
Abstract
RBX2660 (fecal microbiota, live-jslm; REBYOTA®) is an emerging option for the prevention of recurrent Clostridioides difficile infection (CDI) following standard of care (SOC) antibiotics. RBX2660 is a first-in-class, live biotherapeutic product available as a single-dose microbiota suspension for rectal administration. RBX2660 was effective in reducing recurrent CDI following SOC antibiotic therapy in the pivotal, phase 3 PUNCH CD3 trial. In a Bayesian analysis model, RBX2660 was superior to placebo in terms of treatment success, defined as the absence of CDI diarrhea within 8 weeks of study treatment. Most patients with treatment success at 8 weeks remained free of CDI recurrence at 6 months. The effectiveness of RBX2660 has also been demonstrated in the real-world setting. RBX2660 was well tolerated in the PUNCH CD3 trial, with a manageable adverse event (AE) profile. The most common AEs with RBX2660 were gastrointestinal in nature. Most AEs occurred during the first 2 weeks after treatment and were of mild or moderate severity.
Plain Language Summary
Clostridioides difficile is a type of bacteria that can cause infection of the large intestine. It often affects people who have been taking antibiotics. The main symptom of C. difficile infection (CDI) is diarrhea, which can lead to serious complications. Fecal microbiota transplantation is an investigational procedure that transfers feces from a healthy donor into the gut of a patient with CDI. RBX2660 (fecal microbiota, live-jslm; REBYOTA®) is the first FDA-approved fecal microbiota-based live biotherapeutic product that is administered rectally as a single dose to prevent recurrence of CDI after antibiotic therapy for recurrent CDI. In a phase 3 clinical trial, patients with recurrent CDI who received a single dose of RBX2660 had significantly higher rates of treatment success compared with placebo, and this response was sustained through 6 months. RBX2660 was well tolerated, and most adverse events were mild or moderate in severity. RBX2660 is an emerging option for preventing recurrence of CDI after treatment with antibiotics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Digital Features for this Adis Drug Q&A can be found at https://doi.org/10.6084/m9.figshare.24069618 |
First-in-class, fecal microbiota-based live biotherapeutic product |
Supplied as a 150 mL suspension for rectal administration |
Restores the intestinal microbiome |
Reduces recurrent CDI following SOC antibiotic therapy, with a sustained response through 6 months |
Well tolerated, with a manageable AE profile |
What is the rationale for using RBX2660 (REBYOTA®) to prevent recurrence of Clostridioides difficile infection (CDI)?
Clostridioides difficile (formerly Clostridium difficile) is an anaerobic, Gram-positive, spore-producing bacteria responsible for C. difficile infection (CDI) [1, 2]. Clinically, CDI is characterized by diarrhea, abdominal pain, nausea, and fever [1,2,3]. Severe complications of CDI can include sepsis, toxic megacolon, bowel perforation, and death [1]. The Infectious Disease Society of America and Society for Healthcare Epidemiology of America [4] and the American College of Gastroenterology [5] guidelines for the management of CDI recommend the use of antibiotics such as vancomycin, fidaxomicin, and metronidazole as initial therapy. However, the antibiotic selected to treat CDI can also be a contributing factor to the cycle of recurrence [6]. It is estimated that ≈ 20–30% of patients experience CDI recurrence within 1–2 months of the first infection, with the risk of recurrence increasing after each episode [3, 7, 8]. Therefore, new strategies to prevent CDI recurrence are needed.
Fecal microbiota transplantation (FMT), which involves the transplantation of feces from a healthy donor to a recipient, has been used to treat recurrent CDI [3, 6, 8]. FMT is currently recommended as a therapeutic option for second or subsequent CDI recurrence [4, 5]. However, the procedure is not standardized, associated data for FMT is very heterogeneous, and improper donor screening can result in the transfer of pathogens and multidrug-resistant bacteria [2, 6]. This has led to the development of standardized live biotherapeutic products that are regulated as drugs by the FDA [2]. One such product is RBX2660 (fecal microbiota, live-jslm; REBYOTA®), a first-in-class, rectally administered fecal microbiota suspension approved in the USA for the prevention of recurrence of CDI in individuals 18 years of age and older, following antibiotic treatment for recurrent CDI [9]. A summary of the US prescribing information for RBX2660 is provided in Table 1.
