMT > 0.75 mm (a predictor of significant coronary artery disease [53]) were 1.1 (95 % CI 0.2–6.3) and 2.1 (95 % CI 0.4–10.5), comparing middle (12–144 μg/L) and highest (>144 μg/L) with the lowest levels of well-water arsenic (<14 μg/L), respectively. Although not statistically significant, probably due to a small sample size, this increasing trend in ORs with higher arsenic exposure was observed consistently with total urinary arsenic, even after adjusting for total urinary arsenic measured at the time of follow-up. A recent cross-sectional study of 959 subjects nested in the HEALS [54••] confirmed and extended these previous findings in the same population. Arsenic exposure measured in both well water and urine samples at baseline was positively associated with cIMT measured 7 years later, such that a 1-standard-deviation (SD) increase in urinary arsenic (357.9 μg/g creatinine) and well-water arsenic (102 μg/L) were associated with an 11.7-μm (95 % CI 1.8–21.6) and 5.1-μm (95 % CI −0.2 to 10.3) increase in cIMT, respectively. These increases were also seen with an increasing proportion of urinary monomethylarsonic acid (MMA%), the most toxic arsenic metabolite, and every 10 % increase in MMA% was related to an increase of 12.1 μm (95 % CI 0.4–23.8) in cIMT. More recently, in a cross-sectional study of 199 children 3–14 years of age in México, total urinary arsenic was positively associated with increases in cIMT, with a 35-μm and 58-μm increase in cIMT among children exposed to 35–70 μg/L and >70 μg/L total urinary arsenic, respectively, compared with those exposed to <35 μg/L [55]. Although cross-sectional, these data strongly support a role for arsenic exposure in subclinical atherosclerosis, at both high and low-to-moderate levels.
Electrocardiographic Abnormalities
The QT interval on an electrocardiogram (ECG) is defined as the time from the onset of the QRS complex to the end of the T wave. It represents the duration of ventricular electrical systole, including depolarization and repolarization. Its prolongation indicates nonuniform recovery of myocardial excitability and has been shown to lower the ventricular fibrillation threshold and increase susceptibility to ventricular arrhythmia and sudden cardiac death [56]. The prolongation of heart-rate corrected QT interval (QTc) has been associated with an increased risk of cardiovascular and all-cause mortality in a broad range of clinical populations as well as in healthy subjects in population-based studies [57, 58]. The effects of arsenic on QTc prolongation have been the subject of extensive investigation in cross-sectional studies. The first study from Bangladesh showed that the prevalence of prolonged QTc (QTc ≥430 msec) was significantly higher in arsenic-exposed individuals (mean 493 μg/L, range 100–800 μg/L in well water) with skin lesions than in those without skin lesions or not exposed to arsenic [59]. In a study from Turkey, QTc was significantly higher in men (n = 40) chronically exposed to high levels of arsenic in drinking water (mean 659 μg/L, range 422–1,066 μg/L in villages) compared with men (n = 40) not exposed to arsenic [60]. In spite of the significant difference in QTc, the non-corrected QT intervals were actually within normal ranges in both groups [60]. Another study of 313 adults (168 men and 145 women) in Inner Mongolia with well-water arsenic measured at the household level reported a monotonic relationship between well-water arsenic and the prevalence of QTc prolongation (QTc ≥450 msec). However, the exposure categories were wide and incomplete (<21, 100–300, 430–690 μg/L) [61•]. A recent study of 280 men and 355 women in southwestern Taiwan reported a positive association between measures of long-term arsenic exposure and the prevalence of QTc prolongation in a dose–response manner [62]. In a US general population of elderly men with much lower levels of arsenic exposure, an interquartile range (0.059 μg/g) increase in toenail arsenic, a biomarker of long-term arsenic exposure, was associated with a 3.8-msec increase in QT (95 % CI 0.82–6.8) and a 2.5-msec increase in QTc (95 % CI 0.11–4.9) [63]. However, the effects of arsenic exposure on conventional dichotomized QT or QTc prolongation were not examined and therefore it was not clear whether these increases are clinically relevant. Most recently, a larger study of 1,715 adults in the HEALS in Bangladesh with a wide range of arsenic exposure levels (mean 95 μg/L, range 0.1–790 μg/L in well water) found a positive relationship between past and persistent arsenic exposure measured at baseline and QTc prolongation (QTc ≥450 msec in men and ≥460 msec in women) assessed during follow-up [64•]. The adjusted OR for QTc prolongation was 1.17 (95 % CI 1.01–1.35) and 1.18 (95 % CI 0.97–1.43) associated with a 1-SD increase in well-water arsenic (108.7 μg/L) and total urinary arsenic (270.7 μg/g creatinine), respectively. The positive association was stronger in women, with adjusted ORs of 1.24 (95 % CI 1.05–1.47) and 1.24 (95 % CI 1.01–1.53) for a 1-SD increase in well-water arsenic and total urinary arsenic, respectively, compared with 0.99 (95 % CI 0.73–1.33) and 0.86 (95 % CI 0.49–1.51) in men.
QT interval dispersion (QTD) is defined as the inter-lead difference between the longest and shortest QTc intervals in the standard 12-lead ECG. Although QTD provides only an imperfect estimate of the degree of heterogeneity of ventricular repolarization, several studies have suggested that QTD helps identify patients at an increased risk of ventricular arrhythmias or clinical events in a variety of clinical settings as well as predicts all-cause and cardiovascular mortality in the general population [58]. In the Turkey study [60], QTD and transmural dispersion of repolarization (TDR), the interval between the peak and the end of the T wave and a better marker of cardiac repolarization, were significantly higher in the exposed group than the unexposed. In a recent study of 280 men and 355 women conducted in the high-exposed area of Taiwan [65], cumulative arsenic exposure was dose-dependently associated with QTD, and prolongation of QTD, defined as QTD ≥65 msec, predicted all-cause and cardiovascular mortality in this population [65]. Taken together, these data suggest that arsenic exposure may increase QT interval and QTD as one of the underlying mechanisms for increased CVD risk. ECG may be useful for early detection of cardiac toxicity induced by arsenic exposure and for evaluation of populations at high risk for cardiovascular events. Future studies are needed to further examine the associations of lower levels of arsenic exposure with QTD and its prolongation.
