
Abstract
Ovarian cancer is the leading cause of gynecological cancer death. Improved understanding of the biologic pathways and introduction of poly (ADP-ribose) polymerase inhibitors (PARPi) during the last decade have changed the treatment landscape. This has improved outcomes, but unfortunately half the women with ovarian cancer still succumb to the disease within 5 years of diagnosis. Pathways of resistance to PARPi and chemotherapy have been studied extensively, but there is an unmet need to overcome treatment failure and improve outcome. Major mechanisms of PARPi resistance include restoration of homologous recombination repair activity, alteration of PARP function, stabilization of the replication fork, drug efflux, and activation of alternate pathways. These resistant mechanisms can be targeted to sensitize the resistant ovarian cancer cells either by rechallenging with PARPi, overcoming resistance mechanism or bypassing resistance pathways. Augmenting the PARPi activity by combining it with other targets in the DNA damage response pathway, antiangiogenic agents and immune checkpoint inhibitors can potentially overcome the resistance mechanisms. Methods to bypass resistance include targeting non-cross-resistant pathways acting independent of homologous recombination repair (HRR), modulating tumour microenvironment, and enhancing drug delivery systems such as antibody drug conjugates. In this review, we will discuss the first-line management of ovarian cancer, resistance mechanisms and potential strategies to overcome these.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Poly (ADP-ribose) polymerase inhibitors (PARPi) are efficacious in ovarian cancer, but development of resistance has emerged as a critical challenge in improving treatment success. |
Key mechanisms of resistance to PARPi are restoration of homologous recombination repair function, stabilization of the replication fork, changes in PARP function, and activation of alternate pathways. |
Potential strategies to circumvent resistance include boosting the PARPi activity by combining with other drugs or using drugs acting on non-cross-resistant pathways. |
1 Introduction
Ovarian cancer (OC) is the eighth most common malignancy globally, accounting for 313,959 cases and 207,252 deaths annually. Although among gynecologic cancers its incidence is third after cervix and uterine, it is responsible for more deaths [1, 2]. The 5-years cause-specific survival ranges from 20% in stage IV and 40% in stage III to 70% in stage II and 90% in stage I [3]. Although >80% of cases are sporadic and without known hereditary predisposition, about 15–20% of patients have germline mutations. Half of the patients have alterations in genes involved in homologous recombination repair (HRR) pathways, an important DNA damage response (DDR) pathway [4, 5].
Poly (ADP-ribose) polymerase inhibitors (PARPi) have been extensively studied in OC and have demonstrated broad activity in both first-line and recurrent settings. In first-line and platinum-sensitive recurrent OC (PSOC), PARPi have shown efficacy in maintenance (as a single agent and with bevacizumab) as well as in combination with chemotherapy [6,7,8,9,10,11,12,13]. However, due to overlapping toxicities in combination with chemotherapy requiring dose reductions, PARPi are recommended as maintenance therapy only. PARPi have also been evaluated as treatment in recurrent OC with BRCA mutations but this is not the preferred treatment option as recent reports demonstrated a lack of overall survival (OS) benefit [14].
2 DNA Damage Repair (DDR) Pathways
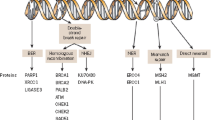
DNA damage occurs constantly in response to internal and environmental factors, and evolution has allowed for robust and overlapping repair mechanisms, to maintain DNA integrity. DNA damage due to various endogenous and exogenous influences is sensed by the cells, and repair pathways are activated based on the type of damage [15, 16]. Mismatch repair (MMR) pathways are activated in response to replication errors, bulky adducts (pyrimidine dimers produced by ultraviolet [UV] exposure) are repaired by nucleotide excision repair (NER), and single-strand DNA breaks (SSB) by base excision repair (BER). Poly (ADP-ribose) polymerase (PARP) is an enzyme involved in the repair of SSBs in the BER pathway (Fig. 1) [17]. Double-stranded DNA breaks (DSB) are produced if cells are exposed to ionizing radiations or chemotherapy drugs, and may lead to genomic instability. These are usually repaired by either the homologous recombination repair (HRR) pathway, which is a high-fidelity, error-free system as it uses sister chromatids as a template, or by the non-homologous end-joining (NHEJ) pathway, which is more efficient but prone to errors (Fig. 1). Impairment in DSB repair due to germline or somatic alterations or epigenetic silencing of genes involved in the HRR pathway is referred to as homologous recombination deficiency (HRD). These pathways are interconnected and work in concert along with cell cycle checkpoints to provide overlapping redundant repair mechanisms to ensure effective repair pathways in case one fails to repair the DNA damage. However, this also allows for an inherent vulnerability as alteration in these pathways by genetic, epigenetic, or other mechanisms can drive malignant transformation in the cells, as in the case of BRCA mutations [18,19,20,21,22]. This simultaneously presents a mechanism of malignant transformation as well as the potential to harness this inherent vulnerability therapeutically by exploiting synthetic lethality in malignant cells.

Mechanisms of DDR. SSB and DSB are introduced in genome in response to external or internal stimuli. A BER is the major pathway for the repair of SSB. It involves recognition of SSB and the recruitment of PARP enzymes at the DNA damage site. PARylation is initiated by the transfer of ADP-ribose residues from NAD+ to PARP, relaxes chromatin. DNA polymerase and DNA ligase III are recruited to repair DNA damage. PARP inhibitors promote ‘PARP trapping’ by blocking PARylation and preventing dissociation of PARP from the SSB site. Unrepaired SSB is converted to DSB. b HRR is the predominant pathway for DSB repair in normal cells. The DSB are sensed by the MRN complex, which activate HRR by recruiting various effector proteins such as ATM, BRCA1, BRCA2, RAD51, and FANCD. It is a high-fidelity error-free system as it uses sister chromatids as a template. NHEJ is a secondary pathway that is less active in normal cells and is error prone. c In the HRD state, NHEJ becomes the more predominant pathway, and DNA damage is accumulated during the repair process, leading to genomic instability. BER base excision repair, DDR DNA damage repair, DSB double-stranded breaks, HRD homologous recombination deficiency, HRR homologous recombination repair, MRN Mre11-Rad50-Nbs1, NAD+ nicotinamide adenine dinucleotide, NHEJ non-homologous end joining, PARP poly (ADP-ribose) polymerase, PARylation poly (ADP-ribosyl)ation, SSB single-stranded breaks. Created with Biorender.com
2.1 Biomarkers of Homologous Recombination Deficiency
HRD is widely used as a prognostic and predictive biomarker in the management of OC. Currently, validated assays such as myChoice HRD and FoundationOne CDx measure genomic scars using next-generation sequencing (NGS) and provide a threshold to quantify HRD. These tests have demonstrated good predictive value for PARPi activity in platinum-sensitive OC. However, their application is limited in the relapse setting as they may not be precise in estimating HRD status in platinum resistance [23,24,25].
HRD status is dynamic and can be influenced by prior therapy and the acquisition of resistance mechanisms. Genomic scars are permanent and tend to persist even when HRR is restored. Therefore, functional assays that can reflect the dynamic HRD status by estimating RAD51 loading or BRCA1 promoter methylation are being developed [26]. RAD51 loading on single-stranded DNA is mediated by BRCA2 and is a crucial step in HRR. This is lost if there is any alteration in the HRR pathway upstream of RAD51. Initial assays, such as the REcombination CAPacity (RECAP) test, induce ex vivo DNA damage in fresh tumour tissue by ionizing radiation. HRR proficiency is assessed by the cell’s ability to repair the damage by staining with RAD51, and RAD51 foci per cell are quantified by fluorescence microscopy. However, the prerequisite of fresh tumour tissue and ex vivo irradiation makes its clinical use difficult [27].
To overcome these challenges, an assay utilizing γH2Ax immunostaining to estimate endogenous DNA damage in formalin-fixed paraffin-embedded (FFPE) tissue has been developed. This can be performed on treatment-naive as well as post-chemotherapy specimens with low tumour content, thereby providing dynamic HRD monitoring. Although the assay predicted response to platinum therapy, it lacks prospective validation and predictive value for PARPi therapy [28].
Another test that can detect dynamic HRD is the BRCA1 promoter methylation assay. It can be performed on FFPE tissue by digital droplet polymerase chain reaction (ddPCR). Methylation status is altered by treatment and loss of BRCA1 methylation has been shown to restore its activity. However, its utility is limited as BRCA1 methylation is seen in only 15% of patients with OC [29]. These functional tests are promising and can extend the predictive utility of genomic scar assays, but require validation in randomized clinical trials.
3 Current Management in Ovarian Cancer
3.1 Surgery and Adjuvant Chemotherapy
Initial management of OC consists of primary debulking surgery (PDS) followed by adjuvant systemic therapy [30]. The goal of surgery is to remove the tumour completely to ideally no residual disease (R0 resection), as this is the most important factor associated with long-term survival [31, 32]. Neoadjuvant chemotherapy (NACT) followed by interval debulking surgery (IDS) is recommended in cases where optimal cytoreduction is not achievable or the patient is deemed unfit due to medical reasons [33,34,35]. Adjuvant chemotherapy is indicated in all patients with advanced OC or early OC with stage IC or II disease, and high-grade histology [36]. Six cycles of carboplatin (area under curve [AUC] 5–6) and paclitaxel (175 mg/m2) administered intravenously every 3 weeks is the preferred regimen [37]. Results from the Gynecologic Oncology Group (GOG) 172 trial favoured adjuvant intraperitoneal chemotherapy in patients with <1 cm residual disease post PDS-based, but more recent data from the GOG 252 trial do not support this approach [38, 39].
3.2 Antiangiogenic Agents
Bevacizumab is a humanized monoclonal antibody that prevents binding of vascular endothelial growth factor (VEGF) to the endothelial receptors and inhibits tumour angiogenesis [40]. It is the first targeted therapy to demonstrate improvement in survival in advanced OC. The role of bevacizumab as maintenance therapy in advanced OC was evaluated in two large, randomized trials. In ICON 7, patients with OC post PDS showed progression-free survival (PFS) improvement with maintenance bevacizumab (19.0 months vs. 17.3 months; hazard ratio [HR] 0.81, 95% confidence interval [CI] 0.7–0.9). High-risk patients (stage III with residual disease >1 cm, and inoperable stage III and IV disease) showed significant improvement in both median PFS (15.9 vs. 10.5 months; HR 0.68) and median OS (36.6 vs. 28.8 months; HR 0.64) with bevacizumab [41, 42]. In GOG-0218, patients with newly diagnosed incompletely resected stage III or IV OC showed improved PFS (HR 0.7, 95% CI 0.62–0.82; p < 0.001) with maintenance bevacizumab. A post hoc analysis showed improved PFS as well as OS in patients who had ascites at onset [43, 44]. Intrinsic tumour chemosensitivity has been suggested as a marker to better define the population who benefit from bevacizumab. It is measured by cancer antigen (CA)-125 ELIMination rate constant K (KELIM) score, calculated using CA-125 kinetics during the initial 100 days of treatment. Chemosensitive tumours have a KELIM score ≥1.0, while chemoresistance tumours have a KELIM score <1.0 [45]. A retrospective analysis of the ICON-7 trial showed that high-risk patients with a KELIM score <1 had better OS (median OS 29.7 vs. 20.6 months; HR 0.78, 95% CI 0.58–1.04: p = 0.09) [46]. Findings were confirmed in the GOG-0218 validation study in which patients with a KELIM score <1 showed improved median PFS (9.8 vs. 6.1 months; HR 0.70, 95% CI 0.59–0.82) and OS (36.3 vs. 31.8 months; HR 0.87, 95% CI 0.73–1.03). The highest benefit was observed in high-risk patients with a KELIM score of <1 [47].
3.3 Poly (ADP-Ribose) Polymerase Inhibitors (PARPi)
3.3.1 Mechanism of PARPi
PARP 1 and 2 are critical components for the repair of SSBs in the cell [48]. These recognize SSBs, bind at the site of DNA damage, and initiate poly (ADP-ribosyl)ation (PARylation), which involves the transfer of ADP-ribose or poly ADP-ribosyl (PAR) residues from nicotinamide adenine dinucleotide (NAD+) and PARP proteins to itself or other proteins [49]. PARylation causes relaxation of the chromatin and the PARP proteins are detached from the site. Various effector proteins such as DNA polymerase-β and DNA ligase III are recruited and damaged DNA is repaired [50].
