
Abstract
The development of new medicines for systemic lupus erythematosus (SLE) has not addressed unmet clinical need, with only three drugs receiving regulatory approval for SLE in the last 60 years, one of which was specifically licensed for lupus nephritis. In the last 20 years it has become clear that activation of type 1 interferons (IFN) is reproducibly detected in the majority of SLE patients, and the actions of IFN in the immune system and on target tissues is consistent with a pathogenic role in SLE. These findings led to considerable drug discovery activity, first with agents directly targeting IFN family cytokines, with results that were encouraging but underwhelming. In contrast, targeting the type I IFN receptor with the monoclonal antibody anifrolumab, thereby blocking all IFN family members, was effective in a phase II clinical trial. This led to a pair of phase III trials, one of which was negative and the other positive, reflecting the difficulty of obtaining outcomes from trials in this complex disease. Nonetheless, the balance of evidence resulted in approval of anifrolumab in multiple jurisdictions from 2021 onwards. Multiple approaches to targeting the type 1 IFN pathway have subsequently had positive phase II clinical trials, including antibodies targeting cells that produce IFN, and small molecules targeting the receptor kinase TYK2, required for IFN signalling. Despite multiple hurdles, it is clear that IFN targeting in SLE is here to stay. The story of IFN-targeting therapy in SLE has lessons for drug development overall in this disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The type I interferons (IFN) are a family of cytokines with a key role in innate immune responses, especially to viral infections. Activation of IFNs has emerged as a key characteristic of multiple autoimmune diseases, especially systemic lupus erythematosus (SLE). |
The production of IFN, IFN proteins themselves, and IFN signalling have each been targeted in clinical trials in SLE. A monoclonal antibody to the common type I IFN receptor, anifrolumab, has been approved for the treatment of SLE, while multiple other agents are in advanced clinical trial stages. |
The lack of success of several IFN-targeting trials, despite robust pre-clinical data, highlight the multiple challenges in SLE drug development and provide lessons to improve outcomes for SLE therapeutics in the future. |
1 Introduction
Systemic lupus erythematosus (SLE, lupus) is a complex and highly clinically heterogenous condition in which the immune system mounts an unprogrammed response to host cells and tissues. As we recently summarized [1], SLE is best considered as a clinically assigned label for a spectrum of health states that have in common autoimmune reactivity to nucleic acids in the form of anti-nuclear antibodies and clinical manifestations related to autoimmunity-driven inflammation. A key characteristic of SLE is the potential for it to manifest in virtually any organ system, but in an unpredictable sequence, such that patients are highly clinically heterogeneous. This creates challenges with measuring changes in disease activity in response to treatment in the setting of a trial. In addition, approaches such as genome-wide transcriptomics have made it clear that biological profiles of patients with SLE are also highly heterogeneous, meaning that in unstratified patients highly targeted therapies may ‘miss the mark’ in substantial subsets. Together these issues have meant that multiple clinical trials of medicines for SLE have failed. Compared with other autoimmune rheumatic diseases such as rheumatoid arthritis in which outcomes have been transformed by the use of targeted biological and small molecule therapies, patients with SLE have high morbidity, low quality of life, and reduced life expectancy.
Despite the clinical and biological heterogeneity of SLE, evidence of aberrant activation of type I interferons (IFN) is highly reproducible across diverse cohorts of patients with SLE, even those with variation in other biomarkers and in clinical profile. This suggests an important role for this family of cytokines in SLE pathogenesis but also that IFN could be a transformative therapeutic target in this disease. Clinical trials of early anti-IFN pathway therapies were encouraging, and despite a clinical development program characterized by inconsistent trial results driven by issues such as those listed above, the anti-IFN receptor monoclonal antibody anifrolumab has received regulatory approval, providing the opportunity to intervene directly on this pathway in patients with SLE. In this article we will review the journey towards approval of IFN-targeting therapies for SLE and consider how this story exemplifies the opportunities and hurdles in SLE drug development.
2 Pathophysiological Rationale for Targeting Type I Interferons (IFN) in Systemic Lupus Erythematosus (SLE)
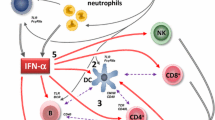
Unravelling the intricacies of SLE pathogenesis is an active and ever-evolving field of research, but understanding the role of type I IFN provides a chance to explore the complexity inherent in the disease. Diverse inflammatory sensor pathways converge on induction of IFN production and initiate multiple pathogenic outcomes, and understanding what is on either side of this bow-shaped concept (Fig. 1) is key to understanding the processes that underlie disease. Of course, type I IFNs are not the sole factors at play, and this concept extends to other effectors in lupus; we delineate a pathway for the selection of new drug targets in SLE later in this review.
The earliest stages of inflammation in SLE involve the activation of the innate immune system, triggered by cellular and nuclear debris. This response is linked to the modification and exposure of normally intracellular antigens, associated with a breakdown of immune self-tolerance to these antigens depending on an individual's genetic and epigenetic background. Clearance of exposed self-antigens to protect against immune auto-activation is facilitated in health by the complement system [2, 3], as well as DNASE1L3, an enzyme crucial for digesting chromatin from apoptotic cells [4,5,6]. Neutrophil extracellular traps (NETs), released during cell death, represent another way through which intracellular molecules are externalized [7], particularly in a subset of neutrophils known as low-density granulocytes, contributing to inflammatory responses and tissue damage [8,9,10].