Markers of Endothelial Dysfunction, Inflammation, and Oxidative Stress
Adhesion of circulating leukocytes to the endothelium and subsequent transmigration into the arterial wall is an early step in atherosclerotic plaque formation. This process is mediated by cell adhesion molecules (CAMs), such as selectins, intercellular adhesion molecule-1 (ICAM-1), and vascular adhesion molecule-1 (VCAM-1), expressed on the surface of activated endothelial cells in response to inflammatory stimuli. The selectins, including E-selectin and P-selectin, mediate transient rolling of the leukocytes along the endothelium, while stronger attachment is mediated by ICAM-1 and VCAM-1 [66]. Circulating markers of systemic inflammation and endothelial dysfunction, such as the soluble parts of CAMs (sE-selectin, sICAM-1, and sVCAM-1), have been associated with risk of fatal and nonfatal cardiovascular events [67–69]. In a cross-sectional analysis of 115 adults (20–65 years of age) with arsenic-related skin lesions from Araihazar, Bangladesh, there was a positive association of arsenic exposure at moderate levels (range 3–864 μg/L), measured as both well-water arsenic and total urinary arsenic, with plasma levels of sICAM-1 and sVCAM-1 [70]. The same group subsequently conducted independent separate cross-sectional analyses and confirmed a positive association between lower arsenic exposure (median 23 μg/L, range 0.1–500.6 μg/L) and plasma levels of sVCAM-1 [71••]. However, no significant association was observed for plasma levels of sE-selectin and sICAM-1. Unlike the study of skin lesion patients, this study consisted of mostly healthy subjects free of skin lesions at baseline with a much lower exposure level and therefore excludes the possibility that the observed association may be a consequence of skin lesions. Although the biological explanation for the variability in associations across these CAMs is not clear, the findings support the link between arsenic exposure and elevated concentrations of certain markers of endothelial dysfunction and suggest a potential mechanism underlying the effect of long-term arsenic exposure on CVD. In another study among adults aged 15–60 years old from a northwest region of Bangladesh, circulating levels of sICAM-1 and sVCAM-1 were significantly higher among participants residing in an arsenic-endemic area (mean 173 μg/L in well water, n = 218) compared with participants from a nonendemic area (mean 2.3 μg/L, n = 106); in addition, sICAM-1 and sVCAM-1 were positively and significantly correlated with arsenic exposure as measured in water, hair, and nail [72]. Additional studies are needed to evaluate the relationship between different CAM molecules and arsenic exposure levels and determine the specificity of the association with some of the CAMs, but not with others.
Studies have also evaluated other intermediate markers. Oxidation of low-density lipoproteins (LDLs) plays a crucial role in the initiation and progression of atherosclerosis [73] and the levels of oxidized LDL (Ox-LDL) in plasma have been used as a biomarker for oxidative stress in vascular systems [74]. Circulating levels of Ox-LDL have been positively related to presence and incidence of CVD [75–77]. The recent Bangladesh study indicated that arsenic exposure in water and hair was dose-dependently related to increases in plasma levels of Ox-LDL, as well as increases in plasma levels of C-reactive protein (CRP) [72], a circulating acute-phase reactant that reflects active systemic inflammation. These markers were increased in concert with increases in plasma levels of sICAM-1 and sVCAM-1 in this study [72]. These findings are consistent with the experimental evidence that CRP and Ox-LDL induces the expression of ICAM-1 and VCAM-1 in endothelial cells, although proper mediation analyses were not conducted [78]. These data provide insight into the interaction of oxidative stress, inflammatory responses, and endothelial dysfunction in the development of arsenic-induced cardiovascular effects.
In the cross-sectional study of Bangladesh [71••], in addition to the CAMs, the associations between arsenic exposure and plasma levels of plasminogen activator inhibitor-1 (PAI-1), matrix metalloproteinase-9 (MMP-9), and myeloperoxidase (MPO) were also assessed. PAI-1 is a member of the plasminogen activator inhibitors that interact with plasminogen activators to keep the balance between the coagulation and fibrinolytic systems of endothelial cells that are involved with thrombosis. In vitro, sodium arsenite has been shown to increase PAI-1 expression of human microvascular endothelial cells and thus lead to reduced fibrinolytic activity [75]. In mice, PAI-1 expression was time- and dose-dependently increased in cardiac tissue with exposure to 50–250 μg/L arsenic in drinking water, and plasma PAI-1 levels were increased in mice exposed to >500 μg/L arsenic [79]. Elevated PAI-1 levels appear to increase the risk of atherothrombotic events and may also promote the progression of CVD [80]. PAI-1 levels were elevated in the highest quartile of well-water arsenic (73.5–500.6 μg/L), and the association between arsenic exposure and PAI-1 levels was stronger among individuals with higher body mass index [71••]. The findings thus do not preclude that high levels of arsenic exposure may be related to increased levels of PAI-1, particularly in susceptible populations. Additional studies are needed to evaluate the association between arsenic exposure and PA1-1, especially at low-to-moderate levels.
MMP-9 is located at the shoulder region of plaques and areas of foam cell accumulation and plays an important role in several stages of atherosclerosis [81]. MMP-9 expression was time-dependently increased in mice chronically exposed to 50–250 μg/L arsenic in drinking water [79]. MPO has been implicated as a catalyst for LDL oxidation [82] and contributes to the overall reactive oxygen species burden in the physiological milieu. Positive associations of circulating levels of MMP-9 and MPO with CVD have been observed in cohort studies [83, 84]. However, there was no association of either well-water arsenic or urinary arsenic with plasma levels of MMP-9 and MPO in 668 participants selected from the HEALS in Araihazar, Bangladesh [71••]. A recent study in West Bengal (n = 91) showed that individuals chronically exposed to arsenic (mean 218 μg/L in well water) had significantly higher MPO activities in serum than the unexposed group; however, no further dose–response relation was found among the exposed group [85]. The only study among children from México found that the proportion of InAs was significantly associated with asymmetric dimethylarginine (ADMA), an endogenous inhibitor of nitric oxide synthase that could lead to endothelial dysfunction and cardiovascular events [55]. However, total arsenic levels were not associated with ADMA. The biological importance of these markers in mediating the cardiovascular effects of arsenic exposure needs further investigation.