PARPi act by multiple mechanisms. The most important among these is ‘PARP trapping’. PARPi block PARylation and prevent the dissociation of PARP from the chromatin, leading to trapping of PARP at the sites of SSBs and preventing its repair. Furthermore, PARPi directly inhibit the activity of PARP by competitively binding to the NAD+ site and blocking the DNA repair [51]. Synthetic lethality between PARPi and defective HRR occurs as inhibition of PARP activity causes collapse of the replication fork (RF) and the consequent generation of DSBs. Repair of DSBs by error prone NHEJ results in accumulation of DNA damage, chromosomal instability, cell cycle arrest and cell death by apoptosis [52, 53].
Mechanism of PARPi activity in HRD-negative OC is not well understood. PARPi-induced immune activation, ribosome biogenesis, and regulation of gene transcription have been suggested alternative mechanisms in such cases [54,55,56].
3.3.2 PARPi Maintenance
Currently, three PARPi (olaparib, niraparib and rucaparib) are available as maintenance therapy after initial platinum-based chemotherapy in newly diagnosed OC. Major trials of PARPi maintenance in first line are summarized in Table 1. In the SOLO1/GOG 3004 trial, patients with advanced OC (CR/PR post platinum-based chemotherapy) with germline or somatic BRCA mutation were randomized to olaparib 300 mg twice daily or placebo up to 2 years. Initial results showed significant improvement in PFS (56.0 vs. 13.8 months; HR 0.33, 95% CI 0.25–0.43; p < 0.001) [57, 58]. A recent update after a follow-up period of 7 years showed significant OS benefit, with 67.0% of patients taking olaparib still alive compared with 46.5% taking placebo (HR 0.55, 95% CI 0.40–0.76; p = 0.0004) [59]. In the PRIMA trial, patients with advanced high-grade serous or endometrioid OC post PR/CR to platinum-based chemotherapy were randomized to niraparib 300 mg once daily (200 mg once daily if body weight <77 kg, platelet count <150,000/mm3, or both) and placebo for up to 3 years. That trial excluded patients who had no visible residual disease after PDS. At a median follow-up of 13.8 months, there was significant improvement in the median PFS in the overall population (13.8 months vs. 8.2 months; HR 0.62, 95% CI 0.50–0.76), HRD-positive (21.9 months vs. 10.4 months; HR 0.43, 95% CI 0.31–0.59) and HRD-negative (8.1 months vs. 5.4 months; HR 0.68, 95% CI 0.49–0.94) subgroups. OS data are not mature yet but there is a trend towards improved OS in all subgroups [7]. In the ATHENA-MONO trial, patients with advanced high-grade OC post PR/CR to platinum-based chemotherapy were randomized to rucaparib 600 mg twice daily for up to 2 years, or placebo. At a median follow-up of 24 months, the median PFS was improved with rucaparib in the overall population (20.2 months vs. 9.2 months; HR 0.52, 95% CI 0.40–0.68) and the HRD-positive (28.7 months vs. 11.3 months; HR 0.47, 95% CI 0.31–0.72), and HRD-negative subgroups (12.1 months vs. 9.1 months; HR 0.65, 95% CI 0.45–0.95) [60].
The PAOLA-1 study was conducted to explore the role of adding olaparib maintenance in the context of standard therapy with chemotherapy plus bevacizumab in advanced OC. In this trial, patients with stage III/IV high-grade serous or endometrioid OC who were in CR/PR post platinum-based chemotherapy and bevacizumab continued bevacizumab and were randomized to olaparib 300 mg twice daily or placebo for up to 2 years. At a median follow-up of 22.9 months, PFS was significantly better in the olaparib with bevacizumab arm in the overall population (22.1 months vs. 16.6 months; HR 0.59, 95% CI 0.49–0.72) and the HRD-positive subgroup (37.2 months vs. 17.7 months; HR 0.33, 95% CI 0.25–0.45), but not in the HRD-negative (16.6 months vs. 16.2 months; HR 1.0, 95% CI 0.75–1.35) subgroup [6, 61]. Although OS data are still immature, a recent update demonstrated improved OS in the HRD-positive subgroup (65.5 months vs. 48.8 months; HR 0.62, 95% CI 0.45–0.85) but not in the overall population (47.3 months vs. 41.5 months; HR 0.92, 95% CI 0.76–1.12) [62]. In this trial, patients potentially had a higher disease burden compared with SOLO1, as stage IV patients constituted 30% (vs. 17% in SOLO1) of the total population, 35% (vs. 22%) had residual macroscopic disease after surgery, and almost half (vs. 37%) of the patients received NACT. Furthermore, as there was no comparator arm with only olaparib maintenance, the role of the addition of bevacizumab to olaparib in this setting is difficult to determine.
PARPi maintenance has consistently shown higher benefit in BRCA-mutated and HRD-positive patients, and even in HRD-proficient patients, albeit this was more modest. Although benefit was observed in the HRD-negative population with niraparib maintenance in the PRIMA trial, the magnitude of benefit is lower compared with that of the HRD-positive population (HR 0.43 vs. 0.68). Similarly, benefit was observed with rucaparib in the ATHENA-MONO trial in the HRD-negative subgroup (HR 0.47 vs. 0.65). It should be noted that in all these trials, patients qualified for participation if they had demonstrable benefit to platinum—this functional predictive biomarker is quite important and may influence the predictive value of HRD status, particularly in the homologous recombination proficient (HRP) setting. Therefore, PARPi as maintenance are recommended in patients with advanced OC who respond to initial platinum-based therapy. Olaparib is recommended in patients with somatic or germline BRCA mutations, while olaparib in combination with bevacizumab is recommended in HRD-positive patients. Niraparib and rucaparib may be used irrespective of BRCA or HRD status. However, the optimal duration of maintenance is unclear. Olaparib ± bevacizumab and rucaparib were studied in maintenance for up to 2 years, while niraparib was administered for 3 years [6, 7, 57, 60]. If patients have not received maintenance PARPi in first-line therapy, they should be considered for PARPi after response to platinum-based therapy in the recurrent setting [9, 10].
It is noteworthy that 70–80% of patients with advanced OC respond to initial platinum-based therapy, while outcomes are poor in the remaining patients who do not respond to first-line platinum-based therapy. As such, options are limited in such patients because of a lack of data due to exclusion from clinical trials. Bevacizumab concurrently and in maintenance is an important drug in patients with suboptimally debulked and advanced disease, as well as patients who are HRP. Based on current data, patients who have advanced disease or who are at high risk should ideally be able to access both bevacizumab and PARPi, particularly if they have BRCA mutations or HRD.
4 Resistance to PARPi
With increasing use of PARPi as maintenance in the first-line setting, PARPi resistance has emerged as a major challenge in the management of OC. Many patients progress during the maintenance phase while others progress after discontinuation of PARPi. Two years of maintenance with olaparib was completed in 47.3% (123/260) of patients in SOLO1 (19.6% discontinued due to progression) and only 27.5% (148/537) of patients in PAOLA1 (36.5% discontinued due to progression). Similarly, in the PRIMA trial at a median follow-up of 13.8 months, 47% of patients were continuing niraparib (45% discontinued due to progression). Therefore, 35–45% of patients discontinue PARPi due to the development of resistance. Potential mechanisms of PARPi resistance (Fig. 2) include restoration of HRR activity, alteration of PARP function, stabilization of the RF, drug efflux by multidrug resistance (MDR) pumps, and activation of alternate pathways [63].

Mechanisms of PARPi resistance. (1) Restoration of homologous recombination repair. a Somatic reversion mutation restores the open reading frame and activity of HRR genes. b Epigenetic reversion by loss of methylation can restore BRCA1/RAD51 function. c Hypomorphic proteins, viz. BRCA1D11q isoform, retains partial activity due to an intact functional domain. d Restoration of end resection through loss of 53BP1 and Shieldin also promote PARPi resistance. (2) Alteration in PARP function due to a PARP1 mutations (R519C), or b Loss of PARG can inhibit PARylation and PARP trapping. (3) Stabilization of the RF. a HRD leads to collapse of the replication fork due to unchecked activity of nucleases such as MRE11, DNA2 and MUS81. Downregulation of these nucleases by loss of RF remodelers (MLL, EZH2, PTIP, SMARCAL1, HLTF) and FANCD2 overexpression promotes fork stabilization and promotes PARPi resistance. b RADX depletion restores fork stabilization by regulating RAD51. RF can also be stabilized by loss of regulation of G1/S cell cycle checkpoint secondary to E2F7 depletion. (4) Drug efflux and activation of alternate pathways can also cause PARPi resistance. 53BP1 TP53-binding protein, HRD homologous recombination deficiency, HRR homologous recombination repair, PARG poly (ADPribose) glycohydrolase, PARPi poly (ADP-ribose) polymerase inhibitor, PARylation poly (ADP-ribosyl)ation, RF replication fork. Created with Biorender.com
4.1 Restoration of Homologous Recombination (HR) Activity
PARPi act by blocking DNA damage repair via homologous recombination, and restoration of this activity is one of the common pathways of resistance. This can occur due to restoration of the functional homologous recombination repair pathway via reversion mutation in BRCA, loss of BRCA promoter methylation, presence of BRCA splice variants, or amplification of wild-type BRCA. Alternatively, homologous recombination repair activity may also be restored by activation of alternative DNA repair pathways, epigenetic changes and re-ignition of end resection due to loss of 53BP1.
Secondary mutations are be observed in 20–30% of patients with PARP resistance in BRCA-positive OCs [64]. These may restore the amino acid sequence similar to wild-type BRCA, restore the reading frame, or reverse epigenetic silencing. Most reversion mutations involve deletions of more than one base pair. These are usually subclonal and patients may demonstrate a heterogenous pattern of progression. Multiple mutations may be detected simultaneously by cell-free DNA analysis, which may result due to the emergence of multiple subclones due to the heterogenous nature of progression [65]. It occurs due to the inherent mutagenic nature of BRCA-mutated cancers as a result of dependence on error-prone DNA repair by NHEJ coupled with heterogeneity leading to multiple sites undergoing change, or as a result of evolutionary adaptation to therapy-induced alterations [64,65,66]. Furthermore, epigenetic changes such as methylation may cause silencing of HRR genes. Loss of promoter methylation may occur during the course of therapy and can also promote resistance to PARPi by activating silenced genes [67, 68]. Methylation status may predict response to PARPi therapy in recurrent OC [69].
Hypomorphic proteins with partial loss of activity may result due to alternative splicing of BRCA1. The BRCA1- Δ11q alternative splice isoform contains all functional domains except for exon 11 and is associated with resistance to platinum and PARPi [70]. Similarly, BRCA1 with mutations in the RING domain retain function and are associated with PARPi resistance [71].
Resection of break ends is a crucial step in HRR. In BRCA1 mutation, failure of resection of break ends prevents recruitment of RAD51 and further DNA repair. This is mediated by the TP53-binding protein (53BP1) pathway, which also involves the Shieldin and RIF1 proteins. In vitro loss of 53BP1 and Shieldin have been shown to be associated with resistance to PARPi [72, 73].
Mutation in the BRCT domain of BRCA1 leads to formation of the truncated form, which is degraded by proteosomes. Inhibition of proteasomal degradation by Hsp90-mediated stabilization promotes accumulation of these truncated proteins. As these truncated proteins retain some activity, accumulation may promote PARPi resistance [74].
4.2 Stabilization of the Replication Fork
In addition to BER, PARP1 plays an important role in stabilization of the RF during replication. The MRE11-RAD50-NBS1 (MRN) senses DSB and binds the break site to initiate HRR. MRE11 possess endonuclease activity, RAD50 is an ATPase and has a DNA-binding site, while NBS1 is a regulatory protein that activates other proteins involved in HRR, viz. ATM and ATR [75]. Endonuclease activity of MRE11 is responsible for initiating end resection to create single-stranded DNA (ssDNA), which are essential for HRR. However, unchecked activity may lead to the collapse of the RF. BRCA prevents MRE11-mediated degradation of nascent DNA at RF [76]. In the presence of BRCA mutations, PARPi promote RF instability and collapse by nuclease (MRE11, DNA2 and MUS81)-mediated DNA degradation [77]. Fork stabilization by other pathways can therefore promote PARPi resistance.

