
Abstract
In the recent past, a plethora of drugs have been approved for the treatment of multiple sclerosis (MS). These therapeutics are mainly confined to immunomodulatory or immunosuppressive strategies but do not sufficiently address remyelination and neuroprotection. However, several neuroregenerative agents have shown potential in pre-clinical research and entered Phase I to III clinical trials. Although none of these compounds have yet proceeded to approval, understanding the causes of failure can broaden our knowledge about neuroprotection and neuroregeneration in MS. Moreover, most of the investigated approaches are characterised by consistent mechanisms of action and proved convincing efficacy in animal studies. Therefore, learning from their failure will help us to enforce the translation of findings acquired in pre-clinical studies into clinical application. Here, we summarise trials on MS treatment published since 2015 that have either failed or were interrupted due to a lack of efficacy, adverse events, or for other reasons. We further outline the rationale underlying these drugs and analyse the background of failure to gather new insights into MS pathophysiology and optimise future study designs. For conciseness, this review focuses on agents promoting remyelination and medications with primarily neuroprotective properties or unconventional approaches. Failed clinical trials that pursue immunomodulation are presented in a separate article.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Innovative study designs with integrated clinical scores and longer observation periods are needed to depict neuroregenerative and neuroprotective effects. |
In particular, add-on trials combining promising neuroregenerative compounds with highly effective monoclonal antibody therapies could be favourable approaches. |
Age-dependent effects, as well as the influence of disease duration, need to be taken more into account in the design of studies evaluating neuroprotective and neuroregenerative agents. |
1 Introduction
The therapy of multiple sclerosis (MS) has experienced breakthroughs during the past 25 years, with many new agents being approved and multiplying therapeutic options for patient care [1]. The vast majority of these drugs were designed to modulate immune cell functions, predominantly benefitting patients with relapsing-remitting MS (RRMS). However, about 10–15% of patients show a primary chronic-progressive disease course (PPMS), and also in RRMS, a transition into secondary progressive MS (SPMS) frequently occurs [2]. The conversion to progressive MS is accompanied by a shift from neuroinflammation-driven disease pathogenesis towards neurodegeneration [3]. Consequently, the efficacy of immunomodulatory treatment is attenuated in patients with progressive disease courses [4, 5]. Thus, therapeutics supporting neuroregeneration are urgently required. These approaches likely include strategies that promote remyelination through inducing proliferation and differentiation of oligodendrocyte precursor cells (OPCs) as well as the survival of mature oligodendrocytes [6]. Beyond remyelination, neuroregeneration can also be achieved by agents providing neuroprotection. These agents target a broad spectrum of detrimental processes such as excitotoxicity, mitochondrial dysfunction, or oxidative stress [7]. Unfortunately, at present none of these neuroregenerative compounds has made its way into clinical use [8].
Nonetheless, drugs that are tested in clinical trials are generally based on a conclusive rationale and were excessively evaluated in animal experiments. The reasons underlying their failure are diverse.
In some studies, insights gained from animal models were just not translatable to clinical trials, even though they did not lack a thoughtful pathophysiological rationale. In other cases, unexpected adverse events (AEs) occurred or methodological insufficiencies obscured the potential beneficial effects of the investigated compounds. These limitations in trial design involve inappropriate study endpoints, insufficient follow-up durations, and samples biases. Therefore, analysis of these studies is crucial for advances in both pre-clinical and clinical research.
This review assesses neuroregenerative and alternative treatment strategies in MS since 2015 that entered clinical Phase I–III trials and either failed or were interrupted. First, we describe the mechanisms of action based on (pre-)clinical data underlying each approach. After delineating the respective trial, we identify key reasons for failure and outline consequences for future research on autoimmune neuroinflammation and MS.
2 Methods
To identify relevant trials, we conducted a MEDLINE search for eligible articles as previously described [9]. We included articles that were published between 01 January 2015 and 31 December 2020. The terms used in Medical Subject Headings were ‘multiple sclerosis’, ‘trial’, and ‘therapy’ or ‘treatment’. Since trials with negative outcomes are merely published as abstracts or not at all, we additionally searched for relevant studies sourced from international conferences (Annual Meeting of the American Academy of Neurology [AAN], European/Americas Committee for Treatment and Research in Multiple Sclerosis [ECTRIMS/ACTRIMS]), consultation of national and international registries for clinical trials (United States National Library of Medicine; clinicaltrials.gov; European Union Drug Regulating Authorities Clinical Trials Database [EudraCT]), and personal communications with the authors. After removal of duplicates, we performed abstract or full text screening to further exclude trials on compounds that either:
-
i.
reached the primary study endpoint (pSE)
-
ii.
were intended to treat secondary complications of MS (e.g., spasticity, fatigue)
-
iii.
that were investigated in combination with other drugs if the other drug was not approved by the European Medicines Agency (EMA)/ U.S. Food and Drug Administration (FDA) for the treatment of MS.
If studies were conducted in cohorts with both relapsing MS or acute optic neuritis (AON) and progressive MS patients, the compound was assigned to whichever group predominated in number.
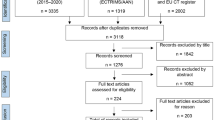
The outlined search returned 6656 records, 21 of which, with a total of 15 distinct agents, met our inclusion criteria (for details on the search strategy see Fig. 1; for details on individual compounds see Table 1).

PRISMA flow diagram of the reviewed literature. AAN American Academy of Neurology, EU CT register European Union Clinical Trials register, ECTRIMS European Committee for Treatment and Research in Multiple Sclerosis
3 Failed Clinical Trials in Acute Optic Neuritis and Relapsing Multiple Sclerosis
3.1 Neuroprotective Approaches
To some degree, MS can be understood as an interplay of neuroinflammation and neurodegeneration [10]. Thus, beyond immunomodulatory treatments, there is an urgent need for neuroprotective therapeutics to ameliorate neurodegenerative processes and to delay disability progression in MS. Neuroprotection involves a plethora of mechanisms ranging from the support of trophic factors and protection against excitotoxicity as well as oxidative stress to restoration of mitochondrial function [7, 8]. Due to this variety of targets, a wide spectrum of neuroprotective agents has been tested in clinical trials.
3.1.1 Atorvastatin
3.1.1.1 Background
Hydroxymethylglutaryl-CoA reductase inhibitors (here referred to as statins) are widely used agents targeting dyslipidaemia. Besides inhibition of the mevalonate pathway, statins were shown to prevent excitotoxicity [11], induce the secretion of neurotrophic factors [12], and reduce the release of free radicals due to suppression of inducible nitric oxide synthase [13]. Beyond neuroprotection, statins exhibit immunomodulatory properties. Among these are the restriction of antigen presentation [14], inhibition of leukocyte migration across the blood-brain barrier (BBB) [15], and induction of a shift of the inflammatory response towards T helper 2 (TH2) cells [16]. Considering these neuroprotective and immunomodulatory effects, statin treatment unsurprisingly proved convincing efficacy in experimental autoimmune encephalomyelitis (EAE) [15, 16]. Moreover, several small studies indicated a benefit of statins in RRMS, either alone or in combination with interferon-β [17,18,19,20,21].
3.1.1.2 Studies
In 2016, the results of the ARIANNA study were published [22]. Enrolling 154 RRMS patients, atorvastatin was evaluated at a dose of 40 mg/day as an add-on therapy to interferon-β1b over 24 months in a double-blind, randomised, placebo-controlled manner. Atorvastatin failed to improve the pSE, i.e., the change in fractional brain volume. Also, secondary endpoints, including clinical (e.g., Expanded Disability Status Scale [EDSS] [23], annualised relapse rate [ARR]), cognitive (Rao battery test [24]), and magnetic resonance imaging (MRI) criteria (e.g., gadolinium-enhanced lesions [GELs]) were not met.
3.1.1.3 Comment
The disappointing outcome of the ARIANNA study is in line with the failure of various other trials investigating the role of statins in RRMS [25,26,27,28, 33]. Foremost, the results of the largest trial in this context, the SIMCOMBIN study, are remarkable [29]. In this Phase IV trial, treatment with simvastatin as an add-on therapy to interferon-β1a failed to show any benefit. In addition, two meta-analyses confirmed that statin treatment does not improve clinical or MRI parameters in RRMS [30, 31]. Some trials even indicated an increased rate of T2 lesions or enhanced clinical disease activity in patients receiving statins [18, 28,29,30, 32, 33]. These observations might depend on statin-induced detrimental processes. These processes might potentially even aggravate neuroinflammation and degeneration. Underlying mechanisms involve enhanced secretion of pro-inflammatory cytokines [34] as well as depletion of OPCs and oligodendrocytes, thereby impairing remyelination [35].
Moreover, statins also mediate effects hampering their combination with interferon-β. Among these are the inhibition of interferon-induced phosphorylation of signal transducer and activator of transcription (STAT) 1 [36] and the counteraction of interferon-mediated suppression of matrix metalloproteinases (MMPs) [37]. Given these antagonistic actions, statins might better be combined with other disease-modifying therapies (DMTs) to create synergistic effects. Since the mechanism of action of interferon-β treatment is multifactorial and still not completely understood [38], the investigation of promising compounds as add-on medications to interferon-β needs to be questioned in general. Instead of interferon-β, the combination with a highly effective immunomodulatory agent characterised by a clear mechanism of action seems to be more suitable to assess the effects of neuroprotection and neuroregeneration.
Notably, simvastatin was shown to reduce brain atrophy by 43% in a double-blind, placebo-controlled trial, including 140 patients with SPMS (MS-STAT) [39]. This promising outcome initiated an ongoing Phase III trial [40]. The convincing results of simvastatin in SPMS raise the question if neuroprotective properties of statins might outweigh their anti-inflammatory capabilities. Noteworthy is a recent study that included patients from the ARIANNA trial and another study that assessed the impact of atorvastatin on RRMS as an add-on medication to interferon-β1a [41]. Forty-two patients who were allocated to placebo and 27 participants who received atorvastatin in the initial studies were followed-up for about 8 years. Remarkably, the atorvastatin group displayed a reduced risk of EDSS progression; however, without significant amelioration of the relapse rate.
In conclusion, statins failed to provide benefits to RRMS patients in the short term. Nevertheless, long-term effects and the use in SPMS need to be further evaluated to confirm the initial positive results.