Lipid Profiles
Few studies have evaluated the effect of arsenic exposure on traditional lipid markers of atherosclerosis, namely, total cholesterol (TC), LDL, high-density lipoprotein (HDL), and triglyceride. Generally, abnormal lipid profiles such as a large amount of TC, especially LDL, and triglyceride, and lower levels of HDL are associated with a higher risk of CVD. In a study of southwestern Taiwan, cumulative arsenic exposure was positively associated with serum levels of TC and LDL; however, this study did not adjust for potential confounding factors such as age [86]. A later study from Bangladesh showed that arsenic-exposed subjects (median 156 μg/L, range 3–875 μg/L) had significantly lower levels of TC, HDL, and LDL as compared with the unexposed subjects [87]. A subsequent Bangladesh study observed that serum levels of TC, LDL, and HDL were lower in arsenic-endemic subjects than those of nonendemic subjects, and all these markers were inversely related to arsenic exposure in well water, nail, and hair [72]. No association with triglyceride was observed in any of these studies. Thus, data on the associations between arsenic exposure and lipid profiles are inconsistent, and whether these lipid markers may serve as causal intermediates between arsenic exposure and CVD are far from clear. For instance, in the study of southwestern Taiwan [86], the prevalence of peripheral vascular disease did not differ by lipid levels. Likewise, another study from the same region found no significant association between these lipids and arsenic-related ischemic heart disease [88]. Conversely, another study found a dose–response relationship between serum levels of TC, HDL, LDL, and triglyceride, and carotid atherosclerosis in this high-exposure area of Taiwan [50•]. In the pediatric population of México, plasma lipids were not correlated with total urinary arsenic [55], indicating that arsenic-mediated early effects on atherogenesis may be independent of the lipid profiles.
Susceptibility to Arsenic-Induced Subclinical Endpoints
The cardiovascular effects of arsenic exposure may be complicated by varied individual susceptibilities. Differences in arsenic methylation capacity are believed to be an important susceptibility factor. InAs in drinking water is present as either AsV or AsIII. AsV is first reduced to AsIII, followed by methylation to monomethylarsonic acid (MMAV). After the reduction of MMAV to MMAIII, a second methylation can occur to generate dimethylarsinic acid (DMAV), which can be further reduced to DMAIII. MMAIII is believed to be more toxic than other metabolites and DMA, being more readily excreted in urine and expelled from body. The composition of arsenic metabolites, which are often expressed as percentages of all arsenic species in urine (i.e., InAs%, MMA%, DMA%) or as ratios (i.e., MMA/InAs, DMA/MMA), is indicative of methylation capacity. Incomplete arsenic methylation capacity, often indicated by a higher urinary MMA% or a lower DMA/MMA, has been associated with an increased risk of skin cancer [89, 90], bladder cancer [91, 92], and skin lesions [93]. In the Bangladesh study [54], the association between well-water arsenic and cIMT was stronger among participants with a higher urinary MMA% and MMA/InAs or a lower DMA/MMA [54]. However, there is no evidence that the positive association between high arsenic exposure (median 700–930 μg/L) and carotid atherosclerosis differed by DMA/MMA or MMA/InAs [88]. The data suggest that susceptibility to subclinical atherosclerosis because of lower methylation capacity may be more critical at lower levels of exposure. However, further studies are needed to confirm this hypothesis.
Evidence is also building that genetic polymorphisms may contribute to individual susceptibility. In this regard, the HEALS group recently conducted the largest and most comprehensive candidate gene-based investigation of gene–environment interactions in arsenic-induced CVD risk. A total of 360 single nucleotide polymorphisms (SNPs) and 18 genes were genotyped, and the investigators identified novel SNPs in ICAM-1 and VCAM-1 that had significant interactions with well-water arsenic in CVD risk, which remained robust after adjustment for multiple testing [94]. Regarding subclinical cardiovascular endpoints, the majority of evidence comes from a series of studies conducted in Taiwan that found the prevalence of arsenic-related carotid atherosclerosis was modifiable by genetic polymorphisms related to oxidative stress, inflammation, and arsenic metabolism [95–99]. However, these studies have included a limited number of genetic variants and did not have enough sample size to evaluate the effect modification at low levels of exposure. Recently, using a candidate gene approach, the HEALS group conducted a cross-sectional study of 1,078 participants to evaluate whether the association between arsenic exposure and cIMT differs by the 360 SNPs in the 18 genes related to arsenic metabolism, oxidative stress, inflammation, and endothelial dysfunction [100]. The investigators identified several novel SNPs in arsenic-3-methyltransferase (AS3MT), a gene encoding the arsenic methyltransferase capable of methylating InAs to MMA and MMA to DMA, which may synergistically interact with arsenic exposure in subclinical atherosclerosis. Importantly, the at-risk genotypes of these SNPs were positively related to urinary MMA% and negatively related to urinary DMA%, indicating that the effect modification of these SNPs may be through its direct effect on levels of MMA% and DMA% in urine. Supporting this, a recent genome-wide association study (GWAS) from the HEALS identified five SNPs in AS3MT that were associated with poor conversion of MMA to DMA, one of which was also associated with an increased risk of skin lesions [101].
Summary and Conclusions
Epidemiologic evidence is accumulating to elucidate the mechanisms by which arsenic causes CVD, and we identified a fairly clear association of arsenic exposure with markers of endothelial dysfunction such as sICAM-1 and sVCAM-1, subclinical atherosclerosis, and QT prolongation — all of which have been implicated as predictors for future cardiovascular events. The association was present both in studies with high levels of arsenic exposure and in studies with low-to-moderate exposure levels. The evidence of a causal effect of arsenic exposure on other intermediate endpoints, such as markers of oxidative stress and inflammation, QTD, and lipid profiles, however, is not as well established by the small number of studies, the heterogeneity in exposure assessment across studies, and inconsistency across studies. In addition, there are other emerging subclinical cardiovascular endpoints, such as F2-isoprostanes (F2-IsoPs), flow-mediated dilation (FMD), peripheral arterial tonometry (PAT), brachial artery distensibility (BAD), and circulating levels of adiponectin that have not been evaluated. Also, the available data are scarce for in utero and early-life exposure, and much remains to be done before we can fully understand susceptibility to the cardiovascular effects of arsenic exposure.