PARPi resistance can be managed by either a PARPi rechallenge, especially in the context of platinum sensitivity; b Overcoming resistance: resistance mechanisms can be overcome through inhibiting angiogenesis, targeting cell cycle regulators, or combining with immune checkpoint inhibitors; or c Bypassing resistance: an alternative strategy is targeting the non-cross-resistant pathways that are independent of HRR. GR glucocorticoid receptors, HRR homologous recombination repair, ICI immune checkpoint inhibitor, PARPi poly (ADP-ribose) polymerase inhibitor. Created with Biorender.com
Multiple remodelers are involved in fork collapse in HRD cells. EZH2 mediates H3K27 methylation and MUS81 recruitment to facilitate restart of the halted fork in BRCA-deficient cells. MELL3/4 triggers H3K4 methylation, which enhances PTIP accumulation and causes MRE11 recruitment. Members of the SNF2-family fork remodellers, including SMARCAL1, HLTF, and ZRANB3, are involved in MRE11-induced RF collapse, but is suppressed by FANCD2. Loss or depletion of these remodelers (EZH2, PTIP, SMARCAL1, HLTF, and ZRANB3) and FANCD2 overexpression promotes fork stabilization and causes resistance to PARPi [77,78,79,80,81,82].
RADX is an ssDNA-binding protein recruited to RFs and prevents fork collapse by regulating RAD51. In cancer cells, loss of RADX allows unchecked RAD51 activity, leading to the formation of DSB, while RADX depletion restores fork protection without restoring HRD in BRCA2-deficient cells [83].
E2F7 is a transcription factor regulating G1/S cell cycle checkpoint and is induced by DNA damage. It causes suppression of HRR by interfering with BRCA/RAD51 action. Depletion of E2F7 in BRCA-deficient cell lines has shown to confer resistance to platinum as well as PARPi [84].
4.3 Alteration of PARP Function
PARP1 attaches to the damaged DNA through its zinc finger DNA-binding domain. This interaction is also modified by allosteric effects of PARPi joining at the catalytic site. PARP1 function can be affected due to mutations or post-translational modifications in PARP1. Mutations within the DNA-binding domain are known to alter PARP trapping by preventing binding to DNA damage sites and resulting in resistance, while mutations in other sites may also cause resistance due to the allosteric effects of these mutations on DNA binding [85, 86].
PARylation is crucial for DNA repair by the homologous recombination pathway and involves the transfer of PAR residues from NAD+ and PARP. This leads to relaxation of the chromatin and recruitment of effector proteins (DNA polymerase-β and DNA ligase) at the site of DNA damage [49, 50]. The enzyme poly (ADPribose) glycohydrolase (PARG) can break PAR strands and reverse PARylation. PARP1-dependent DNA damage signalling is maintained and eventually results in PARPi resistance [87, 88]. PARG overexpression has been observed in OC cell lines and inhibition of PARG can augment the therapeutic effect of PARPi in OC cells [89, 90].
4.4 Drug Efflux
Another well-known mechanism of drug resistance is activation of drug efflux pumps. Multidrug-resistance protein 1 (MDR1), or P-glycoprotein (P-gp), is encoded by the ABCB1 gene. Long-term PARPi treatment can stimulate ABCB1 upregulation as a result of fusions or translocations [91]. Drug efflux as a mechanism of resistance may be observed in up to 15% of patients post progression on PARPi [92].
5 Managing PARP Resistance and Therapies Beyond PARPi
Understanding the mechanism of resistance is the key step before deciding further therapies. The resistant mechanisms discussed above can be targeted to resensitize the resistant OC cells to further therapy. Various modalities (Fig. 3) have been clinically evaluated, including the combination of PARPi with antiangiogenic agents, immune checkpoint inhibitors (ICIs), signal transduction pathway inhibitors, and targeting cell cycle checkpoints. These act by either overcoming resistance by enhancing the activity of PARPi using a combination with other targets in the DDR pathway, or bypassing the resistance mechanism through non-cross-resistant therapies. Alternatively, patients who had received PARPi as maintenance may still respond to PARPi rechallenge as monotherapy or as maintenance after chemotherapy.
5.1 Rechallenge PARPi
Many patients progress after completion of PARPi maintenance therapy. Thus, the rechallenge strategy has been tried in platinum-sensitive patients who respond to platinum-based chemotherapy, as these patients may not be truly resistant to PARPi. In the Quadra trial, niraparib monotherapy was evaluated in recurrent OC post three or more lines of therapy. Among 37 patients who had received prior PARPi therapy, one patient had a confirmed partial response (3%), and the clinical benefit rate at 16 weeks was 20% [93]. In the OReO trial, relapsed PSOC patients who had received one prior line of PARPi (irrespective of BRCA/HRD status), were randomized to either olaparib or placebo maintenance. PFS was significantly better in the BRCA-mutated cohort (median PFS 4.3 months vs. 2.8 months; HR 0.57, 95% CI 0.37–0.87; p = 0.022) as well as the non-BRCA-mutated cohort (median PFS 5.3 months vs. 2.8 months; HR 0.43, 95% CI 0.26–0.71; p = 0.002) [94]. The results were statistically significant but, clinically, of modest value. The trial suggests that PARPi retain activity in platinum-sensitive relapsed OC, and prior PARPi exposure does not essentially indicate complete resistance to therapy.
In the phase II MOLTO study, relapsed high-grade serous OC (HGSOC) patients with germline BRCA mutations were administered with two courses of olaparib maintenance. Patients who did not receive PARPi therapy were treated with platinum-based chemotherapy, followed by olaparib maintenance if they achieved CR/PR after platinum therapy. Patients who had previously received PARPi or who relapsed after receiving initial olaparib were retreated (if they had CR/PR or stable disease to platinum-based therapy) with olaparib (if platinum-free interval ≥ 6 months after initial PARPi therapy) or olaparib with cediranib (if platinum-free interval < 6 months after initial PARPi therapy). Of the 27 patients who began trial treatment (17 PARPi-naive and 10 prior PARPi), 19 started platinum-based therapy at progression on prior PARPi. Among these patients, 12 (63%) received a second course of olaparib ± cediranib, and the duration of second olaparib maintenance was >6 months in 4 (33%) patients. No new safety concerns were identified with the olaparib rechallenge. There was a significant difference in the duration of first and second olaparib maintenance (12.1 months vs. 4.4 months; p < 0.001). Functional HRD evaluation and somatic copy-number alteration (SCNA) assays did not predict PFS after platinum therapy. This study showed that PARPi rechallenge is feasible but it would be difficult to conclude on its efficacy due to small numbers and the lack of a comparator placebo group [95].
Current evidence suggests that PARPi rechallenge is possible in some patients. Fresh tumour biopsy and liquid biopsy may detect BRCA reversion mutation when PARPi are ineffective. Further research is ongoing to select appropriate patients and is crucial to optimizing this strategy.
5.2 Overcoming PARPi Resistance
Enhancing the PARPi activity by combining it with other agents that can overcome the resistance mechanisms has been extensively studied. This includes targeting angiogenic pathways, cell cycle checkpoints, immune system, and using next-generation PARPi.
5.2.1 Antiangiogenic Agents
Antiangiogenic agents inhibit tumour angiogenesis and cause hypoxia in the tumour. Hypoxic changes in the tumour microenvironment (TME) exert multiple effects, including abnormal DNA damage and repair signals, leading to genetic instability. Inhibition of VEGFR3 also downregulates BRCA1/2 gene expression in the tumour cells. Thus the combination of PARPi with antiangiogenic agents may help overcome resistance [96,97,98].
In PSOC, a combination of niraparib and bevacizumab was evaluated in the AVANOVA2 trial. Patients were randomized to either niraparib and bevacizumab or niraparib alone. Improvement in median PFS was observed in the intention-to-treat (ITT) population (11.9 months vs. 5.5 months; HR 0.35, 95% CI 0.21–0.57), bevacizumab-naive patients (14.4 months vs. 6.0 months; HR 0.39, 95% CI 0.22–0.68) and patients without BRCA mutation (11.3 months vs. 4.2 months; HR 0.32, 95% CI 0.17–0.578), but not in patients who were previously exposed to bevacizumab (5.9 months vs. 3.1 months; HR 0.51, 95% CI 0.21–1.26) [99]. Cediranib is an oral multikinase inhibitor of VEGF receptors 1–3, and c-kit has been evaluated in recurrent OC but failed to show much promise. In a phase II trial in PSOC or presence of deleterious germline BRCA1/2 mutation, 90 patients were randomized to olaparib with or without cediranib. The median PFS was significantly improved with the combination (16.5 vs. 8.2 months; HR 0.50; p = 0.007) but OS failed to reach statistical significance (44.2 vs. 33.3 months; HR 0.64; p = 0.11) [100]. No benefit was seen in a phase III trial evaluating olaparib with or without cediranib and platinum-based chemotherapy in platinum-sensitive relapsed OC [87]. In the EVOLVE trial, a translational phase II study in recurrent OC, patients (irrespective of platinum sensitivity) who had progressed on any prior PARPi received olaparib in combination with cediranib. Although responses were seen in only 8.8% of patients, about 50% were progression free at 16 weeks. Reversion mutations (19%), CCNE1 amplification (16%), and ABCB1 upregulation (15%) were common genomic alterations after prior PARPi exposure [92].
5.2.2 Targeting Cell Cycle
Dividing cells transverse through various phases of the cell cycle, viz G1, S, G2 and M. There are certain checkpoints that ensure genetic integrity of the cell while it passes from one phase to another through proteins that act in synergy to regulate this process of transition. The first checkpoint when the resting cell (G1 phase) commits to division is G1/S and is dependent on retinoblastoma (Rb) gene phosphorylation, which in turn is controlled by various cyclins (D and E), cyclin-dependent kinases (CDK) 2 and 4, p16/INK4 and p53. In the S phase, genetic material is duplicated, and in the G2 phase, cells prepare for mitosis by forming various proteins and organelles required in the M phase. G2/M transition is dependent on cyclin B and CDK1 phosphorylation, which is controlled by WEE1, checkpoint kinase (CHK) 1, polo-like kinase 1 (PLK1) and Aurora A. As p53 is universally mutated in serous OC, there is increased reliance on G2/M checkpoint, and blockage at this checkpoint can prevent cell cycle progression and growth of the tumour. Several cell cycle proteins, including cyclins, CDKs, CHK1 and 2, PLK1, and aurora kinases (Aurora A and Aurora B), are overexpressed in malignancies and are involved in carcinogenesis. Agents that act to inhibit these regulators of cell cycle have been shown to prevent tumour progression [101].
5.2.2.1 WEE1 Inhibition
WEE1 is a tyrosine kinase that regulates G2/M checkpoint by inhibiting CDK1 and regulates DNA synthesis in the S phase by inhibiting CDK2. Inhibition of WEE1 by adavosertib (AZD1775) promotes unchecked transition through the G2/M checkpoint, accumulation of damaged DNA, and sensitization to chemotherapy in p53-deficient cells [102]. In a randomized, phase II trial, patients with recurrent platinum-resistant/refractory OC were treated with gemcitabine plus either oral adavosertib or placebo. The adavosertib arm demonstrated significant improvement in objective response rate (ORR; 23 vs. 6%) and median PFS (4.6 vs. 3.0 months; HR 0.55, 95% CI 0.35–0.90). There was also a significant improvement in median OS (11.4 vs. 7.2 months; HR 0.56, 95% CI 0.35–0.91) [103]. In another four-arm, phase II study, two doses of adavosertib were combined with either gemcitabine, paclitaxel, carboplatin, or pegylated liposomal doxorubicin. In all patients, the ORR was 32% and 66.7% in combination with paclitaxel [104]. Both these trials showed that responders were enriched with high CCNE1-amplified tumours. The IGNITE trial investigated the efficacy of single-agent adavosertib in CCNE1-amplified (FISH) or overexpressed (IHC) platinum-resistant OC. Results from the cohort with overexpressed CCNE1 showed an ORR of 53% [105].