The exposed intracellular proteins, either alone or bound to autoantibodies in immune complexes, are processed and presented to T cells by dendritic cells, macrophages, and other antigen-presenting cells, initiating adaptive immune responses to intracellular autoantigens resulting in the classical autoantibodies that characterize SLE. In parallel, dysfunction in the innate immune system, particularly dysregulated type I IFN responses, is crucial in SLE initiation and perpetuation. Nucleic acids recognized through endosomal Toll-like receptors (TLRs) and cytosolic DNA and RNA sensors are potent inducers of inflammatory responses. TLRs, particularly TLR7 and TLR9, and the cytosolic nucleic acid recognition system, including the cGAS/STING pathway [11,12,13], converge to stimulate type I IFN production. Type I IFNs are in fact a family of diverse proteins that signal through a common receptor complex [14]. Various cell types, including plasmacytoid dendritic cells (pDCs) [14,15,16,17,18], monocytes/macrophages, follicular dendritic cells, and keratinocytes [19,20,21,22,23], have been implicated as potential sources of excess type I IFN activity in SLE, as we reviewed elsewhere [1]. Exaggerated type I IFN responses are reproducibly observed in SLE patients, with elevated circulating IFN levels and IFN gene expression signatures associated with disease severity [24]. As well as having direct effects on immune cells and target tissues, type I IFN impedes glucocorticoid effectiveness [25,26,27], and targeting IFN therefore potentially provides a critically needed opportunity to reduce glucocorticoid use in SLE, in line with the latest treatment guidelines [28].
Ongoing investigations aim to uncover whether the different origins of innate immune overactivation correlate with the diverse clinical phenotypes within the broad SLE diagnostic category, potentially leading to distinct therapeutic approaches. Regardless, the success of targeting type I IFN in lupus serves as a prime example of how heeding patient data can steer the development of new therapeutics that offer the most advantage to the most patients. While acknowledging that this approach may not be universally effective, it still stands as the most beneficial strategy to have emerged in decades. Indeed, a suite of new drugs targeting type I IFN is now in development after a prolonged hiatus in the landscape of lupus treatment.

The ‘bow-shaped’ concept of type I interferon production and action in systemic lupus erythematosus. Multiple sources result in the availability of ligands, especially nucleic acids, for cell surface and cytoplasmic sensors that detect innate immune system activating stimuli. These converge by inducing the expression and release of type I interferons by several cell types, but especially plasmacytoid dendritic cells. The many type I interferon family proteins signal through a common receptor, that is in turn expressed on a wide range of immune and resident cells, resulting in a wide range of downstream effects. DCs dendritic cells, IFN interferon, NK natural killer, PMN polymorphonuclear leukocytes, TLR Toll-like receptor
3 Pharmacological Routes Attempted and Early Clinical Trial Results
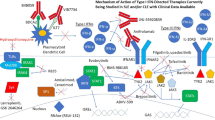
Multiple points along the IFN pathway can potentially be targeted therapeutically, including IFN production, IFN proteins themselves, and IFN signalling (Fig. 2). In the following sections we address each of these opportunities and the results of interventions designed to impact on them.

Points of intervention on the interferon (IFN) pathway in systemic lupus erythematosus (SLE). The type I IFN pathway can be addressed therapeutically at multiple levels, including (i) at the level of production through antibodies to BDCA2 found on the surface of plasmacytoid dendritic cells (pDCs), or small molecules that inhibit signalling through Toll-like receptors (TLRs) 7 and 8; (ii) at the level of IFN cytokine proteins themselves through therapeutic monoclonal antibodies or endogenous antibodies induced by vaccination, or (iii) at the level of signalling by IFN in target cells via blockade of the common IFN receptor (IFNAR) or the kinases such as TYK2 and JAK1 involved in IFNAR signalling
3.1 Targeting the Production of IFN
The encouraging but ultimately negative results of IFN-alpha targeting (see below) were followed by approaches which sought instead to address the source of IFN. In fact, debate continues as to whether the classically considered primary source of IFN-alpha, the pDC, is indeed the primary source of type I IFNs in SLE [14]. Nonetheless, targeting pDCs directly with antibodies against the cell surface marker BDCA2 has been the subject of clinical trials. The non-depleting monoclonal anti-BDCA2 antibody litifilimab, formerly known as BIIB059, was shown in a phase I study to suppress peripheral blood IFN signatures and IFN-induced proteins in cutaneous lupus lesions [18]. Two subsequent phase II trials with primary endpoints in cutaneous lupus [16] and lupus arthritis [17] were positive (Table 1), and a phase III program is underway (Table 2). An alternative approach to targeting pDCs is via CD123, the receptor for interleukin (IL)-3, a cell surface marker for pDCs and eosinophils. A depleting anti-CD123 antibody showed encouraging suppression of IFN pathway activation in ex vivo studies of human SLE patient cells stimulated with immune complexes [29, 30].
An alternative to targeting cells that produce IFN is to target mechanisms involved in IFN production, an approach potentially addressable with small molecules. As we noted earlier, there are many pathways which can entrain IFN production, but evidence including convincing genetic associations and even monogenic index cases based on TLR7 polymorphisms strongly suggest this pathway as a candidate [31]. As well as activating IFN, TLR7 signalling results in the production of multiple other pro-inflammatory cytokines, and impacts on an important subset of double-negative effector B cells that may be involved in aberrant autoantibody production in SLE [32]. Although clinical trials are yet to report, the small molecule TLR7/8 inhibitor enpatoran has been shown to have efficacy in vitro and in murine lupus models [33]; importantly, given evidence mentioned earlier that IFN in general and TLR7 in particular is involved in glucocorticoid resistance [27], enpatoran was shown to increase glucocorticoid sensitivity both in vitro and in vivo [33].
3.2 Targeting IFN Proteins Directly
Direct targeting of cytokines using monoclonal antibodies is an approach that has yielded enormous benefit in other diseases; consider the broad efficacy and safety of anti-TNF antibodies in rheumatic disease, inflammatory bowel disease, and psoriatic disease. Not surprisingly, early efforts at targeting type I IFN focused on antibodies to IFN-alpha. After a successful phase I trial which showed well-tolerated suppression of peripheral blood IFN signatures in SLE patients [34], the anti-IFN-alpha antibody sifalimumab was the subject of a phase II trial [35]. Increased herpes zoster, a not unexpected adverse event that has been observed in many subsequent IFN-targeting studies, was observed but otherwise treatment was relatively well tolerated. The trial confirmed suppression of the peripheral blood IFN signature, but was a negative trial in terms of the primary outcome of reduction in disease activity (Table 1). Analysis after adjustment for protocol violating glucocorticoid use were consistent with efficacy, a finding that was even more persuasive when analysis was restricted to the patients with a high IFN signature at baseline [35]. As we shall see, these considerations impacted on the outcome of other trials in SLE.