How does RBX2660 (REBYOTA®) work?
RBX2660 is a live fecal microbiota suspension for rectal use [9]. It is manufactured from donated human fecal matter that has been screened for transmissible pathogens. The exact mechanism by which RBX2660 exerts its efficacy in preventing recurrence of CDI is unknown [9].
Prevention of recurrent CDI with RBX2660 was associated with restoration of the intestinal microbiome, with responders’ microbiomes showing greater microbiome diversity and becoming more similar to RBX2660 [10, 11]. Administration of RBX2660 was correlated with taxonomic and functional pathway composition convergence of patients’ microbiota to the donor microbiota [11, 12]. Specifically, Bacteroidia and Clostridia increased after treatment, while Gammaproteobacteria and Bacilli decreased [10, 13]. RBX2660 also reduced the abundance of antibiotic-resistant organisms and antibiotic-resistance genes [11, 12], with fecal antibiotic resistance gene carriage decreasing in direct relationship to the degree to which donor microbiota engrafted [12]. Introduction of some strains of antibiotic-resistant organisms was observed during the transplantation process; however, most of these were Escherichia coli commonly found in healthy populations, and none were correlated with clinical infection [11]. Of note, this trial enrolled patients from December 2014 to November 2015, prior to recognition of extended-spectrum beta-lactamase (ESBL) as an important aspect of donor screening. During this time, donor stools were screened for carbapenem-resistant Enterobacteriaceae (CRE) but not ESBL, whereas all donor stools are now screened for both CRE and ESBL [11]. To date, there have been no adverse infection events due to bacterial transmission from RBX2660 in any clinical trials [11, 14].
Accumulation of primary bile acids (i.e., cholic acid and chenodeoxycholic acid) due to the disruption of intestinal microbiota may promote recurrence of CDI, while secondary bile acids (i.e., lithocholic acid and deoxycholic acid) dominate the healthy fecal bile acid profile [15]. RBX2660 altered bile acid composition by significantly reducing fecal levels of primary bile acids and concurrently increasing fecal levels of secondary bile acids [15].
Clinical response to RBX2660 in a phase 3 trial (PUNCH CD3) was associated with clonal engraftment of species into the patients’ microbiome [16] and restoration of bile acid compositions from less to more healthy [17].
What is the clinical efficacy of RBX2660 (REBYOTA®) in preventing recurrence of CDI?
RBX2660 is effective in reducing recurrent CDI following standard of care (SOC) antibiotics for recurrent CDI. The efficacy of RBX2660 was demonstrated in a pivotal, randomized, double-blind, placebo-controlled, multicentre, phase 3 trial (PUNCH CD3) [18] and supported by data from phase 2 trials (PUNCH CD2 [19], PUNCH CD [20], and PUNCH CD open-label [13]) and an open-label phase 3 study (PUNCH CD3 OLS) [21, 22].
PUNCH CD3
Patients eligible for enrolment in PUNCH CD3 were adults aged ≥ 18 years with documented recurrent CDI (defined as one or more recurrences after a primary episode) who had completed one or more courses of SOC antibiotics or had two or more episodes of severe CDI resulting in hospitalization within the previous year [18]. Within the previous 30 days, patients were required to have a positive stool test for the presence of C. difficile with the capability to produce toxins assessed by polymerase chain reaction, enzyme immunoassay, or other assays. All patients were taking antibiotics to control recurrent CDI symptoms. Exclusion criteria included a known history of refractory CDI, inflammatory bowel disease (IBD), and irritable bowel syndrome (IBS). After completing a full course of antibiotics and following a 24–72 h washout period, patients were randomized to receive a single dose of RBX2660 (microbiota suspension) or placebo (normal saline). Randomization was stratified by antibiotics used for the qualifying CDI event (vancomycin alone, vancomycin plus another antibiotic, fidaxomicin alone, or other). Both study treatments were administered rectally as per instructions for use and standard site procedures. Patients who experienced treatment failure within the first 8 weeks were eligible to receive a second treatment course with open-label RBX2660 [18].
