
Abstract
Purpose
Ovarian cancer is the most lethal gynaecological malignancy. Despite the introduction of bevacizumab, standard chemotherapy has remained largely unchanged and the vast majority of patients will relapse within the first two years of diagnosis. However, results from recent clinical trials demonstrating clinical benefits of PARP inhibitor treatment are rapidly changing therapeutic options for many patients with ovarian cancer.
Methods
Given the introduction of new therapeutic options in the treatment of ovarian cancer, we critically review key clinical trials, areas of scientific research and its clinical relevance.
Results
Most notably, patients with BRCA1/2 mutant ovarian cancer benefit from maintenance treatment with PARP inhibitors after (complete or partial) response to platinum-based chemotherapy. Here, we discuss the mechanism of PARP inhibition, multiple drug resistance mechanisms, including BRCA reverse mutations, altered PARP expression, changes in DNA repair pathways, kinase activation and additional drug targets that may augment PARP inhibition.
Conclusion
Although the use of PARP inhibitors is a huge step forward, it is apparent that patients, both with and without BRCA-mutant ovarian cancer, will eventually become resistant to PARP inhibitors. Therefore, novel combination therapies may enhance PARP inhibitor efficacy and overcome resistance mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
While being the fifth most common gynaecological malignancy, ovarian cancer is the leading cause of death from gynaecological malignancies; about 7500 women are newly diagnosed and about 5500 die from the disease in Germany each year [1]. Epithelial ovarian cancer accounts for about 90% of the disease [2], of which high-grade serous ovarian cancer (HGSOC) shows the lowest average 5-year survival rates of only about 40% for advanced stages of the disease [3, 4]. The mainstay of treatment consists of surgical debulking with macroscopic complete resection and platinum-based chemotherapy. It has been demonstrated that surgical debulking is the only modifiable prognostic factor after diagnosis of HGSOC. If macroscopic complete resection can be achieved, 5-year survival rates may improve from around 20% to up to 60% for advanced HGSOC [3]. However, most patients (80%) will relapse within the first 2 years [5]. Despite the burden of the disease, standard chemotherapy regimens have remained largely unchanged for decades [6, 7].
However, the most notable exception to the dearth of new treatment options has been the introduction of the anti-angiogenesis drug bevacizumab for patients with advanced ovarian cancer. Until recently, it had been the only new approved therapy for the last decade. Maintenance therapy with bevacizumab showed an improved PFI [8, 9]. Although it failed to reduce overall survival in the ITT population, the subgroup analysis of high-risk patients showed an overall survival benefit, with high-risk being FIGO stage III with > 1 cm residual disease, FIGO stage IV or inoperable disease [8]. The other very recent milestone and most intriguing new therapeutic option consists of polyADP-ribose polymerase inhibitors (PARPis). In the last few years, several clinical trials have shown benefits for primary as well as recurrent ovarian cancer when used as maintenance treatment after initial complete response (CR) or partial response (PR) to platinum-based chemotherapy [10,11,12,13,14,15,16,17,18] (Table 1). This can partially be explained by the high prevalence of tumours with homologous recombination deficiency (HRD), which is found in about 50% of all ovarian cancers. BRCA1/2 deficiency accounts for about 20% of these cases [19]. This underlies the molecular rationale and the significant clinical benefit of PARPis in BRCA1/2 mutation carriers and patients with HR-deficient tumours. Although it also shows a significant prolonged PFS in patients with BRCA1/2wt and HR-proficient tumours, this substantial clinical benefit is less compared to the other subgroups mentioned above [14]. Due to the lack of a widely available (EMA approved) HRD test with a reproducible threshold, one of the main clinical challenges is the identification of patients that are most likely to benefit from PARP inhibitors. This is particularly true for patients with BRCA1/2wt tumours. Although the most recent first-line clinical trials in ovarian cancer used the same HRD test (myChoice®, Myriad Genetics), thresholds to define patients’ tumours as HRD differed, which greatly hinders comparison of clinical response and prevents smooth translation into common clinical practice [10,11,12]. Nonetheless, this HRD test has been FDA-approved to determine HRD status in tumours.
It is critical to understand the underlying mechanisms of resistance to enhance the clinical use of PARP inhibition in ovarian cancer. Here, we discuss the mechanisms of PARP inhibition, potential drug resistance mechanism, and strategies to enhance efficacy of PARPis in the setting of ovarian cancer. Given the new therapeutic options in the treatment of ovarian cancer, we critically review key areas of scientific research and its clinical relevance.
The role of PARP
The enzyme PARP belongs to a family of proteins that catalyse the polymerisation of ADP-ribose (PARylation) to its target proteins or itself [20]. PARylation uses NAD + as a substrate, releasing nicotinamide as the by-product. PARP-1 and PARP-2 are the only members of the family that have been shown to be involved in DNA single-strand break (DNA SSB) repair [21]. Other functions include cell death and cell cycle regulation [20].
