
Abstract
Apoptosis, or programmed cell death, maintains tissue homeostasis by eliminating damaged or unnecessary cells. However, cells can evade this process, contributing to conditions such as cancer. Escape mechanisms include anoikis, mitochondrial DNA depletion, cellular FLICE inhibitory protein (c-FLIP), endosomal sorting complexes required for transport (ESCRT), mitotic slippage, anastasis, and blebbishield formation. Anoikis, triggered by cell detachment from the extracellular matrix, is pivotal in cancer research due to its role in cellular survival and metastasis. Mitochondrial DNA depletion, associated with cellular dysfunction and diseases such as breast and prostate cancer, links to apoptosis resistance. The c-FLIP protein family, notably CFLAR, regulates cell death processes as a truncated caspase-8 form. The ESCRT complex aids apoptosis evasion by repairing intracellular damage through increased Ca2+ levels. Antimitotic agents induce mitotic arrest in cancer treatment but can lead to mitotic slippage and tetraploid cell formation. Anastasis allows cells to resist apoptosis induced by various triggers. Blebbishield formation suppresses apoptosis indirectly in cancer stem cells by transforming apoptotic cells into blebbishields. In conclusion, the future of apoptosis research offers exciting possibilities for innovative therapeutic approaches, enhanced diagnostic tools, and a deeper understanding of the complex biological processes that govern cell fate. Collaborative efforts across disciplines, including molecular biology, genetics, immunology, and bioinformatics, will be essential to realize these prospects and improve patient outcomes in diverse disease contexts.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Apoptosis, or programmed cell death, is essential for maintaining tissue homeostasis by eliminating damaged or unnecessary cells. The ability of cells to evade apoptosis can contribute to conditions such as cancer. Mechanisms of evasion include anoikis, mitochondrial DNA depletion, c-FLIP, ESCRT, mitotic slippage, anastasis, and blebbishield formation. |
Anoikis is crucial in cellular survival and metastasis, triggered by cell detachment or aberrant adhesion. |
Mitochondrial DNA depletion is linked to cellular dysfunction and various diseases, including cancer. |
The c-FLIP protein family, particularly CFLAR, regulates apoptosis and cellular death processes. |
The ESCRT complex helps cells escape apoptosis by repairing damages through exocytosis, particularly by increasing intracellular Ca2+. |
Antimitotic agents induce mitotic arrest but can lead to mitotic slippage, creating tetraploid cells. |
Anastasis allows cells to escape apoptosis by resurrecting after various triggers. |
Blebbishield formation aids in apoptosis suppression indirectly in cancer stem cells. |
Understanding these mechanisms is crucial for developing targeted therapies for diseases where apoptosis regulation is compromised, considering variations among cancer types and disease contexts. |
1 Introduction
Cellular homeostasis, the delicate balance of cell proliferation and death, is a cornerstone of multicellular organism survival. Among the intricate processes contributing to this equilibrium, apoptosis, or programmed cell death, plays a pivotal role in eliminating damaged or surplus cells. The precise orchestration of apoptosis is essential for maintaining tissue integrity and preventing the development of various pathological conditions. However, the escape of cells from apoptosis has emerged as a significant challenge, particularly in the context of diseases such as cancer.
This review delves into the intricate landscape of cellular homeostasis, exploring the mechanisms through which cells can elude apoptosis. Beyond its role as a safeguard mechanism, apoptosis evasion has been implicated in the progression and persistence of various diseases, posing a formidable obstacle to therapeutic interventions. The elucidation of these escape mechanisms is fundamental for the development of targeted therapies aimed at reinstating apoptosis in cells where its regulation has faltered.
The mechanisms identified include anoikis, a form of apoptosis triggered by cell detachment; mitochondrial DNA (mtDNA) depletion, associated with disruptions in mitochondrial function; cellular FLICE inhibitory protein (c-FLIP), an apoptosis-inhibiting protein; the endosomal sorting complexes required for transport (ESCRT) machinery; mitotic slippage, allowing cells to bypass the mitotic phase; anastasis, the process of cell recovery following initiation of apoptosis; and the formation of blebbishields, protective structures shielding cells from apoptotic signals.
Intriguingly, these mechanisms exhibit a dynamic interplay across various cellular contexts, and their prominence can vary among different types of cancer and disease scenarios. By unravelling the intricacies of apoptosis escape routes, this review aims to contribute to the comprehensive understanding of cellular dynamics and provide a foundation for the development of targeted therapeutic strategies. The exploration of these mechanisms not only sheds light on the complexity of cellular decision-making, but also opens avenues for innovative approaches to reinstate cellular homeostasis and counteract the escape from apoptosis in diverse pathological conditions.
2 Methodology
The search methodology for the review article on apoptosis escape mechanisms was systematically designed to identify relevant studies elucidating the intricate pathways, regulatory factors, and clinical implications associated with cell survival and resistance to apoptosis. Searches were conducted in key databases, including PubMed, Scopus, Web of Science, and Embase, using predefined search strings and keywords related to each identified apoptosis escape mechanism and associated cellular processes. We have compiled the data about the mechanisms of evasion (anoikis, mitochondrial DNA depletion, c-FLIP, ESCRT, mitotic slippage, anastasis, and blebbishield formation) in order from the most researched to the least in these databases in this review. The search was mostly limited to studies published between 2010 and 2023 to ensure the inclusion of the most recent and relevant literature on the topic. However, this time interval was sometimes exceeded when especially critical informatory data was needed. Titles and abstracts were initially screened to identify potentially relevant studies, followed by a full-text screening of selected articles against inclusion and exclusion criteria. Data extraction aimed to capture detailed information on the molecular and cellular mechanisms, signaling pathways, and regulatory factors involved in apoptosis escape, as well as the clinical implications and therapeutic strategies targeting these mechanisms. The review employed a narrative synthesis approach to integrate and interpret the complex interactions between apoptosis escape mechanisms, providing insights into the pathological and physiological processes underlying cell survival, disease progression, and potential therapeutic interventions. The review also highlighted the variability in apoptosis escape strategies among different types of cancer and disease contexts, emphasizing the importance of understanding these mechanisms for developing targeted therapies to induce apoptosis in cancer cells and other diseases where apoptosis regulation is compromised.
3 Anoikis
3.1 Mechanism
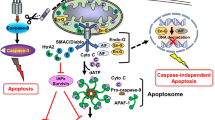
Anoikis (“homelessness” in Greek) is a form of apoptosis that is activated to eliminate cells that have detached from the extracellular matrix (ECM) or exhibited inappropriate cell adhesion [1]. Anoikis is a regulated cell death mechanism that uses both intrinsic and extrinsic pathways [2]. BCL-2 family proteins play key roles in both of these processes. In cells detached from ECM, integrins form complexes with growth factor receptors and cytoskeletal systems are linked through plectin. Instability of cytoskeletal elements can neutralize BCL-2 by causing the release of BMF from the actin and inducing the release of cytochrome c from mitochondria. At this point, plectin cleavage contributes to effector caspase activation and additional cytochrome c release. This is because caspase-8 is activated [3]. Activation of caspase-8 at this point facilitates further cytochrome c release through BID, plectin cleavage, and effector caspase activation. Consequently, flamin and cytokeratin intermediate filaments become separated. The process that actin polymers undergo after separation is not clear. However, the breakdown of anoikis contributes to neoplasia [3].
3.2 Implications on cancer
Recent studies have associated anoikis with different types of cancer, such as lung [4], stomach [5], colorectal [6], breast [7], prostate [8], epithelial ovarian [7, 8] and oral squamous cell carcinoma [9]. Determining the interactions between cells, the tumor microenvironment (TME), and extrinsic factors is essential to understanding the role of anoikis in cancer cells [10]. ECM remodeling results in an important change that produces the anoikis-resistant state, such as decreased expression of fibronectin, collagen IV, and hyaluronic acid and increased expression of laminin; perlecan; integrin subunits (av, β3, α5, and β1); hyaluronidase 1, 2, and 3; and metalloproteinases 2 and 9 [11]. This leads to generation of anoikis-resistant cells. The critical role of extrinsic factors in cells acquiring anoikis properties is explained by the effect of fibronectin on oncogene expression in the ECM. Increased fibronectin in the ECM can direct cells to anoikis by modulating the expression of p53 [12]. According to the study of Sanchez-Ruderisch et al., integrins initiate the anoikis cascade by activating caspase-8, which induces TME leading to anoikis. In their study, the administration of Gal-1 led to swift and steady activation of caspase-8 in HepG2 suspension cultures, as compared with untreated samples. To ascertain the importance of this caspase-8 activation in Gal-1-induced anoikis, a specific inhibitor of caspase-8 (Z-IETD-FMK) was introduced at various intervals subsequent to Gal-1 stimulation. The inhibitor effectively thwarted Gal-1-triggered anoikis when applied within 5 min of commencing Gal-1 treatment. Consequently, they concluded that caspase-8 activation was essential for Gal-1-induced anoikis [13]. Negative TME and cellular stress can promote autophagy, providing the cell with the opportunity to survive [14]. Although autophagy and anoikis resistance collaborate in the metastatic TME, it is unclear which process precedes the other [10].
The resistance to anoikis has been determined to be modulated by various factors including pH, signaling pathways, cell adhesion, growth, and apoptotic proteins [15]. The pathways activated by anoikis resistance can be divided into two categories; the first is activation of survival pathways, and the second is suppression of apoptotic pathways in detached cells [10]. Metabolic intermediates produce the energy required for cellular growth and survival. Oncogenic activation of signaling pathways also reprograms cellular metabolism to increase anoikis resistance. Dysregulation of genes encoding rate-limiting enzymes that maintain cellular homeostasis leads to metabolic perturbation that triggers anoikis resistance before tumor metastasis [15]. High metabolite levels are a common feature in both autophagy and anoikis resistance phenomena. In anoikis-resistant cells, high activity of the SRC/AKT/ERK signaling pathway, which is a necessary and sufficient marker, is associated with high aggressiveness of cancer cells [16]. The activation of Src and the upregulation of various autophagy factors increase glucose metabolism and ATP production, enhancing the survival of cancer cells under conditions where they are detached from the ECM [16].
The main question related to anoikis is how the caspase cascade is activated by the simple detachment of cells from the ECM. The prevalent hypothesis is that death receptors are activated, either through their inherent tendency to self-associate, which might be adequate for signaling, or through interaction with internal death ligands [3]. Pro-apoptotic signals such as caspase-8 in cells detached from the ECM will induce death receptor activation [17]. The second question is how detached cells can escape from death receptors and pro-apoptotic signals while resisting anoikis. A possible mechanism is that in detached cells undergoing apoptosis, anoikis blockade could be associated with cell survival pathways. The cleavage of AKT/protein kinase B (PKB) by caspase-3 under the control of TNF death receptors demonstrates the anoikis-resistant death receptor/caspase/survival cascade. To put it more clearly, a critical level of Akt/PKB activity must be maintained for cell survival and caspases induce anoikis by reducing Akt/PKB activity below this threshold. Moreover, the caspase-mediated proteolysis of Akt/PKB may make cells resistant to secondary survival signals [18]. Dynamic interactions between death receptors that mediate cell contact with the ECM are also important for anoikis resistance. The TRAF2 and FAK collaboration determines to what extent a cell can overcome anoikis [10].