Of the 289 patients who were randomized to treatment, 267 were treated with RBX2660 (n = 180) or placebo (n = 87) and comprised the intention-to-treat (ITT) population [18]. Baseline characteristics were generally comparable between treatment groups, although the proportion of patients aged < 65 years was higher in the placebo group than in the RBX2660 group (62 vs 51%). Patients had a median age of 63 years and a median ATLAS (age, treatment with systemic antibiotics, leukocyte count, albumin, and serum creatinine [23]) score for the qualifying CDI episode of 3.0 (scores range from 0 to 10, with higher scores correlating with a lower cure rate [23]). The most common antibiotic used to treat the qualifying CDI episode was vancomycin (88%). The primary endpoint was treatment success, defined as the absence of CDI diarrhea within 8 weeks of study treatment. The primary analysis population [i.e., the modified ITT (mITT) population] comprised all randomized patients who successfully completed treatment and did not discontinue the trial during the first 8 weeks for reasons unrelated to CDI (n = 262). The primary endpoint was analyzed using a Bayesian hierarchical model that formally integrated information about the treatment effect from the earlier phase 2b PUNCH CD2 trial. This model provides estimates of the treatment success rates for each treatment group in PUNCH CD3 as well as the estimated treatment effect and the associated posterior probability of superiority [18].
RBX2660 was effective at reducing recurrent CDI, as evidenced by a significantly higher treatment success rate with RBX2660 than with placebo in the mITT population (Table 2) [18]. Similar results were seen in the ITT and per-protocol populations (Table 2). Prespecified subgroup analyses of the primary endpoint demonstrated consistent efficacy of RBX2660 across subgroups based on sex, age (< 65 vs ≥ 65 years), race (white vs non-white), and number of previous episodes of CDI (≤ 3 vs > 3) [18]. In post hoc analyses, RBX2660 treatment success rates were consistent regardless of comorbidities [i.e., baseline Charlson Comorbidity Index score of 0–2 (mild), 3–4 (moderate), or 5+ (severe)] [24, 25] and the presence of underlying cardiac, renal, or gastrointestinal (GI) disorders [24]. Among patients with a first CDI recurrence (n = 86), 79% of RBX2660 recipients and 61% of placebo recipients achieved treatment success at week 8 [26].
The proportion of patients with treatment success at 8 weeks who remained free of CDI recurrence at 6 months was 92% with RBX2660 and 91% with placebo (mITT population) [18]. Of the 41 RBX2660 recipients with confirmed treatment failure who received a second treatment course with open-label RBX2660, 22 (54%) achieved treatment success within 8 weeks; of these, 19 (86%) had a sustained response through 6 months. Of the 24 placebo recipients with confirmed treatment failure who were subsequently treated with open-label RBX2660, 15 (63%) achieved treatment success within 8 weeks and all 15 had sustained response through 6 months [18].
RBX2660 improved health-related quality of life (HR-QoL) in patients with recurrent CDI, as assessed using the 32-item C. difficile HR-QoL (Cdiff32) instrument (total scores range from 0 to 100, with 100 being the best possible score) [27]. Cdiff32 total scores improved significantly from baseline at all time points (i.e., weeks 1, 4, and 8) in both treatment groups (all p < 0.001). At week 8, RBX2660 was associated with a significantly (p < 0.05) greater improvement in Cdiff32 mental domain score than placebo. In multivariable adjusted analyses, there were statistically significant differences between RBX2660 and placebo at week 8 for Cdiff32 total score, physical domain score, and mental domain score (all p < 0.05). Among responders (i.e., no recurrence of CDI), improvements from baseline to week 8 were statistically significant (all p < 0.001) for Cdiff32 total score and all three domain scores (i.e., physical, mental, and social), for both RBX2660 and placebo. Non-responders showed numerical improvements from baseline with RBX2600 but not placebo [27].