In contrast to these encouraging findings, a trial of another antibody to IFN-alpha, rontalizumab, was not only negative but also perplexing [36]. After a phase I study in which suppression of IFN signatures by rontalizumab was demonstrated [37], the phase II trial in SLE patients with active disease, ~ 75% of whom had baseline high IFN gene signatures, showed no difference in efficacy between rontalizumab and placebo [36] and in fact suggested superior efficacy in IFN signature negative patients, a finding that remains unexplained. It is important to consider that while gene expression signatures of IFN activation are highly reproducible overall across diverse cohorts of SLE patients [14], no common standard for measurement has been agreed upon [38], and variations in measurement and in the genes that comprise the signatures used in different studies make direct comparison challenging. It is also conceivable that the unconvincing efficacy of antibodies to IFN-alpha imply important contributions from other members of the type I IFN cytokine family.
A fascinating alternative approach to the use of antibodies to target type I IFN arose from a vaccination approach based on an ‘IFN-kinoid’ molecule which induced endogenous anti-IFN antibodies that neutralized IFN gene signatures [39]. A phase II trial of IFN kinoid was negative for the primary endpoint (Table 1), but positive for several secondary endpoints including glucocorticoid tapering and the key treat-to-target endpoint known as the lupus low disease activity state (LLDAS) [39]. Development has not been further pursued, and subsequent evidence that endogenous anti-IFN antibodies and other defects in IFN biology are associated with worse outcomes in patients with COVID-19 [40, 41] may give pause to future developments of such ‘permanent’ approaches.
3.3 Targeting IFN Signalling
3.3.1 IFNAR
All type I IFN family members signal through a common receptor complex known as IFNAR1, which in fact categorizes this cytokine family. Targeting the receptor presents the opportunity to block all type I IFNs, avoiding the potential limitations of targeting only IFN-alpha [35, 36]. Initial in vitro studies demonstrated that targeting IFNAR1 interrupted the positive feedback loop entrained by IFN-alpha as well as IFN gene signature expression induced by SLE patient serum [42]. As the clinical trials of anifrolumab to target IFNAR led to its approval, we will deal with it in detail in a subsequent section, but note here that the ability of IFNAR blockade to prevent signalling by all type I IFN cytokines, and the effect of this to shut positive pro-inflammatory feedback loops entrained by IFNs, may have contributed to the ultimately successful clinical development of anifrolumab. Side-by-side biomarker studies of blocking IFNAR versus individual IFN family cytokines would be extremely informative in this regard, but are unlikely to be done.
3.3.2 TYK2 and JAK1
Signalling through IFNAR is transduced via the receptor kinases TYK2 and JAK1, which in turn phosphorylate STAT1 and STAT2 resulting in direct effects on gene transcription through STAT binding sites in gene regulatory and promoter regions. Although not specific to the IFN pathway, the JAK1/2 inhibitor baricitinib (which is approved in several other inflammatory disease indications) showed clinical effects in a phase II trial that were encouraging [43]. These clinical results were supported by post-hoc biomarker studies showing convincing inhibition of pathways activated by IFN [44]. However, these findings did not translate to convincing results in two subsequent large phase III trials, with one trial positive and another negative for the primary outcome [45, 46] (Table 1), and with neither showing evidence of benefit in secondary outcome measures. Importantly, these trials did not include mandatory glucocorticoid tapering, an approach suggested by some of the earliest IFN targeting trial data as noted earlier [35], and the development of baricitinib in SLE has been abandoned. Another JAK inhibitor, upadacitinib, has had positive phase II trial results reported in abstract form only [47] (Table 1) but has proceeded into phase III trials (Table 2). Interestingly, in that study, upadacitinib was studied both alone and in comparison with and in combination with the Bruton’s tyrosine kinase (BTK) inhibitor evobrutinib, which appeared to have no effect either alone or in combination [47].
TYK2 transduces signalling by type I IFN, IL-12, IL-23, and IL-10, a set of signals distinct from those transduced by JAK1, JAK2, and JAK3 [48]. Polymorphisms in TKY2 have been associated with SLE and other autoimmune diseases [49]; in addition, TYK2 is not involved in growth factor signalling, suggesting the potential for a safety profile differentiated from that of JAK inhibitors. An oral small molecule TYK2 inhibitor, deucravacitinib, has been approved for the treatment of psoriasis, and a phase II clinical trial in SLE was positive with suppression of IFN gene signature accompanied by significant responses across multiple endpoints including LLDAS for the 3-mg twice-daily dose [50] (Table 1). As for litifilimab, confirmation of the efficacy in SLE of deucravacitinib in phase III is required, and while this is ongoing it is challenging as we discuss below.
4 Clinical Development of Anifrolumab
As noted above, anifrolumab is a human monoclonal antibody to IFNAR1, the receptor for all type I IFNs. After in vitro studies demonstrating comprehensive blockade of INFAR signalling, anifrolumab was first tested in phase I studies in systemic sclerosis [51]. The phase II ‘MUSE’ trial in SLE compared two doses of anifrolumab, 300 mg and 1000 mg, both administered intravenously on a monthly basis, in a study using a primary outcome measure that required glucocorticoid tapering as well as reduced disease activity [52]. This trial showed a significant difference in efficacy between anifrolumab and placebo for this outcome measure (Table 1), as well as for other secondary measures including the SLE responder index (SRI) and BILAG-based composite lupus assessment (BICLA), while post-hoc analysis also showed higher attainment of LLDAS with anifrolumab treatment [53]. These findings led to two phase III trials of anifrolumab 300 mg/month, and here ‘the plot thickens’. The first trial to report, TULIP-1, was negative, with similar numbers of patients achieving SRI response with anifrolumab and placebo [54]. In this trial, concomitant medication rules were found to have been applied to the analysis in a way that did not reflect the clinical intent; for example, over-the-counter NSAID use for an intercurrent headache could result in non-responder classification, although post-hoc analysis altering these rules did not affect the primary outcome. Although not formally analysed because the primary outcome measure was negative, odds ratios and confidence intervals for endpoints other than the primary outcome measure appeared positive, especially the BICLA endpoint which had also been positive in the phase II trial [52].