3.1.2 Lipoic Acid
3.1.2.1 Background
Lipoic acid (LA) is an endogenously produced antioxidant, that is used, for instance, for the treatment of diabetic peripheral neuropathy [42]. Anti-oxidative functions depend on a variety of processes. With its reduced form, dihydrolipoic acid, LA forms a potent redox couple [43]. Together, they regenerate other anti-oxidative agents [44], serve as chelators of metal ions [45], and improve mitochondrial activity [43]. Through the scavenging of free radicals [44], LA prevents reactive oxygen species-mediated dysfunction of the BBB and transendothelial monocytic migration [46]. Additionally, LA reduces cytokine secretion, proliferation, and central nervous system (CNS) infiltration of T cells [47, 48]. The impact on T cells seems to be mediated by an intracellular increase of cyclic adenosine monophosphate (cAMP) [49]. Several of the mentioned effects were already observed in MS patients receiving LA [50,51,52]. Given the impact on inflammation and oxidative processes, it is not surprising that EAE experiments demonstrated efficacy of LA in neuroinflammation and neurodegeneration [47, 48].
3.1.2.2 Studies
Completed in 2016, a double-blind, placebo-controlled, randomised Phase I trial analysed the effects of LA in 31 patients diagnosed with experimental AON [53]. Treatment with 1200 mg/day LA or placebo was initiated within 14 days after diagnosis and lasted 6 weeks, followed by an 18-week observation period. The pSE evaluated the influence of LA on retinal nerve fibre layer (RNFL) thickness after 24 weeks compared to baseline. LA failed to improve RNFL regeneration. Strikingly, the LA group even displayed a trend of enhanced RNFL loss compared to placebo. On top of that, the secondary outcome criteria (e.g., RNFL thickness after 12 weeks, changes in low- and high-contrast visual acuity [VA]) were not met.
3.1.2.3 Comment
The results from the reviewed study need to be interpreted with caution due to the small number of participants and the short follow-up period [53]. Nevertheless, the disenchanting outcome of this trial is contrary to the promising pre-clinical results. Of the many EAE studies conducted, however, only two investigated effects on AON. First, Chaudhary et al demonstrated an alleviation of axonal injury both after prophylactic and therapeutic treatment with LA [54]. Protection of retinal ganglion cells (RGCs) was also observed by Dietrich et al [55]. However, the latter study reported that this protection only improved visual functions when LA was administered prior to clinical manifestation but failed to do so when applied after disease onset. Further, unlike prophylactic treatment, therapeutic administration also failed to protect the inner retinal layers from degeneration and ameliorate inflammatory infiltration into the optic nerve. Therefore, LA treatment in AON might only be suitable when administered immediately after symptom onset. Unfortunately, data on the mean duration to the first LA dose are not given in the reviewed trial [53].
Next, pharmacokinetic characteristics of LA also impede its use in clinical care. Not only a short half-life but, even more important, strong interindividual variabilities in the concentration maximum and bioavailability complicate therapy management [52, 56, 57]. Apart from interindividual variations, the application of different formulations of LA results in variable responses [50, 56]. Unfortunately, no data are given concerning the formulation used in the described study [53].
Beyond pharmacokinetics, the consideration of pharmacodynamics is relevant in this context. The response to LA administration seems to vary between the different types of MS [51]. An increased cAMP response of peripheral blood mononuclear cells (PBMCs) upon LA administration was observed in SPMS patients. However, the treatment of RRMS patients led to reduced cAMP levels. The response of patients with AON to LA in terms of cAMP is unknown. Given the LA-induced increase of cAMP in SPMS, it should be mentioned that LA application reduced the annualised percentage brain volume change (PBVC) by 68% in patients with SPMS compared to placebo in a double-blind trial [58]. Consequently, a Phase II study in patients with progressive MS was started in 2018 [59]. Thus, research on LA seems to be more encouraging in patients suffering from progressive MS rather than AON.
3.1.3 Flupirtine Maleate
3.1.3.1 Background
Flupirtine maleate (here referred to as flupirtine) is a non-opioid analgesic drug [60]. Results obtained from animal models of Parkinson’s disease (PD) [61] and Alzheimer’s disease (AD) [62] as well as a clinical trial in patients diagnosed with Creutzfeldt-Jakob disease [63] indicate neuroprotective properties of this agent. Flupirtine seems to stabilise the membrane potential and reduce excitability by activation of KCNQ-type- [64] and G protein-coupled inwardly rectifying potassium channels [65]. Indirect antagonistic effects on N-Methyl-D-aspartate (NMDA) receptor activity [66] and gating of γ-aminobutyric acid (GABA)A receptors [64] seem to further contribute to neuroprotection. Last, flupirtine-induced neuroprotection was also demonstrated in animal models. During experimental AON, for instance, flupirtine treatment alleviated degeneration of RGCs and improved visual functions when used as an add-on medication to interferon-β1a [67].
3.1.3.2 Studies
Including 30 RRMS patients, the FLORIMS study was conducted as a double-blind Phase II trial [60]. Participants received either flupirtine at a daily dosage of 300 mg or placebo for 1 year, both as an add-on treatment to interferon-β1b. While the trial was originally planned with 80 patients, only 30 patients were randomised due to unspecified recruitment problems. Finally, only 12 patients per group completed the trial. Due to the exploratory character of this study, no pSE was defined. Treatment with flupirtine resulted in advantages over placebo concerning clinical parameters such as the number of relapses or EDSS progression. Moreover, the active treatment group also displayed a reduced occurrence of GELs. However, none of these observations was significant. With regard to the safety profile, five patients in the flupirtine group displayed elevated liver enzymes leading to discontinuation in two cases.
3.1.3.3 Comment
Although some non-significant trends indicated a mild benefit, the FLORIMS trial did not add reliable insights into the neuroprotective potential of flupirtine due to the small number of patients attending. This shortage of participants was caused by problems of patient recruitment and reduced the power to only 59% in terms of changes in EDSS [60]. Unfortunately, reasons underlying hampered recruitment were not further defined but may be related to reports of severe cases of flupirtine-induced hepatotoxicity [68]. The drug-induced liver injury resulted in an endorsed withdrawal of flupirtine by the EMA in 2018. The mechanisms of flupirtine-mediated hepatic damage are not completely understood. A genetic association with a class II human leukocyte antigen haplotype was observed, suggesting an inappropriate T cell response [69]. On the other hand, the formation of highly reactive and hepatotoxic metabolites has been discussed [70].
Moreover, there are still uncertainties regarding the role of flupirtine in neuroinflammation. Additional treatment with flupirtine during EAE, for instance, did not show any advantage compared to sole interferon-β1a administration [67]. In line with this observation, flupirtine did not ameliorate EAE symptoms when applied alone. Also, the mentioned protective impact of flupirtine treatment on RGCs during experimental AON was mediated independently from inflammatory processes. Thus, the selection of a RRMS cohort for the reviewed trial needs to be scrutinised. However, following the endorsed withdrawal by EMA, further research on flupirtine treatment in MS is impeded.
3.1.4 Uric Acid—Inosine
3.1.4.1 Background
Uric acid (UA) is a purine metabolite with anti-oxidative capacities [71]. Since oral supplementation does not increase serum concentrations of UA adequately, its precursor inosine can be used to raise UA levels [72]. UA-mediated anti-oxidation is mainly attributed to the scavenging of peroxynitrite [73]. Peroxynitrite is a neurotoxic agent that causes damage to oligodendrocytes [74] and contributes to BBB disruption [75]. Moreover, peroxynitrite is associated with plaque formation in MS [76]. Of note, inosine and UA proved efficacy in several EAE experiments, as treatment alleviated disease severity [77, 78].
Also, clinical data suggest an involvement of UA in MS. First, even though there are contradictory reports, a meta-analysis showed that serum levels of UA are reduced in MS [79]. Interestingly, the lowest concentrations were found in SPMS patients. Second, twin studies demonstrated decreased UA levels in the twin diagnosed with MS [80]. Last, an analysis of more than 20 million patient records revealed that the number of patients suffering both from MS and gout, which is characterised by an increased concentration of UA, was much lower than assumed, indicating a preventive role of UA in neuroinflammation [77].
3.1.4.2 Studies
Safety and efficacy of inosine were evaluated in 36 patients with RRMS in a Phase II trial [71]. Inosine at a daily dose of 3 g or placebo was administered in a double-blind manner as an add-on medication to interferon-β1a. Patients were followed-up for 12 months. The pSE evaluated AEs and laboratory results related to UA. Unsurprisingly, inosine treatment elevated UA levels compared to placebo. Two patients treated with inosine experienced UA concentrations above 10 mg/dL while suffering from renal colic. Ten additional patients showed asymptomatic hyperuricaemia. Secondary objectives analysed clinical (e.g., number of relapses) and radiological disease activity (e.g., number of new MRI lesions). However, inosine failed to show any impact on these criteria.
3.1.4.3 Comment
Besides the mentioned study, a benefit of inosine treatment in MS must be further doubted, given the results from a double-blind, placebo-controlled trial in 159 RRMS patients [81]. There, inosine failed to ameliorate the relapse rate and disability progression. Strikingly, both this and the reviewed study evaluated inosine as an add-on treatment to interferon-β, although the latter is known to increase UA levels by itself [82]. Therefore, the simultaneous interferon-β application might have covered the beneficial effects of inosine.
Further, the selection of RRMS for evaluating inosine treatment remains questionable. Inosine might be more efficient in an SPMS cohort, given that UA levels are most depressed in these patients [79]. Also, considering the neuroprotective capabilities as an anti-oxidant and the lack of immunomodulation by UA in EAE [83], patients with progressive MS might profit more from inosine treatment than RRMS patients.
Since higher dosages than applied might be necessary to induce significant neuroprotection, side effects interfere with the clinical use of inosine. For instance, increased risks for cardiovascular events [84] and nephrolithiasis [85] associated with higher UA concentrations limit the dosage of inosine.
Outside the study design, the propagated mode of action also needs to be questioned. In light of the complexity of MS pathology, addressing a single anti-oxidant might not meet the needs required for manifest clinical improvement. Therefore, it seems unlikely that inosine monotherapy can effectively benefit RRMS patients.
3.2 Neuroregenerative Approaches
In MS lesions, remyelination is a naturally occurring repair mechanism, restoring neuroaxonal functions [86]. Through re-establishment of saltatory conduction, remyelination induces recovery from clinical symptoms [6]. Of note, insufficient myelin repair has been described in all courses of MS, making remyelination-promoting therapies a hopeful approach for both relapsing and progressive forms of MS [87].
3.2.1 LINGO-1—Opicinumab
3.2.1.1 Background
With regard to pre-clinical data, one of the most promising remyelination-influencing targets is leucine-rich repeat and immunoglobulin-like domain-containing Nogo receptor-interacting protein-1 (LINGO-1). LINGO-1 is a highly conserved transmembrane protein expressed selectively on neurons, oligodendrocytes, and OPCs [88].