Anchorage-dependent growth and epithelial-mesenchymal transition (EMT), two critical features during cancer progression and metastatic colonization, have been associated with anoikis [5, 8, 19]. Some possible epigenetic changes enable cells to have phenotypic features, contributing to anchorage-independent cell growth. This hypothesis is supported by the data on c-Myc, Akt, β-catenin, and class I histone deacetylases [10]. Anoikis resistance under loss of ECM contact allows cancer cells to leave the primary tumor site, migrate to a distant site, and form a metastatic lesion. While down-regulation of cell adhesion proteins such as protein tyrosine phosphatase 1B can promote anoikis [20], aggregation in circulating cancer cell clusters promotes anoikis resistance. Many of the signaling pathways associated with anoikis resistance, such as MAPK and AKT, are regulated by cell–cell and cell–ECM interactions of detached and aggregated cells. Consequently, cancer cells resistant to anoikis ensure their survival throughout the circulation [21]. Bone morphogenetic protein 9 (BMP9), or growth and differentiation factor 2 (GDF2), are two of the epigenetic regulators of the anoikis process [22]. One of the most efficient molecules for inducing anoikis resistance is neurotrophic tyrosine kinase receptor B (TRKB), which is frequently overexpressed in tumors [23].
3.3 Treatment Strategies
Overcoming anoikis resistance is thought to have important therapeutic benefits for cancers. Metastatic cancer cells exhibit anoikis resistance, indicating a potential target for treatment. To halt metastasis, further research should be conducted to enhance anoikis sensitivity through death receptors, determining whether they can act as an activator or inhibitor of death receptor-mediated apoptosis [15]. However, targeting death receptors could help effectively treat cancer because they play an important role in escaping anoikis, one of the main causes of their poor prognosis. Several studies have shown that small molecules (such as salinomycin, brevilin A, and synthesized flavonoid derivative GL-V9) inhibit anoikis resistance, invasion, migration, and metastasis by inducing reactive oxygen species (ROS) production in cancer cells [24,25,26,27]. Drugs that can induce oxidative stress may be considered for new therapeutic approaches because they can limit anchorage-independent growth (AIG) in cancer cells while protecting healthy cells [15]. In fact, the shift of the metabolic pathway to glycolysis provides an alternative source of energy production under AIG conditions for cancer cell survival. Blocking or targeting the main pathway of ATP acquisition in anoikis-resistant cells may be a powerful approach. Hypoxia and low pH, the main characteristics of the tumor environment, also enhance the capacity to avoid anoikis and treatment resistance. Reprogramming the metabolic structure of the TME has been observed to increase resistance to anoikis and metastasis, as an acidic environment is essential for the growth of solid tumors [28]. Recent research has shown that some pharmaceuticals (for example, metformin and piplartine) can induce anoikis by regulating oncogenic or metabolic signaling pathways. However, these drugs are not specific for targeting anoikis-resistant cells [15]. There are two possible treatment strategies that have been associated with anoikis in metastasis: (i) inducing anoikis and (ii) preventing phenotypes that are resistant to anoikis [10]. Powerful therapeutic targets to induce anoikis include targeting inhibition of fatty acid β-oxidation (FAO) in specific cancer types, such as high-grade serous ovarian cancer [29], focusing on the FAO pathway in metastasis of colorectal cancer cells [30], disruption of integrin signaling pathways in a mouse model of colorectal cancer [30]. In breast cancer, suppression of thromboxane A2 synthase 1 (TBXAS1) and unregulated thromboxane A2 receptor (TP) prevents metastasis by making cells more sensitive to anoikis [31]. Targeting anoikis resistance may improve the efficacy of anticancer drugs or reduce tumor metastasis after surgery, as suggested by the overexpression of MUC1 and the reduction in oncogene signaling pathways in pancreatic cancer [32]. Targeting the Circ-0004585/miR-1248/transmembrane 9 superfamily member 4 axis, which activates autophagy in patient tissue samples and pancreatic cancer cell lines, may reduce anoikis resistance and metastasis [8]. The use of immunotherapy to induce antibody-dependent cellular cytotoxicity is a new treatment plan to inhibit the main drivers of metastasis-like suppressed anoikis in highly metastatic cancer types [33].
In conclusion, although anoikis has only been reported relatively recently in the literature, it is being extensively studied with findings increasing each day. The determination of the function of anoikis in the processes of survival signal activation and suppression of the apoptotic death pathway in cells detached from the extracellular matrix (ECM) is critically important for understanding cancer cells. A better understanding of the metabolic changes leading to anoikis resistance could open the door to new approaches in addressing metastasis, the primary cause of cancer-related deaths.
4 Mitochondrial DNA Depletion
4.1 Mechanism
A decrease in the copy number of mtDNA [34] content by up to 30% is referred to mtDNA depletion, which causes mitochondrial dysfunction. It has been associated with various diseases, including breast [35], prostate [36], and many other cancer types [37]. Several studies have shown that reduction in mtDNA content in breast and prostate cancer cells promotes disease progression and plays a crucial role in tumor development [38, 39]. Therefore, mtDNA depletion has been a subject of research for many years, and its relationship with diseases has been investigated. MtDNA depletion and resistance to apoptosis are two interconnected concepts that play roles in pathological and physiological processes. It has been shown that metastasis and invasion are induced in cancer cells carrying mtDNA depletion [40]. The difference here could be that cells exhibit different sensitivities to apoptosis-inducing agents [41].
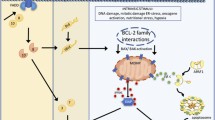
The mitochondrion, with its 16.6 kb DNA, is an autonomous organelle that plays a crucial role in cellular metabolism. MtDNA encodes 13 polypeptides that are essential for oxidative phosphorylation and ATP production, making it the central hub for oxidative phosphorylation [42]. MtDNA is sensitive to mutations due to its proximity to toxic metabolites and inefficient repair mechanisms [43]. Mutations in mtDNA, therefore, are crucial, particularly in terms of the enzymes and proteins required for oxidative phosphorylation, as they can lead to mitochondrial dysfunction and inefficient ATP production [44]. Mitochondrial dysfunction has been associated with various diseases, including cancer, diabetes, and neurodegenerative diseases [45], and dysfunctional or loss of mitochondria can affect gene expression, cell morphology, and function. Dysfunction or loss of function of mitochondria can disrupt calcium homeostasis, apoptosis, and many other pathways (Fig. 1).

Relationship between mtDNA depletion and resistance to apoptosis. This schematic illustrates potential mechanisms underlying the relationship between mitochondrial DNA (mtDNA) depletion and resistance to apoptosis. As depicted, mtDNA depletion may trigger survival error signaling activation, resulting in decreased levels of reactive oxygen species (ROS), retention of pro-apoptotic factors on the membrane, disruption of mitochondrial membrane potential, and an increase in antioxidant regulation. These events collectively promote antiapoptotic pathways, thereby conferring resistance to apoptosis in affected cells
4.2 Implications on Different Cellular Conditions
It is quite intriguing that cells with mtDNA depletion exhibit resistance to apoptosis and invasive behavior, it appears that these cells inhibit apoptosis by promoting survival signaling pathways [46]. To investigate the relationship between mtDNA depletion and apoptosis, cells lacking mtDNA were created by exposing them to ethidium bromide, which damages mtDNA. In one study, it was shown that cells with no mtDNA had lower levels of reactive oxygen species (ROS) compared with the control cells. It was also observed that UV-induced cytochrome c release was lower in cells lacking mtDNA compared with controls [47]. Apoptosis is induced by increased ROS levels. Mitochondrial dysfunction leads to inefficient ROS production, making it difficult for the cell to enter the apoptosis process. This can affect cell proliferation and survival in variable ways depending on the cell type. Additionally, the disruption of mitochondrial membrane potential has also been mentioned as a reason for cells showing resistance to apoptosis with mtDNA depletion [41]
Furthermore, a study showed that cells with mtDNA depletion could contain more reduced glutathione (GSH) [48]. Similarly, an increase in the amount of antioxidants such as GPx and MnSOD has been noted in cells showing mtDNA depletion. This increase in antioxidants has been indicated to play a role in reducing cellular oxidative stress, leading to a decrease in ROS levels and inducing the suppression of p53 [49]. In another study, an important point in the resistance mechanisms to apoptosis was identified in cells showing mtDNA depletion. It was shown that cytochrome c release was lower in cells with mtDNA depletion compared with controls, and morphological changes did not correspond to cytochrome c release patterns [47]. It is known that cells with depleted mtDNA exhibit problems in the release of pro-apoptotic proteins such as BAD, BAX, and BID [50]. However, studies have shown that mtDNA depletion, cytochrome c release, and disruption of mitochondrial membrane potential are not sufficient steps for the initiation and execution of apoptosis. In a study by Biswas et al., mtDNA depletion induced mitochondrial stress in C2C12 rhabdomyoblasts and A549 human lung carcinoma cells, activating the mitochondrial stress signaling cascade from mitochondria to the nucleus, which resulted in a cascade of gene expression changes, morphological changes, and invasiveness of these cells. It was also noted that etoposide-induced apoptosis was inhibited in cells with mtDNA depletion due to a lack of active caspase-8 and pro-caspase-3, contributing to resistance to apoptosis [51]. In another study, it was suggested that the increased expression of antiapoptotic genes such as BCL-2 could be responsible for resistance to apoptosis in cells with reduced mtDNA content [45]. It was shown that cells carrying mtDNA depletion promoted the activation of survival enzymes such as AKT and inhibited TNF-mediated apoptosis [46]. On the basis of all these findings, the decreased ROS production in cells with mtDNA depletion, activation of antiapoptotic gene expression, and inhibition of apoptotic signaling explain why these cells cannot undergo apoptosis.
Cells with mtDNA depletion have been shown to be more resistant to apoptosis, induced by certain agents compared with parental cells. Biswas et al. reported that C2C12 myoblasts with partially depleted mtDNA were less sensitive to staurosporine (STS)-induced apoptosis. It was suggested that the inhibition of apoptosis was due to the retention of BAX, BID, and BAD in the mitochondrial inner membrane; increased expression of Bcl-2 and Bcl-X(L); and the inability to process active Bid, despite the induction of Bax and Bad [50]. Jacques et al. found that cells with mtDNA depletion preserved their apoptosis capacity but exhibited higher resistance to STS-induced apoptosis compared to parental cells [52]. In another study, Dey and Moraes showed that cells with mtDNA depletion were less sensitive to STS-induced apoptosis. Normally, caspases 2, caspase 9, and later executioner caspase 3 are released upon an apoptotic stimulus. In this study, the release of these factors from the mitochondria is blocked by cyclosporin A, an inhibitor of mitochondrial permeability transition pore (MPTP). Therefore, the study indicated that the reason for resistance to apoptosis in osteosarcoma cells with mtDNA depletion was a decrease in caspase 3 activation, without changes in mitochondrial permeability and cytochrome c release [53]. Therefore, it appears there are different mechanisms at play.