As a result, and with prior approval from regulatory agencies and prior to database lock, the primary endpoint for TULIP-2 was changed from SRI to BICLA and the revised medication rules applied. TULIP-2 was positive, with multiple secondary outcome measures also positive [55]. Despite one of the pivotal trials being formally negative, regulators have approved anifrolumab as add-on therapy for moderate to severe active SLE, presumably on the basis of the totality of evidence. Since then, numerous post-hoc analyses have been reported, showing evidence of benefit in skin, joint, and haematological domains of lupus [56], supporting a glucocorticoid-sparing effect [57], reducing flares [58], demonstrating increased attainment of LLDAS and remission [59], and demonstrating acceptable safety [60]. Importantly, increased rates of herpes zoster reactivation, first noted in the first IFN targeting studies nearly 10 years earlier, were observed in anifrolumab-treated patients. A total of 34 patients had herpes zoster-related adverse events in the two TULIP trials; two patients ceased therapy, one of whom had confirmed herpes zoster transverse myelitis. All other events were of mild or moderate severity, all responded to antiviral therapy, and patients continued therapy. Anifrolumab was also associated with increased vial respiratory infections but was otherwise well tolerated [60]. Most recently, a 3-year long-term extension study, which, unusually, was placebo controlled, showed enduring effects of anifrolumab on disease activity and glucocorticoid reduction, and acceptable safety with lower rates of herpes zoster reactivation in years 2–4 [61]. Several cases of COVID-19 were reported in anifrolumab-treated patients; the study was mostly completed in the pre-vaccination era and no cases were reported in vaccinated subjects [61].
Development of anifrolumab in lupus nephritis is at an earlier stage. A phase II trial in lupus nephritis was completed, yielding a negative result for the primary outcome in the 300-mg/month dose group but showing more encouraging results for a regimen including 900-mg doses in the first 3 months [62]. A long-term analysis of data from this study suggested that this anifrolumab regimen was associated with higher rates of both complete renal response and glucocorticoid tapering at year 2 [63]; these data provided the rationale for a phase III trial in LN which is underway (Table 2).
5 Lessons Learned
Although the journey to approval of any new medicine is long and complex, the approval of a new class of medicine for SLE after a long hiatus provides the opportunity to reflect on what can be learnt from that journey. First, we believe the efficacy of anifrolumab makes it clear that IFN is important in the pathogenesis of SLE, with a ‘Koch’s postulate’ triad of evidence including the presence of IFN in disease, induction of disease by exogenous IFN in humans [64, 65], and the benefit of targeting IFN therapeutically. Case reports have begun to emerge of impressive clinical responses to anifrolumab even in patients resistant to B-cell–targeting therapies such as rituximab or belimumab [66, 67]. Although responses to IFNAR blockade are clearly superior to placebo, attainment of validated treat-to-target states such as low disease activity [68] and remission [69] are less than optimal, with post-hoc analysis of the two phase III trials showing that only around one third of patients achieve either state [59]. That this is the case despite clear suppression of IFN activity, as evidenced in IFN gene signatures, is a reminder that pathways which are independent of IFN remain important in SLE and that alternative approaches are still needed. Nonetheless, consideration of IFN blockade with anifrolumab is recommended in the recently updated European treatment guidelines for all SLE patients other than those with mild disease responsive to antimalarials and low-dose glucocorticoids [28].
In relation to IFN gene signatures, a further question is posed but the lessons to be learnt are unclear. Each of the IFN-targeting drugs we have reviewed here apart from upadacitinib had been reported to have suppressive effects on peripheral blood IFN gene signature expression in SLE patients. Direct comparison between agents studied in different trials and responses measured using different combinations of genes is not possible, but it may be that blockade of all type I IFN signalling via targeting the common receptor is more powerfully suppressive of IFN signatures; without a head-to-head comparison it is not possible to definitively conclude that this explains the overall positive results of clinical trials of anifrolumab that led to its approval. A recent review has highlighted the many issues that need to be addressed before IFN signature testing can become routine practice [70]. In the case of anifrolumab, the difference between response to drug and placebo was greater in IFN signature ‘high’ patients than in IFN ‘low’ patients, but this was driven by higher placebo responses in IFN ‘low’ patients while responses to anifrolumab were similar [71]. In our centre, IFN signature testing is not used in clinical practice and it is not generally recommended in recently updated SLE management guidelines [28].
Thirdly, the long list of negative or conflicting phase II and III trials of SLE treatments continues to signify that drug development in this disease is challenging; witness the discordant baricitinib phase III trial results [45, 46] and the ‘narrow escape’ for anifrolumab approval. What are the factors that lead to these discordant outcomes in parallel phase III trials in essentially identical study cohorts? Biological heterogeneity among patients classified clinically has having SLE is well described; notably, the SLE classification criteria, revised in 2019, do not refer to any biological concepts [72], meaning different biologies resulting in similar clusters of clinical manifestations are currently classified together. In turn, this means that therapies that specifically target a given biology may fail to impact on many patients in a given trial, and even large cohort sizes may be insufficient to balance this heterogeneity between studies. Parallel phase III trials are often performed in non-overlapping countries or centres, and the well described variation in SLE phenotype between ancestries [73], and even between countries of different national wealth [74], may impact on trial outcomes too.