Inhibition of LINGO-1 is associated with axonal regeneration, terminal OPC differentiation, and increased survival of neurons as well as oligodendrocytes [89]. Consequently, blockade of LINGO-1-mediated pathways led to clinical improvement and remyelination in several animal models that cover distinct pathophysiological aspects of MS [90,91,92]. Among these are AON, EAE as well as non-inflammatory models such as cuprizone-induced demyelination. Apart from myelin repairing processes, results from adoptive transfer EAE experiments indicate that anti-LINGO-1-induced beneficial effects (e.g., preservation of neuroaxonal integrity) are independent of immunomodulation [92].
To reinforce remyelination, the human monoclonal IgG antibody opicinumab (BIIB003) was designed to suppress LINGO-1-mediated pathways via direct binding [93]. Two Phase I studies assessing opicinumab treatment in healthy volunteers and relapsing MS patients indicated a tolerable safety profile [94].
3.2.1.2 Studies
Opicinumab in AON Started in 2012, a double-blind Phase II study (RENEW) was conducted in 82 patients with first unilateral AON [95]. Following standard high-dose treatment with intravenous methylprednisolone (IVMPS), participants were randomised to receive either 100 mg/kg opicinumab or placebo within 28 days after symptom onset. Treatment was administered every four weeks up to Week 20, followed by a 12-week observation period. The pSE assessed the recovery of the affected optic nerve conduction in terms of visually evoked potentials (VEPs) after 24 weeks. Although opicinumab treatment ameliorated P100 latency, this difference was not significant. Significance was only reached in the per-protocol population at Week 32. Moreover, opicinumab treatment did not improve secondary endpoints such as VA or preservation of RNFL thickness assessed by optical coherence tomography.
Thirty-nine patients were further enrolled in a substudy in which multifocal VEP measurements were used to examine optic nerve repair [96]. Non-significant trends towards reduced latency prolongation and increased recovery of VEP amplitude were observed in the active treatment group.
Recently, the first data of a 2-year follow-up study of the RENEW trial were announced (RENEWED) [97]. Of all participants of the RENEW trial who received at least one dose of opicinumab or placebo, and thus could join this trial, 52 attended the study. Investigation of the pSE (VEP latency) indicated that the observed positive trend in the opicinumab group was maintained over 2 years. However, this trend was not significant.
Opicinumab in RRMS and SPMS The double-blind Phase II SYNERGY trial included 330 RRMS and 89 SPMS patients with relapses who were randomly assigned in a 1:2:2:2:2 ratio to either 3, 10, 30, or 100 mg/kg opicinumab or placebo [98]. Patients received active treatment or placebo every four weeks for 72 weeks as an add-on medication to interferon-β1a. The pSE comprised the proportion of participants with confirmed improvement in EDSS score, Timed 25-Foot Walk (T25FW) [99], Nine-Hole Peg Test (9HPT) [100], or 3s Paced Auditory Serial Addition Test (PASAT-3) [101]. No significant benefit was demonstrated regarding treatment with 3, 10, and 100 mg/kg opicinumab [98]. A significantly higher proportion of participants with an improvement of disability was detected only at a dose of 30 mg/kg opicinumab compared to placebo. However, a significant dose-linear improvement could not be observed. Evaluation of the secondary (percentage of patients with confirmed disease worsening measured by the same criteria) and tertiary endpoint (overall response score comprised of the mentioned criteria) showed several mild effects, favouring opicinumab treatment with 10 or 30 mg/kg.
In 2017, 263 RRMS and SPMS patients were enrolled in a placebo-controlled, randomised, double-blind Phase II trial (AFFINITY) [102, 103]. This study evaluated the administration of 750 mg opicinumab, corresponding to a dose of 10 mg/kg [104], as an add-on to DMTs. The pSE included the integrated response score already used in the SYNERGY study. In October 2020, Biogen announced that the AFFINITY trial failed to meet the pSE [105].
3.2.1.3 Comment
Inhibition of LINGO-1 showed impressive results concerning OPC differentiation and remyelination in animal models [90,91,92] but so far failed to effectively improve clinical parameters in patients. The example of opicinumab prototypically depicts the challenges regarding the translation of remyelination-promoting approaches into human application. First, the appropriate timing for initiating these therapies is still elusive. It seems that remyelination starts immediately after the onset of demyelination and finishes within several weeks or months [6]. Therefore, it can be speculated that remyelination-promoting strategies are most effective when administered instantly after demyelination, and thus at symptom onset. This hypothesis is further supported by a post hoc analysis of the RENEW trial, revealing that patients who started opicinumab treatment earlier after AON onset gained more benefit [106]. In the same trial, the mean duration between the beginning of visual impairment and the first application of opicinumab comprised 24 days [95]. At this time, however, most retinal thinning has already occurred, limiting potential improvements induced by anti-LINGO-1 treatment [95].
Moreover, it is still not clear how remyelination can be reliably measured both clinically and para-clinically. Therefore, a new pSE was used in the SYNERGY study. Strikingly, this endpoint assessed the improvement of disability rather than a delay or arrest of disease progression [98]. Thus, an integrated score as used as the tertiary objective in the SYNERGY study and as pSE in the AFFINITY trial seems to be more suitable [102]. In addition, the SYNERGY study made use of innovative imaging techniques such as magnetisation transfer ratio (MTR) or diffusion tensor imaging (DTI) for specific assessment of remyelination as exploratory objectives [98]. However, the validity of these methods has not been adequately verified. As a consequence of the aforementioned lack of experience with (para-)clinical parameters for remyelination and uncertainties regarding the effect size, it is not surprising that the discussed studies (SYNERGY and RENEW) were also underpowered [95, 98].
Last, there is a lack of experience concerning the appropriate selection of patients who might profit from remyelination-promoting approaches. In the RENEW study, post hoc analysis revealed that opicinumab provided benefit to older AON patients (aged ≥ 33 years) and those with more severe visual impairment at baseline [106]. Most likely, this is due to a diminished intrinsic recovery potential without treatment in these subpopulations. Remyelination capacities, for instance, decline with age [107]. This decline seems to be caused by impaired differentiation of OPCs into sheath-forming oligodendrocytes rather than a lack in the number of OPCs [108]. Therefore, opicinumab potentially restores differentiation of still available OPCs that might become insufficient more frequently in older patients. However, the observation that opicinumab treatment provided most benefit to patients with severe visual impairment at baseline, may also result from a regression to the mean artifact.
Concerning relapsing MS, subpopulation analyses of the SYNERGY trial identified predictors for opicinumab efficacy in RRMS and SPMS patients [98, 109]. These analyses suggest that especially patients with shorter disease duration (≤ 20 years since symptom onset) and MRI criteria, indicating preserved brain integrity at baseline (lower DTI radial diffusivity), benefit from opicinumab. The mentioned predictors are in line with reports defining functional intact axons as a prerequisite for remyelination [6]. Furthermore, restoration of neuronal functionality not only depends on the degree of remyelination but also on region-specific factors as well as the extent of irreversible neuronal network damage [110]. Most likely, patients with a shorter disease duration have a higher integrity of neuronal networks compared to those in advanced disease stages [111] and might therefore benefit from remyelination-promoting approaches. However, in the case of opicinumab, even a long observation period and the inclusion of patients with a short duration of disease did not lead to a positive outcome in the AFFINITY trial. The AFFINITY study included those patients who were shown to have the greatest benefit during the SYNGERY trial. Therefore, the failure of this study raises the question of whether the positive results demonstrated in the subgroup analyses may be rather related to statistical errors in terms of multiple testing.
Given the failure of opicinumab in the mentioned clinical trials, a beneficial effect of opicinumab in relapsing MS is very unlikely. In line, Biogen announced the discontinuation of the development of opicinumab in MS in 2020 [105].
3.2.2 Histamine H3 Receptor—GSK239512
3.2.2.1 Background
The histamine H3 receptor (H3R) is an inhibitory G protein-coupled receptor predominantly expressed in the CNS [112]. Activation of H3R inhibits the release of histamine as well as other neurotransmitters such as acetylcholine, norepinephrine, dopamine, and serotonin [113]. Based on this function, H3R became an attractive target for the treatment of neuropsychiatric disorders [114]. Due to the high level of spontaneous signalling of H3R, (pre-)clinical research focused on inverse agonists of this receptor [115]. Inverse agonists are agents that not only inhibit the binding of agonists but, even more, induce opposite effects by inhibiting the constitutive, i.e., spontaneous receptor activity [116]. Thus, the selective H3R inverse agonist GSK239512 entered clinical Phase II trials for the therapy of cognitive alterations in schizophrenia [117] and AD [118].
In the field of neuroinflammatory research, H3R gained attention, as Chen et al demonstrated that inverse agonists of this receptor promote OPC differentiation [115]. Moreover, they observed an increased expression of H3R on oligodendrocytes in demyelinated human MS lesions. Interestingly, the administration of an H3R inverse agonist resulted in enhanced remyelination accompanied by preserved axonal integrity in a murine model of demyelination. Further support for an involvement of H3R in MS pathogenesis derives from the association between an exonic single nucleotide polymorphism (SNP) in the human gene coding for H3R and an increased susceptibility to MS [115].
3.2.2.2 Studies
After showing an acceptable safety profile in a Phase I study [119], a double-blind Phase II trial on GSK239512 was performed in RRMS [120]. The study included 131 patients with disease activity within the previous year (≥ 1 GELs on MRI or reported relapses). GSK239512 was tested as an add-on treatment to interferon-β1a or glatiramer acetate (GA). Once daily, patients received either placebo or GSK239512, up-titrated within 4–5 weeks to a maximum tolerable dose of up to 80 µg/day. The final dose corresponded to an H3R occupancy in the brain of more than 90% [121]. The co-pSEs assessed the mean changes in MTR in newly developed GELs or Delta-MTR-defined lesions. After 43–44 weeks of treatment, however, GSK239512 missed the target effect size of 0.5. Moreover, there were no relevant improvements of secondary clinical endpoints (e.g., EDSS score, performance at CogState battery [122], ARR). Analysis of MRI scans even favoured placebo regarding the development of new or enlarging T2 lesions. Finally, while only three patients receiving placebo discontinued the study (none were due to AEs), 14 of the treatment group dropped out (seven due to AEs, two at the investigator’s discretion).
3.2.2.3 Comment
Besides a short observation time, one explanation for the small effect sizes might be the small number of participants completing the trial. One underlying reason was the high drop-out frequency in the active treatment group [120]. Also, considering that only patients with a high baseline disease activity were included, an unexpectedly high number of patients did not develop new MRI lesions. Only 55 out of 114 participants contributed lesions to the analysis. Therefore, disease activity in the study cohort was insufficient to evaluate the potential effects of GSK239512 treatment adequately. Taking together the high drop-out rate and the low rate of new MRI lesions, the study was most likely underpowered. Furthermore, the target effect size of 0.5 needs to be questioned in general. Given the complexity of the underlying mechanisms, the reduced sample size, and the heterogeneity of the cohort, the target effect size may have been overestimated. Therefore, GSK239512 should not be discarded solely on the basis of failing to meet this effect size.