PUNCH CD3 OLS
Results of PUNCH CD3 were supported by those of PUNCH CD3 OLS, an ongoing, open-label, phase 3 study of RBX2660 for the reduction of recurrent CDI in patients, including those with common comorbidities [21, 22]. At the time of analysis, the study had enrolled 483 patients aged ≥ 18 years with medically documented recurrent CDI. Following treatment with SOC antibiotics, all patients received a single dose of rectally administered RBX2660. The median age of patients at baseline was 63 years. Some of the GI comorbidities included IBS (13%), ulcerative colitis (7%), Crohn’s disease (4%), and IBD unspecified (1%) [21, 22].
RBX2660 reduced CDI recurrence at 8 weeks, with a sustained clinical response through 6 months [21, 22]. Overall, 75% of patients achieved treatment success (i.e., recurrence-free) at week 8, and most patients (84%) remained recurrence-free after 6 months of follow-up [21, 22]. In ad hoc subgroup analyses, RBX2660 consistently reduced recurrent CDI in patients with underlying GI comorbidities (i.e., gastroesophageal reflux disease, IBS, diverticulitis, IBD, and unspecified colitis) [28] and in patients with immunocompromising conditions (i.e., malignant tumors, end-stage renal disease, immunodeficiency syndromes, HIV, and congenital hemoglobinopathies) [29].
In the real-world setting
Real-world experience with RBX2660 supports the efficacy results observed during the PUNCH CD3 trial [30]. Because an enforcement discretion policy was in place for FMT, RBX2660 was administered off-study via this mechanism for patients who were ineligible/unable to participate in a clinical trial or who required additional treatment beyond a clinical trial. Patient experience was then assessed in a retrospective study of 94 patients aged ≥ 18 years with recurrent CDI who received one or two doses of RBX2660. The mean age of patients was 60 years, and 45% of patients were aged ≥ 65 years. Comorbid conditions included IBS (22%), microscopic colitis (11%), Crohn’s disease (8%), and ulcerative colitis (6%). The overall rate of treatment success (i.e., absence of CDI recurrence within 8 weeks of treatment) was 83% in the primary safety set (PSS; n = 64) and 70% in the full analysis set (FAS; n = 94). Among patients who achieved treatment success at week 8, 89% of those in the PSS and 88% of those in the FAS had a sustained clinical response through 6 months. In both study populations, treatment success rates were similar in patients who received one or two doses of RBX2660 [30].
What is the tolerability of RBX2660 (REBYOTA®) in preventing recurrence of CDI?
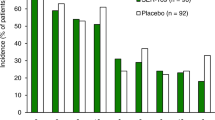
RBX2660 is well tolerated with a manageable adverse event (AE) profile, based on data from the pivotal PUNCH CD3 trial [18]. Through 6 months after blinded treatment, AEs occurred in 56% of RBX2660 recipients and 45% of placebo recipients. The most common (incidence ≥ 5%) AEs with RBX2660 were diarrhea (20% vs 19% with placebo), abdominal pain (19% vs 9%), nausea (11 vs 5%), and abdominal distension (6 vs 5%). Most AEs occurred during the first 2 weeks after treatment and were mild or moderate in severity. Serious AEs occurred in 4% of RBX2660 recipients and 2% of placebo recipients. One patient in the RBX2660 group discontinued because of an AE [18].