PARP-1 is the most studied protein of the family and contains multiple domains with well-characterised functions, including binding to DNA break sites, nuclear recruitment and the enzymatic domain [20]. Specifically, PARP-1 is upstream of the DNA SSB repair mechanism called base excision repair [21]. This allows the identification of a damaged part of DNA, followed by the synthesis of new DNA at the site of the DNA SSB. Here, PARP-1/2 act as a sensor for DNA SSBs, recruiting other key players, such as XRCC1 and DNA ligase 3 [20]. Typically, DNA SSBs are repaired in G1. If unsuccessful, DNA double strand breaks (DNA DSBs) accumulate upon DNA replication [22]. Cells have evolved mechanisms to repair such DNA DSBs, namely HR and non-homologous end-joining (NHEJ). HR and NHEJ are important to maintain genomic integrity, but differ significantly in their mechanism, accuracy, and time of repair within the cell cycle. HR acts mainly in S phase after DNA replication, because it requires DNA sequence homology of sister chromatids as a template to repair DNA DSBs [23]. It is initiated upon DNA 5′-end resection and demonstrates a high-fidelity mechanism that conserves genomic stability. Key players are BRCA1/2, RAD51, and PALB2 [24]. NHEJ acts throughout the cell cycle by directly ligating the ends of DNA DSBs, which increases the chance of deletions and mutations since sister chromotids are not used as template [24]. An important player for NHEJ is 53BP1 which acts in combination with other factors in determining DNA DSB repair [25].
The mechanism(s) of PARP inhibitors
PARPis compromise adequate DNA SSB repair, leading to the persistence of DNA SSBs. If unrepaired in G1, this will lead to the accumulation of DNA DSBs upon DNA replication during S phase. The mechanism of action of PARPis is as follows: PARPis block the NAD + binding site on PARP, effectively inhibiting PARylation. This prevents PARP dissociation from DNA SSBs, resulting in both accumulation of unrepaired DNA SSBs and PARP trapping [26]. If the DNA SSB is not fully repaired and the cell enters S phase, DNA DSBs occur, which, in turn, require an effective repair mechanism to ensure genomic stability [26, 27]. Therefore, cells rely disproportionally on DNA DSB repair in the presence of PARP inhibition. In the case of HRD, as seen in BRCA1/2 mutations carriers, cells repair their DNA DSBs via NHEJ, resulting in deleterious genomic instability. This mechanism is termed synthetic lethality, because PARPis exploit the Achilles’ heel of BRCA1/2-deficient tumour cells. Additionally, PARP trapping occurs because of the lack of autoPARylation which normally initiates DNA dissociation of PARP [28]. Interestingly, the most commonly used PARPi show different potencies of PARP trapping, which is (partly) independent of the inhibitory effect of PARP [26,27,28]. However, it has been shown that the exact extent of PARP trapping potency depends on both the experimental method and whether it is tested as monotherapy or combination therapy, making comparisons difficult. Typically, talazoparib commonly shows the greatest potency and veliparib the lowest, with olaparib, niraparib, and rucaparib in-between [28,29,30]. In summary, PARPis exhibit two main mechanisms of action, by compromising DNA SSB repair and by PARP trapping. Many clinical trials have shown the efficacy of PARPi in patients with ovarian cancer [10,11,12,13,14,15,16,17,18].
Key clinical trials of PARP inhibitors in ovarian cancer
PARPis are licensed as maintenance treatment for recurrent ovarian cancer after response (CR/PR) to platinum-based chemotherapy independent of BRCA1/2 status and also as maintenance treatment for primary high-grade serous ovarian, fallopian tube, or peritoneal cancer after response (CR/PR) to platinum-based chemotherapy in BRCA1/2 mutation carriers. In addition, rucaparib is also licenced as a monotherapy in platinum-sensitive relapsed/progressive BRCAm (somatic or germline) ovarian cancer, if the patient had ≥ 2 lines of platinum-based chemotherapy and is unable to tolerate another platinum-based chemotherapy. There have been many recent clinical trials assessing the efficacy of PARPis in patients with recurrent or primary ovarian cancer [10,11,12,13,14,15,16,17,18]. This showed that PARPis significantly prolong the PFS as a maintenance treatment after response (CR/PR) to platinum-based chemotherapy, most notably in patients with BRCAm and/or HR-deficient ovarian cancers (Table 1).
Olaparib was one of the first PARPis, which was introduced into clinical practice for (recurrent) ovarian cancer. The randomised, double-blind phase 2 clinical trial with olaparib (Study 19) showed an improved PFS of 8.4 months versus 4.8 months in patients with platinum-sensitive recurrent ovarian cancer (Table 1) [15]. The further subgroup analysis of BRCA mutation carriers (about 50% of the randomised patients) showed a significantly improved PFS of 11.2 versus 4.3 months (HR 0.18 [95% CI 0.10–0.31], p < 0.0001) [15, 31]. The overall survival benefit was most pronounced in patients with BRCA1/2 mutations with a median of 34.9 months (29.2–54.6 months) versus 30.2 months (23.1–40.7 months), HR 0.62 [95% CI 0.41–0.94], nominal p = 0.025) [32]. This did not meet the statistical significance threshold set in this clinical trial, but demonstrated that BRCA1/2 mutations carriers are the patients that most likely benefit from olaparib maintenance treatment after response (CR/PR) to platinum-based chemotherapy [32]. The randomised, double-blinded, placebo-controlled SOLO-2 trial confirmed these results in olaparib-treated patients with BRCA-mutated platinum-sensitive recurrent ovarian cancer, showing an improved PFS of 19.1 months versus 5.5 months (HR 0.30 [95% CI 0.22–0.41]) [16].