Another perspective concerning mtDNA depletion and resistance to apoptosis is related to epigenetics. The copy number of mtDNA affects CpG methylation in the nuclear genome and alters the epigenetic profile. Studies suggest that mtDNA depletion can lead to epigenetic changes in the nuclear DNA, which, in turn, modify gene expression with the aim of conserving energy due to mitochondrial dysfunction [54]. Considering the significant role of epigenetics in many pathways, its involvement in this process is possible.
4.3 Treatment Perspectives
The elucidation of the mechanisms of mtDNA depletion and resistance to apoptosis can be considered significant in regard to new treatment approaches. Therapeutic strategies aimed at correcting mitochondrial function and enabling the regulation of apoptosis by cells hold great promise. Gene therapy holds promise by introducing functional copies of mitochondrial genes to address depleted mtDNA levels. Mitochondrial replacement therapy (MRT) offers another avenue by replacing defective mitochondria with healthy ones from donors, potentially preventing the transmission of mitochondrial diseases [55]. Pharmacological interventions targeting mitochondrial dysfunction and apoptosis resistance include mitochondrial-targeted antioxidants, biogenesis inducers, apoptosis inducers, and metabolic modulators [56, 57]. Stem cell therapy offers the potential to replenish tissues affected by mitochondrial dysfunction, while nutritional interventions and lifestyle modifications support mitochondrial health through dietary components, exercise, and stress reduction [58, 59]. Additionally, targeted molecular therapies tailored to specific molecular targets implicated in mitochondrial dysfunction are under investigation for more precise treatment options [60]. These diverse approaches highlight the complexity of addressing mitochondrial disorders and the need for personalized strategies to optimize therapeutic outcomes.
5 c-FLIP
5.1 Mechanism
c-FLIP is a family of proteins that play a critical role in the regulation of cellular death processes, and is referred to as Casper, iFLICE, FLAME-1, CASH, CLARP, MRIT, usurpin, or CFLAR truncated form of caspase-8 lacking enzymatic activity. It shows general homology with truncated pro-caspase-8 and pro-caspase-10. It suppresses extrinsic apoptosis by blocking caspase-8 activation by competing with caspase-8 for binding to Fas-associating protein with death domain (FADD) in the death-inducing signaling complex (DISC) [61]. Thus, c-FLIP functions as an important inhibitor of the extrinsic apoptotic pathway induced by death receptor activation such as tumor necrosis factor-related apoptosis-inducing ligand (TRAIL)/death receptor ligation. Moreover, among the 13 different alternative splicing variants, three are expressed as proteins. c-FLIP has multiple isoforms, among which only two forms, the short form (FLIPS) and the long form (FLIPL), are well characterized at the protein level in human cells [62]. There is also the c-FLIP-R isoform produced by alternative splicing. By comparison, c-FLIPS lacks the caspase-like domain and contains two DEDs with similarity to procaspase-8 followed by a short C-terminal extension containing ∼ 20 amino acids.
Thome et al. identified viral FLICE-inhibitory proteins (v-FLIPs) as virus-induced apoptotic regulatory proteins containing DED (death domain) [63]. They identified six different v-FLIPs and showed that cells expressing them are resistant to apoptosis initiated by FAS (CD95/APO-1), TRAILR1, and TNFR1.
C-FLIPS inhibits apoptosis by preventing the recruitment of caspase-8 to DISC, whereas the role of c-FLIPL is controversial. Indeed, c-FLIPL has been reported as both an inhibitor and inducer of apoptosis signaling, possibly depending on its expression levels [64, 65]. c-FLIPL is homologous to full-length procaspase-8 and the effects of heterodimerization between procaspase-8 on caspase-8 activity vary depending on c-FLIPL concentration [66]. High levels of c-FLIPL inhibit the release of caspase-8 from DISC, while low levels can lead to partial activation [64, 65]. Both FLIPL and FLIPS are unstable proteins regulated by ubiquitination/proteasome-mediated degradation [67]. All three c-FLIP isoforms can interact with the adaptor protein FADD and procaspase-8 through their DEDs in DISC. When caspase-8 is inhibited by c-FLIPS or c-FLIPL, apoptosis is prevented but necroptosis may occur.
5.2 Implications on Different Cell Death Mechanisms
Studies show that c-FLIP can regulate necroptosis in several ways. More specifically, c-FLIP can control necroptosis by influencing the formation of an intracellular protein complex called ripoptosome. c-FLIPL protects the cell from necroptosis by inhibiting the formation of ripoptosomes containing RIP1 and caspase-8, while c-FLIPS can promote ripoptosome formation [68]. The limited caspase-8 activity of procaspase-8-c-FLIP heterodimers prevents necroptosis, but it is still unclear exactly how this mechanism works [66]. Previous studies have shown that cellular inhibitory proteins (cIAPs) prevent ripoptosome formation. Furthermore, c-FLIPL may protect cIAP antagonist-treated cells from necroptosis in addition to Fas-induced cell death (apoptosis) [69]. Furthermore, c-FLIPL can inhibit ripoptosome formation upon stimulation with TLR3 [68]. Therefore, c-FLIP isoforms in the ripoptosome may determine whether cell death occurs via RIP3-dependent necroptosis or caspase-dependent apoptosis [65]. c-FLIPL suppression may further sensitize cells to RIP1/RIP3-dependent necroptosis when stimulated with TNF [70].
c-FLIP has also been associated with ferroptosis. TRAIL-R2, also referred to as death receptor 5 (DR5), is a receptor found on the surface of cells. It binds to TRAIL and forms a trimer structure. This trimerization process triggers the activation of pro-caspase-8 by bringing together Fas-associating protein with death domain (FADD) and procaspase-8, leading to the formation of the death-inducing signaling complex (DISC). This activated caspase-8 then initiates the activation of downstream executioner caspase-3, caspase-6, and caspase-7, which occurs via mitochondria-dependent and mitochondria-independent apoptotic pathways. However, the activation of DR5 triggered by ferroptotic agents is hindered, as it remains inactive due to its interaction with the antiapoptotic regulator, c-FLIP. c-FLIP can bind to FADD and/or procaspase-8 both in a TRAIL-dependent and TRAIL-independent manner, thereby obstructing the formation of the death-inducing signaling complex (DISC) and subsequent initiation of the caspase cascade. In this case, it is thought that c-FLIP may provide this antagonistic effect by inhibiting apoptosis [71].
C-FLIP also plays an inhibitory role against autophagy by preventing the interaction of autophagy-related gene 3 (ATG3) and light chain 3 (LC3) required for the formation of autophagosomes [72]. It is also known that caspase 8, a c-FLIP antagonist, may play a role in the process of autophagy suppression in T cells. Namely, the Fas-associated protein with death domain (FADD) and caspase 8 complex is localized on the autophagosome and this triggers caspase 8 activation, resulting in inhibition of autophagic cell death [73]. c-FLIP has been shown to interact with Beclin-1 protein in autophagy regulation. Beclin-1 is part of a complex required for autophagosome formation, and one study reveals that c-FLIP positively regulates autophagy flux by inhibiting proteasome-mediated degradation of Beclin-1 [74]. These findings provide an important step in elucidating the complex relationship between autophagy and c-FLIP at the cellular level.
5.3 Treatment Strategies
Controlling the regulation of apoptosis through c-FLIP could present a complex target for drug development targeting this pathway. It has been observed that high levels of c-FLIP are associated with cancer and are often linked to a poor prognosis [62, 75]. These high levels suggest that cancer cells have problems in apoptosis regulation and that these cells lose their ability to respond specifically to death receptors. Thus, it is an important resistance factor and a critical antiapoptotic regulator that inhibits chemotherapy-induced apoptosis in malignant cells [76]. Development of drugs that inhibit the function of c-FLIP protein is difficult due to its homology to caspase-8 since these drugs can unintentionally inhibit apoptosis if they target caspase-8 in addition to c-FLIP. In this regard, the role of c-FLIP isoforms in cancer therapy requires further research. However, many agents that reduce c-FLIP expression and make cancer cells more sensitive to death receptors and anticancer drugs have been studied [77]. Several agents that reduce c-FLIP expression and sensitize cancer cells to TRAIL or anticancer drugs have been studied [78]. These include some traditional anticancer drugs such as cisplatin, doxorubicin, actinomycin D, cycloheximide, camptothecin, 9-NC, and topotecan; histone deacetylase (HDAC) inhibitors; MEK1/2, PKC, and PI3K inhibitors; and numerous other compounds [78].
The presence of c-FLIP in EGFR-mutated lung cancer cells can promote resistance to erlotinib treatment, whereas c-FLIP overexpression can rescue these cells from treatment, possibly by altering NF-κB activity. These studies suggest that c-FLIP has the potential to regulate the response of EGFR-mutated cells to erlotinib [61]. In addition, thioridazine combined with curcumin promoted apoptosis in HNSCC cells by down-regulating c-FLIP and Mcl-1 expression through NOX4-mediated ROS production [79]. By inhibiting the degradation of FoxM1, which is associated with cancer initiation progression and drug resistance, c-FLIP induced its expression, thereby leading to the development of cellular resistance to TST (thiostrepton) and osimertinib [80]. In previous studies, it was observed that c-FLIP was regulated by p53, and c-FLIP (S/L) isoforms were down-regulated in HCT116 p53+/+ colon cancer cell line upon oxaliplatin and CPT11 treatments, while c-FLIP was not affected in p53 mutant cells [75]. In a recent study, p53 was up-regulated while c-FLIP(L) was down-regulated in KMU-191-induced apoptosis [81]. These results suggest that DNA damage agents affect c-FLIP expression via p53 in various cancer cell lines. Furthermore, some cancer therapies such as histone deacetylase inhibitors and topoisomerase I inhibitors have been shown to regulate c-FLIP levels [82, 83]. However, it should be remembered that the effects of these DNA-damaging or modifying therapies on c-FLIP may vary depending on the cell type, and these effects may sometimes affect both c-FLIP isoforms.
In conclusion, the roles of c-FLIP protein in apoptosis, survival signaling, and cellular death pathways are highly complex. This protein plays a critical role in the regulation of the apoptosis signaling pathway and the resistance of cancer cells. However, the full functionality and regulation of c-FLIP requires further investigation beyond cell culture in complex organisms.
6 ESCRT
6.1 Mechanism
The endosomal sorting complexes required for transport (ESCRT) machineries, expressed as the endosomal sorting complexes required for transport, are associated with most cellular functions, and their importance in cell signaling is increasing. ESCRT machineries are located in the cell cytoplasm as a complex. Inside the cell, they are associated with many cellular pathways such as plasma membrane repair [84], cytokinetic abscission [85], nuclear envelope maintenance [86], viral replication [87], programmed or unprogrammed cell death, and immune response [84, 88]. The ESCRT machinery prevents cells from undergoing apoptosis by repairing small membrane wounds or ensuring membrane impermeability in various physiological and pathological processes such as microvesicle formation, autophagosome formation, plasma membrane repair, cytokinetic abscission, viral replication compartment formation, lysosome repair, nuclear pore quality control, and nuclear envelope repair [89].