Moreover, the pathophysiological role of H3R in neuroinflammation is still controversially discussed. Contradicting a beneficial role of H3R inverse agonists in MS, mice deficient for H3R displayed an earlier and more intense disease progression in active EAE [123]. This aggravation of symptoms was accompanied by increased permeability of the BBB, leading to more severe inflammatory infiltration. Correspondingly, data acquired in models of cerebral malaria [124] and spinal cord injury [125] indicate a protective influence of H3R on BBB integrity. These observations might also explain the increased rate of T2 lesions in patients treated with GSK239512 [120]. In line with these results, treatment of mice with an agent activating H3R ameliorated EAE symptoms and reduced CNS infiltration [126]. Thus, even agonists of H3R are considered as therapeutic options in MS [127].
Another striking point is the impact of H3R modulation on glial cells. In microglia, for instance, H3R suppresses the production of pro-inflammatory cytokines, chemotaxis as well as phagocytosis [128]. Regarding the influence on astrocytes, H3R contributes to the astrocyte-induced reduction of pro-inflammatory cytokines and secretion of neurotrophic factors [129]. In addition, application of an H3R agonist reduced the expression of co-stimulatory molecules of dendritic cells (DCs) and inhibited DC-induced differentiation of pathogenic T helper 1 (TH1) and 17 (TH17) cells [126]. Therefore, H3R blockade in glial cells and DCs might enhance rather than alleviate neuroinflammation.
Given the shortcomings in study design and conflicting pre-clinical results, the role of GSK239512 in MS remains elusive.
3.3 Other Approaches
3.3.1 ATP-sensitive K+ Channels—Diazoxide
3.3.1.1 Background
Potassium channels are promising targets ameliorating CNS autoimmunity [130, 131]. In the last years, adenosine triphosphate (ATP)-sensitive potassium (KATP) channels gained increasing interest due to their neuroprotective capacities [132]. KATP channels are located both in the plasma (sarcKATP) and mitochondrial membrane (mitoKATP) [133]. One well-known activator of KATP channels is diazoxide, a selective agonist of mitoKATP channels when administered in low doses [133]. Diazoxide appears to be an attractive agent for treating neurological disorders as it induces anti-inflammatory and neuroprotective processes in animal models of stroke [134], PD [135], and AD [136]. Neuroprotection seems to be mediated through maintenance of mitochondrial homeostasis, reduction of oxidative stress, protection against excitotoxicity as well as the implementation of a favourable energy profile [137,138,139]. Moreover, diazoxide was also evaluated in EAE [140, 141]. There, both the prophylactic and therapeutic application resulted in alleviated signs of neuroinflammation. This amelioration was mediated by inhibition of microglial pro-inflammatory activities, restriction of antigen presentation by DCs, and diminished astrocytic activity.
3.3.1.2 Studies
A multicentre, double-blind Phase IIa study evaluated the efficacy and safety of diazoxide in 102 patients with RRMS [142]. Patients were randomised in a 1:1:1 ratio to receive either placebo or diazoxide in a dose of 0.3 or 4 mg/day. Following 24 weeks of treatment, patients could join an additional 24-week observation period. The cumulative number of new T1 GELs was investigated as pSE. Secondary endpoints included further MRI (e.g., new or enlarging T2 lesions and PBVC) and clinical criteria (e.g., EDSS and relapse-free status). Notably, patients treated with diazoxide showed a significantly higher rate of new GELs and a trend towards more frequent new or enlarging T2 lesions. Also, a slight trend of stronger disease progression related to MRI criteria was observed in the 4 mg/day group compared to patients treated with 0.3 mg/day diazoxide. However, treatment significantly reduced the loss of brain volume and displayed a trend of diminished evolution of T2 lesions into black holes. No differences regarding clinical outcome parameters were observed.
3.3.1.3 Comment
Due to methodological limitations, conclusions drawn from this trial need to be interpreted with caution. Negative results may at least partially result from inhomogeneous baseline characteristics. The treated groups displayed an increased disease activity, indicated by more and enlarged T2 lesions at baseline [142]. On the other side, the placebo group showed a higher volume of black holes. Thus, patients in the placebo group might have been in more advanced stages of the disease, associated with dominant neurodegenerative rather than neuroinflammatory characteristics. However, this might not fully explain the higher rate of new lesions and reduced brain atrophy observed in the active treatment group. Concerning brain atrophy, other explanations might include fluid shifts and a direct vasodilating effect of diazoxide [142]. In addition, diazoxide possibly has a more prominent role in neurodegeneration than neuroinflammation, considering its mentioned impact in models of AD and PD along with extensive effects on neuronal preservation [135, 136].
The authors of this trial also hypothesised a modulation of BBB integrity by diazoxide as a mechanism of action. Strikingly, the beneficial actions of diazoxide related to the BBB were not shown pre-clinically. Moreover, diazoxide-treatment did not affect lymphocytic CNS infiltration in EAE [140]. Also, while an inhibitory influence of diazoxide on succinate dehydrogenase was demonstrated [143], earlier studies suggest a protective effect of the latter on BBB function [144]. Conclusively, especially considering the higher frequency of GELs in the treatment group, a beneficial impact of diazoxide on BBB integrity is highly doubtable.
Furthermore, data from animal studies imply that diazoxide-mediated effects highly depend on the exact dosage. In several experiments, lower doses of diazoxide resulted in a better outcome, potentially due to more selective binding to mitoKATP channels [137, 141]. The slight trend of stronger MRI disease activity observed in patients treated with 4 mg/day compared to those receiving 0.3 mg/day [142] give further rise to reconsider the chosen dosage.
In addition, diazoxide was shown to promote OPC proliferation and oligodendrocyte differentiation, leading to enhanced myelination in vivo [145, 146]. Unfortunately, remyelination was not assessed in the reviewed trial.
As mentioned before, other types of potassium channels such as voltage-gated potassium (KV) channels raised interest as potential mediators of neuroinflammation and neurodegeneration [147]. One well-known blocker of KV channels, 4-aminopyridine (4-AP), is already approved to treat ambulatory disability in MS [148]. Beyond symptomatic treatment, recent data indicate that 4-AP might also prevent neuroaxonal loss. For instance, 4-AP administration seemed to alleviate clinical signs of chronic EAE and retinal neurodegeneration during experimental AON [149]. In a retrospective trial, including 103 MS patients, the same authors reported an amelioration of macular RNFL loss following 2 years of 4-AP intake without a significant benefit in terms of peripapillary RNFL thickness or total retinal preservation [149]. Contradicting a beneficial impact of 4-AP on neuroaxonal injury, however, Göbel et al and Moriguchi et al did not observe an effect of 4-AP treatment in comparable models of chronic EAE [150, 151]. Moreover, Ruck et al and others could not detect a significant improvement in the MS Functional Composite (MSFC) [152] in 4-AP-treated MS patients, further questioning an amelioration of disability progression induced by 4-AP [153,154,155]. These conflicting results and the ambiguous performance of diazoxide highlight the critical need for ongoing research to take advantage of the therapeutic potential of potassium channel modulators in MS.
3.3.2 Minocycline
3.3.2.1 Background
Minocycline is a second-generation antibiotic of the tetracycline class. It first raised interest as a neuroprotective agent in 1998 when it was shown to induce beneficial effects in ischaemic brain injury [156]. Since then, minocycline was tested in various neurodegenerative diseases [157]. In EAE, minocycline treatment resulted in clinical and histological improvements when administered both as a single agent [158] or in combination with approved DMTs such as interferon-β [159] or GA [160]. These improvements are mediated by a plethora of actions. First, minocycline is known to be a potent inhibitor of microglial [161] and astrocytic activation [162]. Second, it reduces the activity of MMPs [163]. Despite other detrimental effects, MMPs degrade extracellular matrix proteins around blood vessels [158, 163]. Thereby, activation of MMPs facilitates BBB breakdown and inflammatory CNS infiltration [158]. Third, minocycline was shown to induce a shift in the immune response from TH1 towards TH2 cells [162]. Apart from the mentioned effects, minocycline is also capable of reducing apoptosis [162], glutamate excitotoxicity [164], and oxidative stress [165].
Further support for a neuroprotective and immunomodulatory role of minocycline derives from clinical studies. These trials show a reduced conversion of clinically isolated syndrome into MS and decreased numbers of GELs in RRMS patients [166,167,168].
3.3.2.2 Studies
The role of minocycline in RRMS was further tested in a double-blind, randomised Phase II trial (RECYCLINE) [169]. Following a run-in period of three months, 149 and 155 patients received minocycline or placebo, respectively, both in combination with interferon-β1a for 96 weeks. The pSE evaluated the time to first qualifying relapse, while secondary endpoints included the ARR, number of new or enlarging T2 lesions as well as changes in brain volume. However, the trial met none of these endpoints. Only a trend towards a reduced ARR in the minocycline group was observed, while results of disability progression and brain volume change tended to favour placebo. Notably, the study was terminated prematurely due to a halt of minocycline production. However, merely two patients were affected by study termination.
3.3.2.3 Comment
Most striking in the methodology of the RECYCLINE study is the unexpectedly low rate of relapses. Only 23% of all patients experienced a relapse within the 96 weeks of treatment [169]. Therefore, evaluation of the pSE, namely the time to first qualifying relapse, is hampered.
Despite this limitation, one could also doubt long-term improvements induced by minocycline. Suppression of astrocytes and microglia as well as reduced MMP activity are beneficial during the acute inflammatory response. However, these cells and proteins have crucial functions in the resolution of inflammation and tissue repair [6]. Removal of myelin, for instance, is a prerequisite for remyelination [170]. Moreover, microglia and astrocytes supply essential factors and create a favourable microenvironment critically required for remyelination [171, 172]. The same is true for MMPs. MMP9, for instance, facilitates remyelination through removing the proteoglycan neural/glial antigen 2 [173]. Thus, unselective inhibition of microglia, astrocytes, and MMPs might counteract neuroregeneration. Impaired tissue repair and remyelination might also partially explain the trend towards increased disability progression following minocycline treatment in the RECYCLINE study [169].
Last, interferon-β might not be the optimal co-medication for minocycline treatment. Just like minocycline, beneficial effects of interferon-β depend at least partially on reduced MMP activity [37] and a shift of T cell differentiation towards a TH2 response [174]. A combination with therapeutics inducing other processes than minocycline might be more suitable to address a variety of targets.