Another study (ENGOT-OV16/NOVA) evaluated the effect of niraparib maintenance treatment after response (CR/PR) to platinum-based chemotherapy. This randomised, double-blinded, placebo-controlled phase 3 clinical trial distinguished three groups of patients in the order of increasing efficacy to niraparib: BRCAwt and HR-proficient tumours < BRCAwt and HR-deficient tumours < BRCA1/2 m tumours [14]. This showed that patients with BRCA1/2 mutant tumours benefited most from maintenance treatment with 21.0 versus 5.5 months (HR 0.27 [95% CI 0.17–0.41]) (Table 1) [14]. Expanding the use of PARPis beyond BRCA mutations and trying to identify predictive biomarkers, the ARIEL-2 (part1) trial further classified tumours according to HRD status. The trial used high versus low loss of heterozygosity (LOH) as a marker, measured by NGS. Although showing an improved PFS in patients with high LOH BRCAwt tumours versus low LOH BRCAwt tumours [5.7 months (5.3–7.6 months) versus 5.2 months] (3.6–5.5 months) (HR 0.62 [95% CI 0.42–0.90]), this single-arm trial could not determine whether or not LOH/HRD status can be used as a predictive biomarker [17]. The use of HRD status as a biomarker for treatment response was further assessed in the double-blind, placebo-controlled, ARIEL-3 trial (phase 3) (Table 1) [18]. This trial also included an exploratory analysis showing that patients with BRCAwt and low LOH tumours have a significantly improved PFS when treated with rucaparib versus placebo (HR 0.58 [95% CI 0.40–0.85]) [18]. Together with the results of the NOVA and Study19 trial [14, 15], the ARIEL-3 trial expanded the use of PARPis to patients with platinum-sensitive recurrent ovarian cancer beyond HRD and BRCA mutation status [18].
These trials were followed by studies investigating the benefit in patients with newly diagnosed ovarian cancer after responding (CR/PR) to first-line platinum-based chemotherapy. A recent clinical trial (SOLO-1) compared olaparib versus placebo in BRCA1/2 mutation carriers with newly diagnosed ovarian cancer after response (CR/PR) to platinum-based chemotherapy [13]. This clinical trial (SOLO-1) showed a significantly improved PFS in the olaparib arm (60% versus 27%) at 3 years (Table 1) [13]. At the time of publication, the median PFS was not reached (i.e. not published) for the treatment arm and the clinical trial has not yet reported on overall survival. Interestingly, the authors estimate that the first subsequent therapy or death would be 51.8 months in the olaparib and 15.1 months in the placebo group (HR 0.30 [95% CI 0.22–0.40] [13]. The most recently published trials (VELIA, PRIMA, and PAOLA-1, Table 1) demonstrated a clear benefit of PARPis (veliparib, niraparib, and olaparib) in first-line treatment in patients with HR-deficient ovarian cancer, showing a significant improved PFS [10,11,12]. In addition to the different PARPis investigated as first-line therapy in these clinical trials, many key parameters also differed: combination treatment (bevacizumab or not), start time of the individual PARPi treatment (only during, throughout or after CR/PR to chemotherapy), patient cohort (high risk of relapse/progression or broad patient cohort), and thresholds for HRD testing of tumour tissue (myChoice®, Myriad Genetics). This requires careful consideration of when and how to translate these findings into common clinical practice (Table 1) [10,11,12]. First, the PAOLA-1 trial included bevacizumab in the maintenance treatment, which was part of the inclusion criteria and had to be started during chemotherapy; the other two trials did not. Given that bevacizumab maintenance is the standard treatment in many countries for advanced ovarian cancer, it would have been most interesting to investigate the clinical efficacy of combining of niraparib and bevacizumab in the patient cohort of the PRIMA trial. Interestingly, a three-arm clinical trial comparing niraparib and bevacizumab (± a checkpoint inhibitor) versus standard of care in patients with platinum-sensitive recurrent ovarian cancer is planned, but not yet recruiting (NCT03806049). This trial is based on the randomised superiority phase 2 trial of niraparib alone versus niraparib and bevacizumab in platinum-sensitive recurrent ovarian cancer, which showed an improved PFS of 11.9 months (8.5–16.7) versus 5.5 months (3.8–6.3), (HR 0.35 [95% CI 0.21–0.57], p < 0.0001) [33]. Second, the VELIA trial was the only trial that was designed with two different experimental arms: PARPi treatment during chemotherapy only, followed by placebo maintenance treatment compared to veliparib throughout chemotherapy and in the maintenance treatment. It demonstrated that PARP inhibition (with veliparib) during chemotherapy only is inferior to PARP inhibition throughout chemotherapy and maintenance treatment. The chemotherapy-only arm did not demonstrate an improved PFS across the different subgroups (BRCAm, HRD, and BRCAwt). The veliparib (during chemotherapy only)-treated ITT patient cohort showed a PFS of 15.2 months (14.1–17.3 months) versus 17.3 months (15.1–19.1 months) in the placebo-treated ITT patient cohort (HR 1.07 [95% CI 0.90–1.29]) [10]. Third, compared to the other two trials, the PRIMA trial included high-risk patients, i.e., FIGO III patients with visible residual tumour