It is a mechanism preserved throughout evolution and initially identified as a component of the vacuolar protein sorting mutant in yeast [90]. The ESCRT complex consists of multimeric components with different functions categorized as ESCRT 0, I, II, III, and VPS4 [91,92,93,94]. In particular, ESCRT- III uniquely plays a role in suppressing various types of regulated cell death, including necroptosis [88], pyroptosis [95], and ferroptosis [96], by repairing damaged plasma membranes.
The intracellular space is composed of a continuous flow of signaling. Therefore, the plasma membrane, which forms the connection of the cell with the surrounding cells, is important for the continuity of cell signaling. For this reason, the plasma membrane has a structure that can form reconstructions in connection with dynamic processes that may occur inside the cell. Any damage to the plasma membrane is a warning for the cell. Therefore, cells have various developed functions that can repair membrane damage [97]. The ESCRT-III complex fulfils this expressed function.
6.2 Implications on Cancer
The perforin-granzyme pathway developed to eliminate tumor cells in the apoptosis mechanism involves perforins released from cytotoxic T lymphocytes or natural killer cells, which create pores in the target cell [98]. Ca2+ and granzyme B release into the cell through these pores’ triggers caspase activation and initiates apoptosis signaling. Loss of plasma membrane integrity activates membrane repair proteins in the cell. In this way, ESCRT machinery delays apoptosis by repairing these pores in the membrane, creating resistance against immunotherapy, and limiting T-cell-based treatments [89].
In particular, the increase in intracellular Ca2+, which is considered to be the initiator of the apoptosis mechanism, forms a bridge between apoptosis and ESCRT. Therefore, ESCRT complex is expressed as one of the escape mechanisms from apoptosis. Intracellular Ca2+ concentration increases as a result of membrane damage, triggering the apoptotic mechanism. Increased intracellular Ca2+ concentration stimulates the accumulation of ESCRT-III components in the plasma membrane. Both macro- and micro-level damages are repaired by the ESCRT-III complex by exocytosis of the damaged membrane section. In addition to the ESCRT-III complex, Ca2+-sensitive annexin proteins are also involved in the membrane repair process [99]. Thus, ESCRT complex components are a protective mechanism that allows for escape from cell death. Studies have shown that programmed cell death occurs in cells lacking ESCRT complex proteins [89]. In particular, the ESCRT-III component is thought to be a new target to increase the sensitivity of the chemotherapeutic agent applied in cancer treatment because in cells with genetic depletion of the ESCRT-III core component, the anticancer agent applied is more effective and promotes cell death compared with cells in which the ESCRT-III component is active.
ESCRT-III consists of more than ten subcomponents. Studies have reported the role of genetic polymorphisms in ESCRT-III components in promoting cancer cells. This shows the determinant role of ESCRT-III components in terms of cell death and survival [100]. The ESCRT-III subunit CHMP-4C plays a decisive role in the last critical phase of cell division, cytokinetic abscission. In the aforementioned study, it was reported that stopping the cytokinetic abscission of the mutant CMHP-4C subunit may cause aneuploidy as a result of improper segregation of chromosomes. In the same study, it has been proven that it has a synergistic effect, especially with the loss of p53, and increases the sensitivity to form tumor cells [100]. A study conducted on endometrial carcinoma, evaluating ESCRT-III subunits in terms of tumor parameters and prognosis, suggests that ESCRT-III subunits could serve as biomarkers associated with the cancer direction [101]. The pores formed by cytotoxic T lymphocytes were repaired by ESCRT components and delayed T-cell-induced cell death. Therefore, it is reported that ESCRT components create resistance against the immune response [89].
6.3 Treatment Perspectives
ESCRT complex components have recently gained increasing importance in terms of cancer and cell death mechanisms. While the role of ESCRT components in intracellular signaling has been demonstrated in many studies, it is worthy of further investigation for therapeutic purposes. In conclusion, ESCRT machineries seem to be candidates to become new generation treatment targets thanks to further research, developing technology, and new approaches.
7 Mitotic slippage
7.1 Mechanism
Antimitotic agents are used in cancer treatment by inducing cells to arrest mitosis [102]. This treatment works by targeting microtubules and prolonged activation of the spindle assembly checkpoint (SAC) [103], which controls correct and complete chromosome segregation [104]. SAC operates through the mitotic checkpoint complex (MCC) [70], which is composed of MAD2, BUB3, BUBR1 (BUB1B or MAD3), and CDC20. The MCC provides mitotic control by inhibiting the ubiquitin-mediated proteolysis activity of the anaphase-promoting complex/cyclosome (APC/C), which targets substrates such as cyclin b and securin. The activity of antimitotic agents not only causes cells to arrest in mitosis through prolonged activation of SAC, but also causes cells to escape apoptosis by resisting mitotic arrest [105]. This process of apoptotic escape is called mitotic slippage, in which cells pass through cytokinesis, leading to the emergence of tetraploid cells [106].
There is no specific mechanism representing mitotic slippage, but in general cyclin B1 degradation is prominent in this regard, even during prolonged mitotic arrest [105]. In the presence of any non-spindle-bound chromosome, MAD2 binds to CDC20, enabling MCC formation and subsequently inhibiting APC/C [107]. Conversely, when cells are arrested in mitosis for prolonged periods of time, MAD2 activity is reduced, resulting in attenuation of MCC, and consequently, mitotic shift occurs with gradual degradation of cyclin B1 [105].
CDC20 and CDH1 proteins act as a adaptor for the activation of APC/C [108]. Knockdown of CDH1 and CDC20 inhibit APC/C activity and the proteolysis of APC/C substrates cannot occur. This finding underlines that the activation of APC/C is an important mechanism for mitotic slippage [102]. While CDC20 is an activator of APC/C in anaphase transition, the role of CDH1 as an APC/C activator has a broad spectrum from late mitosis to the G1/S transition [108]. In a study, it was reported that ATP loss causes mitotic slippage during mitotic arrest and in this mechanism CDH1 is involved in cyclin B1 degradation as an APC/C activator instead of CDC20 [109].
7.2 Implications on Cancer
Besides cyclin B1 degradation, different possible processes for mitotic slippage have also been investigated. One study involves histone deacetylase inhibitors (HDACi), a class of anticancer drugs. HDACi induce mitotic slippage by triggering cells for mitotic exit, even in the case of abnormal segregation of chromosomes. In this mechanism, HDACi treatment prevents the accumulation of chromosomal passenger complex components critical for the regulation of mitosis (protein kinase Aurora B, INCENP, Survivin, and Borealin) at the centromere. This event is the main cause of HDCAi-induced mitotic slippage, as it leads to insufficient BubR1 phosphorylation and fails to maintain mitotic arrest resulting in mitotic exit [110]. Another study indicates that SWR1-chromatin remodeling complex is an important structure for SAC integrity in Saccharomyces cerevisiae. Presumably, SWR1 complex prevents mitotic slippage by inhibiting the expression of some genes that have not been fully determined. Nevertheless, mitotic slippage occurs at the end of the process following SAC activation in cells lacking SWR1-C where the SAGA (SPT-ADA-GCN5 acetyltransferase) complex is essential for mitotic slippage [111]. In different research, PP1 phosphatase was shown to be involved in mitotic slippage process in budding yeast. In this process, PP1 phosphatase accelerates mitotic slippage by making unstable MCC through dephosphorylation of MAD3 (BUBR1) Ser 268 [112].
In nocodazole-treated cells, DNA damage and P21 expression are increased, and cells are arrested in mitosis, i.e., prolonged mitotic arrest leads to DNA damage and ultimately apoptosis. On the contrary, upregulation of the apoptosis inhibitor Triap1, which is regulated by P53, reduces DNA damage and P21 expression and prevents apoptosis by inhibiting cytochrome C release from mitochondria. Here, we suggest that there may be some epigenetic mechanisms underlying the upregulation of Triap1 that promotes mitotic slippage. Accordingly, it has been put forward that Triap1 mRNA is targeted by some miRNAs and Triap expression can be increased by down-regulation of these miRNAs [113].
The fate of cells after mitotic slippage continues in different processes, and accordingly, mitotic slippage can be overcome by different methods [114]. Cells undergoing mitotic slippage can continue as polyploid cells and enter the cell cycle, which can cause genomic instability. Cells undergoing mitotic slippage can enter the process of cellular aging by stalling at the G1 checkpoint, and finally, cells can undergo apoptosis [114].
7.3 Treatment Strategies
DNA damage and p53 induction are increased in cells undergoing mitotic slippage [115]. This is the most important reason why cells go into apoptosis after mitotic slippage [114]. The emergence of cellular senescence and senescence-associated secretory phenotype (SASP) after mitotic slippage is a process linked to autophagy. Inhibition of autophagy during mitotic slippage leads to accelerated cell death bypassing the cellular aging process. These findings suggest strategies involving inhibition of autophagy in combination with antimitotic agent treatment to eliminate the undesirable consequences of cells involved in the senescence process by resisting antimitotic drug treatment with mitotic slippage [116]. One way to overcome cells undergoing mitotic slippage by resisting mitotic arrest is the inhibition of Bcl-xL antiapoptotic proteins. By inhibiting these proteins, mitotic arrest-resistant cells become susceptible to death in mitosis. In this treatment method, which is considered as an anticancer treatment approach created by the combination of antimitotic agents and Bcl-xl inhibitors, it is underlined that the use of Bcl-xL inhibitor will only give results before or immediately after mitotic slippage occurs [117]. One study indicates that following paclitaxel treatment in breast cancer, the activation of apoptosis is suppressed after increased DNA damage caused by mitotic slippage. Herein, miR-125b acts in the suppression of apoptosis after mitotic slippage by repressing the expression of genes encoding pro-apoptotic proteins. In this regard, treatment methods targeting miR-125b can be considered as a therapeutic approach [118].
Mitotic slippage, to be evaluated as a mechanism of escape from apoptosis, leads to emergence of proliferative cells that escape from therapeutic applications. Defining the mitotic slippage mechanism and targeting cells that escape apoptosis with various strategies similar to the approaches mentioned above is an important research topic for the prevention of cancer proliferation and more comprehensive studies are needed for this purpose.
8 Anastasis
8.1 Mechanism
Anastasis is a process in which cells become active and functioning by stopping or reversing cell death by removing the agent that stimulates apoptosis, which means “resurrection, to revive” in Greek [119, 120]. Tang et al. [97] first observed that human cervical cancer HeLa cells could survive after the induction of apoptosis by an apoptotic agent, ethanol. The most important criterion for induction of anastasis is the removal of apoptotic stimuli, which can be both chemical (e.g., ethanol, staurosporine, chemotherapeutic drugs) and physical (e.g., protein deprivation, cold shock) triggers. Anastasis has been detected in both in vitro cell models, such as human cervical cancer HeLa cells, glioma H4 cells, mouse primary liver cells, NIHT3T3 cells, and in vivo in Drosophila melanogaster [121, 122].