Finally, we have come to understand that alongside biological heterogeneity, negative, discordant, or difficult-to-interpret trials are likely also due to a combination of issues relating to patient selection, study design, and outcome measures. Patients need us to be better at these human-assigned aspects of testing the efficacy of drugs in SLE in clinical trials; indeed, the Lupus Foundation of America has identified better outcome measures as a critical need in improving the lives of patients with SLE [75]. We recently reviewed the many issues with current outcome measures that contribute to these challenges [76]. As one example, the use of SRI as a primary outcome measure dictates the use of the SLE disease activity index (SLEDAI) as the disease activity measure, despite SLEDAI not being designed to measure responses to therapy. Spurred by these issues, a major global effort supported by academia, industry, and patient organizations has launched a project to develop a novel primary outcome measure for SLE trials, adhering to contemporary measurement and regulatory principles [77]. In that program, a ‘clean slate’ approach is being taken, beginning with enumerating the domains of disease involvement that are important to patients and then planning novel ways to assess response to therapy in each domain, for example using continuous variables and individual patient baselines, to ultimately yield a multi-domain instrument which can characterize individual patient responses across the heterogeneity of disease.
6 Future Drug Selection in SLE
Advancements in our understanding of SLE continue to reveal the cellular and molecular mechanisms underlying the disease. Such discoveries are made by immunologists, geneticists or clinicians, but synergize when multidisciplinary teams collaborate with a singular focus. The development of type I IFN-targeting therapies showcases the effectiveness of collaboration between basic and translational researchers, clinicians, and industry partners to advance transcriptional and biological observations in humans into treatments that are now utilized in the clinic.
Innovative technologies change the nature and speed at which these advancements are made. Genomic studies unveil genetic variants that can offer insights into potentially aberrant pathways in individual patients [31], guiding the selection for therapeutic strategies in a stride towards personalized medicine for lupus. Beyond the genome, transcriptomic and proteomic studies can identify gene expression signatures, revealing biomarkers, novel pathogenic or protective biological mechanisms, and potential drug targets. SLE lacks biomarkers that are reliable indicators of disease activity, severity, and organ involvement, or could guide treatment selection, predict response, and influence novel target candidate identification. However, the identification of the type I IFN signature was a notable exception, sparking comprehensive enquiries into the contribution of IFN to SLE pathogenesis and facilitating the development of the suite of therapeutics targeting this pathway, as reviewed here.
Novel candidate identification can be expedited using emerging massively parallel techniques such as high-throughput CRISPR screening. This allows identification of key cellular pathways involved in a specific customizable readout, to generate a short list for validation and further experimental testing. Circling back to fundamental biological studies is crucial in this process, as is exploring connections between candidates and clinical phenotypes through in-depth analysis of large gene and protein expression datasets. This is particularly advantageous when clinical data are detailed, allowing for multivariate analyses to pinpoint correlations of the gene expression with disease activity. Machine learning will be especially convenient for this type of study.
As was exemplified for the type I IFN pathways, candidate targets need rigorous validation studies in vitro and in vivo using animal models or preferably ex vivo patient samples. In parallel, the feasibility of selectively modulating candidate targets can be assessed and an appropriate modality chosen, such as small molecules, monoclonal antibodies or nanobodies, gene or cell therapies. The cost, treatment method, and distribution considerations of these modalities vary substantially and dictate future market access, together with safety and efficacy. Multidisciplinary scientific teams working with industry partners from an early point to advise and share knowledge can increase the likelihood that the biology has a pathway towards translation and commercialization, speeding the pathway to market. Likewise, engagement with patient groups leads to incorporation of their perspectives and priorities, a crucial element in the drug development process. Finally, improvements in the way treatment responses are measured, biomarker-based patient selection, and close attention to critical success factors such as glucocorticoid tapering, are needed to ensure that high late-clinical trial failure rates in SLE are a thing of the past [76].
We believe this comprehensive approach, integrating diverse disciplines and perspectives from the outset, offers the most effective strategy to develop more therapies for SLE beyond anifrolumab. As we fill and progress the therapeutic pipeline, it is imperative to learn from the collaborative success exemplified by targeting IFN in lupus and expedite it, mirroring the accelerated approach witnessed in the development of therapeutics and vaccines during the COVID era. Lupus patients have waited long enough—while upholding rigorous standards for safety and efficacy, let us not delay the delivery of innovative therapies.
References
Morand EF, Fernandez-Ruiz R, Blazer A, Niewold TB. Advances in the management of systemic lupus erythematosus. BMJ. 2023;26(383):e073980.
Fernandez-Ruiz R, Belmont HM. The role of anticomplement therapy in lupus nephritis. Transl Res. 2022;245:1–17.
Botto M, Dell’ Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, et al. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat Genet. 1998;19(1):56–9.
Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. 2011;43(12):1186–8.
Hartl J, Serpas L, Wang Y, Rashidfarrokhi A, Perez OA, Sally B, et al. Autoantibody-mediated impairment of DNASE1L3 activity in sporadic systemic lupus erythematosus. J Exp Med. 2021;218(5).
Sisirak V, Sally B, D’Agati V, Martinez-Ortiz W, Özçakar ZB, David J, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell. 2016;166(1):88–101.
Gupta S, Kaplan MJ. Bite of the wolf: innate immune responses propagate autoimmunity in lupus. J Clin Invest. 2021;131(3).
Denny MF, Yalavarthi S, Zhao W, Thacker SG, Anderson M, Sandy AR, et al. A distinct subset of proinflammatory neutrophils isolated from patients with systemic lupus erythematosus induces vascular damage and synthesizes type I IFNs. J Immunol. 2010;184(6):3284–97.
Carlucci PM, Purmalek MM, Dey AK, Temesgen-Oyelakin Y, Sakhardande S, Joshi AA, et al. Neutrophil subsets and their gene signature associate with vascular inflammation and coronary atherosclerosis in lupus. JCI Insight. 2018;3(8).
Silvestre-Roig C, Braster Q, Wichapong K, Lee EY, Teulon JM, Berrebeh N, et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature. 2019;569(7755):236–40.
Skopelja-Gardner S, An J, Tai J, Tanaka L, Sun X, Hermanson P, et al. The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent.