after primary debulking and inoperable FIGO IV disease [11]. This may explain the difference in the median PFS in the overall populations between the clinical trials: 23.8 months (VELIA) ≥ 22.1 months (PAOLA-1) > 13.8 months (PRIMA) [10,11,12]. The PRIMA trial was also the only trial (of the three mentioned above), which showed a significant benefit in PFS for niraparib-treated patients with HR-proficient tumours (incl. unknown HRD status), albeit to a smaller effect than seen in the HRD cohort. Specifically, niraparib significantly improved PFS in patients with HR-proficient tumours with 8.1 months (5.7–9.4 months) versus 5.4 months (4.0–7.3 months) in placebo-treated patients with HR-proficient tumours (HR 0.68 [95% CI 0.49–0.94], p = 0.020) [11]. Finally, the three clinical trials (VELIA, PRIMA, and PAOLA-1) relied on a commercially available assay to assess HRD status (myChoice®, Myriad Genetics); yet agreed standard procedures are lacking, i.e., the thresholds that define “HRD positivity” [10,11,12]. The set threshold for positive result of HRD was ≥ 42 in the PRIMA and PAOLA-1 trials, whereas the threshold was set as ≥ 33 in the VELIA trial, consequently, including more patients with HR-deficient tumours in the latter. Hence, one of the biggest challenges will be establishing clinically reproducible, widely available, and standardised testing for HRD status in the absence of BRCA1/2 mutations. This would identify those patients that will benefit from PARP inhibition after response to chemotherapy. A serological test would be of high clinical interest, if it circumvents the requirement of tissue sampling, and ultimately allowing the stratification of patients with recurrent ovarian cancer, when obtaining fresh tumour tissue is not typically an option.
Although these clinical trials showed promising results by significantly improving PFS, key questions remain. It is obvious that most, if not all, patients will eventually develop PARPi resistance. The time of resistance may dependent on the distinct vulnerability that underlies the efficacy of PARPis, i.e., BRCA mutation and HRD status. One could speculate that the degree of complexity of HRD in tumours underpins the development of resistance. This means that resistance to a PARPi would develop quicker if it does not “require” to counteract a (comparatively) complex mechanism. This assumption is based on the clinical observation that, after response to platinum-based chemotherapy, patients with tumours classed as “HR-proficient” benefit less from PARPis compared to patients with HR-deficient or BRCA1/2 mutant tumours [14]. Genomic analysis of long-term versus short-term responders to olaparib showed that response to olaparib was associated with BRCA1/2 mutations [34]. The authors further speculated that the underlying type of BRCA mutations may allow for a more accurate prediction of long-term responders. Similarly, a more accurate HRD test could give us a better understanding of the factors contributing to a sustained clinical response to PARP inhibition. On one hand, a potential approach to identify novel biomarkers would be to uncover the underlying resistance mechanisms in patients who do not respond to PARP inhibition. On the other hand, it would be essential to identify long-term survivors with BRCAwt and HR-proficient tumours treated with PARPi and characterise the underlying biomarkers that may predict such an exceptional response. This exciting avenue would also shed light onto potential strategies to enhance PARPi efficacy.
Resistance mechanisms and strategies to increase efficacy
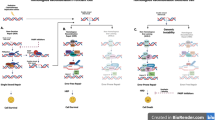
Many potential mechanisms of PARPi resistance have been described (Fig.1). The strongest rationale for the clinical development of PARPis stemmed from the response seen in BRCA1/2-deficient (and HR-deficient) cells. Hence, an intriguing mechanism of resistance involves secondary somatic reversion mutations in the BRCA1/2 genes in tumour cells, which essentially restore HR [35]. This could potentially be explained by tumour heterogeneity and clonal expansion driven by chemotherapy, termed “Darwinian escape” [36]. Comparing primary and recurrent ovarian cancers, it was shown that 13 out of 46 recurrent HGSOCs (28.3%, 95% CI 17.3–42.6%) had a secondary mutation and 2 out of 64 primary HGSOCs (3.1%, 95% CI 1.0–10.7%) in germline BRCA1/2 mutation carriers. This was even more pronounced in platinum-resistant versus platinum-sensitive ovarian cancer, with almost 50% of platinum-resistant cancer (12 out of 26) showing BRCA1/2 reversion mutations [37]. Secondary reverse mutations in BRCA1/2 were also shown to correlate with PARPi resistance in ovarian cancer and other cancers [35, 37]. Most interestingly, secondary reversion mutation of BRCA1/2 alleles have been detected by analysing circulating free DNA (cfDNA) in patients with prostate cancer [38]. This would offer a non-invasive method to more accurately predict response to platinum-based chemotherapy and/or PARPi treatment [38]. Other mechanisms affecting gene expression include BRCA1/2 hypermethylation [27]. Secondary reversion mutations of RAD51, a component of the HR machinery, have been implicated in causing PARPi resistance in rucaparib-treated patients with platinum-sensitive HGSOC [39].