There is limited knowledge about the functional mechanisms of cell viability and recovery in anastasis. Although the exact mechanisms remain unclear, live cell imaging systems and gene expression profiling studies have provided insights into some potential strategies for anastasis mechanisms [123, 124]. On the basis of this information, it has been reported that anastasis can occur at different stages of apoptosis, such as mitochondrial membrane potential disruption (MOMP), cytochrome c release, caspase activation, DNA damage, nuclear condensation, formation of apoptotic bodies, and translocation of phosphatidylserine (PS) to the cell surface [123]. Recent gene expression profiling studies have shown that autophagy proteins (ATG12, SQSTM1), heat shock proteins (HSP27, HSP40, and HSP90), p53 inhibitor MDM2 and anti-proliferative (BTG1, CDKN1A, TRP53INP1, GADD45G) genes are up-regulated during anastasis. It was also observed that there was a change in the expression levels of genes encoding histone proteins [124]. In addition, following the formation of apoptotic bodies and removal of the apoptosis-inducing staurosporine, human cervical cancer HeLa cells were able to establish apparently normal morphology [120, 122]. How apoptotic bodies can restore normal cell function and morphology remains a question mark. Anastase studies have suggested improvement in B lymphoma cells and mouse breast cancer cells after PS exposure [120].
Anastasis may act as an unexpected physiological mechanism involved in embryonic development and homeostasis in multicellular organisms [125]. Furthermore, it may also function as a survival mechanism to protect cells that are challenging to differentiate and proliferate, such as mature neurons and cardiomyocytes. In this context, promoting anastasis could be a therapeutic strategy for the treatment of heart cell injures and brain damage, aiming to preserve and heal damaged heart cells and neurons [121, 124].
8.2 Implications on Cancer Treatment
Anastasis may be a mechanism that cancer cells can use as an escape strategy from anticancer treatment such as chemotherapy and radiation, leading to cancer recurrence [125]. In this way, cancer cells that escape therapies may develop new mutations and acquire more drug resistance. In studies, it has been observed that anastatic breast cells and human cervical cells have increased drug resistance and become more migratory. Furthermore, up-regulation of genes related to cell migration (MMP9, MMP10, MMP13) and strong angiogenic factors such as ANGPTL4 and VEGFA in anastatic cells, suggests that anastasis and cancer metastasis are associated during a relapse. Accordingly, anastasis may contribute to metastasis by promoting tumor cell survival and migration. Therefore, targeting anastasis may be a novel therapeutic strategy to prevent or halt cancer development and progression [97, 120].
During anastasis, different DNA repair systems are activated, but the cell cannot repair all DNA damage. Therefore, micronucleus formation, DNA breaks, chromosomal abnormalities, and genome instability are observed in most anastatic cells. These cells then undergo oncogenic transformation, acquiring permanent genetic changes. This supports the observation that anastasis contributes to tumor formation by allowing the recovery of genetically damaged cells, and that repeated tissue damage increases the risk of cancer [120, 122]. Tang et al. observed that germ cells with DNA damage undergo anastasis in Drosophila under environmental stress conditions such as starvation. Therefore, since anastasis may contribute to the transmission of mutations between generations, understanding its mechanisms is important in preventing the transmission of mutations [126]. Identifying cells that survive apoptosis and monitoring their fate in animals is important for developing new therapies [97, 121]. The development of the in vivo “Caspase Tracker Biosensor” has facilitated the long-term monitoring of anastasis, allowing for the investigation of its undefined functions, mechanisms, and therapeutic effects [121, 126].
EMT plays a critical role in wound healing, embryonic development, tumor progression, and metastasis. This fundamental feature of anastasis is stimulated by antiapoptotic factors. During anastasis, the up-regulated TGFβ signaling and SNAI1 expression are thought to enhance EMT and chemotherapy resistance during tumor progression. Anastasis facilitates the transformation of tumor cells into cancer stem cells, potentially leading to increased resistance to drugs and tumor recurrence [120, 127]. Xu et al. observed that CD44, a stem cell marker, was highly expressed in anastatic breast cancer cells after induction of apoptosis by STS and paclitaxel. There are some indications in the study that epigenetic regulation participates in this process. First, while CD44 was hypermethylated and CD24 was hypomethylated in control cells, CD44 was hypomethylated and CD24 was hypermethylated in anastatic cells. These data suggest that the methylation status of CD44 and CD24 is associated with the expression levels of the genes and induces the emergence of cells with cancer stem cell characteristics by causing epigenetic modifications in CD44/CD24 promoter regions [128].
In conclusion, as mentioned above, it has been proven in studies that the anastasis mechanism has both advantages and disadvantages (Table 1). There are still many issues that need to be investigated about anastasis and studies to understand its mechanism are ongoing. The development of new therapeutic strategies to understand the anastasis mechanism will offer exciting opportunities to prevent the development of diseases.
9 Blebbishield formation
9.1 Mechanism
Cancer stem cells are characterized by different markers in different cancer types. They differ in genetic heterogeneity, sphere formation, immune escape, metastasis, reactive oxygen species production, drug resistance, and tumorigenicity [103, 129,130,131,132]. Tumorigenicity is noteworthy, even if not all of these features are present in every cancer stem cell [133].
In terms of these features, apoptosis is directly suppressed in many cancer cells, whereas in cancer stem cells, this occurs through an indirect mechanism. Cancer stem cells survive through an intrinsic pathway and develop a strategy that forms the last line of defense after apoptosis [134]. This strategy is termed blebbishield formation and is described as a mechanism of escape from apoptosis in cancer stem cells. Critical for cancer stem cells, blebbishield formation is described as a fallback mechanism that occurs following the onset of apoptosis and is a mechanism by which cancer stem cells evolve to resurrect. Blebbishield formations are known as structures formed from apoptotic bodies by initiating cellular transformation of apoptotic cancer cells [135].
9.2 Implications on Cancer
In blebbishield formation, the transformation of apoptotic cancer cells occurs in two separate phases as membrane fusion and the formation of sphere-forming blebbishields. This formation occurs after the formation of apoptotic bodies in cancer stem cells, which morphologically and biochemically initiate apoptosis signaling. These bodies assemble with the nucleus-containing part of the cancer stem cells that will undergo apoptosis, forming a blebbishield structure with a spherical and elongated morphology. Blebbishield structures form stem cell spheres that are transformed by fusion and regenerate cancer stem cells from these spheres. Fusion between blebbishield spheres is also characterized by mitotic cells or immunological cells. In particular, fusion between blebbishield spheres and immunologic cells is advantageous for cancer stem cells to evade immunosuppressors [136]. In this mechanism, formation of serpentine filopodia based on endocytosis and exocytosis is required for the attachment of apoptotic bodies and fusion to occur (Fig. 2). Filopodia are structures that form actin-rich cell membrane protrusions and are associated with many cellular functions such as cell migration, wound healing, growth, and cancer [137]. Thus, each form of blebbishield can combine and transform into cancer cells [134].

Blebbishield formation process.
Blebbishield formation alone is not enough to escape from apoptosis. Secondary necrosis is observed in most cells due to ATP deficiency after blebbishield formation. Blebbishield formation and escape from secondary necrosis that are specific to cancer stem cells have not been fully elucidated in cancer stem cells [138]. Therefore, the transformation phase is thought to play a critical role in cancer recurrence.
Cells derived from blebbishield-derived spheres show tumorigenic properties. They are also associated with drug resistance and serve as markers for cancer cells. RT4, a human bladder cancer cell line, is one of the first cells in which blebbishield spheres were detected. When blebbishield formation and tumorigenicity in bladder cancer cells are evaluated, active nuclei, intact mitochondria, granular ER, and clusters of apoptotic Golgi bodies are observed under transmission electron microscopy [135]. Therefore, the blebbishield serves as an emergency escape program, particularly in the recurrence of bladder cancer, despite the apoptosis induced by chemotherapeutics during the treatment. Blebbishield structures in bladder cancer cells result in an increase in tumor size and have implications for metastasis and prognosis [139].
K-RAS, along with functional survival genes such as VEGF, p19-VHL, and also expressed VEGFR2, which is the receptor associated with VEGF, as well as downstream signaling components such as N-MYC, p70S6K, and SP1, are responsible for the regulation of cellular transformation in the blebbishield mechanism [140, 141]. Glycolysis is promoted especially through the inhibition of apoptosis via IAP proteins and ROS detoxification.
Effectively, K-RAS leads to cellular transformation [134, 140, 141]. The formation of spheres, i.e., transformation, is observed in both low and high tumorigenic cells of bladder cancer through K-RAS [140]. During the blebbishield program, to prevent apoptotic cells from undergoing secondary necrosis, K-RAS oligomerizes with BAD, BAK, BAX, and p27 to increase glycolysis. The aim here is to promote the preference of the glycolytic pathway in cancer cells [140]. K-RAS and BAD are important for cellular transformation due to their roles in both apoptosis and cellular survival [142, 143].
K-RAS plays an active role in blebbishield formation and subsequent cancer recurrence. However, K-RAS alone is not sufficient for tumorigenicity. Therefore, chromosomal instability is also associated with cellular transformation [144]. Studies have reported that during the transformation phase of blebbishield formation, p53 expression is suppressed and severe chromosomal instability is also observed [136]. However, in a different study, mutant p53 expression was shown to induce mitochondrial depolarization, inhibiting transformation and leading the cell to apoptosis [145].
As mentioned, blebbishield-mediated transformation prevents secondary necrosis and regulates glycolysis through K-RAS and related genes [141], and glycolysis metabolite lactate offers a strong survival advantage to transformed cancer stem cells using the blebbishield emergency program. At this stage, pH decreases in the cells, thereby enhancing the bioavailability of VEGF in the microenvironment. In this context, the blebbishield emergency program selects potential cells on the basis of post-apoptotic survival, increased glycolysis, and lactate production [135].
9.3 Treatment Strategies
Detection of blebbishield formation in cancer cells is of therapeutic importance. Targeting the two distinct phases of cellular transformation is reported to be more effective [146]. Recently developed SMAC mimetics at the molecular level target apoptosis resistance in cancer cells. In studies conducted in this respect, it is observed that SMAC mimetics effectively induce cell death, especially in combination with the death receptor TNF-α or other related ligands. However, despite the induction of apoptosis in cancer cells in combination with FAS-L, a subset of apoptotic cells survives apoptosis and undergoes cellular transformation. This is associated with cancer recurrence [134, 135, 141, 147, 148]. When evaluated as a chemotherapeutic, studies support the effectiveness of caspase-3 inhibitor z-DEVD-fmk and PARP inhibitors in preventing blebbishield transformation [140, 149].
Particularly, CF3DODA-Me (a synthetic derivative of triterpenoid glycyrrhetinic acid from liquorice) inhibits K-RAS, thereby preventing transformation. Additionally, in experiments conducted in soft agar, it is observed that Sp1, which leads the blebbishield program, directs the cell toward apoptosis by suppressing VEGF-mediated signaling and activating caspase-3. Sp1, as a transcription factor, regulates the expression of VEGF. In addition, caspase-3 acts as a central link to destroy multiple drivers of the blebbishield emergency program because Sp1 is known as a direct or indirect target of caspase-3 [146].