Studies are lacking on the investigation of arsenic exposure in relation to other emerging subclinical cardiovascular endpoints or markers. F2-IsoPs are specific products of lipid peroxidation that are excreted and detectable in urine and have been positively associated with CVD risk factors in children and adults [102]. Arsenic increases lipid peroxides in cell systems and animal models [103]; in vitro work also observed dose-dependent increases in the levels of 8-isoprostane, the most thoroughly investigated F2-IsoPs, in response to AsIII exposure [104]. Evidence has suggested that dysregulated production of adipokines by adipose tissue may play a role in the pathogenesis of CVD. Adiponectin, one of the most studied adipokines, has anti-inflammatory and anti-oxidative properties and, in contrast to other adipokines, is protective against obesity and obesity-related disorders [105]. Arsenic exposure suppressed differentiation of human mesenchymal stem cells to adipocytes, leading to decreased expression of adiponectin [106]. In addition, arsenic added to differentiated adipocytes inhibited adiponectin generation [107], although no significant effects of the arsenic exposure on circulating adiponectin in vivo was observed. However, epidemiologic studies on arsenic exposure in relation to F2-IsoPs or adiponectin are lacking. Importantly, noninvasive assessment of endothelial dysfunction has recently received increasing attention. Despite evidence of the effect of arsenic exposure on endothelial dysfunction from experimental studies, no epidemiologic studies have evaluated the association between arsenic exposure and measures of endothelial dysfunction, such as PAT, which measures pulsatile arterial volume changes before and after induced ischemia by nonultrasound-based finger plethysmography to determine endothelial function, or ultrasound-based FMD, a method that has been validated for evaluating endothelial function. Taken together, additional studies are needed to systemically evaluate the effects of arsenic exposure on these emerging subclinical endpoints of CVD.
As mentioned, the differences in arsenic exposure across studies may have limited the causal inference. In the studies from southwestern Taiwan, arsenic exposure was measured ecologically using the median level of well-water arsenic in a village where the participants resided. The inaccuracy in exposure assessment could lead to an underestimation of arsenic’s effect. Such a study design may not be appropriate for investigating effects of arsenic exposure at lower levels, as the misclassification of exposure may mask the association and even lead to biased results. In the US, the heterogeneity of drinking water resources, the limited exposure range, and the relatively high migration within the nation pose challenges for epidemiologic studies to track the change in arsenic exposure at the individual level. In rural Bangladesh, typically all members of a household drink water from a single tube well for decades in the absence of interventions. Soon after the HEALS was established, remediation efforts began to reduce the levels of arsenic exposure in the highly exposed cohort participants and their immediate family members. Therefore, it is possible to track changes in exposure in the families. With repeated measurements of arsenic exposure every 2 years since 2000, the HEALS provides an unprecedented opportunity to evaluate the cardiovascular effects of both continuing arsenic exposure and reduction of exposure. Research on the influence of change in exposure status is crucial to elucidate whether remediation is beneficial to arsenic-induced CVD and other chronic diseases.
Importantly, recent evidence of growing concern indicates that dietary intake of arsenic can significantly contribute to an individual’s overall exposure level [3–5], especially in populations with limited exposure to arsenic via drinking water. Children who reported consuming rice had urinary arsenic concentrations that were nearly 40 % higher than those who did not consume rice [4], and rice consumption has been associated with total urinary arsenic, as well as InAs, MMA, and DMA in pregnant women [5]. In the US, an analysis of diet, water, and urinary arsenic data concluded that 75 % of InAs exposure was attributed to diet in households with arsenic in drinking water at concentrations of 10 μg/L or lower [108]. Thus, exposure to InAs from diet should be considered when estimating the health effects of total arsenic exposure. Studies that take advantage of biomarkers of arsenic exposure, such as levels in urine, blood, toenails, and hair, as a measure of exposure from all sources can more precisely examine the effects at lower levels such as those common to the US.
There is highly suggestive evidence that early-life exposure to arsenic causes detrimental pregnancy and birth outcomes, impaired development in infancy and childhood, and increased risks of adverse health effects including cancer and CVD later in life [109]. Importantly, the literature indicates that cardiovascular effects may begin in utero or in early childhood and CVD risk factors measured in adolescence or early adulthood are predictive of CVD up to 50 years later [110]. The examination of the relationship between arsenic exposure in early childhood and intermediate phenotypes for CVD during adolescence and young adulthood may help establish a causal role of early-life arsenic exposure in the development of CVD in later life and provide avenues for early intervention. As indicated above, however, epidemiologic studies on this topic are very limited. A life-course approach offers a way to conceptualize how underlying environmental determinants of health, experienced at different life-course stages, can differentially influence the risk of chronic diseases [111, 112].
Although it is recognized that many common diseases are a result of complex interplays of genetic and environmental factors, studies of gene-environment interactions in CVD and its subclinical surrogates relevant to arsenic exposure are just emerging. The few existing studies have used a candidate gene approach irrespective of the sample sizes and the number of SNPs included. While this approach has such strengths as biological plausibility and clear a priori hypotheses, it is always open to bias given the undefined or even controversial functional significance of some SNPs, the multifaceted nature of mechanistic pathways, and the existence of genes or SNPs that may have important roles in disease etiology but are not yet known. An alternative approach is to use GWAS, which allows for an unbiased screening of genetic variants, or whole genome sequencing that has the advantage of discovering rare variants. Future GWAS with innovative analysis approaches are needed to assess the interaction between arsenic exposure and genetic factors in CVD-related outcomes. Aside from arsenic methylation capacity and genetics, other factors may also influence susceptibility to the health effects of arsenic. For instance, recent experimental findings suggest that gut microbes might be involved in presystemic arsenic metabolism; i.e., arsenic methylation that occurs before arsenic enters the internal environment of our bodies, by converting AsV to AsIII and forming MMAV and MMAIII [113, 114]. Whether differences in the gut microbial composition may influence arsenic methylation capacity and subsequently contribute to susceptibility to the subclinical and clinical cardiovascular consequences of arsenic exposure represents a critical gap in our knowledge. A systemic evaluation of these emerging factors along with other well known factors such as nutrition, smoking, and co-exposures is needed.