The safety of RBX2660 was demonstrated in an integrated analysis of data from three phase 2 trials (PUNCH CD, PUNCH CD2, and PUNCH CD open-label) and two phase 3 trials (PUNCH CD3 and PUNCH CD3 OLS) [14]. In the safety population (n = 1061), treatment-emergent AEs (TEAEs) through 6 months were reported in 507/763 (66%) patients who received RBX2660 only and 50/83 (60%) patients who received placebo only. Most TEAEs were of mild or moderate severity and were related to pre-existing conditions. The most common (incidence ≥ 5%) TEAEs associated with RBX2660 were diarrhea (21% vs 18% with placebo), abdominal pain (15 vs 8%), nausea (8 vs 4%), flatulence (7 vs 1%), abdominal distension (7 vs 4%), and urinary tract infection (6 vs 5%). Serious TEAEs occurred in 12% of RBX2660 recipients and 7% of placebo recipients, most of which were considered related to CDI and pre-existing conditions. There were no unexpected TEAEs and the incidence of potentially life-threatening TEAEs was low. TEAEs leading to death within 6 months after treatment occurred in 18 (2%) RBX2660 recipients. One death (severe CDI recurrence) was considered related to CDI and cardiovascular comorbidities and possibly related to RBX2660; however, the event was subsequently determined to not be a product-related safety concern [14].
A retrospective study has demonstrated the real-world safety of RBX2660 [30]. The primary endpoint of this study was the number of patients with TEAEs, defined as AEs occurring on or after the day of treatment with RBX2660. In the PSS, a total of 144 TEAEs were reported in 40 (63%) RBX2660 recipients. The most common (incidence ≥ 5%) TEAEs were abdominal pain (14%), diarrhea (14%), urinary tract infection (11%), CDI (8%), abdominal distension (6%), flatulence (6%), and nausea (6%). Most TEAEs were mild to moderate in severity. Severe and potentially life-threatening TEAEs (including ileus, organ failure, failure to thrive, and major depression) occurred in 8% of patients, serious TEAEs occurred in 13% of patients, and treatment-related TEAEs occurred in 17% of patients [30].
What is the current clinical position of RBX2660 (REBYOTA®) in preventing recurrence of CDI?
RBX2660, the first fecal microbiota-based live biotherapeutic, is an emerging option for the prevention of recurrent CDI following SOC antibiotic therapy. In the pivotal PUNCH CD3 trial, a single, rectally administered dose of RBX2660 effectively reduced recurrent CDI, with a sustained response through 6 months [18]. One limitation of PUNCH CD3 is the exclusion of participants with IBS and IBD [18]. However, RBX2660 also consistently reduced CDI recurrence in PUNCH CD3-OLS [21, 22] and in a retrospective real-world study [30], both of which enrolled more diverse populations, including patients with underlying GI comorbidities such as IBS and IBD. These populations are likely to be more representative of the general recurrent CDI population.
As per current treatment guidelines, FMT is recommended as a therapeutic option for patients with two or more recurrences of CDI [4, 5]. However, for patients with demographic, pharmacologic, and environmental risk factors for CDI recurrence, earlier usage (i.e., for a first recurrence) should be considered [31]. Indeed, results of a post hoc analysis of PUNCH CD3 support the use of RBX2660 in patients with a first CDI recurrence [26].
Despite its efficacy and inclusion in treatment guidelines, conventional FMT is limited by the lack of standardized manufacturing processes [14]. To ensure patient safety, RBX2660 is subject to standardized screening procedures and testing protocols in accordance with US FDA requirements [14]. As part of the manufacturing process, RBX2660 stool donors are required to undergo thorough screening and routine testing for a wide range of pathogens, including viruses, bacteria, and parasites [18]. Moreover, administration of RBX2660 is well tolerated in patients with recurrent CDI. The AE profile of RBX2660 is manageable, with most AEs occurring during the first 2 weeks after treatment and being of mild or moderate severity.