In conclusion, when evaluating blebbishield formation, it is evident that cell death is not a terminal process in cancer stem cells. Studies suggest that a mechanism unique to cancer stem cells enables the resurrection of cancer cells through transformation. Therefore, it is clear that this issue is critical and still not fully elucidated. In addition, since many genes are involved in this process, blebbishield formation is associated not only with cancer but also with many cellular functions [134, 135]. Since epigenetic mechanisms are associated with many cellular functions, including cancer, it is inevitable that blebbishield formation is also linked to epigenetic mechanisms. Changes in the balance of epigenetic regulation are known to occur in cancer cells. The dual nature of DNA-level alterations, both stimulating apoptosis and exerting epigenetic effects, is noteworthy. Therefore, exploring the common ground between epigenetic mechanisms and blebbishield formation will contribute to the development of targeted therapeutic approaches for cancer.
10 Future Perspective
The investigation into apoptosis rescue mechanisms and their role in cellular homeostasis holds significant promise for shaping the future landscape of cancer therapeutics and disease management. As our understanding of these intricate processes deepens, several future perspectives emerge, offering avenues for research, innovation, and therapeutic development in the context of precision targeting of apoptosis pathways, personalized medicine strategies, combination therapies, exploration of novel biomarkers, unraveling crosstalk between escape mechanisms, integration of systems biology approaches, and clinical translation of findings. In conclusion, the investigation into apoptosis rescue mechanisms represents a dynamic and evolving field with far-reaching implications for cancer treatment and beyond. The future holds exciting opportunities for researchers, clinicians, and pharmaceutical developers to harness this knowledge and revolutionize our approach to inducing apoptosis in pathological conditions.
11 Tweetable Abstract
Unraveling the Escape: Explore how cells defy programmed cell death (apoptosis) through mechanisms such as anoikis, mitochondrial DNA depletion, c-FLIP, ESCRT, mitotic slippage, anastasis, and blebbishield formation.
References
Chiarugi P, Giannoni E. Anoikis: a necessary death program for anchorage-dependent cells. Biochem Pharmacol. 2008;76(11):1352–64.
Grossmann J. Molecular mechanisms of “detachment-induced apoptosis—Anoikis.” Apoptosis. 2002;7:247–60.
Frisch SM, Screaton RA. Anoikis mechanisms. Curr Opin Cell Biol. 2001;13(5):555–62.
Tsai J-Y, Tsai S-H, Wu C-C. The chemopreventive isothiocyanate sulforaphane reduces anoikis resistance and anchorage-independent growth in non-small cell human lung cancer cells. Toxicol Appl Pharmacol. 2019;362:116–24.
Ye G, et al. Nuclear MYH9-induced CTNNB1 transcription, targeted by staurosporin, promotes gastric cancer cell anoikis resistance and metastasis. Theranostics. 2020;10(17):7545.
Zhang X, et al. A miR-124/ITGA3 axis contributes to colorectal cancer metastasis by regulating anoikis susceptibility. Biochem Biophys Res Commun. 2018;501(3):758–64.
Sousa B et al. P-cadherin induces anoikis-resistance of matrix-detached breast cancer cells by promoting pentose phosphate pathway and decreasing oxidative stress. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2020;1866(12): 165964.
Yu Y, et al. CircCEMIP promotes anoikis-resistance by enhancing protective autophagy in prostate cancer cells. J Exp Clin Cancer Res. 2022;41(1):1–17.
Bunek J, Kamarajan P, Kapila YL. Anoikis mediators in oral squamous cell carcinoma. Oral Dis. 2011;17(4):355–61.
Jalilzadeh N et al. Understanding and targeting anoikis in metastasis for cancer therapies. Cell Biol Int. 2022.
de Sousa Mesquita AP, et al. Acquisition of anoikis resistance promotes alterations in the Ras/ERK and PI3K/Akt signaling pathways and matrix remodeling in endothelial cells. Apoptosis. 2017;22:1116–37.
Tafolla E, et al. JNK1 and JNK2 oppositely regulate p53 in signaling linked to apoptosis triggered by an altered fibronectin matrix: JNK links FAK and p53. J Biol Chem. 2005;280(20):19992–9.
Sanchez-Ruderisch H, et al. Galectin-1 sensitizes carcinoma cells to anoikis via the fibronectin receptor α5β1-integrin. Cell Death Differ. 2011;18(5):806–16.
Paoli P, Giannoni E, and Chiarugi P. Anoikis molecular pathways and its role in cancer progression. Biochimica et Biophysica Acta (BBA)-Mol Cell Res. 2013;1833(12):3481–3498.
Adeshakin FO, et al. Mechanisms for modulating anoikis resistance in cancer and the relevance of metabolic reprogramming. Front Oncol. 2021;11: 626577.
Cai Q, Yan L, Xu Y. Anoikis resistance is a critical feature of highly aggressive ovarian cancer cells. Oncogene. 2015;34(25):3315–24.
Rytömaa M, Martins LM, Downward J. Involvement of FADD and caspase-8 signalling in detachment-induced apoptosis. Curr Biol. 1999;9(18):1043–6.
Bachelder RE, et al. The cleavage of Akt/protein kinase B by death receptor signaling is an important event in detachment-induced apoptosis. J Biol Chem. 2001;276(37):34702–7.
Yoon SJ, DeNicola GM. IL1RAP pulls a double shift in the cysteine factory. Cancer Discov. 2021;11(11):2679–81.
Hilmarsdottir B, et al. Inhibition of PTP1B disrupts cell–cell adhesion and induces anoikis in breast epithelial cells. Cell Death Dis. 2017;8(5):e2769–e2769.
Stupack DG, et al. Apoptosis of adherent cells by recruitment of caspase-8 to unligated integrins. J Cell Biol. 2001;155(3):459–70.
Varadaraj A, et al. Epigenetic regulation of GDF2 suppresses anoikis in ovarian and breast epithelia. Neoplasia. 2015;17(11):826–38.
Yu X, et al. Suppression of anoikis by the neurotrophic receptor TrkB in human ovarian cancer. Cancer Sci. 2008;99(3):543–52.
An H, et al. Salinomycin promotes anoikis and decreases the CD44+/CD24-stem-like population via inhibition of STAT3 activation in MDA-MB-231 cells. PLoS One. 2015;10(11): e0141919.
Yang L, et al. Regulation of AMPK-related glycolipid metabolism imbalances redox homeostasis and inhibits anchorage independent growth in human breast cancer cells. Redox Biol. 2018;17:180–91.
Saleem MZ et al. Brevilin A inhibits STAT3 signaling and induces ROS-dependent apoptosis, mitochondrial stress and endoplasmic reticulum stress in MCF-7 breast cancer cells. OncoTargets and Therapy, 2020;p. 435–50.
Schömel N, et al. UGCG influences glutamine metabolism of breast cancer cells. Sci Rep. 2019;9(1):15665.
Peppicelli S, et al. Anoikis resistance as a further trait of acidic-adapted melanoma cells. J Oncol. 2019. https://doi.org/10.1155/2019/8340926.
Sawyer BT, et al. Targeting fatty acid oxidation to promote anoikis and inhibit ovarian cancer progression. Mol Cancer Res. 2020;18(7):1088–98.
Terasaki M, et al. Dietary fucoxanthin induces anoikis in colorectal adenocarcinoma by suppressing integrin signaling in a murine colorectal cancer model. J Clin Med. 2019;9(1):90.
Xu R, et al. Aspirin suppresses breast cancer metastasis to lung by targeting anoikis resistance. Carcinogenesis. 2022;43(2):104–14.
Bose M, et al. Targeting tumor-associated MUC1 overcomes anoikis-resistance in pancreatic cancer. Transl Res. 2023;253:41–56.
Zhang H-F, et al. Proteomic screens for suppressors of anoikis identify IL1RAP as a promising surface target in Ewing sarcoma. Cancer Discov. 2021;11(11):2884–903.
Rötig A and Poulton J. Genetic causes of mitochondrial DNA depletion in humans. Biochimica et Biophysica Acta (BBA)-Mol Basis Dis. 2009;1792(12):1103–8.
Tseng LM, et al. Mitochondrial DNA mutations and mitochondrial DNA depletion in breast cancer. Genes Chromosom Cancer. 2006;45(7):629–38.
Petros JA, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci. 2005;102(3):719–24.
Filograna R, et al. Mitochondrial DNA copy number in human disease: the more the better? FEBS Lett. 2021;595(8):976–1002.
Guha M, et al. Mitochondrial retrograde signaling induces epithelial–mesenchymal transition and generates breast cancer stem cells. Oncogene. 2014;33(45):5238–50.
Li X, et al. MtDNA depleted PC3 cells exhibit Warburg effect and cancer stem cell features. Oncotarget. 2016;7(26):40297.
Cook LS, et al. Endometrial cancer and a family history of cancer. Gynecol Oncol. 2013;130(2):334–9.
Ferraresi R, et al. Resistance of mtDNA-depleted cells to apoptosis. Cytometry A. 2008;73(6):528–37.
Radzak A, et al. Insights regarding mitochondrial DNA copy number alterations in human cancer. Int J Mol Med. 2022;50(2):1–18.
Van Houten B, Hunter SE, Meyer JN. Mitochondrial DNA damage induced autophagy, cell death, and disease. Front Biosci (Landmark edition). 2016;21:42.
Tuppen HA et al. Mitochondrial DNA mutations and human disease. Biochimica et Biophysica Acta (BBA)-Bioenergetics. 2010;1797(2):113–28.
Rommelaere G, et al. Hypersensitivity of mtDNA-depleted cells to staurosporine-induced apoptosis: roles of Bcl-2 downregulation and cathepsin B. Am J Physiol Cell Physiol. 2011;300(5):C1090–106.
Suzuki S, et al. Constitutive activation of AKT pathway inhibits TNF-induced apoptosis in mitochondrial DNA-deficient human myelogenous leukemia ML-1a. Cancer Lett. 2008;268(1):31–7.
Chen H, et al. Mitochondrial DNA depletion causes decreased ROS production and resistance to apoptosis. Int J Mol Med. 2016;38(4):1039–46.
Aghajanova L, et al. Obstetrics and gynecology residency and fertility needs: national survey results. Reprod Sci. 2017;24(3):428–34.
Park SY, et al. Resistance of mitochondrial DNA-depleted cells against cell death: role of mitochondrial superoxide dismutase. J Biol Chem. 2004;279(9):7512–20.
Biswas G, Anandatheerthavarada H, Avadhani N. Mechanism of mitochondrial stress-induced resistance to apoptosis in mitochondrial DNA-depleted C2C12 myocytes. Cell Death Differ. 2005;12(3):266–78.
Biswas G, Guha M, Avadhani NG. Mitochondria-to-nucleus stress signaling in mammalian cells: nature of nuclear gene targets, transcription regulation, and induced resistance to apoptosis. Gene. 2005;354:132–9.