More than 200 million persons worldwide might be chronically exposed to arsenic in drinking water at concentrations above the World Health Organization (WHO) and US Environmental Protection Agency (EPA) standard of 10 μg/L [115, 116]. Millions more are exposed to arsenic through their diet and no standards for arsenic in foods currently exist. Since CVD is the leading cause of mortality worldwide and arsenic exposure at low-to-moderate levels is more prevalent, a small increased risk associated with arsenic exposure can cause a large number of excess deaths. Prospective analyses of populations with available data on both intermediate endpoints and clinical disease outcomes may be useful to further support biological plausibility of the putative causal pathways linking arsenic exposure to CVD.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Nachman KE et al. Arsenic species in poultry feather meal. Sci Total Environ. 2012;417–418:183–8.
Wilson D, Hooper C, Shi X. Arsenic and lead in juice: apple, citrus, and apple-base. J Environ Health. 2012;75(5):14–20. quiz 44.
Jackson BP et al. Arsenic, organic foods, and brown rice syrup. Environ Health Perspect. 2012;120(5):623–6.
Davis MA et al. Rice consumption and urinary arsenic concentrations in U.S. children. Environ Health Perspect. 2012;120(10):1418–24.
Gilbert-Diamond D et al. Rice consumption contributes to arsenic exposure in US women. Proc Natl Acad Sci U S A. 2011;108(51):20656–60.
Morales KH et al. Risk of internal cancers from arsenic in drinking water. Environ Health Perspect. 2000;108(7):655–61.
Chen CJ, Kuo TL, Wu MM. Arsenic and cancers. Lancet. 1988;1(8582):414–5.
Chiou HY et al. Incidence of internal cancers and ingested inorganic arsenic: a seven-year follow-up study in Taiwan. Cancer Res. 1995;55(6):1296–300.
Argos M et al. A prospective study of arsenic exposure from drinking water and incidence of skin lesions in Bangladesh. Am J Epidemiol. 2011;174(2):185–94.
Ahsan H et al. Arsenic exposure from drinking water and risk of premalignant skin lesions in Bangladesh: baseline results from the Health Effects of Arsenic Longitudinal Study. Am J Epidemiol. 2006;163(12):1138–48.
Parvez F et al. Nonmalignant respiratory effects of chronic arsenic exposure from drinking water among never-smokers in Bangladesh. Environ Health Perspect. 2008;116(2):190–5.
Mazumder DN et al. Arsenic in drinking water and the prevalence of respiratory effects in West Bengal, India. Int J Epidemiol. 2000;29(6):1047–52.
Moon K, Guallar E, Navas-Acien A. Arsenic exposure and cardiovascular disease: an updated systematic review. Curr Atheroscler Rep. 2012;14(6):542–55.
Chen CJ et al. Increased prevalence of hypertension and long-term arsenic exposure. Hypertension. 1995;25(1):53–60.
Rahman M et al. Hypertension and arsenic exposure in Bangladesh. Hypertension. 1999;33(1):74–8.
Chen CJ et al. Dose–response relationship between ischemic heart disease mortality and long-term arsenic exposure. Arterioscler Thromb Vasc Biol. 1996;16(4):504–10.
Tseng CH et al. Long-term arsenic exposure and ischemic heart disease in arseniasis-hyperendemic villages in Taiwan. Toxicol Lett. 2003;137(1–2):15–21.
Yuan Y et al. Acute myocardial infarction mortality in comparison with lung and bladder cancer mortality in arsenic-exposed region II of Chile from 1950 to 2000. Am J Epidemiol. 2007;166(12):1381–91.
Chen Y et al. Arsenic exposure from drinking water and mortality from cardiovascular disease in Bangladesh: prospective cohort study. BMJ. 2011;342:d2431.
Chen Y et al. A prospective study of arsenic exposure, arsenic methylation capacity, and risk of cardiovascular disease in Bangladesh. Environ Health Perspect. 2013;121(7):832–8.
Sohel N et al. Arsenic in drinking water and adult mortality: a population-based cohort study in rural Bangladesh. Epidemiology. 2009;20(6):824–30.
Chiou HY et al. Dose–response relationship between prevalence of cerebrovascular disease and ingested inorganic arsenic. Stroke. 1997;28(9):1717–23.
Engel RR, Smith AH. Arsenic in drinking water and mortality from vascular disease: an ecologic analysis in 30 counties in the United States. Arch Environ Health. 1994;49(5):418–27.
Lewis DR et al. Drinking water arsenic in Utah: a cohort mortality study. Environ Health Perspect. 1999;107(5):359–65.
Zierold KM, Knobeloch L, Anderson H. Prevalence of chronic diseases in adults exposed to arsenic-contaminated drinking water. Am J Public Health. 2004;94(11):1936–7.
Meliker JR et al. Arsenic in drinking water and cerebrovascular disease, diabetes mellitus, and kidney disease in Michigan: a standardized mortality ratio analysis. Environ Health. 2007;6:4.
Medrano MA et al. Arsenic in public water supplies and cardiovascular mortality in Spain. Environ Res. 2010;110(5):448–54.
Lisabeth LD et al. Arsenic in drinking water and stroke hospitalizations in Michigan. Stroke. 2010;41(11):2499–504.
Moon KA et al. Association between exposure to low to moderate arsenic levels and incident cardiovascular disease: a prospective cohort study. Ann Intern Med. 2013;159(10):649–59. In this large prospective cohort study in American Indians from the Strong Heart Study, arsenic exposure was assessed using the sum of inorganic and methylated arsenic species in urine at baseline. Arsenic exposure at low levels (<100 μg/L) was associated with increased risks of incident fatal and nonfatal cardiovascular disease, coronary heart disease, and stroke.