The efficacy of adavosertib post progression on PARPi was evaluated in the phase II EFFORT trial. Patients were randomized to either adavosertib alone or adavosertib with olaparib, and there was improvement in ORR (29 vs. 23%) and median PFS (6.8 vs. 5.5 months). BRCAwt patients showed improved ORR compared with BRCAm patients in both arms [106].
5.2.2.2 ATR Inhibition
ATR plays a very important role in cell cycle by inducing RF stalling, and activates CHK1, cell division cycle 25 (CDC25A/C), and WEE1, which prevents the progression of the cell cycle. ATR inhibition can reverse PARPi resistance due to RF protection by promoting the division of cells with DNA damage [107]. In a phase II trial, patients with recurrent OC (≤1 line for platinum resistance) were randomized to gemcitabine with/without berzosertib. The median PFS was significantly better in the berzosertib arm (22.9 vs. 14.7 weeks; HR 0.57, 90% CI 0.33–0.98; p = 0.044) [108]. Authors identified replication stress as an important biomarker of response to gemcitabine. Patients with high replication stress (defined as at least one genomic alteration due to the dysregulated RB pathway and/or oncogene-induced replication stress) [109]. Single-agent activity of the oral ATR inhibitor RP3500 in advanced OC was recently demonstrated in the phase I TRESR trial, with an ORR of 25% in patients with synthetic lethal genomic alterations [110]. In the CAPRI trial, olaparib and ceralasertib combination showed an ORR of 46% in patients with BRCAm or HRD-positive PSOC post progression on PARPi [111].
5.2.2.3 Checkpoint Kinase Inhibition
CHK1 and CHK2 are kinases regulating the G2/M cell cycle checkpoint by phosphorylating CDC25C and CDC25A. In response to DNA damage, these are activated by ATM/ATR and arrest cell cycle at the G2/M checkpoint to permit DNA damage repair [112, 113]. Prexasertib, a selective inhibitor of CHK1/CHK2, prevents activation of the CHK and allows cell cycle progression under persistent replication stress. It was evaluated as monotherapy in recurrent BRCA wild-type OC in a phase II study. Partial response was observed in 8/24 evaluable patients (PR 33%), while grade 4 neutropenia was observed in 79% [114] of patients. In another phase II study in platinum-resistant or -refractory OC, PR was seen in 12.1% and 6.9% of patients, respectively [115]. Prexasertib and olaparib combination was studied in a phase I trial in advanced solid tumours. Among 18 patients with BRCA-mutated OC who had progressed on prior PARPi, 4 (22%) had confirmed PR [116].
5.2.2.4 POLθ Inhibition
DNA polymerase theta (Polθ) is an enzyme involved in theta-mediated end joining (TMEJ), an error-prone backup pathway of DSB repair. Inhibition of Polθ has shown synthetic lethality in BRCA-deficient cells. ART558 is an inhibitor of Polθ and can reverse PARPi resistance secondary to defects in the 53BP1/Shieldin complex [117].
5.2.3 Immune Checkpoint Inhibitors
PARPi have synergistic activity in combination with ICIs, primarily via action on the cGAS-cGAMP-STING pathways. PARPi upregulate STING, which promotes the release of proinflammatory cytokines in the TME and also upregulates expression of programmed cell death-ligand 1 (PD-L1) on OC cells [118, 119]. In the phase I/II TOPACIO/KEYNOTE-162 trial in women with recurrent OC or advanced triple-negative breast cancer (TNBC), niraparib was administered in combination with pembrolizumab. Among patients with OC, responses were modest, with an ORR of 18% and a disease control rate (DCR) of 65%. Furthermore, responses were observed irrespective of BRCA, platinum sensitivity, or prior bevacizumab administration [120].
5.2.4 Next-Generation PARPi
AZD5305 is a specific PARP1 inhibitor with better efficacy and safety profile compared with current PARPi. A phase I/IIa PETRA trial is currently evaluating AZD5305 in patients with advanced metastatic ovarian, breast, pancreatic or prostate cancer with loss-of-function mutation in BRCA1/2, PALB2, RAD51C or RAD51D and prior PARPi treatment. Initial reports showed an ORR of 28% [121]. At present, there are limited data to indicate whether second-line PARPi are effective at overcoming resistance to first-generation agents.
5.3 Bypassing PARPi Resistance
PARPi resistance can be bypassed through targeting non-cross-resistant pathways acting independently of HRR. Several approaches have been investigated, including modulating the TME, enhancing drug delivery to tumour cells, and targeting different pathways.
5.3.1 Targeting the Immune System
ICIs have thus far failed to demonstrate meaningful benefit in OC, both in first-line maintenance and in the treatment of recurrent OC. The mechanism of immune resistance in OC is not universal and several genetic, immune, and metabolic factors contribute to establish an immunosuppressive milieu and lack of response to ICIs [122, 123]. T cells infiltrating OC express inhibitory receptors, such as programmed cell death protein 1(PD-1), cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), and lymphocyte activation gene-3 (LAG-3), leading to impaired function of T cells. Tumour-infiltrating CD8-positive T cells secrete interferon-γ, which further upregulates PD-L1 expression on OC cells and macrophages [124,125,126]. Additionally, the impact of mutational profile and anatomic sites has been suggested to contribute to the immunosuppressed and hostile TME [127]. However, a recent press release from sponsors of the DUO-O trial suggest significant PFS benefit with the addition of durvalumab to platinum-based chemotherapy and bevacizumab followed by durvalumab/olaparib/bevacizumab maintenance in newly diagnosed advanced OC [128]. DUO-O was a three-arm, placebo-controlled, randomized trial evaluating the efficacy of durvalumab in combination with platinum-based chemotherapy and bevacizumab, followed by maintenance with durvalumab and bevacizumab with or without olaparib in newly diagnosed patients with advanced OC.
In Javelin Ovarian 100, patients with newly advanced OC were randomized to chemotherapy (six cycles of carboplatin/paclitaxel) followed by avelumab maintenance, chemotherapy plus avelumab followed by avelumab maintenance (avelumab combination group), or chemotherapy followed by observation (control group). After interim analysis, the trial was stopped as PFS crossed the futility boundary (median PFS 16.6 months vs. 18.1 months vs. not reached) [129]. In Javelin Ovarian 200, recurrent platinum-resistant or -sensitive OC patients were randomized to avelumab, avelumab plus liposomal doxorubicin, or liposomal doxorubicin alone; no benefit was observed in median PFS [130]. In the IMagyn050 trial in newly advanced OC, patients were randomized to atezolizumab or placebo with paclitaxel/carboplatin and bevacizumab; median PFS was 19.5 vs. 18.4 months (HR 0.92, 95% CI 0.79–1.07; p = 0.28) in the overall population and 20.8 vs. 18.5 months (HR 0.80, 95% CI 0.65–0.99; p = 0.038) in the PD-L1-positive population [131].
In the ATALANTE/ov29 trial, patients with platinum-sensitive relapsed OC were randomized to atezolizumab or placebo with chemotherapy and bevacizumab. The median PFS in the overall population was 13.5 vs. 11.2 months (HR 0.83, 95% CI 0.69–0.99; p = 0.041), and 15.2 vs. 13.1 months (HR 0.86, 95% CI 0.63–1.16; p = 0.30) in PD-L1+ patients [132].
Currently, there are many trials exploring adoptive cell therapy, vaccine-based therapies, bispecific antibodies (BiTEs), chimeric antigen receptor therapy (CART), and oncolytic viruses, which may provide opportunities to bypass PARP resistance.
Nemvaleukin alfa is an engineered cytokine that selectively binds to the intermediate-affinity interleukin (IL)-2 receptor to preferentially activate and expand CD8+ T cells and natural killer (NK) cells with minimal expansion of CD4+ Tregs. It does not bind to high-affinity receptors due to steric hinderance, thereby avoiding adverse effects associated with it. In a phase I/II trial in multiple solid tumours, nemvaleukin alfa in combination with pembrolizumab showed an impressive ORR of 28.6% (including two complete responses) and a DCR of 71.4% in heavily pretreated OC patients [133]. ARTISTRY-7 (NCT05092360), a phase III randomized study of nemvaleukin alfa and pembrolizumab versus chemotherapy in platinum-resistant OC is currently recruiting.
Ubamatamab is an MUC16/CD3 bispecific antibody. In this first-in-human phase I study in patients with recurrent platinum-resistant OC and elevated CA125, an ORR of 14.3% was observed in those receiving one or more full doses [134].
Maveropepimut-S (DPX-Survivac) is a T-cell-activating vaccine with T-cell epitopes derived from survivin (tumour-associated antigen). In combination with low-dose cyclophosphamide, Maveropepimut-S showed robust T-cell response in OC patients [135]. In the phase I PESCO trial, maveropepimut-S in combination with pembrolizumab and low-dose cyclophosphamide demonstrated tolerable adverse effects, with a response rate of 16% in the initial 24 patients [136]. Similarly, OSE2101 (a multiple-neoepitope vaccine restricted to HLA-A2-positive patients targeting TP53, MAGE2, MAGE3, CEA and HER2) is under evaluation as maintenance therapy (post platinum-based chemotherapy) alone or in combination with pembrolizumab in the TEDOVA trial [137].
5.3.2 Antibody Drug Conjugates
Antibody drug conjugates (ADCs) are monoclonal antibodies conjugated to a cytotoxic payload that aim to deliver the cytotoxic agents directly to the cancer cells, thereby minimizing the toxicity associated with its systemic exposure. These bind the cell surface antigens and are internalized by endocytosis. The antibodies are selective against tumour-associated antigens and are connected to the cytotoxic agent by a linker that is stable while in circulation but is dissociated after entering the cells [138]. Various antigens of interest in OC that are in clinical trials include folate receptor-α (FRα), NaPi2, tissue factor (TF), mesothelin, MUC16, protein tyrosine kinase 7 (PTK7), and Trop2 [139]. Mirvetuximab soravtansine is an ADC against FRα with soravtansine (microtubule inhibitor), as the cytotoxic payload has been studied as a single agent in a recurrent setting as well in combination with chemotherapy and bevacizumab. In the phase III FORWARD 1 trial, patients with platinum-resistant OC who had received one to three prior lines of therapy and had positive FRα expression (≥50% of tumour cells with any FRα membrane staining visible at ≤10× microscope objective) on their tumours were randomly assigned to receive mirvetuximab (6 mg/kg) or chemotherapy. Although the ORR (24% vs. 10%) and CA-125 responses (53% vs. 25%) were improved, there was no difference in terms of PFS in the ITT population (4.1 vs. 4.4 months) as well as high FRα expression (≥75%) [94]. In the phase II, single-arm SORAYA trial, patients with platinum-resistant OC (PROC) who had received one to three prior lines of therapy and had high FRα expression (≥75% of viable tumour cells exhibiting ≥2+ level membrane staining intensity in the Ventana FOLR1 assay) were enrolled. All patients had received prior bevacizumab and about half of the patients had received prior PARPi therapy. At a median follow-up of 13.4 months, the ORR was 32.4% (95% CI 23.6–42.2%) and the median duration of response (DOR) was 6.9 months (95% CI 5.6–9.7 months). The most common adverse events (all grade) were blurred vision (41%), keratopathy (29%), and nausea (29%) [140].
Mirvetuximab soravtansine was recently granted FDA accelerated approval for platinum-resistant OC patients with high FRα expression [141]. The results differed in the two studies due to variable estimation criteria for FRα expression. In FORWARD1, patients with tumours having any level of expression on tumour cells were included, while in SORAYA, ≥2+ level membrane staining intensity was required. An exploratory analysis demonstrated that the predictive biomarker assay in the FORWARD 1 trial did not sufficiently enrich for high folate expressors. Therefore, careful selection of patients is required before considering them for these agents. MIRASOL, a randomized, phase III trial to evaluate the efficacy of mirvetuximab soravtansine compared with chemotherapy (weekly paclitaxel, pegylated liposomal doxorubicin, or topotecan) in PROC (post one to three lines) has completed accrual. FRα testing was performed by the Ventana FOLR1CDx assay and high expression was defined as ≥75% of cells with PS2+ staining intensity. A recent press release by the sponsors reported improved ORR with mirvetuximab soravtansine (42.3 % vs. 15.9 %). Significant improvement in PFS (5.62 vs. 3.98 months; HR 0.65; p < 0.0001) and OS (16.46 vs. 12.75 months; HR 0.67; p = 0.0046) was also reported [142, 143].