To date, no randomized clinical trials have directly compared the efficacy of RBX2660 with other approved pharmacological therapies for prevention of CDI recurrence. For example, SER-109 (fecal microbiota spores, live-brpk) is an orally administered microbiota-based therapeutic indicated to prevent the recurrence of CDI following antibiotic treatment for recurrent CDI [32]. A recent systematic review and meta-analysis (up to May 2021) comparing interventions added to antibiotic therapy found that RBX2660, bezlotoxumab/actoxumab, and bezlotoxumab were all more effective than placebo at reducing CDI recurrence, with odds ratios of 0.47 (95% CI 0.22–0.99), 0.47 (95% CI 0.37–0.60), and 0.53 (95% CI 0.42–0.68), respectively [33]. Actoxumab and SER-109 were not superior to placebo [33]. Results of such indirect comparisons should be interpreted cautiously. Head-to-head clinical trials comparing the efficacy and tolerability of RBX2660 relative to other agents would be of interest. A multicentre, single-arm, phase 3 trial (CDI-SCOPE) is currently underway to assess the safety and efficacy of RBX2660 when delivered by colonoscopy to adults with recurrent CDI [34].
As the most common healthcare-associated infection in the USA [35], CDI carries a substantial clinical and economic burden [36, 37]. Moreover, recurrent CDI can considerably increase the use of medical resources and related expenses, with direct medical costs related to recurrent CDI in the USA estimated to be $2.8 billion per year [35]. In a recent cost-effectiveness analysis using a Markov model with a lifetime horizon, RBX2660 was found to be cost effective relative to SOC from a US third-party payer perspective, with an incremental cost-effectiveness ratio of $US18,727 per quality-adjusted life-year gained [37]. In a budget impact analysis, RBX2660 was demonstrated to be cost saving from a US third-party payer perspective, with higher initial drug costs being offset by savings in direct medical costs through prevention of CDI recurrence [36].
Change history
24 November 2023
A Correction to this paper has been published: https://doi.org/10.1007/s40267-023-01035-8
References
Alshrari AS, Hudu SA, Elmigdadi F, et al. The urgent threat of Clostridioides difficile infection: a glimpse of the drugs of the future, with related patents and prospects. Biomedicines. 2023;11(426):1–20.
Gonzales-Luna AJ, Carlson TJ, Garey KW. Emerging options for the prevention and management of Clostridioides difficile infection. Drugs. 2023;83(2):105–16.
Chopra T, Hecht G, Tillotson G. Gut microbiota and microbiota-based therapies for Clostridioides difficile infection. Front Med (Lausanne). 2022;9(1093329):1–6.
Johnson S, Lavergne V, Skinner AM, et al. Clinical practice guideline by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA): 2021 focused update guidelines on management of Clostridioides difficile infection in adults. Clin Infect Dis. 2021;73(5):e1029–44.
Kelly CR, Fischer M, Allegretti JR, et al. ACG clinical guidelines: prevention, diagnosis, and treatment of Clostridioides difficile infections. Am J Gastroenterol. 2021;116(6):1124–47.
Buckley AM, Moura IB, Wilcox MH. The potential of microbiome replacement therapies for Clostridium difficile infection. Curr Opin Gastroenterol. 2022;38(1):1–6.
Bainum TB, Reveles KR, Hall RG 2nd, et al. Controversies in the prevention and treatment of Clostridioides difficile infection in adults: a narrative review. Microorganisms. 2023;11(2):1–22.
Hocking L, Ianiro G, Leong RW, et al. Faecal microbiota transplantation for recurrent C. difficile infections: challenges and improvement opportunities for clinical practice and healthcare systems. Aliment Pharmacol Ther. 2023;57(5):549–64.
Ferring Pharmaceuticals Inc. REBYOTA™ (fecal microbiota, live-jslm) suspension, for rectal use: US prescribing information. 2022. https://dailymed.nlm.nih.gov/dailymed/drugInfo.cfm?setid=7af8a7f6-a441-4dc6-a151-138a89166fbb. Accessed 1 Sep 2023.
Blount KF, Shannon WD, Deych E, et al. Restoration of bacterial microbiome composition and diversity among treatment responders in a phase 2 trial of RBX2660: an investigational microbiome restoration therapeutic. Open Forum Infect Dis. 2019;6(4):1–10.
Kwak S, Choi J, Hink T, et al. Impact of investigational microbiota therapeutic RBX2660 on the gut microbiome and resistome revealed by a placebo-controlled clinical trial. Microbiome. 2020;8(125):1–16.