Barchowsky A et al. Stimulation of reactive oxygen, but not reactive nitrogen species, in vascular endothelial cells exposed to low levels of arsenite. Free Radic Biol Med. 1999;27(11–12):1405–12.
Chen YC, Lin-Shiau SY, Lin JK. Involvement of reactive oxygen species and caspase 3 activation in arsenite-induced apoptosis. J Cell Physiol. 1998;177(2):324–33.
Maiti S, Chatterjee AK. Differential response of cellular antioxidant mechanism of liver and kidney to arsenic exposure and its relation to dietary protein deficiency. Environ Toxicol Pharmacol. 2000;8(4):227–35.
Santra A et al. Hepatic damage caused by chronic arsenic toxicity in experimental animals. J Toxicol Clin Toxicol. 2000;38(4):395–405.
Kotyzova D, Bludovska M, Eybl V. Differential influences of various arsenic compounds on antioxidant defense system in liver and kidney of rats. Environ Toxicol Pharmacol. 2013;36(3):1015–21.
Straub AC et al. Arsenic-stimulated liver sinusoidal capillarization in mice requires NADPH oxidase-generated superoxide. J Clin Invest. 2008;118(12):3980–9.
Bunderson M, Coffin JD, Beall HD. Arsenic induces peroxynitrite generation and cyclooxygenase-2 protein expression in aortic endothelial cells: possible role in atherosclerosis. Toxicol Appl Pharmacol. 2002;184(1):11–8.
Bunderson M et al. Arsenic exposure exacerbates atherosclerotic plaque formation and increases nitrotyrosine and leukotriene biosynthesis. Toxicol Appl Pharmacol. 2004;201(1):32–9.
Wu MM et al. Gene expression of inflammatory molecules in circulating lymphocytes from arsenic-exposed human subjects. Environ Health Perspect. 2003;111(11):1429–38.
Tsai SH et al. Arsenite stimulates cyclooxygenase-2 expression through activating IkappaB kinase and nuclear factor kappaB in primary and ECV304 endothelial cells. J Cell Biochem. 2002;84(4):750–8.
Srivastava S et al. Arsenic exacerbates atherosclerotic lesion formation and inflammation in ApoE-/- mice. Toxicol Appl Pharmacol. 2009;241(1):90–100.
Padovani AM, Molina MF, Mann KK. Inhibition of liver x receptor/retinoid X receptor-mediated transcription contributes to the proatherogenic effects of arsenic in macrophages in vitro. Arterioscler Thromb Vasc Biol. 2010;30(6):1228–36.
Kumagai Y, Pi J. Molecular basis for arsenic-induced alteration in nitric oxide production and oxidative stress: implication of endothelial dysfunction. Toxicol Appl Pharmacol. 2004;198(3):450–7.
Balakumar P, Kaur J. Arsenic exposure and cardiovascular disorders: an overview. Cardiovasc Toxicol. 2009;9(4):169–76.
Navas-Acien A et al. Arsenic exposure and cardiovascular disease: a systematic review of the epidemiologic evidence. Am J Epidemiol. 2005;162(11):1037–49.
Navas-Acien A et al. Arsenic exposure and type 2 diabetes: a systematic review of the experimental and epidemiological evidence. Environ Health Perspect. 2006;114(5):641–8.
Abhyankar LN et al. Arsenic exposure and hypertension: a systematic review. Environ Health Perspect. 2012;120(4):494–500.
Lorenz MW et al. Prediction of clinical cardiovascular events with carotid intima-media thickness—a systematic review and meta-analysis. Circulation. 2007;115(4):459–67.
Stein JH, Korcarz CE, Hurst RT. Use of carotid ultrasound to identify subclinical vascular disease and evaluate cardiovascular disease risk: a consensus statement from the American society of Echocardiography carotid intima-media thickness task force (vol 21, pg 93, 2008). J Am Soc Echocardiogr. 2008;21(4):376.
Greenland P et al. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation. 2010;122(25):e584–636.
Wang CH et al. Biological gradient between long-term arsenic exposure and carotid atherosclerosis. Circulation. 2002;105(15):1804–9. This ecological study from southwestern Taiwan used the median level of well-water arsenic in a village where the participants resided and found a dose–response relationship between high levels of arsenic exposure (700–930 μg/L) and carotid atherosclerosis, defined based on carotid artery intima-media thickness and carotid plaque.
Wu MM et al. Effect of plasma homocysteine level and urinary monomethylarsonic acid on the risk of arsenic-associated carotid atherosclerosis. Toxicol Appl Pharmacol. 2006;216(1):168–75.
Chen Y et al. Arsenic exposure from drinking-water and carotid artery intima-medial thickness in healthy young adults in Bangladesh. J Health Popul Nutr. 2006;24(2):253–7.
Holaj R et al. Intima-media thickness of the common carotid artery is the significant predictor of angiographically proven coronary artery disease. Can J Cardiol. 2003;19(6):670–6.
Chen Y et al. Arsenic exposure from drinking water, arsenic methylation capacity, and carotid intima-media thickness in Bangladesh. Am J Epidemiol. 2013;178(3):372–81. In this cross-sectional study nested in the HEALS from Bangladesh, in which >90 % of participants have been exposed to arsenic levels of 0.1–300 μg/L, there was a positive association between arsenic exposure measured at the individual level through well water and urine samples at baseline and cIMT measured on average 7 years later.
Osorio-Yanez C et al. Carotid intima-media thickness and plasma asymmetric dimethylarginine in Mexican children exposed to inorganic arsenic. Environ Health Perspect. 2013;121(9):1090–6.
Algra A et al. QTc prolongation measured by standard 12-lead electrocardiography is an independent risk factor for sudden death due to cardiac arrest. Circulation. 1991;83(6):1888–94.
Goldberg RJ et al. Duration of the QT interval and total and cardiovascular mortality in healthy persons (The Framingham Heart Study experience). Am J Cardiol. 1991;67(1):55–8.