In a phase Ib/II study, mirvetuximab was evaluated in combination with carboplatin and bevacizumab in patients with PSOC with one to two prior lines of therapy. ORR was observed in 81% of patients, with a median DOR of 10.7 months and median PFS of 12.0 months [144]. Mirvetuximab in combination with bevacizumab demonstrated an ORR of 39% and a median PFS of 6.9 months in heavily pretreated patients with PROC [145]. Similarly, an ORR of 43%, with a median PFS of 5.2 months, was observed in combination with pembrolizumab in PROC with two to four prior lines of therapy [146].
NaPi2B, a sodium-dependent phosphate transport protein, is expressed in 80–100% of OC cells, but not on normal ovarian tissue [147]. The anti-NaPi2b ADC lifastuzumab vedotin was evaluated in a phase II randomized trial in PROC. A higher ORR was observed compared with liposomal doxorubicin (34% vs. 15%), but PFS was not significantly improved [148]. Another anti-NaPi2b, upifitamab rilsodotin, showed an ORR of 23% in the ITT population and 34% in patients with high NaPi2b expression in pretreated PROC [149]. TF is involved in the extrinsic pathway of coagulation and aberrant expression may be observed in a variety of solid tumours, including epithelial OC [150]. In a phase I/II trial of tisotumab vedotin (ADC against TF) in multiple advanced solid tumours, the ORR in OC patients was 13.9% [151]. Mesothelin is a glycoprotein important for cellular adhesions and is overexpressed in up to 88% of OC patients [152]. Anetumab ravtansine (AR) is a fully human ADC against mesothelin conjugated to a microtubule inhibitor. In a phase II randomized trial in platinum-resistant/refractory OC, a combination of anetumab with bevacizumab failed to show any benefit over paclitaxel/bevacizumab. The ORR was 55% with paclitaxel/bevacizumab compared with 18% in the experimental arm. The median PFS was 9.6 months with paclitaxel/bevacizumab compared with 5.3 months with anetumab/bevacizumab (HR 1.7, 95% CI 0.9–3.4) [153].
5.3.3 Modulation of Glucocorticoid Receptors
Cortisol binds to glucocorticoid receptors (GRs) and inhibits cell death by apoptosis, leading to chemotherapy resistance. GRs are abundantly expressed on OC cells and high expression is associated with poor outcomes [154]. Relacorilant is a selective modulator of GRs, which compete with cortisol and can reverse the cortisol-induced chemotherapy resistance in multiple solid tumours [155]. In a phase II, randomized study, recurrent PROC patients were randomized to nab-paclitaxel alone or nab-paclitaxel with intermittent (150 mg/day the day before, of, and after nab-paclitaxel administration) or a continuous schedule of relacorilant (100 mg daily). The intermittent schedule was associated with a significantly improved median PFS compared with nab-paclitaxel alone (5.55 vs. 3.76 months; HR 0.6, 95% CI 0.44–0.98; p = 0.038), but OS failed to reach statistical significance (13.9 vs. 12.2 months; HR 0.67, 95% CI 0.43–1.03; p = 0.066) [156, 157].
5.3.4 Gas6/Axl Signalling
Axl is a receptor tyrosine kinase associated with chemotherapy resistance and poor outcomes in various cancers. It is activated by the binding of its ligand Gas6 (growth arrest specific 6), which acts as growth factor and activates signalling pathways to promote cellular proliferation, invasion, migration, epithelial-mesenchymal transition, angiogenesis, immune evasion, and survival [158]. Axl is expressed on serous OC cells but not on healthy ovarian cells [159]. Batiraxcept (AVB-500) is a fusion protein containing the Fc region of heavy chain immunoglobulin (Ig) G1 fused to the extracellular region of Axl. It acts as a decoy receptor and binds Gas6 with 200-fold high affinity. Batiraxcept was evaluated in phase Ib trial in patients with platinum-resistant OC in combination with paclitaxel or pegylated liposomal doxorubicin. In patients receiving paclitaxel, the ORR was 34.8% and the median PFS and OS was 3.1 and 10.3 months, respectively [160]. GOG-3059/ENGOT OV66 (NCT04729608) is a randomized, phase III study comparing the efficacy of batiraxcept (AVB-S6-500) in combination with paclitaxel in patients with platinum-resistant OC, and is currently recruiting.
5.3.5 G-Quadruplex Stabilizers
G-quadruplexes (G4) are transient guanine-rich tertiary structures that are found at promoter sites and telomeric regions in human genome. These are believed to be involved in gene regulation and other processes during cell division, but the precise role of G4 is still undetermined. G4 unwinding proteins/helicases remove these G4 structures in normal cells [161, 162]. Alteration in this process can cause transcriptional changes and DNA breaks, leading to genome instability. HRR is the predominant pathway involved in DNA damage repair in response to G4 alterations [163]. G4-induced genomic instability has been linked to carcinogenesis, although synthetic lethality in this context is being explored as a therapeutic target in HRD cancers [164]. CX-5461 (Pidnarulex) is a G4 stabilizer that promotes tertiary structure and RF arrest, leading to cancer cell death in HRD tumours. It was evaluated in phase I trial in multiple solid tumours and response was observed in 1 (14.2%) patient with BRCA2 mutations among 7 patients with OC. Overall responses were observed in 4/29 patients (ORR 13.8%) with BRCA2 and PALB2 alterations (breast, ovarian, pancreatic cancer) [165]. CX-5461 is currently being evaluated in a phase Ib trial in patients with HRD [166].
5.3.6 Chemotherapy
Chemotherapy remains an option in patients post progression on PARPi. Although there is an overlap between resistance mechanisms of PARPi and platinum, PARPi resistance does not necessarily denote platinum resistance. In ARIEL 4, a phase III study in relapsed OC patients with deleterious germline or somatic BRCA alterations and no prior PARPi, patients were randomized to rucaparib or chemotherapy (based on platinum sensitivity). Fully platinum-sensitive patients (platinum free interval >12 months) were administered platinum-based chemotherapy, while others received weekly paclitaxel. The primary endpoint was investigator-assessed PFS in the efficacy population (deleterious BRCA mutations without BRCA reversion mutations). Median PFS was significantly improved with rucaparib in the efficacy population (7.4 months vs. 5.7 months; HR 0.64, 95% CI 0.49–0.84); however, the median OS was better in patients receiving chemotherapy (25.4 months vs. 19.4 months; HR 1.31, 95% CI 0.99–1.72). This was mainly driven by the platinum-resistant subgroup. Similarly, in patients with BRCA reversion mutations, the median PFS was greater with chemotherapy (5.5 months vs. 2.9 months; HR 2.77, 95% CI 0.99–7.76). As crossover was permitted, 80 (69%) patients randomized to chemotherapy crossed-over to rucaparib, while 42.1% of patients in the rucaparib arm did not receive subsequent anticancer treatment. PFS2 (PFS during the first subsequent anticancer treatment) was better in patients crossing over to rucaparib. Furthermore, translational analysis showed that there was a decrease in BRCA reversion mutations in three of four patients randomized to paclitaxel (in the pre- and post-treatment plasma samples). This suggests that paclitaxel may reverse PARPi resistance due to BRCA reversion mutations [14, 167].
6 Conclusion
PARPi resistance has emerged as a major challenge in the management of OC. Ongoing research has provided valuable insight into the mechanisms of resistance to PARPi. Restoration of HRR and RF stability appears to be the major pathways involved. However, it may be difficult to ascertain the exact mechanism in a single patient due to complex interplay between the various pathways. This presents a challenge to develop newer therapies to overcome or bypass the resistance. Multiple trials are currently evaluating the newer agents and combinations targeting these pathways. Further translational research is warranted in this area, with the incorporation of biomarkers to direct the management strategy.
References
Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin. 2023;73(1):17–48. https://doi.org/10.3322/caac.21763.
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(3):209–49.
Torre LA, Trabert B, DeSantis CE, Miller KD, Samimi G, Runowicz CD, et al. Ovarian cancer statistics, 2018. CA Cancer J Clin. 2018;68(4):284–96.
Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 2016;2(4):482–90.
Zhang S, Royer R, Li S, McLaughlin JR, Rosen B, Risch HA, et al. Frequencies of BRCA1 and BRCA2 mutations among 1342 unselected patients with invasive ovarian cancer. Gynecol Oncol. 2011;121(2):353–7.
Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R, et al. Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med. 2019;381(25):2416–28.
González-Martín A, Pothuri B, Vergote I, DePont CR, Graybill W, Mirza MR, et al. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2019;381(25):2391–402.
Monk BJ, Parkinson C, Lim MC, O’Malley DM, Oaknin A, Wilson MK, et al. A randomized, phase III trial to evaluate rucaparib monotherapy as maintenance treatment in patients with newly diagnosed ovarian cancer (ATHENA-MONO/GOG-3020/ENGOT-ov45). J Clin Oncol. 2022;40(34):3952–64.
Coleman RL, Fleming GF, Brady MF, Swisher EM, Steffensen KD, Friedlander M, et al. Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med. 2019;381(25):2403–15.
Gelmon KA, Tischkowitz M, Mackay H, Swenerton K, Robidoux A, Tonkin K, et al. Olaparib in patients with recurrent high-grade serous or poorly differentiated ovarian carcinoma or triple-negative breast cancer: a phase 2, multicentre, open-label, non-randomised study. Lancet Oncol. 2011;12(9):852–61.
Oza AM, Cibula D, Benzaquen AO, Poole C, Mathijssen RHJ, Sonke GS, et al. Olaparib combined with chemotherapy for recurrent platinum-sensitive ovarian cancer: a randomised phase 2 trial. Lancet Oncol. 2015;16(1):87–97.
Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med. 2016;375(22):2154–64.
Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A, et al. Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet. 2017;390(10106):1949–61.
Oza AM, Lisyanskaya AS, Fedenko AA, de Melo AC, Shparik Y, Bondarenko I, et al. 518O Overall survival results from ARIEL4: a phase III study assessing rucaparib vs chemotherapy in patients with advanced, relapsed ovarian carcinoma and a deleterious BRCA1/2 mutation. Ann Oncol. 2022;33:S780.
Lans H, Hoeijmakers JHJ, Vermeulen W, Marteijn JA. The DNA damage response to transcription stress. Nat Rev Mol Cell Biol. 2019;20(12):766–84.
Choi EH, Yoon S, Koh YE, Seo YJ, Kim KP. Maintenance of genome integrity and active homologous recombination in embryonic stem cells. Exp Mol Med. 2020;52(8):1220–9.
Hoeijmakers JHJ. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–74.
Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8(3):193–204.
Scully R, Panday A, Elango R, Willis NA. DNA double-strand break repair-pathway choice in somatic mammalian cells. Nat Rev Mol Cell Biol. 2019;20(11):698–714.
Tubbs A, Nussenzweig A. Endogenous DNA damage as a source of genomic instability in cancer. Cell. 2017;168(4):644–56.
Xiao X, Cai F, Niu X, Shi H, Zhong Y. Association between P16INK4a promoter methylation and ovarian cancer: a meta-analysis of 12 published studies. PLoS One. 2016;11(9): e0163257.
Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002;3(6):415–28.
Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017;23(4):517–25.
Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, et al. Homologous Recombination Deficiency (HRD) score predicts response to platinum-containing neoadjuvant chemotherapy in patients with triple-negative breast cancer. Clin Cancer Res. 2016;22(15):3764–73.
Stover EH, Fuh K, Konstantinopoulos PA, Matulonis UA, Liu JF. Clinical assays for assessment of homologous recombination DNA repair deficiency. Gynecol Oncol. 2020;159(3):887–98.
Morice PM, Coquan E, Weiswald LB, Lambert B, Vaur D, Poulain L. Identifying patients eligible for PARP inhibitor treatment: from NGS-based tests to 3D functional assays. Br J Cancer. 2021;125(1):7–14.
van Wijk LM, Vermeulen S, Meijers M, van Diest MF, ter Haar NT, de Jonge MM, et al. The RECAP test rapidly and reliably identifies homologous recombination-deficient ovarian carcinomas. Cancers. 2020;12(10):2805.