Jacques C, et al. mtDNA controls expression of the death associated protein 3. Exp Cell Res. 2006;312(6):737–45.
Dey R, Moraes CT. Lack of oxidative phosphorylation and low mitochondrial membrane potential decrease susceptibility to apoptosis and do not modulate the protective effect of Bcl-xL in osteosarcoma cells. J Biol Chem. 2000;275(10):7087–94.
Smiraglia D, et al. A novel role for mitochondria in regulating epigenetic modifications in the nucleus. Cancer Biol Ther. 2008;7(8):1182–90.
Herbert M, Turnbull D. Progress in mitochondrial replacement therapies. Nat Rev Mol Cell Biol. 2018;19(2):71–2.
Apostolova N, Victor VM. Molecular strategies for targeting antioxidants to mitochondria: therapeutic implications. Antioxid Redox Signal. 2015;22(8):686–729.
Valero T. Editorial (thematic issue: mitochondrial biogenesis: pharmacological approaches). Curr Pharm Des. 2014;20(35):5507–9.
Zhao L, et al. Mesenchymal stem cell therapy targeting mitochondrial dysfunction in acute kidney injury. J Transl Med. 2019;17:1–9.
Lippi L, et al. Impact of nutraceuticals and dietary supplements on mitochondria modifications in healthy aging: a systematic review of randomized controlled trials. Aging Clin Exp Res. 2022;34(11):2659–74.
Dhapola R, et al. Recent advances in molecular pathways and therapeutic implications targeting mitochondrial dysfunction for Alzheimer’s disease. Mol Neurobiol. 2022;59(1):535–55.
Shi P, et al. The third-generation EGFR inhibitor, osimertinib, promotes c-FLIP degradation, enhancing apoptosis including TRAIL-induced apoptosis in NSCLC cells with activating EGFR mutations. Transl Oncol. 2019;12(5):705–13.
Shirley S, Micheau O. Targeting c-FLIP in cancer. Cancer Lett. 2013;332(2):141–50.
Thome M, et al. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386(6624):517–21.
Micheau O, et al. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J Biol Chem. 2002;277(47):45162–71.
Chang DW, et al. c-FLIPL is a dual function regulator for caspase-8 activation and CD95-mediated apoptosis. EMBO J. 2002;21(14):3704–14.
Hughes MA, et al. Co-operative and hierarchical binding of c-FLIP and caspase-8: a unified model defines how c-FLIP isoforms differentially control cell fate. Mol Cell. 2016;61(6):834–49.
Chen S, et al. Celecoxib promotes c-FLIP degradation through Akt-independent inhibition of GSK3. Can Res. 2011;71(19):6270–81.
Feoktistova M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43(3):449–63.
He M, He Y. A role for c-FLIPL in the regulation of apoptosis, autophagy, and necroptosis in T lymphocytes. Cell Death Differ. 2013;20(2):188–97.
Oberst A, et al. Catalytic activity of the caspase-8–FLIPL complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363–7.
Lee YS, et al. Ferroptosis-inducing agents enhance TRAIL-induced apoptosis through upregulation of death receptor 5. J Cell Biochem. 2019;120(1):928–39.
Lee J-S, et al. FLIP-mediated autophagy regulation in cell death control. Nat Cell Biol. 2009;11(11):1355–62.
Bell BD, et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci. 2008;105(43):16677–82.
Tomaipitinca L, et al. c-FLIP regulates autophagy by interacting with Beclin-1 and influencing its stability. Cell Death Dis. 2021;12(7):686.
Galligan L, et al. Chemotherapy and TRAIL-mediated colon cancer cell death: the roles of p53, TRAIL receptors, and c-FLIP. Mol Cancer Ther. 2005;4(12):2026–36.
Safa AR, Pollok KE. Targeting the anti-apoptotic protein c-FLIP for cancer therapy. Cancers. 2011;3(2):1639–71.
Zhang T, et al. Cross-talk between TGF-beta/SMAD and integrin signaling pathways in regulating hypertrophy of mesenchymal stem cell chondrogenesis under deferral dynamic compression. Biomaterials. 2015;38:72–85.
Safa AR. Roles of c-FLIP in apoptosis, necroptosis, and autophagy. J Carcinogenesis Mutagenesis. 2013.
Seo SU, et al. NOX4-mediated ROS production induces apoptotic cell death via down-regulation of c-FLIP and Mcl-1 expression in combined treatment with thioridazine and curcumin. Redox Biol. 2017;13:608–22.
Wang W-D, et al. c-FLIP promotes drug resistance in non-small-cell lung cancer cells via upregulating FoxM1 expression. Acta Pharmacol Sin. 2022;43(11):2956–66.
Kim S, et al. KMU-191 Induces Apoptosis in human clear cell renal cell carcinoma caki cells through modulation of Bcl-xL, Mcl-1 (L), c-FLIP (L), and p53 proteins. J Cancer. 2023;14(12):2224.
Park S-J, et al. Trichostatin A sensitizes human ovarian cancer cells to TRAIL-induced apoptosis by down-regulation of c-FLIPL via inhibition of EGFR pathway. Biochem Pharmacol. 2009;77(8):1328–36.
Pathil A, et al. HDAC inhibitor treatment of hepatoma cells induces both TRAIL-independent apoptosis and restoration of sensitivity to TRAIL. Hepatology. 2006;43(3):425–34.
Scheffer LL, et al. Mechanism of Ca2+-triggered ESCRT assembly and regulation of cell membrane repair. Nat Commun. 2014;5(1):5646.
Morita E, et al. Human ESCRT and ALIX proteins interact with proteins of the midbody and function in cytokinesis. EMBO J. 2007;26(19):4215–27.
Güttinger S, Laurell E, Kutay U. Orchestrating nuclear envelope disassembly and reassembly during mitosis. Nat Rev Mol Cell Biol. 2009;10(3):178–91.
Snyder JC, et al. Functional interplay between a virus and the ESCRT machinery in archaea. Proc Natl Acad Sci. 2013;110(26):10783–7.
Yoon S, et al. MLKL, the protein that mediates necroptosis, also regulates endosomal trafficking and extracellular vesicle generation. Immunity. 2017;47(1):51–65 (e7).
Ritter AT, et al. ESCRT-mediated membrane repair protects tumor-derived cells against T cell attack. Science. 2022;376(6591):377–82.
Liu J, Kang R, Tang D. ESCRT-III-mediated membrane repair in cell death and tumor resistance. Cancer Gene Ther. 2021;28(1–2):1–4.
Babst M, et al. Endosome-associated complex, ESCRT-II, recruits transport machinery for protein sorting at the multivesicular body. Dev Cell. 2002;3(2):283–9.
Babst M, et al. Escrt-III: an endosome-associated heterooligomeric protein complex required for mvb sorting. Dev Cell. 2002;3(2):271–82.
Schöneberg J, et al. Reverse-topology membrane scission by the ESCRT proteins. Nat Rev Mol Cell Biol. 2017;18(1):5–17.
Katzmann DJ, Babst M, Emr SD. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell. 2001;106(2):145–55.
Rühl S, et al. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. 2018;362(6417):956–60.
Dai E, et al. ESCRT-III–dependent membrane repair blocks ferroptosis. Biochem Biophys Res Commun. 2020;522(2):415–21.
Tang D, et al. PAMP s and DAMP s: signal 0s that spur autophagy and immunity. Immunol Rev. 2012;249(1):158–75.
Gregor L, Stock S, Kobold S. ESCRT machinery: role of membrane repair mechanisms in escaping cell death. Signal Transduct Target Ther. 2022;7(1):238.
Sønder SL, et al. Annexin A7 is required for ESCRT III-mediated plasma membrane repair. Sci Rep. 2019;9(1):6726.
Sadler JB, et al. A cancer-associated polymorphism in ESCRT-III disrupts the abscission checkpoint and promotes genome instability. Proc Natl Acad Sci. 2018;115(38):E8900–8.
Yang Y, Wang M. Genomic analysis of the endosomal sorting required for transport complex III pathway genes as therapeutic and prognostic biomarkers for endometrial carcinoma. Transl Cancer Res. 2022;11(9):3108–27.
Lee J, et al. Substrate degradation by the anaphase promoting complex occurs during mitotic slippage. Cell Cycle. 2010;9(9):1792–801.
Maugeri-Saccà M, Vigneri P, De Maria R. Cancer stem cells and chemosensitivity. Clin Cancer Res. 2011;17(15):4942–7.
Lara-Gonzalez P, Westhorpe FG, Taylor SS. The spindle assembly checkpoint. Curr Biol. 2012;22(22):R966–80.
Lok TM, et al. Mitotic slippage is determined by p31comet and the weakening of the spindle-assembly checkpoint. Oncogene. 2020;39(13):2819–34.
Nakayama Y, Inoue T. Antiproliferative fate of the tetraploid formed after mitotic slippage and its promotion; a novel target for cancer therapy based on microtubule poisons. Molecules. 2016;21(5):663.
Yu H. Structural activation of Mad2 in the mitotic spindle checkpoint: the two-state Mad2 model versus the Mad2 template model. J Cell Biol. 2006;173(2):153–7.
Qiao X, et al. APC/C-Cdh1: from cell cycle to cellular differentiation and genomic integrity. Cell Cycle. 2010;9(19):3904–12.
Park YY, et al. ATP depletion during mitotic arrest induces mitotic slippage and APC/CCdh1-dependent cyclin B1 degradation. Exp Mol Med. 2018;50(4):1–14.
Stevens FE, et al. Histone deacetylase inhibitors induce mitotic slippage. Oncogene. 2008;27(10):1345–54.
Caydasi AK, et al. SWR1 chromatin remodeling complex prevents mitotic slippage during spindle position checkpoint arrest. Mol Biol Cell. 2023;34(2):ar11.
Ruggiero A, et al. The phosphatase PP1 promotes mitotic slippage through Mad3 dephosphorylation. Curr Biol. 2020;30(2):335–43.
Pavani M, et al. Triap1 upregulation promotes escape from mitotic-slippage-induced G1 arrest. Cell Rep. 2023;42(3): 112215.
Cheng B, Crasta K. Consequences of mitotic slippage for antimicrotubule drug therapy. Endocr Relat Cancer. 2017;24(9):T97–106.
Zhu Y, Zhou Y, Shi J. Post-slippage multinucleation renders cytotoxic variation in anti-mitotic drugs that target the microtubules or mitotic spindle. Cell Cycle. 2014;13(11):1756–64.
Jakhar R, et al. Autophagy governs protumorigenic effects of mitotic slippage–induced senescence. Mol Cancer Res. 2018;16(11):1625–40.
Suleimenov M, et al. Bcl-xL activity influences outcome of the mitotic arrest. Front Pharmacol. 2022;13: 933112.
Matuszyk J, Klopotowska D. miR-125b lowers sensitivity to apoptosis following mitotic arrest: implications for breast cancer therapy. J Cell Physiol. 2020;235(10):6335–44.
Raj AT, et al. Potential role of anastasis in cancer initiation and progression. Apoptosis. 2019;24(5–6):383–4.