Resistance mechanisms and potential targets of combination therapies. A summary of resistance mechanisms and potential drug targets with a list of corresponding inhibitors that can be used in combination with PARPi
However, PARPi efficacy can also be impaired by BRCA-independent mechanisms that restore HR. The TP53-binding protein 1 (53BP1) is another important player in deciding the pathway by which DNA DSBs are repaired. It antagonises BRCA1 and inhibits end resection of DNA DSBs, thus favouring NHEJ [40]. In turn, the loss of 53BP1 has shown to favour HR in the absence of BRCA by potentially promoting DNA DSB end resection and RAD51 recruitment [41]. In tumour samples, low expression levels of 53BP1 correlated with a poorer response to PARPis in ovarian cancer with HRD [42]. In triple-negative breast cancer models, loss of 53BP1 rescues BRCA deficiency, reduces chemotherapy hypersensitivity, and is associated with a poorer prognosis [43]. This suggests clinical significance in testing for 53BP1 status in patients to assess efficacy of PARP inhibition. Similarly, the loss of a downstream factor of 53BP1, Rev7, has also shown to restore HR in BRCA-deficient cells, leading to PARPi resistance [44]. It remains to be seen how this mechanism of resistance could be alleviated. One could speculate that yet to be discovered factors that activate or stabilise 53BP1 could be used to restore drug efficacy in this context.
Another possibility is the loss or diminished expression of PARP-1 in tumour cells, because they essentially lack the drug target. This is particularly true for the mechanism of PARP trapping, which relies on the formation of DNA-bound complexes [26]. This suggests that stable PARP–DNA complex (at least) contributes to the cytotoxic effect of PARP inhibition [28]. Other resistance mechanisms are linked to the dynamic equilibrium between PARP-1 and PARG. The latter inhibits PARylation and effectively works as a physiological PARPi, which is needed to fully block PARylation in PARPi-treated cells [45].
Besides general drug efflux mechanisms involving p-glycoproteins [27], PARPi resistance could rely on other potentially ‘drugable’ factors that impair PARPi efficacy. Hence, PARPi sensitivity could be restored using combination therapy if the resistance mechanisms could be circumvented.
An interesting target could be the receptor tyrosine kinase c-Met, mesenchymal–epithelial transition factor, which is encoded by the c-met protooncogen. It is known that high c-Met expression is associated with a poor prognosis in ovarian cancer [46], which hints at a potential therapeutic exploitation of this pathway. More recently, c-Met has been linked to PARPi resistance in breast cancer cells, as well as restoring HR in a BRCA-independent manner to impair PARPi function. A small phase 2 clinical trial assessing the monoclonal antibody rilotumumab in ovarian cancer has shown no benefits as a single agent [47]. However, patient selection has been (at least) questionable, because c-Met expression has not been an inclusion criterion. Given that one patient had a complete response and two patients (out 31) had a 6-month PFS, it would be interesting to look for aberrant c-Met expression in this subgroup. This would be consistent with the previous studies demonstrating high expression levels of c-Met in about 10% of HGSOC [48]. Another c-Met inhibitor cabozantinib has demonstrated some clinical benefits in phase 2 clinical trials as a single agent in patients with platinum-resistant or -refractory ovarian cancer [49]. Interestingly, c-Met inhibition was shown to impair HR in vitro [50, 51], and it was recently shown that c-Met-mediated PARP phosphorylation confers PARPi resistance in preclinical breast cancer models [52]. However, it remains to be seen whether targeting c-Met in combination with PARP inhibition can show substantial clinical benefit in ovarian cancer.
The inhibition of cyclin-dependent kinases (CDK) has been suggested to sensitise BRCA-proficient cells to PARPis. They play a crucial role in cell cycle progression and DNA damage control, also affecting BRCA1/2 directly. CDK1 promotes mitotic progression by binding cyclinB1 [53]. Phosphorylation of BRCA1 by CDK1 is also important for activation of downstream signalling and foci formation [54]. In turn, loss of inhibition of CDK1 impairs BRCA function, creating a state of “BRCAness” [55]. It was shown in breast cancer cells that compromising CDK1 activity (by depletion or inhibition) sensitises cells to PARPis in BRCA-proficient tumours. This offers an attractive drug target, because a plethora of commercially available CDK inhibitors exists. Given CDK4/6 inhibitors, such as palbociclib or ribociclib, are already licensed for hormone-receptor positive breast cancer [52], this might facilitate early clinical translation, given the vast existing clinical experience with a comparatively similar class of drugs [56]. CDK1 inhibitors are thought to be particularly useful in combination therapy with PARPis, as this would create a state of ‘BRCAness’ [57].
The combination of PI3K inhibitors with PARPi is a well-studied and comparatively advanced approach of extending the use of PARPi. Similar to CDK1 inhibitors, PI3K inhibitors create a state of ‘BRCAness’ in BRCA-proficient cells. It has been shown that the PI3K inhibitor (BKM120) downregulates BRCA expression in cell lines and in patient-derived xenografts [58]. This was mediated by ERK signalling via the transcription factor ETS1 [58]. Other mechanisms of PI3K inhibition might be the impaired recruitment of RAD51 to sites of DNA DSBs, thus reducing HR [59]. Recent phase 1 clinical trials of combining PI3K and PARPis are promising and warrant further clinical evaluation [60, 61]. In a dose escalation trial of BKM120 in combination with olaparib, it was shown that the median duration of stable disease in patients (n = 45) without progressive disease as best overall response was 6.9 months (90% CI 5.5–7.5 months) [60].