Okin PM et al. Assessment of QT interval and QT dispersion for prediction of all-cause and cardiovascular mortality in American Indians: the Strong Heart Study. Circulation. 2000;101(1):61–6.
Ahmad SA et al. Electrocardiographic abnormalities among arsenic-exposed persons through groundwater in Bangladesh. J Health Popul Nutr. 2006;24(2):221–7.
Yildiz A et al. Effect of chronic arsenic exposure from drinking waters on the QT interval and transmural dispersion of repolarization. J Int Med Res. 2008;36(3):471–8.
Mumford JL et al. Chronic arsenic exposure and cardiac repolarization abnormalities with QT interval prolongation in a population-based study. Environ Health Perspect. 2007;115(5):690–4. This is the first study from Inner Mongolia that evaluated the effect of low-to-moderate levels of arsenic exposure on QTc prolongation and found a monotonic relationship between well-water arsenic measured at the household level and QTc prolongation. However, the exposure categories were wide (≤21, 100–300, 430–690 μg/L).
Wang CH et al. Increased risk of QT prolongation associated with atherosclerotic diseases in arseniasis-endemic area in southwestern coast of Taiwan. Toxicol Appl Pharmacol. 2009;239(3):320–4.
Mordukhovich I et al. Association between low-level environmental arsenic exposure and QT interval duration in a general population study. Am J Epidemiol. 2009;170(6):739–46.
Chen Y et al. Arsenic exposure from drinking water and QT-interval prolongation: results from the Health Effects of Arsenic Longitudinal Study. Environ Health Perspect. 2013;121(4):427–32, 432e1-7. The most recent large study nested in the HEALS that found a positive association between arsenic exposure and QTc prolongation, especially among women.
Wang CH et al. Arsenic-induced QT dispersion is associated with atherosclerotic diseases and predicts long-term cardiovascular mortality in subjects with previous exposure to arsenic: a 17-year follow-up study. Cardiovasc Toxicol. 2010;10(1):17–26.
Adams DH, Shaw S. Leucocyte-endothelial interactions and regulation of leucocyte migration. Lancet. 1994;343(8901):831–6.
Blankenberg S et al. Circulating cell adhesion molecules and death in patients with coronary artery disease. Circulation. 2001;104(12):1336–42.
Hwang SJ et al. Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: the Atherosclerosis Risk In Communities (ARIC) study. Circulation. 1997;96(12):4219–25.
Ridker PM et al. Plasma concentration of soluble intercellular adhesion molecule 1 and risks of future myocardial infarction in apparently healthy men. Lancet. 1998;351(9096):88–92.
Chen Y et al. Association between arsenic exposure from drinking water and plasma levels of soluble cell adhesion molecules. Environ Health Perspect. 2007;115(10):1415–20.
Wu F et al. Association between arsenic exposure from drinking water and plasma levels of cardiovascular markers. Am J Epidemiol. 2012;175(12):1252–61. This cross-sectional study nested in the HEALS assessed the relation between arsenic exposure and plasma levels of six biomarkers of systemic inflammation and endothelial dysfunction and found a positive association of well-water arsenic and total urinary arsneic with plasma levels of soluble vascular adhesion molecule-1.
Karim MR et al. Increases in oxidized low-density lipoprotein and other inflammatory and adhesion molecules with a concomitant decrease in high-density lipoprotein in the individuals exposed to arsenic in Bangladesh. Toxicol Sci. 2013;135(1):17–25.
Steinberg D. Role of oxidized LDL and antioxidants in atherosclerosis. Adv Exp Med Biol. 1995;369:39–48.
Heinecke JW. Oxidants and antioxidants in the pathogenesis of atherosclerosis: implications for the oxidized low density lipoprotein hypothesis. Atherosclerosis. 1998;141(1):1–15.
Holvoet P et al. Circulating oxidized LDL is a useful marker for identifying patients with coronary artery disease. Arterioscler Thromb Vasc Biol. 2001;21(5):844–8.
Tsimikas S et al. Oxidized phospholipids predict the presence and progression of carotid and femoral atherosclerosis and symptomatic cardiovascular disease: five-year prospective results from the Bruneck study. J Am Coll Cardiol. 2006;47(11):2219–28.
Meisinger C et al. Plasma oxidized low-density lipoprotein, a strong predictor for acute coronary heart disease events in apparently healthy, middle-aged men from the general population. Circulation. 2005;112(5):651–7.
Wadham C et al. High-density lipoproteins neutralize C-reactive protein proinflammatory activity. Circulation. 2004;109(17):2116–22.
Soucy NV et al. Neovascularization and angiogenic gene expression following chronic arsenic exposure in mice. Cardiovasc Toxicol. 2005;5(1):29–41.
Vaughan DE. PAI-1 and atherothrombosis. J Thromb Haemost. 2005;3(8):1879–83.
Galis ZS et al. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. 1994;94(6):2493–503.
Zhang R et al. Association between myeloperoxidase levels and risk of coronary artery disease. JAMA. 2001;286(17):2136–42.
Meuwese MC et al. Serum myeloperoxidase levels are associated with the future risk of coronary artery disease in apparently healthy individuals: the EPIC-Norfolk Prospective Population Study. J Am Coll Cardiol. 2007;50(2):159–65.
Hansson J et al. Biomarkers of extracellular matrix metabolism (MMP-9 and TIMP-1) and risk of stroke, myocardial infarction, and cause-specific mortality: cohort study. PLoS One. 2011;6(1):e16185.
Banerjee M et al. Evaluation of the serum catalase and myeloperoxidase activities in chronic arsenic-exposed individuals and concomitant cytogenetic damage. Toxicol Appl Pharmacol. 2010;249(1):47–54.
Tseng CH et al. Lipid profile and peripheral vascular disease in arseniasis-hyperendemic villages in Taiwan. Angiology. 1997;48(4):321–35.
Nabi AH, Rahman MM, Islam LN. Evaluation of biochemical changes in chronic arsenic poisoning among Bangladeshi patients. Int J Environ Res Public Health. 2005;2(3–4):385–93.
Hsueh YM et al. Low serum carotene level and increased risk of ischemic heart disease related to long-term arsenic exposure. Atherosclerosis. 1998;141(2):249–57.