Langdon A, Schwartz DJ, Bulow C, et al. Microbiota restoration reduces antibiotic-resistant bacteria gut colonization in patients with recurrent Clostridioides difficile infection from the open-label PUNCH CD study. Genome Med. 2021;13(28):1–18.
Orenstein R, Dubberke ER, Khanna S, et al. Durable reduction of Clostridioides difficile infection recurrence and microbiome restoration after treatment with RBX2660: results from an open-label phase 2 clinical trial. BMC Infect Dis. 2022;22(245):1–13.
Lee C, Louie T, Bancke L, et al. Safety of fecal microbiota, live-jslm (REBYOTA™) in individuals with recurrent Clostridioides difficile infection: data from five prospective clinical trials. Therap Adv Gastroenterol. 2023. https://doi.org/10.1177/17562848231174277.
Papazyan R, Ferdyan N, Srinivasan K, et al. Human fecal bile acid analysis after investigational microbiota-based live biotherapeutic delivery for recurrent Clostridioides difficile infection. Microorganisms. 2023;11(135):1–14.
Blount K, Claypool J, Myers PN, et al. Significant and durable microbiome compositional changes and clonal engraftment in a phase 3 trial of fecal microbiota, live-jslm for recurrent Clostridioides difficile infection [oral presentation]. In: 33rd European Congress of Clinical Microbiology & Infectious Diseases. 2023.
Papazyan R, Fuchs B, Blount K, et al. Rapid restoration of bile acid compositions after treatment with RBX2660 for recurrent Clostridioides difficile infection: results from the PUNCH CD3 phase 3 trial [abstract no 1039]. Open Forum Infect Dis. 2021;8(Suppl 1):S610.
Khanna S, Assi M, Lee C, et al. Efficacy and safety of RBX2660 in PUNCH CD3, a phase III, randomized, double-blind, placebo-controlled trial with a Bayesian primary analysis for the prevention of recurrent Clostridioides difficile infection. Drugs. 2022;82(15):1527–38.
Dubberke ER, Lee CH, Orenstein R, et al. Results from a randomized, placebo-controlled clinical trial of a RBX2660: a microbiota-based drug for the prevention of recurrent Clostridium difficile infection. Clin Infect Dis. 2018;67(8):1198–204.
Orenstein R, Dubberke E, Hardi R, et al. Safety and durability of RBX2660 (microbiota suspension) for recurrent Clostridium difficile infection: results of the PUNCH CD study. Clin Infect Dis. 2016;62(5):596–602.
Khanna S, Dubberke ER, Knapple WL, et al. An interim analysis of a phase 3, open-label study indicates efficacy and safety of RBX2660 in patients with recurrent Clostridioides difficile infection [abstract no. S132]. Am J Gastroenterol. 2022;117(Suppl 2, 10S):S96.
Khanna S, Dubberke ER, Knapple WL, et al. An ad hoc analysis of a phase 3, open-label study indicates efficacy and safety of RBX2660 in patients with recurrent Clostridioides difficile infection [oral presentation]. In: American College of Gastroenterology (ACG) Annual Scientific Meeting and Postgraduate Course. 2022.
Miller MA, Louie T, Mullane K, et al. Derivation and validation of a simple clinical bedside score (ATLAS) for Clostridium difficile infection which predicts response to therapy. BMC Infect Dis. 2013;13:148.
Tillotson G, Archbald-Pannone L, Johnson S, et al. Microbiota-based live biotherapeutic RBX2660 for the reduction of recurrent Clostridioides difficile infection in older adults with underlying comorbidities. Open Forum Infect Dis. 2023;10(1):1–9.
Tillotson GS, Ando M, Ng S, et al. Treatment success of RBX2660 in reducing recurrent Clostridioides difficile infection in patients with underlying comorbidities [abstract no. Su1600]. Gastroenterology. 2022;162(7 Suppl):S647–8.