There are promising strategies that may enhance the clinical use of PARPi, given the variety of (pre-)clinically tested combination therapies. There is particular need to increase clinical efficacy in patients with BRCAwt tumours because of the comparatively small(er) effect of PARPi in this patient cohort [10,11,12, 14, 17, 18, 32]. On the other hand, it will be crucial to more accurately predict PARPi efficacy in patients with HRD and (to a lesser extend) BRCA mutation carriers.
Future prospects
Although different combination therapies could potentially enhance PARPi efficacy and circumvent resistance mechanisms, it will be pivotal to identify those patients that are most likely to benefit. Therefore, adequate predictive biomarkers will need to be identified to allow for accurate patient selection.
To screen for HRD, rather than relying soley on the clinical response to platinum-based chemotherapy, would offer a more accurate patient stratification. This clearly depends on whether further characterisation could lead to improved treatment options for patients, as well as the availability and reproducibility of such a test. Nonetheless, it would give more patients the chance to be treated with a PARPi, and potentially additional targeted therapies.
Ongoing clinical trials are assessing the maintenance treatment of PARPi in combination with bevacizumab or checkpoint inhibitors. There are several current clinical trials investigating the use of PARPi in combination with checkpoint inhibitors in ovarian cancer (NCT03806049, NCT03574779, NCT03598270, NCT02657889, NCT02953457, NCT03522246, and NCT03737643). It remains to be seen whether those combinations show substantial clinical efficacy and improve overall survival.
References
Barnes B, Kraywinkel K, Nowossadeck E, Schönfeld I, Starker A, Wienecke A, Wolf U (2016) Bericht zum Krebsgeschehen in Deutschland 2016. Robert Koch-Institut. Berlin, Germany
Prat J (2012) New insights into ovarian cancer pathology. Ann Oncol 23(Suppl 10):x111–x117
Wimberger P, Wehling M, Lehmann N, Kimmig R, Schmalfeldt B, Burges A et al (2010) Influence of residual tumor on outcome in ovarian cancer patients with FIGO stage IV disease: an exploratory analysis of the AGO-OVAR (Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group). Ann Surg Oncol 17:1642–1648
Wimberger P, Lehmann N, Kimmig R, Burges A, Meier W, du Bois A et al (2007) Prognostic factors for complete debulking in advanced ovarian cancer and its impact on survival. An exploratory analysis of a prospectively randomized phase III study of the Arbeitsgemeinschaft Gynaekologische Onkologie Ovarian Cancer Study Group (AGO-OVAR). Gynecol Oncol. 106:69–74
du Bois A, Reuss A, Pujade-Lauraine E, Harter P, Ray-Coquard I, Pfisterer J (2009) Role of surgical outcome as prognostic factor in advanced epithelial ovarian cancer: a combined exploratory analysis of 3 prospectively randomized phase 3 multicenter trials: by the Arbeitsgemeinschaft Gynaekologische Onkologie Studiengruppe Ovarialkarzinom (AGO-OVAR) and the Groupe d'Investigateurs Nationaux Pour les Etudes des Cancers de l'Ovaire (GINECO). Cancer 115:1234–1244
Ledermann JA (2018) First-line treatment of ovarian cancer: questions and controversies to address. Ther Adv Med Oncol 10:1758835918768232
Klotz DM, Wimberger P (2017) Cells of origin of ovarian cancer: ovarian surface epithelium or fallopian tube? Arch Gynecol Obstet 296:1055–1062
González Martín A, Oza AM, Embleton AC, Pfisterer J, Ledermann JA, Pujade-Lauraine E et al (2019) Exploratory outcome analyses according to stage and/or residual disease in the ICON7 trial of carboplatin and paclitaxel with or without bevacizumab for newly diagnosed ovarian cancer. Gynecol Oncol 152:53–60
Perren TJ, Swart AM, Pfisterer J, Ledermann JA, Pujade-Lauraine E, Kristensen G et al (2011) A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med 365:2484–2496
Coleman RL, Fleming GF, Brady MF, Swisher EM, Steffensen KD, Friedlander M et al (2019) Veliparib with first-line chemotherapy and as maintenance therapy in ovarian cancer. N Engl J Med. 381:2403–2415
Ray-Coquard I, Pautier P, Pignata S, Pérol D, González-Martín A, Berger R et al (2019) Olaparib plus bevacizumab as first-line maintenance in ovarian cancer. N Engl J Med 381:2416–2428
González-Martín A, Pothuri B, Vergote I, DePont Christensen R, Graybill W, Mirza MR et al (2019) Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med. 381:2391–2402
Moore K, Colombo N, Scambia G, Kim BG, Oaknin A, Friedlander M et al (2018) Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 379:2495–2505
Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A et al (2016) Niraparib maintenance therapy in platinum-sensitive, recurrent ovarian cancer. N Engl J Med 375:2154–2164
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G et al (2012) Olaparib maintenance therapy in platinum-sensitive relapsed ovarian cancer. N Engl J Med 366:1382–1392