An J, Durcan L, Karr RM, Briggs TA, Rice GI, Teal TH, et al. Expression of cyclic GMP-AMP synthase in patients with systemic lupus erythematosus. Arthritis Rheumatol. 2017;69(4):800–7.
Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis. 2018;77(10):1507–15.
Psarras A, Wittmann M, Vital EM. Emerging concepts of type I interferons in SLE pathogenesis and therapy. Nat Rev Rheumatol. 2022;18(10):575–90.
Swiecki M, Colonna M. The multifaceted biology of plasmacytoid dendritic cells. Nat Rev Immunol. 2015;15(8):471–85.
Werth VP, Furie RA, Romero-Diaz J, Navarra S, Kalunian K, Vollenhoven RFV, et al. Trial of anti-BDCA2 antibody litifilimab for cutaneous lupus erythematosus. N Engl J Med. 2022;387(4):321–31.
Furie RA, van Vollenhoven RF, Kalunian K, Navarra S, Romero-Diaz J, Werth VP, et al. Trial of anti-BDCA2 antibody litifilimab for systemic lupus erythematosus. N Engl J Med. 2022;387(10):894–904.
Furie R, Werth VP, Merola JF, Stevenson L, Reynolds TL, Naik H, et al. Monoclonal antibody targeting BDCA2 ameliorates skin lesions in systemic lupus erythematosus. J Clin Investig. 2019;129(3):1359–71.
Sontheimer C, Liggitt D, Elkon KB. Ultraviolet B irradiation causes stimulator of interferon genes-dependent production of protective type I interferon in mouse skin by recruited inflammatory monocytes. Arthritis Rheumatol. 2017;69(4):826–36.
Sarkar MK, Hile GA, Tsoi LC, Xing X, Liu J, Liang Y, et al. Photosensitivity and type I IFN responses in cutaneous lupus are driven by epidermal-derived interferon kappa. Ann Rheum Dis. 2018;77(11):1653–64.
Das A, Heesters BA, Bialas A, O’Flynn J, Rifkin IR, Ochando J, et al. Follicular dendritic cell activation by TLR ligands promotes autoreactive B cell responses. Immunity. 2017;46(1):106–19.
Caielli S, Cardenas J, de Jesus AA, Baisch J, Walters L, Blanck JP, et al. Erythroid mitochondrial retention triggers myeloid-dependent type I interferon in human SLE. Cell. 2021;184(17):4464-79.e19.
Psarras A, Alase A, Antanaviciute A, Carr IM, Md Yusof MY, Wittmann M, et al. Functionally impaired plasmacytoid dendritic cells and non-haematopoietic sources of type I interferon characterize human autoimmunity. Nat Commun. 2020;11(1):6149.
Northcott M, Jones S, Koelmeyer R, Bonin J, Vincent F, Kandane-Rathnayake R, et al. Type 1 interferon status in systemic lupus erythematosus: a longitudinal analysis. Lupus Sci Med. 2022;9(1):e000625.
Northcott M, Gearing LJ, Nim HT, Nataraja C, Hertzog P, Jones SA, et al. Glucocorticoid gene signatures in systemic lupus erythematosus and the effects of type I interferon: a cross-sectional and in-vitro study. Lancet Rheumatol. 2021;3(5):e357–70.
Dankers W, Northcott M, Bennett T, D’Cruz A, Sherlock R, Gearing LJ, et al. Type 1 interferon suppresses expression and glucocorticoid induction of glucocorticoid-induced leucine zipper (GILZ). Front Immunol. 2022;13:1034880.
Guiducci C, Gong M, Xu Z, Gill M, Chaussabel D, Meeker T, et al. TLR recognition of self nucleic acids hampers glucocorticoid activity in lupus. Nature. 2010;465(7300):937–41.
Fanouriakis A, Kostopoulou M, Andersen J, Aringer M, Arnaud L, Bae SC, et al. EULAR recommendations for the management of systemic lupus erythematosus: 2023 update. Ann Rheum Dis. 2023.
Monaghan K, Jordan J, Sato T, Cesaroni M, Benson J, Ng M, et al. Depletion of plasmacytoid dendritic cells with JNJ-56022473 minimises induction of an interferon gene signature in response to TLR9 and SLE immune complex stimulation. Lupus Sci Med. 2017;4(Suppl 1):A42-A.
Monaghan KA, Hoi A, Gamell C, Tai TY, Linggi B, Jordan J, et al. CSL362 potently and specifically depletes pDCs in vitroand ablates SLE-immune complex-induced IFN responses. iScience. 2023;26(7):107173.
Brown GJ, Canete PF, Wang H, Medhavy A, Bones J, Roco JA, et al. TLR7 gain-of-function genetic variation causes human lupus. Nature. 2022;605(7909):349–56.
Jenks SA, Cashman KS, Zumaquero E, Marigorta UM, Patel AV, Wang X, et al. Distinct effector B cells induced by unregulated toll-like receptor 7 contribute to pathogenic responses in systemic lupus erythematosus. Immunity. 2018;49(4):725-39 e6.
Deshmukh A, Pereira A, Geraci N, Tzvetkov E, Przetak M, Catalina MD, et al. Preclinical evidence for the glucocorticoid-sparing potential of a dual toll-like receptor 7/8 inhibitor in autoimmune diseases. J Pharmacol Exp Ther. 2023.
Merrill JT, Wallace DJ, Petri M, Kirou KA, Yao Y, White WI, et al. Safety profile and clinical activity of sifalimumab, a fully human anti-interferon α monoclonal antibody, in systemic lupus erythematosus: a phase I, multicentre, double-blind randomised study. Ann Rheum Dis. 2011;70(11):1905–13.
Khamashta M, Merrill JT, Werth VP, Furie R, Kalunian K, Illei GG, et al. Sifalimumab, an anti-interferon-α monoclonal antibody, in moderate to severe systemic lupus erythematosus: a randomised, double-blind, placebo-controlled study. Ann Rheum Dis. 2016;75(11):1909–16.