Khanna S, Tillotson G, Ando M, et al. Efficacy and safety of RBX2660 in patients after first recurrence of Clostridioides difficile infection: results from a phase 3, randomized, placebo-controlled study [abstract no. S189 plus poster D0100]. Am J Gastroenterol. 2022;117(Suppl 2, 10S):S138.
Garey KW, Dubberke ER, Guo A, et al. Effect of fecal microbiota, live-jslm (RBL) on health-related quality of life in patients with recurrent Clostridioides difficile infection: results from the PUNCH CD3 clinical trial. Open Forum Infect Dis. 2023;10(8):ofad383.
Khanna S, Dubberke ER, Knapple WL, et al. Efficacy and safety of RBX2660 in reducing recurrent Clostridioides difficile infection in patients with underlying gastrointestinal comorbidities [abstract no. S159]. Am J Gastroenterol. 2022;117(Suppl 2, 10S):S113–4.
Dubberke E, Fischer M, Tillotson G, et al. Safety and efficacy of fecal microbiota, live-jslm in reducing recurrent Clostridioides difficile infection in immunocompromised participants [poster no. PJK0609]. In: 33rd European Congress of Clinical Microbiology & Infectious Diseases (ECCMID). 2023.
Feuerstadt P, Harvey A, Yoho DS, et al. Retrospective analysis of the safety and efficacy of fecal microbiota, live-jslm (REBYOTA™) administered under enforcement discretion to patients with Clostridioides difficile infection. Open Forum Infect Dis. 2023;10(5):ofad171.
Feuerstadt P. Where does RBX2660 fit within clinical practice? Expert Rev Anti Infect Ther. 2023;21(3):225–6.
Aimmune Therapeutics Inc. VOWST (fecal microbiota spores, live-brpk) capsules, for oral administration. 2023. https://dailymed.nlm.nih.gov/dailymed/getFile.cfm?setid=99e5a37a-930c-4641-bcdd-7013ec1c15fe&type=pdf. Accessed 1 Sep 2023.
Paschos P, Ioakim K, Malandris K, et al. Add-on interventions for the prevention of recurrent Clostridioides difficile infection: a systematic review and network meta-analysis. Anaerobe. 2021;71(102441):1–10.
US National Institutes of Health. ClinicalTrials identifier NCT05831189. 2023. https://clinicaltrials.gov. Accessed 1 Sep 2023.
Feuerstadt P, Nelson WW, Drozd EM, et al. Mortality, health care use, and costs of Clostridioides difficile infections in older adults. J Am Med Dir Assoc. 2022;23(10):1721–8.
Lodise T, Guo A, Yang M, et al. Budget impact analysis of REBYOTA™ (fecal microbiota, live-jslm [FMBL]) for preventing recurrent Clostridioides difficile infection in the US. Adv Ther. 2023;40:2801–19.
Lodise T, Guo A, Yang M, et al. Cost-effectiveness analysis of REBYOTA™ (fecal microbiota, live-jslm [FMBL]) versus standard of care for the prevention of recurrent Clostridioides difficile infection in the USA. Adv Ther. 2023;40:2784–800.
Acknowledgments
The article was reviewed by: E. J. C. Goldstein, R. M. Alden Research Laboratory, UCLA School of Medicine, Santa Monica, CA, USA; N. Mehta, Emory University School of Medicine, Atlanta, GA, USA. During the peer review process, the marketing authorization holder of RBX2660 (REBYOTA®) was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Authorship and conflict of interest
Hannah A. Blair is a salaried employee of Adis International Ltd/Springer Nature, and declares no relevant conflicts of interest. All authors contributed to this article and are responsible for its content.
Ethical approval, consent to participate, consent to publish, availability of data and material, code availability
Not applicable.
Open Access
This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
Additional information
The original article has been revised due to retrospective open choice order.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Blair, H.A. RBX2660 (REBYOTA®) in preventing recurrence of Clostridioides difficile infection: a profile of its use in the USA. Drugs Ther Perspect 39, 331–338 (2023). https://doi.org/10.1007/s40267-023-01023-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40267-023-01023-y