Pujade-Lauraine E, Ledermann JA, Selle F, Gebski V, Penson RT, Oza AM et al (2017) Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): a double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol 18:1274–1284
Swisher EM, Lin KK, Oza AM, Scott CL, Giordano H, Sun J et al (2017) Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): an international, multicentre, open-label, phase 2 trial. Lancet Oncol 18:75–87
Coleman RL, Oza AM, Lorusso D, Aghajanian C, Oaknin A, Dean A et al (2017) Rucaparib maintenance treatment for recurrent ovarian carcinoma after response to platinum therapy (ARIEL3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 390:1949–1961
Network CGAR (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474:609–615
Amé JC, Spenlehauer C, de Murcia G (2004) The PARP superfamily. BioEssays 26:882–893
Ström CE, Johansson F, Uhlén M, Szigyarto CA, Erixon K, Helleday T (2011) Poly (ADP-ribose) polymerase (PARP) is not involved in base excision repair but PARP inhibition traps a single-strand intermediate. Nucleic Acids Res 39:3166–3175
Kuzminov A (2001) Single-strand interruptions in replicating chromosomes cause double-strand breaks. Proc Natl Acad Sci USA 98:8241–8246
Nicolay NH, Carter R, Hatch SB, Schultz N, Prevo R, McKenna WG et al (2012) Homologous recombination mediates S-phase-dependent radioresistance in cells deficient in DNA polymerase eta. Carcinogenesis 33:2026–2034
Lord CJ, Ashworth A (2012) The DNA damage response and cancer therapy. Nature 481:287–294
Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C et al (2018) 53BP1 cooperation with the REV7-shieldin complex underpins DNA structure-specific NHEJ. Nature 560:122–127
Pommier Y, O'Connor MJ, de Bono J (2016) Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 8:362ps17
D'Andrea AD (2018) Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst) 71:172–176
Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH et al (2012) Trapping of PARP1 and PARP2 by clinical PARP inhibitors. Cancer Res 72:5588–5599
Hopkins TA, Ainsworth WB, Ellis PA, Donawho CK, DiGiammarino EL, Panchal SC et al (2019) PARP1 trapping by PARP inhibitors drives cytotoxicity in both cancer cells and healthy bone marrow. Mol Cancer Res 17:409–419
Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S et al (2014) Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther 13:433–443
Ledermann J, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G et al (2014) Olaparib maintenance therapy in patients with platinum-sensitive relapsed serous ovarian cancer: a preplanned retrospective analysis of outcomes by BRCA status in a randomised phase 2 trial. Lancet Oncol 15:852–861
Ledermann JA, Harter P, Gourley C, Friedlander M, Vergote I, Rustin G et al (2016) Overall survival in patients with platinum-sensitive recurrent serous ovarian cancer receiving olaparib maintenance monotherapy: an updated analysis from a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Oncol 17:1579–1589
Mirza MR, Åvall Lundqvist E, Birrer MJ, dePont CR, Nyvang GB, Malander S et al (2019) Niraparib plus bevacizumab versus niraparib alone for platinum-sensitive recurrent ovarian cancer (NSGO-AVANOVA2/ENGOT-ov24): a randomised, phase 2, superiority trial. Lancet Oncol 20:1409–1419
Lheureux S, Lai Z, Dougherty BA, Runswick S, Hodgson DR, Timms KM et al (2017) Long-term responders on olaparib maintenance in high-grade serous ovarian cancer: clinical and molecular characterization. Clin Cancer Res 23:4086–4094
Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA et al (2008) Resistance to therapy caused by intragenic deletion in BRCA2. Nature 451:1111–1115
Greaves M, Maley CC (2012) Clonal evolution in cancer. Nature 481:306–313
Norquist B, Wurz KA, Pennil CC, Garcia R, Gross J, Sakai W et al (2011) Secondary somatic mutations restoring BRCA1/2 predict chemotherapy resistance in hereditary ovarian carcinomas. J Clin Oncol 29:3008–3015
Quigley D, Alumkal JJ, Wyatt AW, Kothari V, Foye A, Lloyd P et al (2017) Analysis of circulating cell-free DNA identifies multiclonal heterogeneity of. Cancer Discov 7:999–1005
Kondrashova O, Nguyen M, Shield-Artin K, Tinker AV, Teng NNH, Harrell MI et al (2017) Secondary somatic mutations restoring. Cancer Discov 7:984–998
Gupta A, Hunt CR, Chakraborty S, Pandita RK, Yordy J, Ramnarain DB et al (2014) Role of 53BP1 in the regulation of DNA double-strand break repair pathway choice. Radiat Res 181:1–8
Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A et al (2010) 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 141:243–254
Hurley RM, Wahner Hendrickson AE, Visscher DW, Ansell P, Harrell MI, Wagner JM et al (2019) 53BP1 as a potential predictor of response in PARP inhibitor-treated homologous recombination-deficient ovarian cancer. Gynecol Oncol 153:127–134
Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H et al (2010) 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 17:688–695
Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P et al (2015) REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature 521:541–544
Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R et al (2019) Selective loss of PARG restores PARylation and counteracts PARP inhibitor-mediated synthetic lethality. Cancer Cell 35:950–952
Sawada K, Radjabi AR, Shinomiya N, Kistner E, Kenny H, Becker AR et al (2007) c-Met overexpression is a prognostic factor in ovarian cancer and an effective target for inhibition of peritoneal dissemination and invasion. Cancer Res 67:1670–1679
Martin LP, Sill M, Shahin MS, Powell M, DiSilvestro P, Landrum LM et al (2014) A phase II evaluation of AMG 102 (rilotumumab) in the treatment of persistent or recurrent epithelial ovarian, fallopian tube or primary peritoneal carcinoma: a Gynecologic Oncology Group study. Gynecol Oncol 132:526–530
Liu S, Zheng Y, Volpi D, El-Kasti M, Klotz D, Tullis I et al (2015) Toward operative in vivo fluorescence imaging of the c-Met proto-oncogene for personalization of therapy in ovarian cancer. Cancer 121:202–213
Vergote IB, Smith DC, Berger R, Kurzrock R, Vogelzang NJ, Sella A et al (2017) A phase 2 randomised discontinuation trial of cabozantinib in patients with ovarian carcinoma. Eur J Cancer 83:229–236
Medová M, Aebersold DM, Blank-Liss W, Streit B, Medo M, Aebi S et al (2010) MET inhibition results in DNA breaks and synergistically sensitizes tumor cells to DNA-damaging agents potentially by breaching a damage-induced checkpoint arrest. Genes Cancer 1:1053–1062
Medová M, Aebersold DM, Zimmer Y (2012) MET inhibition in tumor cells by PHA665752 impairs homologous recombination repair of DNA double strand breaks. Int J Cancer 130:728–734
Du Y, Yamaguchi H, Wei Y, Hsu JL, Wang HL, Hsu YH et al (2016) Blocking c-Met-mediated PARP1 phosphorylation enhances anti-tumor effects of PARP inhibitors. Nat Med 22:194–201
Lin ZP, Zhu YL, Ratner ES (2018) Targeting cyclin-dependent kinases for treatment of gynecologic cancers. Front Oncol 8:303
Johnson N, Cai D, Kennedy RD, Pathania S, Arora M, Li YC et al (2009) Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Mol Cell 35:327–339
Johnson N, Li YC, Walton ZE, Cheng KA, Li D, Rodig SJ et al (2011) Compromised CDK1 activity sensitizes BRCA-proficient cancers to PARP inhibition. Nat Med 17:875–882
Preusser M, De Mattos-Arruda L, Thill M, Criscitiello C, Bartsch R, Ruhstaller T et al (2018) CDK4/6 inhibitors in the treatment of patients with breast cancer: summary of a multidisciplinary round-table discussion. ESMO Open 3:e000368
Xia Q, Cai Y, Peng R, Wu G, Shi Y, Jiang W (2014) The CDK1 inhibitor RO3306 improves the response of BRCA-proficient breast cancer cells to PARP inhibition. Int J Oncol 44:735–744
Ibrahim YH, García-García C, Serra V, He L, Torres-Lockhart K, Prat A et al (2012) PI3K inhibition impairs BRCA1/2 expression and sensitizes BRCA-proficient triple-negative breast cancer to PARP inhibition. Cancer Discov 2:1036–1047
Juvekar A, Burga LN, Hu H, Lunsford EP, Ibrahim YH, Balmañà J et al (2012) Combining a PI3K inhibitor with a PARP inhibitor provides an effective therapy for BRCA1-related breast cancer. Cancer Discov 2:1048–1063
Matulonis UA, Wulf GM, Barry WT, Birrer M, Westin SN, Farooq S et al (2017) Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann Oncol 28:512–518
Konstantinopoulos PA, Barry WT, Birrer M, Westin SN, Cadoo KA, Shapiro GI et al (2019) Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: a dose-escalation and dose-expansion phase 1b trial. Lancet Oncol 20:570–580
Acknowledgement
Open Access funding provided by Projekt DEAL. We thank Dr. M. Stevense (TU Dresden) for critical comments on the manuscript.
Funding
DMK is funded by the Else-Kröner Fresenius Stiftung (Grant number = 060_5217).
Author information
Authors and Affiliations
Contributions
DMK: project development, data collection, manuscript writing, and editing. PW: supervision and manuscript editing.
Corresponding author
Ethics declarations
Conflict of interest
DMK is a clinician-scientist and works as a medical doctor at the Carl Gustav Carus University Hospital Dresden, TU Dresden. PW is the director of the Department of Gynecology and Obstetrics at the Carl Gustav Carus University Hospital Dresden, TU Dresden. PW reports receiving consulting fees, grant, and travel support from Amgen, AstraZeneca, MSD, Novartis, Pfizer, PharmaMar, Roche, Clovis, and Tesaro, and travel support and consulting fees from Eisai and TEVA.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Klotz, D.M., Wimberger, P. Overcoming PARP inhibitor resistance in ovarian cancer: what are the most promising strategies?. Arch Gynecol Obstet 302, 1087–1102 (2020). https://doi.org/10.1007/s00404-020-05677-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00404-020-05677-1