Yu RC et al. Arsenic methylation capacity and skin cancer. Cancer Epidemiol Biomarkers Prev. 2000;9(11):1259–62.
Chen YC et al. Arsenic methylation and skin cancer risk in southwestern Taiwan. J Occup Environ Med. 2003;45(3):241–8.
Tseng CH et al. Long-term arsenic exposure and incidence of non-insulin-dependent diabetes mellitus: a cohort study in arseniasis-hyperendemic villages in Taiwan. Environ Health Perspect. 2000;108(9):847–51.
Parvez F et al. A prospective study of respiratory symptoms associated with chronic arsenic exposure in Bangladesh: findings from the Health Effects of Arsenic Longitudinal Study (HEALS). Thorax. 2010;65(6):528–33.
Ahsan H et al. Arsenic metabolism, genetic susceptibility, and risk of premalignant skin lesions in Bangladesh. Cancer Epidemiol Biomarkers Prev. 2007;16(6):1270–8.
Wu F et al. Interaction between arsenic exposure from drinking water and genetic susceptibility in cardiovascular disease risk in Bangladesh. Submitted. 2013.
Hsieh YC et al. Risk of carotid atherosclerosis associated with genetic polymorphisms of apolipoprotein E and inflammatory genes among arsenic exposed residents in Taiwan. Toxicol Appl Pharmacol. 2008;227(1):1–7.
Hsueh YM et al. Genetic polymorphisms of oxidative and antioxidant enzymes and arsenic-related hypertension. J Toxicol Environ Health A. 2005;68(17–18):1471–84.
Wu MM et al. GT-repeat polymorphism in the heme oxygenase-1 gene promoter and the risk of carotid atherosclerosis related to arsenic exposure. J Biomed Sci. 2010;17:70.
Hsieh YC et al. Significantly increased risk of carotid atherosclerosis with arsenic exposure and polymorphisms in arsenic metabolism genes. Environ Res. 2011;111(6):804–10.
Wang YH et al. Effects of arsenic exposure and genetic polymorphisms of p53, glutathione S-transferase M1, T1, and P1 on the risk of carotid atherosclerosis in Taiwan. Atherosclerosis. 2007;192(2):305–12.
Wu F et al. Interaction between arsenic exposure from drinking water and genetic susceptibility in carotid intima-media thickness in Bangladesh. Toxicol Appl Pharmacol. 2014; pii: S0041–008X(14)00059-3. doi:10.1016/j.taap.2014.02.014.
Pierce BL et al. Genome-wide association study identifies chromosome 10q24.32 variants associated with arsenic metabolism and toxicity phenotypes in Bangladesh. PLoS Genet. 2012;8(2):e1002522.
Patrono C, FitzGerald GA. Isoprostanes: potential markers of oxidant stress in atherothrombotic disease. Arterioscler Thromb Vasc Biol. 1997;17(11):2309–15.
Ramos O et al. Arsenic increased lipid peroxidation in rat tissues by a mechanism independent of glutathione levels. Environ Health Perspect. 1995;103 Suppl 1:85–8.
Han SG, Castranova V, Vallyathan V. Heat shock protein 70 as an indicator of early lung injury caused by exposure to arsenic. Mol Cell Biochem. 2005;277(1–2):153–64.
Balagopal PB et al. Nontraditional risk factors and biomarkers for cardiovascular disease: mechanistic, research, and clinical considerations for youth: a scientific statement from the American Heart Association. Circulation. 2011;123(23):2749–69.
Klei LR, Garciafigueroa DY, Barchowsky A. Arsenic activates endothelin-1 Gi protein-coupled receptor signaling to inhibit stem cell differentiation in adipogenesis. Toxicol Sci. 2013;131(2):512–20.
Garciafigueroa DY et al. Arsenic-stimulated lipolysis and adipose remodeling is mediated by G-protein-coupled receptors. Toxicol Sci. 2013;134(2):335–44.
Kurzius-Spencer M et al. Contribution of diet to aggregate arsenic exposures—an analysis across populations. J Expo Sci Environ Epidemiol. 2013.
Farzan SF, Karagas MR, Chen Y. In utero and early life arsenic exposure in relation to long-term health and disease. Toxicol Appl Pharmacol. 2013;272(2):384–90.
Whitlock G et al. Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet. 2009;373(9669):1083–96.
Ben-Shlomo Y, Kuh D. A life course approach to chronic disease epidemiology: conceptual models, empirical challenges and interdisciplinary perspectives. Int J Epidemiol. 2002;31(2):285–93.
Lynch J, Smith GD. A life course approach to chronic disease epidemiology. Annu Rev Public Health. 2005;26:1–35.
Pinyayev TS et al. Preabsorptive metabolism of sodium arsenate by anaerobic microbiota of mouse cecum forms a variety of methylated and thiolated arsenicals. Chem Res Toxicol. 2011;24(4):475–7.
Van de Wiele T et al. Arsenic metabolism by human gut microbiota upon in vitro digestion of contaminated soils. Environ Health Perspect. 2010;118(7):1004–9.
United States Environmental Protection Agency. Fact sheet: drinking water standard for arsenic. EPA 815-F-00-015. Available: http://www.epa.gov/safewater/arsenic/regulations_factsheet.html Accessed 6 March 2012.
WHO (World Health Organization). Guidelines for Drinking-water Quality: Incorporating First and Second Addenda to Third Edition. Vol. 1—Recommendations. Geneva: WHO Press. Available: http://www.who.int/water_sanitation_health/dwq/gdwq3/en/index.html Accessed 1 October 2012.
Acknowledgments
Yu Chen has received grant support from the National Institutes of Health (R01ES017541 and P30ES000260).
Compliance with Ethics Guidelines
ᅟ
Conflict of Interest
Fen Wu, Peter Molinaro, and Yu Chen declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Wu, F., Molinaro, P. & Chen, Y. Arsenic Exposure and Subclinical Endpoints of Cardiovascular Disease. Curr Envir Health Rpt 1, 148–162 (2014). https://doi.org/10.1007/s40572-014-0011-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-014-0011-2