4 Failed Clinical Trials in Progressive Multiple Sclerosis
4.1 Neuroprotective Approaches
4.1.1 Acid-Sensing Ion Channels—Amiloride
4.1.1.1 Background
A certain group of ion channels linked to Na+- and Ca2+-dependent neuroaxonal injury is the family of acid-sensing ion channels (ASICs). These are proton-gated cationic channels activated by an acidic pH leading to an influx of Na+ and Ca2+ ions [175]. Amiloride, well-known as a diuretic, turned out to be an unspecific inhibitor of ASICs and already proved efficacy in animal models of PD [176], stroke [177], and Huntington’s disease [178]. Consistently, amiloride-induced neuroprotection alleviated axonal degeneration as well as clinical symptoms of acute EAE [179, 180]. More importantly, amiloride treatment also ameliorated clinical signs of EAE performed in Biozzi ABH mice, a model resembling characteristics of progressive MS [179, 180]. Although ASICs are also expressed on several immune cells (e.g., B cells, T cells, and macrophages), compelling evidence suggests that beneficial effects of amiloride in EAE are mediated independently from immunomodulation [180].
Beyond insights gathered from animal experiments, there are also clinical data supporting the hypothesis of an involvement of ASICs in MS pathophysiology. First, SNPs located in a gene coding for ASICs are associated with enhanced susceptibility to MS [181]. Moreover, axonal ASIC expression is upregulated at the border of acute MS lesions [179]. Importantly, amiloride treatment was already tested in an uncontrolled pilot study in PPMS [182]. There, it slowed brain atrophy and alleviated worsening of tissue damage related to markers of DTI.
4.1.1.2 Studies
Amiloride in AON In the ACTION trial, amiloride was evaluated in patients with a first episode of unilateral AON [183]. In this double-blind Phase II study, 48 patients were randomly assigned to either daily treatment with 10 mg amiloride or placebo. Treatment was initiated within 28 days after symptom onset and continued for five months. Steroid treatment after AON onset was allowed but not mandatory. The pSE evaluated the change of the peripapillary RNFL thickness after six months. However, this endpoint was not reached. In addition, analysis of secondary endpoints, including structural, visual, and electrophysiological criteria, did not show an advantage of amiloride treatment. Patients of the active treatment group even displayed a significantly prolonged peak time of VEPs.
Amiloride in SPMS Following the positive outcome of the mentioned pilot study in PPMS [182], the efficacy of amiloride treatment in SPMS was tested in a multi-arm, double-blind Phase IIb trial, in parallel to fluoxetine and riluzole (MS-SMART) [184]. For a period of 96 weeks, 223 patients were included to receive either 10 mg/day amiloride or placebo. However, amiloride treatment failed to induce any difference concerning the pSE, i.e., the PBVC after 96 weeks. Moreover, secondary MRI (PBVC after 24 weeks, new or enlarging T2 lesions after 96 weeks) and clinical endpoints (e.g., changes in EDSS) were not met.
4.1.1.3 Comment
Besides the negative outcomes of the ACTION and MS-SMART trials, large registry-based cohort studies could not detect an association between amiloride treatment and a decreased risk for MS or MS-related hospitalisation, further questioning the efficacy of amiloride in MS [185].
Moreover, as the pathophysiology of AON is mainly mediated by inflammatory processes [186], selectively targeting the neuroaxonal loss without affecting the immunological response might be insufficient. Notably, 20% of patients in the placebo but only 5% in the treatment group received steroids after AON onset in the ACTION study [183]. Considering the improvement of visual functions by corticosteroid treatment in AON [187], the higher frequency of steroid administration might have influenced the outcome in favour of the placebo group. This connection is especially important given the extended time frame of treatment within 28 days after onset, as crucial damage might have already occurred before the initiation of therapy [183]. Future studies should, therefore, investigate amiloride treatment in obligate combination with high-dose IVMPS rapidly after AON onset. Concomitant immunosuppressive strategies might, for instance, enhance the efficacy of amiloride by ameliorating the inflammatory milieu and immune-mediated neuroaxonal injury.
Notably, there are also uncertainties related to the mechanism of action of amiloride in MS. ASIC opening is mediated by an acidic environment. Strikingly, extracellular acidosis was solely demonstrated in inflammatory lesions in EAE [180]. In MS, acidosis was merely assumed, as inflammatory lesions exhibited a higher lactate concentration [188]. However, this increase of lactate seemed to correlate with the degree of inflammation and was already reduced in inactive plaques, further doubting the impact of amiloride particularly in progressive MS. In line with this, ASIC expression in MS lesions was predominantly observed at the lesion border, which is associated with a more inflammatory milieu [179]. A recent study evaluates imaging-based measurements of pH in the brain tissue of MS patients and might thus shed light on this issue [189].
4.1.2 Fluoxetine
4.1.2.1 Background
Selective serotonin reuptake inhibitors (SSRIs) are well-known therapeutics for the treatment of psychiatric disorders [190]. One of these SSRIs is fluoxetine. Despite its traditional use, a report in 1991 mentioned 20 MS patients who experienced clinical improvement under fluoxetine treatment [191]. Therefore, SSRIs were repurposed in the field of neuroinflammation and neurodegeneration. Repurposing of SSRIs was further supported by insights derived from EAE experiments. There, both the prophylactic and therapeutic application of fluoxetine alleviated clinical EAE progression and enhanced disease remission [192, 193]. Several mechanisms underlying EAE amelioration are discussed. Foremost, a neuroprotective mode of action is considered. Fluoxetine was shown to stimulate astrocytic glycogenolysis and lactate release, thereby providing energy supply to neurons [194, 195]. In addition, fluoxetine enhances the production of neurotrophic factors [196] and inhibits voltage-gated calcium [197] as well as sodium channels [198]. In this way, fluoxetine prevents Ca2+- and Na+-induced neurotoxicity [197, 198]. Beyond neuroprotection, several models indicate a stabilising impact of fluoxetine on the BBB through restoring tight junction molecules [199, 200]. Finally, fluoxetine has several implications concerning the inflammatory response. Among these are an inhibited microglial activation [200], impaired glial antigen presentation [201], and subsequently, a reduced release of pro-inflammatory cytokines by T cells [202].
4.1.2.2 Studies
Started in 2012, a randomised, double-blind Phase II trial evaluated the impact of fluoxetine on disease progression in 77 PPMS and 55 SPMS patients (FLUOX-PMS) [203]. Participants received either 40 mg/day fluoxetine or placebo over 108 weeks. The pSE assessed the time to a 12-week confirmed 20% increase in the T25FW or 9HPT. However, no significant improvement was observed in the analysis of primary and secondary objectives (e.g., cognitive tests and MRI criteria).
Fluoxetine was further investigated in the already described multi-arm Phase IIb MS-SMART trial at a dose of 40 mg/day [184]. One hundred and eleven SPMS patients were allocated to the fluoxetine group, and 112 received placebo. However, fluoxetine failed to affect the pSE, namely the PBVC, after 96 weeks. To the contrary, PBVC was even enhanced compared to placebo after 24 weeks. In terms of secondary objectives, fluoxetine treatment reduced the number of new or enlarging T2 lesions. Other secondary endpoints (e.g., changes in EDSS) were not met.
4.1.2.3 Comment
In conclusion, fluoxetine treatment did not lead to positive outcomes in two Phase II trials. While the MS-SMART study was sufficiently powered [184], an unexpectedly low rate of disease progression was observed in the FLUOX-PMS trial, strongly reducing its power [203]. Additionally, a double-blind, placebo-controlled trial in 42 patients with PPMS or SPMS did not demonstrate an advantage of fluoxetine treatment [204]. Taking together the failure of fluoxetine in all three trials, a convincing beneficial effect in progressive MS is unlikely.
When looking at the mode of action, the initially assumed predominant neuroprotective role of fluoxetine might be outweighed by its anti-inflammatory impact. Fluoxetine treatment results in a variety of immunomodulatory processes, which were observed in both animal and human studies [193, 205]. Along the same lines, fluoxetine proved efficacy in animal models characterised by strong inflammatory pathophysiology [192, 193]. The use of animal models hallmarked by a pronounced inflammatory pathophysiology may also explain why promising data of fluoxetine treatment in pre-clinical experiments were not translatable into clinical trials including patients with progressive MS. Possibly, an animal model of progressive MS, such as EAE in non-obese diabetic mice, would have been more appropriate to study the effects of fluoxetine on the pathophysiology of progressive disease courses.
Further support for a predominant anti-inflammatory role of fluoxetine derives from two clinical studies. First, escitalopram, another SSRI, reduced the cumulative risk for relapses compared to controls in an open-label trial [206]. Also, in a double-blind, placebo-controlled study in RRMS and SPMS patients, fluoxetine itself decreased the formation of GELs [207]. In line with a putative use of fluoxetine in relapsing MS is the reduction of T2 lesions in the MS-SMART trial, although this observation needs to be confirmed considering that post-gadolinium scans at baseline were missing [184]. Taken together, fluoxetine potentially provides benefits to patients with relapsing rather than progressive MS.
4.1.3 Riluzole
4.1.3.1 Background
Glutamate excitotoxicity is a widely discussed mechanism contributing to the pathophysiology of neurodegeneration [208]. Underlying processes of glutamate excitotoxicity involve axonal damage and neuronal cell death [209, 210]. Furthermore, glutamate excitotoxicity leads to oligodendrocyte damage and subsequent demyelination [208]. Therefore, glutamate antagonists gained interest as neuroprotective therapies.
One well-known glutamate antagonist is riluzole, which is commonly used in the therapy of amyotrophic lateral sclerosis [211]. Besides suppressing the release of glutamate from nerve terminals, riluzole stabilises sodium channels in an inactivated state [211]. In line, EAE experiments indicated neuroprotective effects induced by riluzole application [210]. Moreover, a pilot study performed in 16 patients with progressive MS showed a reduction in the rate of cervical cord atrophy due to treatment with riluzole [212].
4.1.3.2 Studies
Following the mentioned pilot study in progressive MS, riluzole was further investigated in the multi-arm Phase IIb MS-SMART trial [184]. Two hundred and twenty-three SPMS patients were included to receive either 100 mg/day riluzole or placebo for a period of 96 weeks. However, the active treatment group failed to meet the pSE (PBVC after 96 weeks) and all secondary outcome parameters (e.g., changes in EDSS or MSFC).
4.1.3.3 Comment
Beside the MS-SMART trial and the pilot study, riluzole was further evaluated in combination to weekly administered interferon-β1a [213]. This randomised placebo-controlled Phase II trial included 43 patients with RRMS or clinically isolated syndrome. Of note, treatment with riluzole did not reduce brain atrophy in this study. In line with the results of riluzole, treatment with the NMDA antagonist memantine failed to improve cognitive impairment in relapsing and progressive MS patients [214]. Given the lack of an effect of riluzole and memantine treatment on both CNS atrophy and clinical parameters, the question arises to what extent glutamate excitotoxicity is relevant to neurodegenerative processes in MS. On the other hand, the high number of glutamate-dependent cell targets and mechanisms of action lead to pleiotropic effects of these glutamate antagonists [208]. Eventually, further identification of relevant signalling pathways involved, together with the design of highly specific compounds, could advance our understanding of the role of glutamate excitotoxicity in neurodegeneration during MS.