Kalunian KC, Merrill JT, Maciuca R, McBride JM, Townsend MJ, Wei X, et al. A Phase II study of the efficacy and safety of rontalizumab (rhuMAb interferon-α) in patients with systemic lupus erythematosus (ROSE). Ann Rheum Dis. 2015;75(1):196–202.
McBride JM, Jiang J, Abbas AR, Morimoto A, Li J, Maciuca R, et al. Safety and pharmacodynamics of rontalizumab in patients with systemic lupus erythematosus: results of a phase I, placebo-controlled, double-blind, dose-escalation study. Arthritis Rheum. 2012;64(11):3666–76.
Rodriguez-Carrio J, Burska A, Conaghan PG, Dik WA, Biesen R, Eloranta ML, et al. Association between type I interferon pathway activation and clinical outcomes in rheumatic and musculoskeletal diseases: a systematic literature review informing EULAR points to consider. RMD Open. 2023;9(1):e002864.
Houssiau FA, Thanou A, Mazur M, Ramiterre E, Gomez Mora DA, Misterska-Skora M, et al. IFN-α kinoid in systemic lupus erythematosus: results from a phase IIb, randomised, placebo-controlled study. Ann Rheumat Dis. 2019:annrheumdis-2019-216379.
Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn errors of type I IFN immunity in patients with life-threatening COVID-19. Science (New York, NY). 2020;370(6515).
Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science (New York, NY). 2020;370(6515).
Riggs JM, Hanna RN, Rajan B, Zerrouki K, Karnell JL, Sagar D, et al. Characterisation of anifrolumab, a fully human anti-interferon receptor antagonist antibody for the treatment of systemic lupus erythematosus. Lupus Sci Med. 2018;5(1):e000261-e311.
Wallace DJ, Furie RA, Tanaka Y, Kalunian KC, Mosca M, Petri MA, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2018;392(10143):222–31.
Dorner T, Tanaka Y, Petri MA, Smolen JS, Wallace DJ, Dow ER, et al. Baricitinib-associated changes in global gene expression during a 24-week phase II clinical systemic lupus erythematosus trial implicates a mechanism of action through multiple immune-related pathways. Lupus Sci Med. 2020;7(1):e000424.
Petri M, Bruce IN, Dorner T, Tanaka Y, Morand EF, Kalunian KC, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 3 trial (SLE-BRAVE-II). Lancet. 2023;401:1011–9.
Morand EF, Vital EM, Petri M, van Vollenhoven R, Wallace DJ, Mosca M, et al. Baricitinib for systemic lupus erythematosus: a double-blind, randomised, placebo-controlled, phase 3 trial (SLE-BRAVE-I). Lancet. 2023;401(10381):1001–10.
Merrill JT, Tanaka Y, D’cruz D, Vila K, Siri D, Zeng X, et al. Efficacy and safety of abbv-599 high dose (elsubrutinib 60 mg and upadacitinib 30 mg) and upadacitinib monotherapy for the treatment of systemic lupus erythematosus: a phase 2, double-blind, placebo-controlled trial. Ann Rheum Dis. 2023;82(Suppl 1):91–2.
Strobl B, Stoiber D, Sexl V, Mueller M. Tyrosine kinase 2 (TYK2) in cytokine signalling and host immunity. Front Biosci (Landmark Ed). 2011;16(9):3214–32.
Dendrou CA, Cortes A, Shipman L, Evans HG, Attfield KE, Jostins L, et al. Resolving TYK2 locus genotype-to-phenotype differences in autoimmunity. Sci Transl Med. 2016;8(363):363ra149.
Morand E, Pike M, Merrill JT, van Vollenhoven R, Werth VP, Hobar C, et al. Deucravacitinib, a tyrosine kinase 2 inhibitor, in systemic lupus erythematosus: a phase II, randomized, double-blind, placebo-controlled trial. Arthritis Rheumatol. 2023;11(75):242–52.
Guo X, Higgs BW, Bay-Jensen AC, Karsdal MA, Yao Y, Roskos LK, et al. Suppression of T cell activation and collagen accumulation by an anti-IFNAR1 mAb, anifrolumab, in adult patients with systemic sclerosis. J Invest Dermatol. 2015;135(10):2402–9.
Furie R, Khamashta M, Merrill JT, Werth VP, Kalunian K, Brohawn P, et al. Anifrolumab, an anti-interferon-alpha receptor monoclonal antibody, in moderate to severe SLE. Arthritis Rheum. 2017;69(2):376–86.
Morand EF, Trasieva T, Berglind A, Illei GG, Tummala R. Lupus Low Disease Activity State (LLDAS) attainment discriminates responders in a systemic lupus erythematosus trial: post-hocanalysis of the Phase IIb MUSE trial of anifrolumab. Ann Rheum Dis. 2018;77(5):706–13.
Furie RA, Morand EF, Bruce IN, Manzi S, Kalunian KC, Vital EM, et al. Type I interferon inhibitor anifrolumab in active systemic lupus erythematosus (TULIP-1): a randomised, controlled, phase 3 trial. Lancet Rheumatol. 2019;1(4):e208–19.
Morand EF, Furie R, Tanaka Y, Bruce IN, Richez C, Bae S-C, et al. Trial of anifrolumab in active systemic lupus erythematosus. N Engl J Med. 2020;382(3):211–21.
Morand EF, Furie RA, Bruce IN, Vital EM, Dall’Era M, Maho E, et al. Efficacy of anifrolumab across organ domains in patients with moderate-to-severe systemic lupus erythematosus: a post-hoc analysis of pooled data from the TULIP-1 and TULIP-2 trials. Lancet Rheumatol. 2022;4(4):e282–92.
Bruce IN, van Vollenhoven RF, Morand EF, Furie RA, Manzi S, White WB, et al. Sustained glucocorticoid tapering in the phase 3 trials of anifrolumab: a post-hoc analysis of the TULIP-1 and TULIP-2 trials. Rheumatology (Oxford). 2022;62(4):1526–34.