Pikkusaari S, Tumiati M, Virtanen A, Oikkonen J, Li Y, Perez-Villatoro F, et al. Functional homologous recombination assay on FFPE specimens of advanced high-grade serous ovarian cancer predicts clinical outcomes. Clin Cancer Res. 2023;29(16):3110–23.
Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J Natl Cancer Inst. 2000;92(7):564–9.
du Bois A, Quinn M, Thigpen T, Vermorken J, Avall-Lundqvist E, Bookman M, et al. 2004 consensus statements on the management of ovarian cancer: final document of the 3rd International Gynecologic Cancer Intergroup Ovarian Cancer Consensus Conference (GCIG OCCC 2004). Ann Oncol. 2005;16:viii7–12.
du Bois A, Reuss A, Pujade-Lauraine E, Harter P, Ray-Coquard I, Pfisterer J. Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: a combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: by the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d’Investigateurs Nationaux Pour les Etudes des Cancers de l’Ovaire (GINECO). Cancer. 2009;115(6):1234–44.
Wimberger P, Wehling M, Lehmann N, Kimmig R, Schmalfeldt B, Burges A, et al. Influence of residual tumor on outcome in ovarian cancer patients with FIGO stage IV disease: an exploratory analysis of the AGO-OVAR (Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group). Ann Surg Oncol. 2010;17(6):1642–8.
Vergote I, du Bois A, Amant F, Heitz F, Leunen K, Harter P. Neoadjuvant chemotherapy in advanced ovarian cancer: on what do we agree and disagree? Gynecol Oncol. 2013;128(1):6–11.
Wright AA, Bohlke K, Armstrong DK, Bookman MA, Cliby WA, Coleman RL, et al. Neoadjuvant chemotherapy for newly diagnosed, advanced ovarian cancer: Society of Gynecologic Oncology and American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2016;34(28):3460–73.
Vergote I, Coens C, Nankivell M, Kristensen GB, Parmar MKB, Ehlen T, et al. Neoadjuvant chemotherapy versus debulking surgery in advanced tubo-ovarian cancers: pooled analysis of individual patient data from the EORTC 55971 and CHORUS trials. Lancet Oncol. 2018;19(12):1680–7.
Chan JK, Tian C, Monk BJ, Herzog T, Kapp DS, Bell J, et al. Prognostic factors for high-risk early-stage epithelial ovarian cancer: a Gynecologic Oncology Group study. Cancer. 2008;112(10):2202–10.
Chan JK, Tian C, Fleming GF, Monk BJ, Herzog TJ, Kapp DS, et al. The potential benefit of 6 vs. 3 cycles of chemotherapy in subsets of women with early-stage high-risk epithelial ovarian cancer: an exploratory analysis of a Gynecologic Oncology Group study. Gynecol Oncol. 2010;116(3):301–6.
Armstrong DK, Bundy B, Wenzel L, Huang HQ, Baergen R, Lele S, et al. Intraperitoneal cisplatin and paclitaxel in ovarian cancer. N Engl J Med. 2006;354(1):34–43.
Walker JL, Brady MF, Wenzel L, Fleming GF, Huang HQ, DiSilvestro PA, et al. Randomized trial of intravenous versus intraperitoneal chemotherapy plus bevacizumab in advanced ovarian carcinoma: an NRG oncology/gynecologic oncology group study. J Clin Oncol. 2019;37(16):1380–90.
Presta LG, Chen H, O’Connor SJ, Chisholm V, Meng YG, Krummen L, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res. 1997;57(20):4593–9.
Oza AM, Cook AD, Pfisterer J, Embleton A, Ledermann JA, Pujade-Lauraine E, et al. Standard chemotherapy with or without bevacizumab for women with newly diagnosed ovarian cancer (ICON7): overall survival results of a phase 3 randomised trial. Lancet Oncol. 2015;16(8):928–36.
Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med. 2011;365(26):2484–96.
Ferriss JS, Java JJ, Bookman MA, Fleming GF, Monk BJ, Walker JL, et al. Ascites predicts treatment benefit of bevacizumab in front-line therapy of advanced epithelial ovarian, fallopian tube and peritoneal cancers: an NRG Oncology/GOG study. Gynecol Oncol. 2015;139(1):17–22.
Burger RA, Brady MF, Bookman MA, Fleming GF, Monk BJ, Huang H, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med. 2011;365(26):2473–83.
You B, Freyer G, Gonzalez-Martin A, Lheureux S, McNeish I, Penson RT, et al. The role of the tumor primary chemosensitivity relative to the success of the medical-surgical management in patients with advanced ovarian carcinomas. Cancer Treat Rev. 2021;100: 102294.
Colomban O, Tod M, Peron J, Perren TJ, Leary A, Cook AD, et al. Bevacizumab for newly diagnosed ovarian cancers: best candidates among high-risk disease patients (ICON-7). JNCI Cancer Spectr. 2020;4(3): pkaa026.
You B, Purdy C, Copeland LJ, Swisher EM, Bookman MA, Fleming G, et al. Identification of patients with ovarian cancer experiencing the highest benefit from bevacizumab in the first-line setting on the basis of their tumor-intrinsic chemosensitivity (KELIM): the GOG-0218 Validation Study. J Clin Oncol. 2022;40(34):3965–74.
Langelier MF, Planck JL, Roy S, Pascal JM. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science. 2012;336(6082):728–32.
Pascal JM, Ellenberger T. The rise and fall of poly(ADP-ribose): an enzymatic perspective. DNA Repair. 2015;32:10–6.
Caldecott KW, Aoufouchi S, Johnson P, Shall S. XRCC1 polypeptide interacts with DNA polymerase beta and possibly poly (ADP-ribose) polymerase, and DNA ligase III is a novel molecular “nick-sensor” in vitro. Nucleic Acids Res. 1996;24(22):4387–94.
Kim C, Wang XD, Yu Y. PARP1 inhibitors trigger innate immunity via PARP1 trapping-induced DNA damage response. Elife. 2020;9: e60637.
Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7.
Farmer H, McCabe N, Lord CJ, Tutt ANJ, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21.
Brenner JC, Ateeq B, Li Y, Yocum AK, Cao Q, Asangani IA, et al. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 2011;19(5):664–78.
Lee EK, Konstantinopoulos PA. PARP inhibition and immune modulation: scientific rationale and perspectives for the treatment of gynecologic cancers. Ther Adv Med Oncol. 2020;12:1758835920944116.
Kim DS, Camacho CV, Nagari A, Malladi VS, Challa S, Kraus WL. Activation of PARP-1 by snoRNAs controls ribosome biogenesis and cell growth via the RNA helicase DDX21. Mol Cell. 2019;75(6):1270-1285.e14.
Banerjee S, Moore KN, Colombo N, Scambia G, Kim BG, Oaknin A, et al. Maintenance olaparib for patients with newly diagnosed advanced ovarian cancer and a BRCA mutation (SOLO1/GOG 3004): 5-year follow-up of a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2021;22(12):1721–31.
Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M, et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 2018;379(26):2495–505.
DiSilvestro P, Banerjee S, Colombo N, Scambia G, Kim BG, Oaknin A, et al. Overall survival with maintenance Olaparib at a 7-year follow-up in patients with newly diagnosed advanced ovarian cancer and a BRCA mutation: the SOLO1/GOG 3004 trial. J Clin Oncol. 2023;41(3):609–17.
Monk BJ, Parkinson C, Lim MC, O’Malley DM, Oaknin A, Wilson MK, et al. A randomized, phase III trial to evaluate rucaparib monotherapy as maintenance treatment in patients with newly diagnosed ovarian cancer (ATHENA–MONO/GOG-3020/ENGOT-ov45). J Clin Oncol. 2022;40(34):3952–64.
Nagao S, Harter P, Leary A, Cropet C, Pignata S, Fujiwara K, et al. 176O Final overall survival (OS) results from the phase III PAOLA-1/ENGOT-ov25 trial evaluating maintenance olaparib (ola) plus bevacizumab (bev) in patients (pts) with newly diagnosed advanced ovarian cancer (AOC). Ann Oncol. 2022;33:S1503–4.
Ray-Coquard IL, Leary A, Pignata S, Cropet C, Martin AJG, Bogner G, et al. LBA29 Final overall survival (OS) results from the phase III PAOLA-1/ENGOT-ov25 trial evaluating maintenance olaparib (ola) plus bevacizumab (bev) in patients (pts) with newly diagnosed advanced ovarian cancer (AOC). Ann Oncol. 2022;33:S1396–7.
Wakefield MJ, Nesic K, Kondrashova O, Scott CL. Diverse mechanisms of PARP inhibitor resistance in ovarian cancer. Biochim Biophys Acta BBA. 2019;1872(2): 188307.
Tobalina L, Armenia J, Irving E, O’Connor MJ, Forment JV. A meta-analysis of reversion mutations in BRCA genes identifies signatures of DNA end-joining repair mechanisms driving therapy resistance. Ann Oncol. 2021;32(1):103–12.
Lin KK, Harrell MI, Oza AM, Oaknin A, Ray-Coquard I, Tinker AV, et al. BRCA reversion mutations in circulating tumor DNA predict primary and acquired resistance to the PARP inhibitor rucaparib in high-grade ovarian carcinoma. Cancer Discov. 2019;9(2):210–9.
Christie EL, Fereday S, Doig K, Pattnaik S, Dawson SJ, Bowtell DDL. Reversion of BRCA1/2 germline mutations detected in circulating tumor DNA from patients with high-grade serous ovarian cancer. J Clin Oncol. 2017;35(12):1274–80.
Nesic K, Kondrashova O, Hurley RM, McGehee CD, Vandenberg CJ, Ho GY, et al. Acquired RAD51C promoter methylation loss causes PARP inhibitor resistance in high-grade serous ovarian carcinoma. Cancer Res. 2021;81(18):4709–22.
Kondrashova O, Topp M, Nesic K, Lieschke E, Ho GY, Harrell MI, et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat Commun. 2018;9(1):3970.
Swisher EM, Kwan TT, Oza AM, Tinker AV, Ray-Coquard I, Oaknin A, et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat Commun. 2021;12(1):2487.
Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, et al. The BRCA1-Δ11q alternative splice isoform bypasses germline mutations and promotes therapeutic resistance to PARP inhibition and cisplatin. Cancer Res. 2016;76(9):2778–90.
Wang Y, Krais JJ, Bernhardy AJ, Nicolas E, Cai KQ, Harrell MI, et al. RING domain-deficient BRCA1 promotes PARP inhibitor and platinum resistance. J Clin Invest. 2016;126(8):3145–57.
Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, et al. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49(5):858–71.
Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–54.
Johnson N, Johnson SF, Yao W, Li YC, Choi YE, Bernhardy AJ, et al. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc Natl Acad Sci. 2013;110(42):17041–6.
Williams GJ, Lees-Miller SP, Tainer JA. Mre11–Rad50–Nbs1 conformations and the control of sensing, signaling, and effector responses at DNA double-strand breaks. DNA Repair. 2010;9(12):1299–306.
Stracker TH, Petrini JHJ. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol. 2011;12(2):90–103.
Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535(7612):382–7.
Michl J, Zimmer J, Buffa FM, McDermott U, Tarsounas M. FANCD2 limits replication stress and genome instability in cells lacking BRCA2. Nat Struct Mol Biol. 2016;23(8):755–7.
Rondinelli B, Gogola E, Yücel H, Duarte AA, van de Ven M, van der Sluijs R, et al. EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol. 2017;19(11):1371–8.
Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang JW, et al. Restoration of replication fork stability in BRCA1- and BRCA2-deficient cells by inactivation of SNF2-family fork remodelers. Mol Cell. 2017;68(2):414-430.e8.
Kim TM, Son MY, Dodds S, Hu L, Luo G, Hasty P. RECQL5 and BLM exhibit divergent functions in cells defective for the Fanconi anemia pathway. Nucleic Acids Res. 2015;43(2):893–903.
Kais Z, Rondinelli B, Holmes A, O’Leary C, Kozono D, D’Andrea AD, et al. FANCD2 maintains fork stability in BRCA1/2-deficient tumors and promotes alternative end-joining DNA repair. Cell Rep. 2016;15(11):2488–99.