4.1.4 Ubiquinone—Idebenone
4.1.4.1 Background
Ubiquinone (CoQ) is an anti-oxidant and lipophilic electron carrier in the mitochondrial electron transport chain. However, low water solubility makes CoQ impracticable for clinical application [215]. Idebenone, a water-soluble short-chain analogue of CoQ [216], is therapeutically used in the treatment of Friedreich’s ataxia and Leber’s hereditary optic neuropathy, both caused by mitochondrial dysfunction [217]. Just like CoQ, idebenone administration was shown to detoxify free radicals and inhibit lipid peroxidation [218]. Moreover, treatment with idebenone regenerates mitochondrial function potentially through restoring the electron flow using a bypass mechanism [219, 220]. As oxidative stress and mitochondrial dysfunction also contribute to neuroaxonal damage in neuroinflammation and neurodegeneration [8], idebenone is considered a putative agent in MS [215].
Beyond its neuroprotective features, idebenone also has anti-inflammatory capabilities. In microglia, it suppressed the production of pro-inflammatory factors (e.g., interleukin (IL)-1β, tumour necrosis factor-α) and induced a shift towards a M2 phenotype that is related to anti-inflammation and regeneration [221]. Given the neuroprotective and immunomodulatory properties, idebenone could be a promising medication in MS.
4.1.4.2 Studies
In a double-blind, placebo-controlled Phase I/II trial, idebenone was evaluated in 77 PPMS patients (IPPoMS) [222]. A 1-year pre-treatment period was followed by an active phase of 2 years with a daily application of 2250 mg idebenone. The pSE investigated the change in the Combinatorial Weight-Adjusted Disability Score (CombiWISE) consisting of EDSS, T25FW, 9HPT, and Scripps Neurological Disability Scale [223]. However, treatment had no impact on the CombiWISE. Additionally, evaluation of secondary objectives, including changes in the enlargement of ventricular volume as well as differences in the single categories of the CombiWISE, did not reveal notable improvements induced by idebenone.
Afterwards, all patients who had completed the IPPoMS trial were invited to an open-label extension study [224]. There, all patients should receive idebenone for 1 additional year. Unfortunately, publication of the extension study is missing.
4.1.4.3 Comment
The shortage of available clinical data hampers the analysis of the role of idebenone in MS. Moreover, pre-clinical insights are sparse. Idebenone was once tested in EAE, failing to affect disease incidence, onset, and severity [225]. Further, idebenone had no impact on neuroinflammation or axonal damage.
It can be speculated that the failure of idebenone was related to the dependency of its function on cytoplasmic NAD(P)H:quinone oxidoreductase 1 (NQO1). NQO1 catalyses the reduction of idebenone into idebenol [220]. While idebenone itself exhibits oxidative properties [226] and impairs the electron transport chain [227], the favourable actions are attributed to idebenol [215]. Unfortunately, NQO1 expression is mainly restricted to astrocytes and a subset of oligodendrocytes, while it is rare in neurons [228]. Therefore, the potential for direct neuroprotection is limited [229]. However, as NQO1 expression is inducible, combination with agents selectively upregulating NQO1 in neurons might enforce idebenone treatment in neurodegeneration [229].
Conclusively, the paucity of data impedes an assumption of the impact of idebenone on neuroinflammation. Nonetheless, the underwhelming performance in the IPPoMS trial and insights derived from animal experiments contradict a beneficial role of idebenone in PPMS, at least when used as monotherapy.
4.2 Neuroregenerative Approaches
4.2.1 Erythropoietin
4.2.1.1 Background
Erythropoietin (EPO) is an internal body hormone stimulating erythropoiesis. Interestingly, tissue expression studies identified EPO and its receptor (EPO-R) in neurons, glial, and also endothelial cells of the CNS [230,231,232]. Emerging evidence describes the EPO-EPO-R-axis as an endogenous neuroprotective system getting activated in response to neuronal damage in terms of hypoxia, metabolic stress, or inflammation, just as observed in EAE [233, 234]. Accordingly, exogenously administered EPO ameliorated clinical signs of neuroinflammation in mice [235,236,237]. Clinical improvement depends at least partly on the induction of remyelination. In vitro, for instance, EPO promotes OPC differentiation and oligodendrocyte maturation [230, 238]. Further, it enhances the expression of myelin markers after cuprizone-induced demyelination and proliferation of OPCs during EAE [239, 240].
Besides remyelination, EPO prevents different hallmarks of autoimmune neuroinflammation, such as BBB breakdown or axonal injury [235,236,237]. The underlying mechanisms of neuroprotection involve neurotrophic [241], anti-oxidative [242], and anti-apoptotic processes [243]. Even more, EPO modulates the inflammatory response, leading to increased expansion of regulatory T cells and suppressed differentiation of TH17 cells [236].
In addition to the aforementioned pre-clinical observations, a small exploratory open-label Phase I/IIa trial on high-dose EPO treatment demonstrated a long-lasting significant increase in walking distance and improvements of cognitive impairment in PPMS and SPMS [244]. Prolonged EPO treatment was further assessed in a small study in a SPMS cohort, indicating beneficial effects of EPO in terms of neurophysiological criteria (e.g., increased intracortical facilitation) and a trend of ameliorated fatigue [245].
4.2.1.2 Studies
Following the first promising clinical results, a randomised, double-blind Phase II trial was performed in 18 PPMS and 34 SPMS patients (without relapses within the last 2 years) [246]. The mean disease duration varied between 16.7 years in the EPO and 14.9 years in the placebo group. Participants received either 48,000 IU EPO or placebo for 24 weeks (in the first half weekly, afterwards biweekly) followed by a 24-week observation period. IVMPS 1 g was applied prior to the first and second infusion of EPO or placebo. The pSE investigated the change of a composite score consisting of the maximum gait distance, hand dexterity, and cognition after 24 weeks. Patients treated with EPO demonstrated a slight increase in the composite score, indicating clinical improvement. However, this trend was not significant and could not be observed after 48 weeks. Analysis of clinical parameters (e.g., EDSS and MSFC), MRI assessment (e.g., T2 lesion volume and PBVC) as well as patient-reported outcomes (36-Item Short-Form Health Survey [247]) did not reveal any advantage of EPO treatment. Concerning the safety profile, most patients in the active treatment group needed blood-lettings due to increased haematocrit and experienced hypertensive episodes more frequently.
4.2.1.3 Comment
While a first exploratory pilot study indicated improvements under EPO treatment, especially in terms of motor functions [244], these results could not be reproduced in the reviewed Phase II trial, even though a positive trend was observed [246]. An underlying reason for failure might be the mean disease duration of over 16 years. Indeed, remyelination declines with MS duration and is less present in chronic lesions [6, 87]. Thus, agents promoting remyelination might be more effective in earlier disease courses, since an effect on long-lasting, inactive lesions is doubtful. In line with this assumption, a small double-blind study supported efficacy of EPO-treatment in RRMS patients with an acute relapse [248]. The first promising results of EPO in AON in a placebo-controlled Phase II trial further support this hypothesis [249]. Conclusively, the selection of patients in advanced stages of disease progression might have masked potentially existing beneficial effects of EPO treatment.
Nonetheless, the potential use of EPO in MS is hampered by side effects. Administration of EPO not only induces erythropoiesis but has several other implications such as vasoconstriction or thrombocythemia, thereby enhancing the risk for thromboembolic events and hypertension [233]. Thus, patients with cardiovascular risk factors, a history of thromboembolic events, or immobility were excluded from clinical trials [246, 249, 250]. Moreover, EPO can cause rare but potentially fatal pure red cell aplasia [251]. Due to the expression of EPO and EPO-R on several malignant tumours [252] and reports of tumour expansion under EPO treatment [253, 254], patients with a history of malignancy should not receive this drug either. Taken together, a remarkable proportion of patients, especially those suffering from multimorbidity, would not be eligible for treatment even if EPO were to be effective in MS.
On the other hand, tissue-protective processes may not only be mediated via the classical EPO-R. Foremost, these effects seem to be induced by the interaction of a distinct EPO region with a heteromeric receptor consisting of one EPO-R subunit and the β-common chain shared by members of the IL-3 receptor family [233, 255]. Thus, derivates of EPO were shown to exhibit neuroprotection without affecting erythropoiesis [235]. Among these derivates is JM-4 [256]. This peptide protected mice from demyelination and alleviated the pro-inflammatory response, thereby ameliorating clinical signs of EAE [256]. Of note, an early Phase I study was recently initiated to evaluate the safety and efficacy of JM-4 in the context of MS [257].
4.2.2 Carboxylase Enzymes—Biotin
4.2.2.1 Background
Biotin is a ubiquitous water-soluble vitamin, acting as an essential co-enzyme for several carboxylases [258]. Based on a case report on the efficacy of biotin in SPMS, the first studies were introduced to evaluate biotin as a potential target in progressive MS [259]. Reduced levels of biotin observed in the cerebrospinal fluid (CSF) and serum of MS patients gave further rise to repurpose biotin in the context of neuroinflammation [260]. Additionally, deficiency of biotinidase, an enzyme required for biotin recycling [261], leads to demyelination and neurological symptoms that are comparable to MS [262].
The rationale behind the use of biotin is based on the hypothesis of a ‘virtual hypoxia’ in MS [258]. It is assumed that mitochondrial dysfunction and an increased need for energy due to demyelination result in a mismatch of energy supply and demand. Consequently, this mismatch would lead to neuroaxonal injury. Biotin potentially addresses both reduced energy production and increased demand. First, being a critical co-enzyme of three carboxylases involved in the tricarboxylic acid cycle, increased concentrations of biotin might foster neuronal ATP supply [263]. Second, biotin is required in oligodendrocytes for the activity of two carboxylases that produce a substrate of fatty acid synthesis [263]. In this way, biotin might enhance remyelination due to increased levels of these fatty acids, which are needed for myelin production [264]. Through restoring saltatory conduction and thus, restricting membrane excitation to the nodes of Ranvier, biotin might reduce the energy demand [265].
Further evidence supporting a beneficial role of biotin in MS derives from a small, uncontrolled study in 23 PPMS and SPMS patients, reporting improved visual and motor functions in almost all patients [266]. Following this pilot study, a first Phase III trial in 154 PPMS and SPMS patients (SPI) showed alleviation of disability through biotin treatment [267].