Furie R, Morand EF, Askanase AD, Vital EM, Merrill JT, Kalyani RN, et al. Anifrolumab reduces flare rates in patients with moderate to severe systemic lupus erythematosus. Lupus. 2021;30(8):1254–63.
Morand EF, Abreu G, Furie RA, Golder V, Tummala R. Lupus low disease activity state attainment in the phase 3 TULIP trials of anifrolumab in active systemic lupus erythematosus. Ann Rheum Dis. 2023;82:639–45.
Tummala R, Abreu G, Pineda L, Michaels MA, Kalyani RN, Furie RA, et al. Safety profile of anifrolumab in patients with active SLE: an integrated analysis of phase II and III trials. Lupus Sci Med. 2021;8(1).
Kalunian KC, Furie R, Morand EF, Bruce IN, Manzi S, Tanaka Y, et al. A randomized, placebo-controlled phase III extension trial of the long-term safety and tolerability of anifrolumab in active systemic lupus erythematosus. Arthritis Rheumatol. 2023;75(2):253–65.
Jayne D, Rovin B, Mysler EF, Furie RA, Houssiau FA, Trasieva T, et al. Phase II randomised trial of type I interferon inhibitor anifrolumab in patients with active lupus nephritis. Ann Rheum Dis. 2022;81(4):496–506.
Jayne D, Rovin B, Mysler E, Furie R, Houssiau F, Trasieva T, et al. Anifrolumab in lupus nephritis: results from second-year extension of a randomised phase II trial. Lupus Sci Med. 2023;10(2).
Buchanan S, Rosemergy I, Healy P. Drug-induced subacute cutaneous lupus erythematosus due to treatment with interferon beta-1a. N Z Med J. 2013;126(1379):98–101.
Sladkova V, Mares J, Lubenova B, Hlustik P, Kanovsky P. Drug-induced systemic lupus erythematosus in interferon beta-1b therapy. Neuro Endocrinol Lett. 2011;32(1):4–6.
Carter LM, Wigston Z, Laws P, Vital EM. Rapid efficacy of anifrolumab across multiple subtypes of recalcitrant cutaneous lupus erythematosus parallels changes in discrete subsets of blood transcriptomic and cellular biomarkers. Br J Dermatol. 2023;189(2):210–8.
Khan MA, Khan FH, Khan HB, Saadeh C, Davey N. Role of anifrolumab in refractory cutaneous manifestations of lupus erythematosus: a case series and literature review. Cureus. 2023;15(5): e39553.
Golder V, Kandane-Rathnayake R, Huq, PhD HTN, MD PWL, MD PSFL, et al. Lupus low disease activity state as a treatment endpoint for systemic lupus erythematosus: a prospective validation study. Lancet Rheumatol. 2019;1(2):e95–102.
van Vollenhoven RF, Bertsias G, Doria A, Isenberg D, Morand E, Petri MA, et al. 2021 DORIS definition of remission in SLE: final recommendations from an international task force. Lupus Sci Med. 2021;8(1).
Burska A, Rodriguez-Carrio J, Biesen R, Dik WA, Eloranta ML, Cavalli G, et al. Type I interferon pathway assays in studies of rheumatic and musculoskeletal diseases: a systematic literature review informing EULAR points to consider. RMD Open. 2023;9(1):e002876.
Vital EM, Merrill JT, Morand EF, Furie RA, Bruce IN, Tanaka Y, et al. Anifrolumab efficacy and safety by type I interferon gene signature and clinical subgroups in patients with SLE: post hoc analysis of pooled data from two phase III trials. Ann Rheum Dis. 2022;81(7):951–61.
Aringer M, Costenbader K, Daikh D, Brinks R, Mosca M, Ramsey-Goldman R, et al. 2019 European League Against Rheumatism/American College of Rheumatology classification criteria for systemic lupus erythematosus. Ann Rheum Dis. 2019;78(9):1151–9.
Catalina MD, Bachali P, Yeo AE, Geraci NS, Petri MA, Grammer AC, et al. Patient ancestry significantly contributes to molecular heterogeneity of systemic lupus erythematosus. JCI Insight. 2020;5(15).
Kandane-Rathnayake R, Golder V, Louthrenoo W, Chen YH, Cho J, Lateef A, et al. Association of lupus low disease activity state and remission attainment and reduced mortality in systemic lupus erythematosus: a prospective multi-national, longitudinal cohort study. Lancet Rheumatol. 2022 (in press).
Tse K, Sangodkar S, Bloch L, Arntsen K, Bae SC, Bruce IN, et al. The ALPHA Project: establishing consensus and prioritisation of global community recommendations to address major challenges in lupus diagnosis, care, treatment and research. Lupus Sci Med. 2021;8(1):e004433.
Connelly K, Golder V, Kandane-Rathnayake R, Morand EF. Clinician-reported outcome measures in lupus trials: a problem worth solving. Lancet Rheumatol. 2021;3:e595.
Connelly K, Eades LE, Koelmeyer R, Ayton D, Golder V, Kandane-Rathnayake R, et al. Towards a novel clinical outcome assessment for systemic lupus erythematosus: first outcomes of an international taskforce. Nat Rev Rheumatol. 2023;19(9):592–602.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
SJ has received research grants from EMD Serono. EM has received research grants and/or consulting fees from AstraZeneca, Biogen, Bristol Myers Squibb, EMD Serono and Neovacs.
Funding
Open Access funding enabled and organized by CAUL and its Member Institutions. EFM is funded by the National Health and Medical Research Council of Australia and the Lupus Research Alliance, US. SAJ is funded by the National Health and Medical Research Council of Australia.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
Author contributions
EFM and SAJ co-wrote the manuscript, responded to peer review, and approved the final submission.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Jones, S.A., Morand, E.F. Targeting Interferon Signalling in Systemic Lupus Erythematosus: Lessons Learned. Drugs 84, 625–635 (2024). https://doi.org/10.1007/s40265-024-02043-2
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-024-02043-2