Mohammed RN, et al. Anastasis: cell recovery mechanisms and potential role in cancer. Cell Commun Signal. 2022;20(1):81.
Sun G, Montell DJ. Q&A: cellular near death experiences—What is anastasis? BMC Biol. 2017;15(1):1–5.
Tang HM, Tang HL. Anastasis: recovery from the brink of cell death. R Soc Open Sci. 2018;5(9): 180442.
Tang HL, et al. In vivo CaspaseTracker biosensor system for detecting anastasis and non-apoptotic caspase activity. Sci Rep. 2015;5(1):9015.
Tang HM, et al. Molecular signature of anastasis for reversal of apoptosis. F1000Research. 2017;6:43.
Tang HM, et al. Transcriptomic study of anastasis for reversal of ethanol-induced apoptosis in mouse primary liver cells. Sci Data. 2022;9(1):418.
Tang HM, Fung MC, Tang HL. Detecting anastasis in vivo by CaspaseTracker biosensor. J Visualiz Exp. 2018;132: e54107.
Zakharov I, Savitskaya M, Onishchenko G. The problem of apoptotic processes reversibility. Biochem Mosc. 2020;85:1145–58.
Xu Y, et al. Apoptosis reversal promotes cancer stem cell-like cell formation. Neoplasia. 2018;20(3):295–303.
Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5(4):275–84.
Golebiewska A, et al. Critical appraisal of the side population assay in stem cell and cancer stem cell research. Cell Stem Cell. 2011;8(2):136–47.
Klonisch T, et al. Cancer stem cell markers in common cancers–therapeutic implications. Trends Mol Med. 2008;14(10):450–60.
Pece S, et al. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140(1):62–73.
Cho, R.W. and M.F. Clarke, Recent advances in cancer stem cells. Current Opinion in Genetics & d+Development. 2008;18(1):48–53.
Jinesh G, Kamat A. Blebbishield emergency program: an apoptotic route to cellular transformation. Cell Death Differ. 2016;23(5):757.
Jinesh G, et al. Blebbishields, the emergency program for cancer stem cells: sphere formation and tumorigenesis after apoptosis. Cell Death Differ. 2013;20(3):382–95.
Jinesh GG, Kamat AM. The blebbishield emergency program overrides chromosomal instability and phagocytosis checkpoints in cancer stem cells. Can Res. 2017;77(22):6144–56.
Mattila PK, Lappalainen P. Filopodia: molecular architecture and cellular functions. Nat Rev Mol Cell Biol. 2008;9(6):446–54.
Silvera D, Formenti SC, Schneider RJ. Translational control in cancer. Nat Rev Cancer. 2010;10(4):254–66.
Jinesh GG, Kamat AM. Blebbishields and mitotic cells exhibit robust macropinocytosis. BioFactors. 2017;43(2):181–6.
Jinesh G, et al. Mitochondrial oligomers boost glycolysis in cancer stem cells to facilitate blebbishield-mediated transformation after apoptosis. Cell Death Discovery. 2016;2(1):1–12.
Jinesh GG, Kamat AM. Endocytosis and serpentine filopodia drive blebbishield-mediated resurrection of apoptotic cancer stem cells. Cell Death Discovery. 2016;2(1):1–10.
Cox AD, Der CJ. The dark side of Ras: regulation of apoptosis. Oncogene. 2003;22(56):8999–9006.
Danial N. BAD: undertaker by night, candyman by day. Oncogene. 2008;27(1):S53–70.
Tang FH, et al. KRAS mutation coupled with p53 loss is sufficient to induce ovarian carcinosarcomas in mice. Int J Cancer. 2017;140(8):1860–9.
Jinesh GG, et al. Mutant p53s and chromosome 19 microRNA cluster overexpression regulate cancer testis antigen expression and cellular transformation in hepatocellular carcinoma. Sci Rep. 2021;11(1):12673.
Taoka R, et al. CF 3 DODA-Me induces apoptosis, degrades Sp1, and blocks the transformation phase of the blebbishield emergency program. Apoptosis. 2017;22:719–29.
Jinesh GG, Levi Willis D, Kamat AM. Bladder cancer stem cells: biological and therapeutic perspectives. Curr Stem Cell Res Ther. 2014;9(2):89–101.
Jinesh GG, et al. Novel PKC-ζ to p47phox interaction is necessary for transformation from blebbishields. Sci Rep. 2016;6(1):23965.
Milo GE, et al. Inhibition of carcinogen-induced cellular transformation of human fibroblasts by drugs that interact with the poly (ADP-ribose) polymerase system: Initial evidence for the development of transformation resistance. FEBS Lett. 1985;179(2):332–6.
Morozevich G, et al. Implication of α2β1 integrin in anoikis of MCF-7 human breast carcinoma cells. Biochem Mosc. 2015;80:97–103.
Bianchi ME, Mezzapelle R. The chemokine receptor CXCR4 in cell proliferation and tissue regeneration. Front Immunol. 2020;11: 560971.
Kochetkova M, Kumar S, McColl S. Chemokine receptors CXCR4 and CCR7 promote metastasis by preventing anoikis in cancer cells. Cell Death Differ. 2009;16(5):664–73.
Li Y, et al. VSTM2L contributes to anoikis resistance and acts as a novel biomarker for metastasis and clinical outcome in ovarian cancer. Biochem Biophys Res Commun. 2023;658:107–15.
McGriff SC, et al. Optimal endocrine evaluation and treatment of male infertility. Urol Clin. 2020;47(2):139–46.
Assidi M. Infertility in men: advances towards a comprehensive and integrative strategy for precision theranostics. Cells. 2022;11(10):1711.
Kiserud CE, et al. Gonadal function after cancer treatment in adult men. Tidsskrift for den Norske laegeforening: tidsskrift for praktisk medicin, ny raekke. 2008;128(4):461–5.
Bhasin S, Salehian B. Gonadotropin therapy of men with hypogonadotropic hypogonadism. Curr Ther Endocrinol Metab. 1997;6:349–52.
Brinton LA, Sahasrabuddhe VV and Scoccia B. Fertility drugs and the risk of breast and gynecologic cancers. In: Seminars in reproductive medicine. 2012. Thieme Medical Publishers.
Gasparri ML, et al. Biological impact of unilateral oophorectomy: does the number of ovaries really matter? Geburtshilfe Frauenheilkd. 2021;81(03):331–8.
Barry JA, Azizia MM, Hardiman PJ. Risk of endometrial, ovarian and breast cancer in women with polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod Update. 2014;20(5):748–58.
Fukazawa T, et al. Accelerated degradation of cellular FLIP protein through the ubiquitin-proteasome pathway in p53-mediated apoptosis of human cancer cells. Oncogene. 2001;20(37):5225–31.
Borrelli S, et al. p63 regulates the caspase-8-FLIP apoptotic pathway in epidermis. Cell Death Differ. 2009;16(2):253–63.
Benoit V, et al. Caspase-8-dependent HER-2 cleavage in response to tumor necrosis factor α stimulation is counteracted by nuclear factor κB through c-FLIP-L expression. Can Res. 2004;64(8):2684–91.
Kreuz S, et al. NF-κB inducers upregulate cFLIP, a cycloheximide-sensitive inhibitor of death receptor signaling. Mol Cell Biol. 2001;21(12):3964–73.
Micheau O, et al. NF-κB signals induce the expression of c-FLIP. Mol Cell Biol. 2001;21(16):5299–305.
Lin Y, et al. Involvement of c-FLIP and survivin down-regulation in flexible heteroarotinoid-induced apoptosis and enhancement of TRAIL-initiated apoptosis in lung cancer cells. Mol Cancer Ther. 2008;7(11):3556–65.
Ricci MS, et al. Direct repression of FLIP expression by c-myc is a major determinant of TRAIL sensitivity. Mol Cell Biol. 2004;24(19):8541–55.
Li W, Zhang X, Olumi AF. MG-132 sensitizes TRAIL-resistant prostate cancer cells by activating c-Fos/c-Jun heterodimers and repressing c-FLIP (L). Can Res. 2007;67(5):2247–55.
Zhang X, et al. c-Fos as a proapoptotic agent in TRAIL-induced apoptosis in prostate cancer cells. Can Res. 2007;67(19):9425–34.
Nam SY, et al. Upregulation of FLIPS by Akt, a possible inhibition mechanism of TRAIL-induced apoptosis in human gastric cancers. Cancer Sci. 2003;94(12):1066–73.
Skurk C, et al. The Akt-regulated forkhead transcription factor FOXO3a controls endothelial cell viability through modulation of the caspase-8 inhibitor FLIP. J Biol Chem. 2004;279(2):1513–25.
Hsu T-S, et al. c-FLIP is a target of the E3 ligase deltex1 in gastric cancer. Cell Death Dis. 2018;9(2):135.
Schäfer JA, et al. ESCRT machinery mediates selective microautophagy of endoplasmic reticulum in yeast. EMBO J. 2020;39(2): e102586.
Strack B, et al. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114(6):689–99.
Chittenden B, et al. Polycystic ovary syndrome and the risk of gynaecological cancer: a systematic review. Reprod Biomed Online. 2009;19(3):398–405.
Concepción-Zavaleta MJ, et al. Thyroid disfunction and female infertility. A comprehensive review. Diabet Metab Syndrome. 2023;17(11): 102876.
Jiang YT, et al. Infertility and ovarian cancer risk: Evidence from nine prospective cohort studies. Int J Cancer. 2020;147(8):2121–30.
Tang HM, et al. Molecular signature of anastasis for reversal of apoptosis. F1000Res. 2017;6:43.
Seervi M, et al. Molecular profiling of anastatic cancer cells: potential role of the nuclear export pathway. Cell Oncol (Dordr). 2019;42(5):645–61.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open access funding provided by the Scientific and Technological Research Council of Türkiye (TÜBİTAK).
Availability of Data and Materials
The datasets used and/or analyzed during the current study are available from the corresponding author on upon reasonable request.
Authors' Contributions
S.E. conceived the study and wrote abstract, introduction, and all the parts other than sections, and also reviewed the manuscript. S.A. wrote sections 4 and 5 and reviewed the manuscript. D.D. wrote sections 5 and 8 and drew Fig. 2. M.S. wrote section 3 and drew Fig. 1. S.K. wrote section 7 and Table 1. Y.K. wrote section 2. D.K. wrote section 6. N.T.H. and S.G. reviewed the manuscript. All the authors participated in performing the literature review. All authors have read and approved the final manuscript.
Conflict of Interest
The authors (S.E., S.A., D.D., S.K., M.S., Y.K., D.K., N.T.H., S.G.) declare that they have no conflict of interest.
Ethics Approval and Consent to Participate
Not applicable.
Patient Consent for Publication
Not applicable.
Code Availability
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Ergün, S., Aslan, S., Demir, D. et al. Beyond Death: Unmasking the Intricacies of Apoptosis Escape. Mol Diagn Ther 28, 403–423 (2024). https://doi.org/10.1007/s40291-024-00718-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40291-024-00718-w