Dungrawala H, Bhat KP, Le Meur R, Chazin WJ, Ding X, Sharan SK, et al. RADX promotes genome stability and modulates chemosensitivity by regulating RAD51 at replication forks. Mol Cell. 2017;67(3):374-386.e5.
Clements KE, Thakar T, Nicolae CM, Liang X, Wang HG, Moldovan GL. Loss of E2F7 confers resistance to poly-ADP-ribose polymerase (PARP) inhibitors in BRCA2-deficient cells. Nucleic Acids Res. 2018;46(17):8898–907.
Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH, et al. Blocking c-Met–mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med. 2016;22(2):194–201.
Pettitt SJ, Krastev DB, Brandsma I, Dréan A, Song F, Aleksandrov R, et al. Genome-wide and high-density CRISPR-Cas9 screens identify point mutations in PARP1 causing PARP inhibitor resistance. Nat Commun. 2018;9(1):1849.
Francica P, Rottenberg S. Mechanisms of PARP inhibitor resistance in cancer and insights into the DNA damage response. Genome Med. 2018;10(1):101.
O’Sullivan J, Tedim Ferreira M, Gagné JP, Sharma AK, Hendzel MJ, Masson JY, et al. Emerging roles of eraser enzymes in the dynamic control of protein ADP-ribosylation. Nat Commun. 2019;10(1):1182.
Chen SH, Yu X. Targeting dePARylation selectively suppresses DNA repair–defective and PARP inhibitor–resistant malignancies. Sci Adv. 2019;5(4): eaav4340.
Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, et al. Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell. 2018;33(6):1078-1093.e12.
Christie EL, Pattnaik S, Beach J, Copeland A, Rashoo N, Fereday S, et al. Multiple ABCB1 transcriptional fusions in drug resistant high-grade serous ovarian and breast cancer. Nat Commun. 2019;10(1):1295.
Lheureux S, Oaknin A, Garg S, Bruce JP, Madariaga A, Dhani NC, et al. EVOLVE: a multicenter open-label single-arm clinical and translational phase II trial of cediranib plus olaparib for ovarian cancer after PARP inhibition progression. Clin Cancer Res. 2020;26(16):4206–15.
Rimel B, Secord AA, Geller MA, Miller D, Cloven N, Fleming GF, et al. Safety and efficacy results of retreatment with a PARP inhibitor monotherapy in late-line recurrent ovarian cancer: results from a subset of the QUADRA trial. Gynecol Oncol. 2020;156:e4-5.
Pujade-Lauraine E, Selle F, Scambia G, Asselain B, Marmé F, Lindemann K, et al. LBA33 Maintenance olaparib rechallenge in patients (pts) with ovarian carcinoma (OC) previously treated with a PARP inhibitor (PARPi): phase IIIb OReO/ENGOT Ov-38 trial. Ann Oncol. 2021;32:S1308–9.
Morgan RD, Clamp AR, White DJ, Price M, Burghel GJ, Ryder WDJ, et al. Multi-maintenance olaparib therapy in relapsed, germline BRCA1/2-mutant high-grade serous ovarian cancer (MOLTO): a phase II trial. Clin Cancer Res. 2023;29(14):2062–611.
Kaplan AR, Gueble SE, Liu Y, Oeck S, Kim H, Yun Z, et al. Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci Transl Med. 2019;11(492): eaav4508.
Chan N, Koritzinsky M, Zhao H, Bindra R, Glazer PM, Powell S, et al. Chronic hypoxia decreases synthesis of homologous recombination proteins to offset chemoresistance and radioresistance. Cancer Res. 2008;68(2):605–14.
Lim J, Yang K, Taylor-Harding B, Wiedemeyer WR, Buckanovich RJ. VEGFR3 inhibition chemosensitizes ovarian cancer stemlike cells through down-regulation of BRCA1 and BRCA2. Neoplasia. 2014;16(4):343–53 (e2).
Mirza MR, Åvall Lundqvist E, Birrer MJ, dePont CR, Nyvang GB, Malander S, et al. Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): a randomised, phase 2, superiority trial. Lancet Oncol. 2019;20(10):1409–19.
Liu JF, Barry WT, Birrer M, Lee JM, Buckanovich RJ, Fleming GF, et al. Overall survival and updated progression-free survival outcomes in a randomized phase II study of combination cediranib and olaparib versus olaparib in relapsed platinum-sensitive ovarian cancer. Ann Oncol. 2019;30(4):551–7.
Otto T, Sicinski P. Cell cycle proteins as promising targets in cancer therapy. Nat Rev Cancer. 2017;17(2):93–115.
Vriend LEM, De Witt Hamer PC, Van Noorden CJF, Würdinger T. WEE1 inhibition and genomic instability in cancer. Biochim Biophys Acta. 2013;1836(2):227–35.
Lheureux S, Cristea MC, Bruce JP, Garg S, Cabanero M, Mantia-Smaldone G, et al. Adavosertib plus gemcitabine for platinum-resistant or platinum-refractory recurrent ovarian cancer: a double-blind, randomised, placebo-controlled, phase 2 trial. Lancet. 2021;397(10271):281–92.
Moore KN, Chambers SK, Hamilton EP, Chen L, Oza AM, Ghamande SA, et al. Adavosertib with chemotherapy in patients with primary platinum-resistant ovarian, fallopian tube, or peritoneal cancer: an open-label, four-arm, Phase II Study. Clin Cancer Res. 2022;28(1):36–44.
Au-Yeung G, Bressel M, Prall O, Surace D, Andrews J, Mongta S, et al. IGNITE: a phase II signal-seeking trial of adavosertib targeting recurrent high-grade, serous ovarian cancer with cyclin E1 overexpression with and without gene amplification. J Clin Oncol. 2022;40(16 Suppl):5515.
Westin SN, Coleman RL, Fellman BM, Yuan Y, Sood AK, Soliman PT, et al. EFFORT: efficacy of adavosertib in parp ResisTance: a randomized two-arm non-comparative phase II study of adavosertib with or without olaparib in women with PARP-resistant ovarian cancer. J Clin Oncol. 2021;39(15 Suppl):5505.
Ngoi NYL, Westin SN, Yap TA. Targeting the DNA damage response beyond poly(ADP-ribose) polymerase inhibitors: novel agents and rational combinations. Curr Opin Oncol. 2022;34(5):559.
Konstantinopoulos PA, Cheng SC, Wahner Hendrickson AE, Penson RT, Schumer ST, Doyle LA, et al. Berzosertib plus gemcitabine versus gemcitabine alone in platinum-resistant high-grade serous ovarian cancer: a multicentre, open-label, randomised, phase 2 trial. Lancet Oncol. 2020;21(7):957–68.
Konstantinopoulos PA, da Costa AABA, Gulhan D, Lee EK, Cheng SC, Hendrickson AEW, et al. A Replication stress biomarker is associated with response to gemcitabine versus combined gemcitabine and ATR inhibitor therapy in ovarian cancer. Nat Commun. 2021;12(1):5574.
Yap T, Lee E, Spigel D, Fontana E, Højgaard M, Lheureux S, et al. Abstract CC04-01: first-in-human biomarker-driven phase I TRESR trial of ataxia telangiectasia and Rad3-related inhibitor (ATRi) RP-3500 in patients (pts) with advanced solid tumors harboring synthetic lethal (SL) genomic alterations. Mol Cancer Ther. 2021;20(12 Suppl):CC04-CC11.
Wethington SL, Shah PD, Martin LP, Tanyi JL, Latif NA, Morgan MA, et al. Combination of PARP and ATR inhibitors (olaparib and ceralasertib) shows clinical activity in acquired PARP inhibitor-resistant recurrent ovarian cancer. J Clin Oncol. 2021;39(15 Suppl):5516.
Zannini L, Delia D, Buscemi G. CHK2 kinase in the DNA damage response and beyond. J Mol Cell Biol. 2014;6(6):442–57.
Patil M, Pabla N, Dong Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell Mol Life Sci. 2013;70(21):4009–21.
Lee JM, Nair J, Zimmer A, Lipkowitz S, Annunziata CM, Merino MJ, et al. Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild-type recurrent high-grade serous ovarian cancer: a first in class, proof-of-concept, single arm phase 2 study. Lancet Oncol. 2018;19(2):207–15.
Konstantinopoulos PA, Lee J, Gao B, Miller R, Lee JY, Colombo N, et al. A Phase 2 study of prexasertib (LY2606368) in platinum resistant or refractory recurrent ovarian cancer. Gynecol Oncol. 2022;167(2):213–25.
Do KT, Kochupurakkal B, Kelland S, de Jonge A, Hedglin J, Powers A, et al. Phase 1 combination study of the CHK1 inhibitor prexasertib and the PARP inhibitor olaparib in high-grade serous ovarian cancer and other solid tumors. Clin Cancer Res. 2021;27(17):4710–6.
Zatreanu D, Robinson HMR, Alkhatib O, Boursier M, Finch H, Geo L, et al. Polθ inhibitors elicit BRCA-gene synthetic lethality and target PARP inhibitor resistance. Nat Commun. 2021;12(1):3636.
Ding L, Kim HJ, Wang Q, Kearns M, Jiang T, Ohlson CE, et al. PARP inhibition elicits STING-dependent antitumor immunity in Brca1-deficient ovarian cancer. Cell Rep. 2018;25(11):2972-2980.e5.
Li T, Chen ZJ. The cGAS-cGAMP-STING pathway connects DNA damage to inflammation, senescence, and cancer. J Exp Med. 2018;215(5):1287–99.
Konstantinopoulos PA, Waggoner S, Vidal GA, Mita M, Moroney JW, Holloway R, et al. Single-arm phases 1 and 2 trial of niraparib in combination with pembrolizumab in patients with recurrent platinum-resistant ovarian carcinoma. JAMA Oncol. 2019;5(8):1141–9.
Yap TA, Im SA, Schram AM, Sharp A, Balmana J, Baird RD, et al. Abstract CT007: PETRA: first in class, first in human trial of the next generation PARP1-selective inhibitor AZD5305 in patients (pts) with BRCA1/2, PALB2 or RAD51C/D mutations. Cancer Res. 2022;82(12 Suppl):CT007.
Johnson RL, Cummings M, Thangavelu A, Theophilou G, de Jong D, Orsi NM. Barriers to immunotherapy in ovarian cancer: metabolic, genomic, and immune perturbations in the tumour microenvironment. Cancers. 2021;13(24):6231.
Wu JWY, Dand S, Doig L, Papenfuss AT, Scott CL, Ho G, et al. T-cell receptor therapy in the treatment of ovarian cancer: a mini review. Front Immunol. 2021;12: 672502.
Wang X, Teng F, Kong L, Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. OncoTargets Ther. 2016;9:5023–39.
Abiko K, Matsumura N, Hamanishi J, Horikawa N, Murakami R, Yamaguchi K, et al. IFN-γ from lymphocytes induces PD-L1 expression and promotes progression of ovarian cancer. Br J Cancer. 2015;112(9):1501–9.
Rodriguez GM, Galpin KJC, McCloskey CW, Vanderhyden BC. The tumor microenvironment of epithelial ovarian cancer and its influence on response to immunotherapy. Cancers. 2018;10(8):242.
Vázquez-García I, Uhlitz F, Ceglia N, Lim JLP, Wu M, Mohibullah N, et al. Ovarian cancer mutational processes drive site-specific immune evasion. Nature. 2022;612(7941):778–86.
LYNPARZA® and IMFINZI® combination improved progression-free survival in newly diagnosed patients with advanced ovarian cancer without tumor BRCA mutations in DUO-O Phase III trial. 2023. https://www.astrazeneca-us.com/media/press-releases/2023/lynparza-and-imfinzi-combination-improved-progression-free-survival-in-newly-diagnosed-patients-with-advanced-ovarian-cancer-without-tumor-brca-mutations-in-duo-phase-iii-trial.html
Monk BJ, Colombo N, Oza AM, Fujiwara K, Birrer MJ, Randall L, et al. Chemotherapy with or without avelumab followed by avelumab maintenance versus chemotherapy alone in patients with previously untreated epithelial ovarian cancer (JAVELIN Ovarian 100): an open-label, randomised, phase 3 trial. Lancet Oncol. 2021;22(9):1275–89.