4.2.2.2 Studies
A second double-blind Phase III trial was initiated in 2016 (SPI2) [268], in which 227 PPMS and 415 SPMS patients without relapses in the previous 2 years were randomly assigned to either 300 mg/day biotin or placebo. All patients remained in the double-blind, placebo-controlled phase of the trial until the last-entered participant reached Month 15. At that time, all patients switched to biotin treatment at the next scheduled visit. Therefore, the placebo-controlled part of the study lasted between 15 and 27 months (mean duration: 20.1 months). Continuation of treatment with DMTs was permitted throughout the whole trial. The pSE evaluated differences in the proportion of patients with a confirmed improvement in EDSS score or T25FW after 12 months. Unfortunately, biotin failed to provide significant benefits in terms of the pSE. Furthermore, the study did not meet any secondary endpoint (e.g., time to EDSS progression, change in T25FW).
In addition to the SPI2 trial, two studies were conducted in real-world settings. The first was performed in seven PPMS and 36 SPMS patients, including also relapsing patients [269]. Using an open-label design, all patients received 300 mg/day biotin for 1 year as an add-on medication in the case of a pre-existing DMT. One-third of the patients reported subjective worsening of MS, leading to an increased EDSS score in two cases. Only two participants experienced clinical improvements, which were, however, not sufficient to decrease EDSS. Strikingly, only 24 of 43 patients completed the whole study duration. The main reasons for withdrawal included a lack of efficacy and worsening of symptoms.
The second open-label study within a real-word setting included 84 PPMS and 94 SPMS patients [270]. Concomitant intake of DMTs was permitted. The pSE assessed the improvement of disability after 12 months of high-dose biotin treatment, measured by a decrease in EDSS. A reduction of the EDSS score was observed in only six patients. Secondary outcome parameters, analysing disability, processing speed, and radiological activity, revealed no benefits other than a significant improvement in the pain and discomfort dimension in a patient questionnaire. Although the ARR was not enhanced compared to the time prior to biotin treatment, MRI assessment demonstrated radiological activity in about 30% of patients with MRI scans.
4.2.2.3 Comment
Not only failure in clinical trials but also inconclusiveness of the propagated role of biotin in autoimmune neuroinflammation question further application. Clinical improvement through biotin substitution in the case of biotinidase deficiency appears consistent in the light of severe shortness of biotin. However, it seems unlikely that the mere increase of fatty acid supply enhances the highly complex processes of remyelination in MS. Moreover, recent pre-clinical data provide evidence contradicting a beneficial impact of biotin on autoimmune neuroinflammation. Buonvicino et al reported that biotin failed to increase ATP levels in murine cortical neurons [271]. Further, biotin was unable to protect neurons from glutamate-induced excitotoxicity and oligodendrocytes from cuprizone-mediated injury. Even more, biotin treatment did not affect EAE progression in non-obese diabetic mice, resembling progressive MS [271]. To the best of our knowledge, biotin was also never shown to promote remyelination in vitro or in vivo. Given the lack of beneficial effects of biotin treatment on brain atrophy and serum neurofilament levels in the SPI2 trial [268], a neuroprotective effect is also highly unlikely.
Besides the doubts about its mechanism of action, some studies reported increased rates of relapses and MRI lesions under high-dose biotin treatment even in patients without previous relapses [267, 269, 270, 272,273,274,275,276]. A possible biotin-mediated influence on BBB integrity has not been assessed. Furthermore, immunomodulatory effects of high-dose biotin are not sufficiently clarified. Investigation of PBMCs obtained from biotin-treated patients with clinical or MRI worsening indicated changes in immune cell frequencies [277]: a decrease in the overall number of lymphocytes, a reduction of CD4+ as well as CD8+ T cells, and an increase of class-switched memory IgD-CD27+ B cells were reported. Unfortunately, subpopulations of CD4+ T cells were not further characterised, limiting conclusions about biotin-mediated effects on the inflammatory response.
However, the SPI2 trial did not confirm the fear of a higher relapse rate [268]. Additionally, a large retrospective study (IPBio-SeP) investigates the incidence of relapses in biotin-treated patients [278]. Intermediate analysis of 1279 biotin-receiving patients and 483 controls did not show a difference in the frequency of relapses after 16 months.
One reason for the different outcomes of biotin treatment in the SPI compared to the SPI2 trial may have been the markedly higher placebo response rate in the SPI2 trial strongly reducing the power of this study [268]. However, given the much higher number of patients in the SPI2 trial, it is more likely that the beneficial effects observed in the SPI trial resulted from a type 1 error [268]. In addition, the rate of disability progression was reduced in both groups of the SPI2 trial. This, in turn, may have caused the observation period to be too short to depict the neuroregenerative effects. A striking point in the study design of the SPI2 trial is the pSE as it evaluated the improvement of disability. However, a delay of disease progression seems to be much more realistic than a significant improvement of clinical symptoms.
Altogether, given the negative results of the SPI2 trial and two studies in real-world settings, a beneficial effect of biotin in progressive MS in unlikely.
4.3 Other Approaches
4.3.1 Glycogen Synthase Kinase-3—Lithium
4.3.1.1 Background
Being one of the first FDA approved drugs, lithium is a mood-stabiliser essential for the treatment of bipolar disorders [279]. Pre-clinical data obtained from several EAE models give rise to the use of lithium in neuroinflammatory conditions [280, 281]. Although lithium affects diverse targets, amelioration of neuroinflammation seems to be mainly mediated by suppression of glycogen synthase kinase-3 (GSK-3) [280]. Inhibition of GSK-3, for instance, interferes with the generation of TH1 [282] and TH17 cells [283]. Suppression of GSK-3 might also preserve BBB integrity through upregulation of the WNT/β-catenin pathway [284]. There is further evidence for the involvement of lithium and GSK-3 in MS as serum lithium levels are reduced in RRMS patients [285], while expression of GSK-3β was upregulated in patients suffering from PPMS [286].
4.3.1.2 Studies
Following promising pre-clinical results, an open-label, rater-blinded Phase I/II trial was performed in 3 PPMS and 20 SPMS patients [287]. Disease duration varied from 3 to 43 years. The study was conducted in a cross-over design, with random assignment to lithium treatment in either the first or second year. Lithium doses varied between 150 and 300 mg/day. Simultaneous treatment with DMTs (natalizumab, interferon-β, GA) was permitted. The application of lithium resulted in positive but non-significant trends for the pSE, i.e., the PBVC. Moreover, lithium treatment failed to affect secondary clinical objectives (e.g., change in EDSS, MSFC) but significantly improved patient-reported outcomes in terms of the mental domains of the MS Quality of Life-54 questionnaire [288].
4.3.1.3 Comment
Besides the open-label design, heterogenous baseline characteristics in terms of divergent disease durations and simultaneous application of DMTs in some patients hinder interpretation of the results. Unfortunately, there are no data on correlations between outcome results and DMT intake or patients’ lithium serum levels, which might have been interesting given the low doses administered.
Further, due to the variety of affected targets, lithium-induced aggravation of neuroinflammation cannot be excluded. Among the possibly involved mechanisms is the WNT/β-catenin-mediated dysfunction of remyelination [289]. Another example is the activation of the protein kinase Akt-1 by lithium, as recent data demonstrate a detrimental role of Akt-1 in EAE [290]. In line with this, a retrospective analysis of lithium treatment including 101 US veterans with all types of MS revealed increased relapse rates. The worsening of EDSS, however, was significantly slowed [291].
Given the small number of participants, methodological limitations, and the lack of other prospective trials, the role of lithium in MS remains elusive.
5 Discussion
In this review, we give an update on clinical trials evaluating remyelination-promoting strategies, neuroprotective treatments, and other approaches that either failed or were interrupted for other reasons. Thereby we continue our series of previous reviews on failed trials in the context of MS [292,293,294,295]. Of note, a current update on failed or interrupted studies of immunomodulatory agents has been recently given in a separate article [9].
The most important lesson learned from the failed trials is the demand for further research. Many of the pathways involved are known. However, we do not ultimately understand their interplay resulting in remyelination and, more important, the mechanisms underlying its insufficiency [6]. There is a lack of comprehension of why remyelination fails in some patients, while others display a high proportion of remyelinated lesions [87]. Further insights into the crucial processes of remyelination are required to address them sufficiently. Accordingly, given the heterogeneity of lesions, the variety of disease courses, and the complexity of MS, it seems unlikely that targeting single pathways will be efficient [296]. Therefore, a combination of approaches that promote not only OPC proliferation and differentiation but also establish a favourable microenvironment seems to be more promising [297]. Moreover, the combination with immunomodulatory drugs might create a less inflammatory but more favourable milieu enhancing remyelination and providing further benefit by tackling MS pathophysiology more widely. Of the reviewed drugs that were tested in combination with immunomodulatory agents, most agents were examined as add-on therapeutics to interferon-β [22, 60, 71, 98, 120, 169]. Due to the plethora of interferon-mediated mechanisms, however, there is a high risk for (unforeseen) interactions [38]. Considering these unpredictable interactions, the use of compounds in combination with interferon-β might lead to antagonistic effects. In the case of atorvastatin, for instance, combined use with interferon-β has potentially antagonised interferon-mediated suppression of MMPs and phosphorylation of STAT 1 [36, 37]. This may have attenuated beneficial clinical effects in the active treatment group. On the other hand, the combination of interferon-β with compounds that induce comparable processes might cover differences between the active treatment and the control group, as described for inosine (see 3.1.4) and minocycline (see 3.3.2). In contrast, monoclonal antibodies such as natalizumab could be a more suitable alternative, as they provide several advantages over interferon-β. First, the more selective mechanisms of action facilitate combinations with promising compounds since interactions at the molecular level are easier to predict [298]. Second, monoclonal antibody therapies provide a higher degree of disease control as well as a stronger suppression of the inflammatory activity making them a favourable partner in add-on studies with neuroregenerative agents [299].
We also observed a trend to repurpose well-known therapeutics that are licensed for other diseases in the context of autoimmune neuroinflammation. Thereby, investigators not only minimise the risk of unforeseen adverse reactions but also reduce costs and accelerate the progression through the different phases of clinical trials to enter the market. However, none of these drugs has been resoundingly successful [22, 142, 169, 183, 184, 203, 222, 246, 287]. Along with a lack of efficacy, in some cases, the initial indication for approval turns out to cause side effects limiting the use in MS, as seen for EPO [233]. Nevertheless, the latter example shows that designing new and more specific derivates might be a way to reduce side effects but still benefit from the initial rationale.
Given the frequent failure of approaches that proved efficacy in pre-clinical experiments, it becomes obvious that animal models help study specific facets of CNS autoimmunity but cannot resemble the whole intricacy of MS. The cuprizone model, for instance, gives valuable insights into processes of remyelination but neglects the inflammatory impact [110]. Another useful tool for studying the neuroregenerative abilities is a model using injections of the white matter gliotoxin lysolecithin [300]. This acute injury in a localised area allows remyelination to be assessed in the absence of ongoing tissue damage [301]. However, this injury occurs without chronic involvement of lymphocytes [296]. EAE, on the other hand, shares many immunological and histological characteristics with MS. Still, the intensity of the ongoing inflammatory response might impede the investigation of remyelination [296].