Pujade-Lauraine E, Fujiwara K, Ledermann JA, Oza AM, Kristeleit R, Ray-Coquard IL, et al. Avelumab alone or in combination with chemotherapy versus chemotherapy alone in platinum-resistant or platinum-refractory ovarian cancer (JAVELIN Ovarian 200): an open-label, three-arm, randomised, phase 3 study. Lancet Oncol. 2021;22(7):1034–46.
Moore KN, Bookman M, Sehouli J, Miller A, Anderson C, Scambia G, et al. Atezolizumab, bevacizumab, and chemotherapy for newly diagnosed stage III or IV ovarian cancer: placebo-controlled randomized phase III trial (IMagyn050/GOG 3015/ENGOT-OV39). J Clin Oncol. 2021;39(17):1842–55.
Kurtz JE, Pujade-Lauraine E, Oaknin A, Belin L, Tsibulak I, Cibula D, et al. LBA30 Phase III ATALANTE/ov29 trial: atezolizumab (Atz) versus placebo with platinum-based chemotherapy (Cx) plus bevacizumab (bev) in patients (pts) with platinum-sensitive relapse (PSR) of epithelial ovarian cancer (OC). Ann Oncol. 2022;33:S1397.
Vaishampayan UN, Tomczak P, Muzaffar J, Winer IS, Rosen SD, Hoimes CJ, et al. Nemvaleukin alfa monotherapy and in combination with pembrolizumab in patients (pts) with advanced solid tumors: ARTISTRY-1. J Clin Oncol. 2022;40(16 Suppl):2500.
Nieuwenhuysen EV, O’Malley D, O’Cearbhaill RE, Moore KN, Hamilton EP, Yeku O, et al. 523MO Ubamatamab (REGN4018, MUC16xCD3 bispecific antibody) monotherapy in patients with recurrent ovarian cancer (OC): phase I dose-escalation analysis. Ann Oncol. 2022;33:S784.
Dorigo O, Fiset S, MacDonald LD, Bramhecha Y, Hrytsenko O, Dirk B, et al. DPX-Survivac, a novel T-cell immunotherapy, to induce robust T-cell responses in advanced ovarian cancer. J Clin Oncol. 2020;38(5 Suppl):6.
Veneziani A, Lheureux S, Alqaisi H, Bhat G, Colombo I, Gonzalez E, et al. Pembrolizumab, maveropepimut-S, and low-dose cyclophosphamide in advanced epithelial ovarian cancer: results from phase 1 and expansion cohort of PESCO trial. J Clin Oncol. 2022;40(16 Suppl):5505.
Leary A, Deluche E, Favier L, Paoletti X, Mansi L, Tredan O, et al. TEDOVA/GINECO-OV244b/ENGOT-ov58 trial: neo-epitope based vaccine OSE2101 alone or in combination with pembrolizumab versus best supportive care (BSC) as maintenance in platinum-sensitive recurrent ovarian cancer with disease control after platinum. J Clin Oncol. 2022;40(16 Suppl):TPS5614.
Drago JZ, Modi S, Chandarlapaty S. Unlocking the potential of antibody-drug conjugates for cancer therapy. Nat Rev Clin Oncol. 2021;18(6):327–44.
Calo CA, O’Malley DM. Antibody-drug conjugates for the treatment of ovarian cancer. Expert Opin Biol Ther. 2021;21(7):875–87.
Matulonis UA, Lorusso D, Oaknin A, Pignata S, Dean A, Denys H, et al. Efficacy and safety of mirvetuximab soravtansine in patients with platinum-resistant ovarian cancer with high folate receptor alpha expression: results from the SORAYA study. J Clin Oncol. 2023;41(13):2436–45.
US FDA, Center for Drug Evaluation and Research. FDA grants accelerated approval to mirvetuximab soravtansine-gynx for FRα positive, platinum-resistant epithelial ovarian, fallopian tube, or peritoneal cancer. FDA. 2022. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-mirvetuximab-soravtansine-gynx-fra-positive-platinum-resistant
Moore K, Konecny G, Martin L, Floquet A, O’Malley D, Colombo N, et al. MIRASOL: a randomized, open-label, phase 3 study of mirvetuximab soravtansine vs. investigator’s choice of chemotherapy in advanced high-grade epithelial ovarian, primary peritoneal, or fallopian tube cancers with high folate-alpha (FRα) expression (297). Gynecol Oncol. 2022;166:S156–7.
ELAHERE® Demonstrates Overall Survival Benefit in the Phase 3 MIRASOL Trial in Patients with FRα-Positive Platinum-Resistant Ovarian Cancer | ImmunoGen, Inc. https://investor.immunogen.com/news-releases/news-release-details/elaherer-demonstrates-overall-survival-benefit-phase-3-mirasol
O’Malley DM, Richardson DL, Vergote IB, Gilbert L, Castro C, Provencher D, et al. 833P Mirvetuximab soravtansine (MIRV), a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with carboplatin (CARBO) and bevacizumab (BEV): Final results from a study in patients (pts) with recurrent platinum sensitive ovarian cancer. Ann Oncol. 2020;31:S626–7.
O’Malley DM, Matulonis UA, Birrer MJ, Castro CM, Gilbert L, Vergote I, et al. Phase Ib study of mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), in combination with bevacizumab in patients with platinum-resistant ovarian cancer. Gynecol Oncol. 2020;157(2):379–85.
Matulonis UA, Moore KN, Martin LP, Vergote IB, Castro C, Gilbert L, et al. Mirvetuximab soravtansine, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC), with pembrolizumab in platinum-resistant ovarian cancer (PROC): initial results of an expansion cohort from FORWARD II, a phase Ib study. Ann Oncol. 2018;29:viii339.
Levan K, Mehryar M, Mateoiu C, Albertsson P, Bäck T, Sundfeldt K. Immunohistochemical evaluation of epithelial ovarian carcinomas identifies three different expression patterns of the MX35 antigen, NaPi2b. BMC Cancer. 2017;17(1):303.
Banerjee S, Oza AM, Birrer MJ, Hamilton EP, Hasan J, Leary A, et al. Anti-NaPi2b antibody-drug conjugate lifastuzumab vedotin (DNIB0600A) compared with pegylated liposomal doxorubicin in patients with platinum-resistant ovarian cancer in a randomized, open-label, phase II study. Ann Oncol. 2018;29(4):917–23.
Richardson D, Hamilton E, Barve M, Anderson C, Taylor S, Lakhani N, et al. Updated results from the phase 1 expansion study of Upifitamab Rilsodotin (UpRi; XMT-1536), a NaPi2b-directed Dolaflexin Antibody Drug Conjugate (ADC) in Ovarian Cancer (076). Gynecol Oncol. 2022;166:S48.
Cocco E, Varughese J, Buza N, Bellone S, Lin KY, Bellone M, et al. Tissue factor expression in ovarian cancer: implications for immunotherapy with hI-con1, a factor VII-IgGF(c) chimeric protein targeting tissue factor. Clin Exp Metastasis. 2011;28(7):689–700.
de Bono JS, Concin N, Hong DS, Thistlethwaite FC, Machiels JP, Arkenau HT, et al. Tisotumab vedotin in patients with advanced or metastatic solid tumours (InnovaTV 201): a first-in-human, multicentre, phase 1–2 trial. Lancet Oncol. 2019;20(3):383–93.
Ordóñez NG. Application of mesothelin immunostaining in tumor diagnosis. Am J Surg Pathol. 2003;27(11):1418–28.
Lheureux S, Alqaisi H, Cohn DE, Chern JY, Duska LR, Jewell A, et al. A randomized phase II study of bevacizumab and weekly anetumab ravtansine or weekly paclitaxel in platinum-resistant or refractory ovarian cancer NCI trial#10150. J Clin Oncol. 2022;40(16 Suppl):5514.
Veneris JT, Darcy KM, Mhawech-Fauceglia P, Tian C, Lengyel E, Lastra RR, et al. High glucocorticoid receptor expression predicts short progression-free survival in ovarian cancer. Gynecol Oncol. 2017;146(1):153–60.
Munster PN, Greenstein AE, Fleming GF, Borazanci E, Sharma MR, Custodio JM, et al. Overcoming Taxane resistance: preclinical and phase 1 studies of Relacorilant, a selective glucocorticoid receptor modulator, with Nab-Paclitaxel in Solid Tumors. Clin Cancer Res. 2022;28(15):3214–24.
Colombo N, Van Gorp T, Matulonis UA, Oaknin A, Grisham RN, Fleming GF, et al. Overall survival data from a 3-arm, randomized, open-label, phase 2 study of relacorilant, a selective glucocorticoid receptor modulator, combined with nab-paclitaxel in patients with recurrent platinum-resistant ovarian cancer. J Clin Oncol. 2022;40(17 Suppl):LBA5503.
Colombo N, Nguyen DD, Fleming GF, Grisham RN, Lorusso D, Gorp TV, et al. 721O Relacorilant, a selective glucocorticoid receptor modulator, in combination with nab-paclitaxel improves progression-free survival in patients with recurrent platinum-resistant ovarian cancer: a 3-arm, randomized, open-label, phase II study. Ann Oncol. 2021;32:S725.
Rankin EB, Giaccia AJ. The receptor tyrosine kinase AXL in cancer progression. Cancers. 2016;8(11):103.
Rankin EB, Fuh KC, Taylor TE, Krieg AJ, Musser M, Yuan J, et al. AXL is an essential factor and therapeutic target for metastatic ovarian cancer. Cancer Res. 2010;70(19):7570–9.
Fuh KC, Bookman MA, Liu JF, Coleman RL, Herzog TJ, Thaker PH, et al. Phase 1b study of AVB-500 in combination with paclitaxel or pegylated liposomal doxorubicin platinum-resistant recurrent ovarian cancer. Gynecol Oncol. 2021;163(2):254–61.
Spiegel J, Adhikari S, Balasubramanian S. The structure and function of DNA G-quadruplexes. Trends Chem. 2020;2(2):123–36.
Chambers VS, Marsico G, Boutell JM, Di Antonio M, Smith GP, Balasubramanian S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat Biotechnol. 2015;33(8):877–81.
Bryan TM. Mechanisms of DNA replication and repair: insights from the study of G-quadruplexes. Mol Basel Switz. 2019;24(19):3439.
Zimmer J, Tacconi EMC, Folio C, Badie S, Porru M, Klare K, et al. Targeting BRCA1 and BRCA2 deficiencies with G-quadruplex-interacting compounds. Mol Cell. 2016;61(3):449–60.
Hilton J, Gelmon K, Bedard PL, Tu D, Xu H, Tinker AV, et al. Results of the phase I CCTG IND.231 trial of CX-5461 in patients with advanced solid tumors enriched for DNA-repair deficiencies. Nat Commun. 2022;13:3607.
Alqaisi H, Lheureux S, McMullen M, Knox JJ, Bowering V, Lee C, et al. OZM-114: Phase Ib expansion study of CX-5461in patients with solid tumors and BRCA2 and/or PALB2 mutation. J Clin Oncol. 2022;40(16 Suppl):TPS5621.
Kristeleit R, Lisyanskaya A, Fedenko A, Dvorkin M, de Melo AC, Shparyk Y, et al. Rucaparib versus standard-of-care chemotherapy in patients with relapsed ovarian cancer and a deleterious BRCA1 or BRCA2 mutation (ARIEL4): an international, open-label, randomised, phase 3 trial. Lancet Oncol. 2022;23(4):465–78.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The authors are supported by the Princess Margaret Cancer Foundation.
Conflicts of Interest
Amit M. Oza: PI and Steering Committees with AstraZeneca and GSK; advisory boards for Merck (uncompensated) and Morphosys; and Chief Executive Officer at Ozmosis Research (uncompensated). Vikas Garg reports no conflicts of interest.
Ethics Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Data Availability
No datasets were generated or analysed during the current study.
Authors’ Contributions
VG made substantial contributions to the concept and design of the work and approved the final version to be published. AMO revised the work critically for important intellectual content and approved the final version to be published.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Garg, V., Oza, A.M. Treatment of Ovarian Cancer Beyond PARP Inhibition: Current and Future Options. Drugs 83, 1365–1385 (2023). https://doi.org/10.1007/s40265-023-01934-0
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-023-01934-0