Moreover, it is highly relevant to select animal models according to the presumed mechanism of action of the tested compounds. For example, agents treating neurodegeneration should be studied in models that share pathophysiological features of progressive MS, such as EAE in non-obese diabetic MS mice [271, 302]. Nevertheless, promising agents need to be evaluated in not just one animal model but different in vivo and in vitro approaches, covering a variety of aspects involved in MS.
Another important aspect involves frequently observed problems regarding the reproducibility of pre-clinical data between different laboratories. These problems are partly based on to the strong dependence of experiments on factors such as the strain or genetic background of the animals, the environment, or the diet [303]. A hopeful approach to improve the reproducibility of insights gained from animal experiments may be pre-clinical randomised controlled trials (pRCTs) that are conducted in a number of different laboratories under standardised conditions [304]. These pRCTs could provide a higher degree of comparability and validity of pre-clinical data. Therefore, these pRCTs could be an important step to bridge the gap between pre-clinical studies and Phase I clinical trials.
Furthermore, it needs to be discussed to what extent compounds in early clinical trials should necessarily be evaluated solely on the basis of the achievement of statistical significance of study endpoints. In some cases, such as GSK239512 [120], the effect size may have been overestimated. Therefore, possibly also in light of the already high costs, many studies were likely underpowered. Nevertheless, these studies can provide important insights into the mechanisms of action of the evaluated compounds. More importantly, subgroup analyses can be used to select patients who may benefit from treatment in subsequent clinical trials. Therefore, promising candidates should not be discarded just because they failed to meet the pSE.
6 Conclusions
The key conclusion is the critical requirement of appropriate study designs. Most importantly, there is still a need for reliable and sensitive outcome parameters in trials investigating neuroregenerative therapeutics. To date, there is no marker available that can distinguish between pre-existing and remyelinated myelin [6]. In addition, it is not clear how to properly quantify remyelination. A variety of imaging techniques, including MTR, DTI, or positron emission tomography, have emerged and have already been proven to depict the extent of remyelination [305,306,307]. However, these methods are not widely used or sufficiently validated [308]. In the trials reviewed, only the study on GSK239512 made use of one of these techniques as pSE [120]. Another approach to detect remyelinating effects is the measurement of multifocal VEPs, a technique that uses simultaneous stimulations of the visual field [96]. In this way, multifocal VEPs provide a higher degree of sensitivity and specificity in detecting regeneration of the anterior visual pathway compared to conventional VEPs [309].
In terms of studies investigating neuroprotective approaches, future trials might also include neurofilament light chain (NfL) measurement. Neurofilaments are part of the neuronal cytoskeleton and particularly enriched in axons. Thus, neurofilaments are released into the CSF and serum upon neuroaxonal injury [310] making NfL measurement a promising tool elucidating neuroprotective effects. Although not being MS-specific, NfL was shown to be a biomarker of disease activity and predictor of the long-term outcome in MS tracking both neuroinflammatory and neurodegenerative damage [310,311,312].
As well as these parameters, the demand for adequate clinical outcome criteria is even more critical. Compounds will only be successful therapeutic options for patient care if they not only improve paraclinical parameters, but provide a relevant benefit to patients. Therefore, sensitive clinical endpoints that depict the effects of neuroregeneration (e.g., improvements of cognitive symptoms) are urgently required. In particular, integrated clinical scores reflecting different clinical dimensions, as used in the SYNERGY [98] and IPPoMS trials [222], seem to be favourable to assess the clinical effects of neuroprotective and neuroregenerative agents.
Further, the beneficial effects of neuroprotection are mainly attributed to regeneration from axonal injury and reduced accumulation of neuronal damage over time [6]. Thus, clinical effects are unlikely to be observed after a single relapse. They might rather be seen after years in terms of delayed disability and disease progression, thereby claiming for long-term trials [308]. Yet, most of the current trials do not meet this need for an extensive follow-up period.
Another important aspect with regard to the study design, especially of remyelinating therapies, is the age of the patients included. Taking the subgroup analysis of the RENEW study as an example, age-dependent differences in the efficacy of remyelinating approaches become clear [106]. Remyelination capacities, for instance, decline with age [107]. Future studies need to show, whether this decline leads to a decreased success of remyelinating therapies or possibly, on the contrary, to an improved outcome of remyelinating therapies due to a wider margin of improvement. Therefore, the age of the participants needs to be considered in the study design of these therapies in form of stricter age restrictions and age matching approaches. In addition to the influence of age, the disease duration also appears to have a major impact on the efficacy of neuroregenerative as well as neuroprotective therapies. Underlying reasons seem to involve advanced damage of neuroaxonal structures. First, the presence of functionally intact axons is a mandatory prerequisite for remyelination [6]. Second, the integrity of neuronal networks seems to be significantly reduced in advanced stages of the disease, reducing the potential success of neuroprotective approaches [111]. Hence, a more accurate consideration of disease duration is essential for the study design of future trials.
Contrasting results of the same agent in different disease entities, as observed in trials on atorvastatin (see 3.1.1) or EPO (see 4.2.1), underline the importance of a strict separation of these entities in clinical studies. Together with the relevance of the different pathophysiological concepts underlying the disease courses [3], this observation highlights the importance of a careful selection of the appropriate patient population for the success of clinical trials.
Finally, his review further emphasises the need for publication of failed clinical studies. Although these trials did not measure up to the expectations, they nevertheless provide essential information on MS pathophysiology. In addition, they help to understand issues concerning the translation of insights gained from pre-clinical studies and are indispensable for advancements in study design. Therefore, investigators and journals should be further encouraged to publish negative outcomes.
References
Tintore M, Vidal-Jordana A, Sastre-Garriga J. Treatment of multiple sclerosis—success from bench to bedside. Nat Rev Neurol. 2019;15(1):53–8. https://doi.org/10.1038/s41582-018-0082-z.
Miller DH, Leary SM. Primary-progressive multiple sclerosis. Lancet Neurol. 2007;6(10):903–12. https://doi.org/10.1016/S1474-4422(07)70243-0.
Mahad DH, Trapp BD, Lassmann H. Pathological mechanisms in progressive multiple sclerosis. Lancet Neurol. 2015;14(2):183–93. https://doi.org/10.1016/S1474-4422(14)70256-X.
Faissner S, Plemel JR, Gold R, Yong VW. Progressive multiple sclerosis: from pathophysiology to therapeutic strategies. Nat Rev Drug Discov. 2019;18(12):905–22. https://doi.org/10.1038/s41573-019-0035-2.
Vaughn CB, Jakimovski D, Kavak KS, Ramanathan M, Benedict RHB, Zivadinov R, et al. Epidemiology and treatment of multiple sclerosis in elderly populations. Nat Rev Neurol. 2019;15(6):329–42. https://doi.org/10.1038/s41582-019-0183-3.
Stangel M, Kuhlmann T, Matthews PM, Kilpatrick TJ. Achievements and obstacles of remyelinating therapies in multiple sclerosis. Nat Rev Neurol. 2017;13(12):742–54. https://doi.org/10.1038/nrneurol.2017.139.
Yong H, Chartier G, Quandt J. Modulating inflammation and neuroprotection in multiple sclerosis. J Neurosci Res. 2018;96(6):927–50. https://doi.org/10.1002/jnr.24090.
Correale J, Gaitan MI, Ysrraelit MC, Fiol MP. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain. 2017;140(3):527–46. https://doi.org/10.1093/brain/aww258.
Rolfes L, Pawlitzki M, Pfeuffer S, Huntemann N, Wiendl H, Ruck T, et al. Failed, interrupted, or inconclusive trials on immunomodulatory treatment strategies in multiple sclerosis: update 2015–2020. BioDrugs. 2020. https://doi.org/10.1007/s40259-020-00435-w.
Frischer JM, Bramow S, Dal-Bianco A, Lucchinetti CF, Rauschka H, Schmidbauer M, et al. The relation between inflammation and neurodegeneration in multiple sclerosis brains. Brain. 2009;132(Pt 5):1175–89. https://doi.org/10.1093/brain/awp070.
Zacco A, Togo J, Spence K, Ellis A, Lloyd D, Furlong S, et al. 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors protect cortical neurons from excitotoxicity. J Neurosci. 2003;23(35):11104–11.
Wu H, Lu D, Jiang H, Xiong Y, Qu C, Li B, et al. Simvastatin-mediated upregulation of VEGF and BDNF, activation of the PI3K/Akt pathway, and increase of neurogenesis are associated with therapeutic improvement after traumatic brain injury. J Neurotrauma. 2008;25(2):130–9. https://doi.org/10.1089/neu.2007.0369.
van der Most PJ, Dolga AM, Nijholt IM, Luiten PG, Eisel UL. Statins: mechanisms of neuroprotection. Prog Neurobiol. 2009;88(1):64–75. https://doi.org/10.1016/j.pneurobio.2009.02.002.
Peng X, Jin J, Giri S, Montes M, Sujkowski D, Tang Y, et al. Immunomodulatory effects of 3-hydroxy-3-methylglutaryl coenzyme-A reductase inhibitors, potential therapy for relapsing remitting multiple sclerosis. J Neuroimmunol. 2006;178(1–2):130–9. https://doi.org/10.1016/j.jneuroim.2006.06.005.
Greenwood J, Walters CE, Pryce G, Kanuga N, Beraud E, Baker D, et al. Lovastatin inhibits brain endothelial cell Rho-mediated lymphocyte migration and attenuates experimental autoimmune encephalomyelitis. FASEB J. 2003;17(8):905–7. https://doi.org/10.1096/fj.02-1014fje.
Youssef S, Stuve O, Patarroyo JC, Ruiz PJ, Radosevich JL, Hur EM, et al. The HMG-CoA reductase inhibitor, atorvastatin, promotes a Th2 bias and reverses paralysis in central nervous system autoimmune disease. Nature. 2002;420(6911):78–84. https://doi.org/10.1038/nature01158.
Vollmer T, Key L, Durkalski V, Tyor W, Corboy J, Markovic-Plese S, et al. Oral simvastatin treatment in relapsing-remitting multiple sclerosis. Lancet. 2004;363(9421):1607–8. https://doi.org/10.1016/S0140-6736(04)16205-3.
Sena A, Pedrosa R, Graca MM. Therapeutic potential of lovastatin in multiple sclerosis. J Neurol. 2003;250(6):754–5. https://doi.org/10.1007/s00415-003-1070-8.
Paul F, Waiczies S, Wuerfel J, Bellmann-Strobl J, Dorr J, Waiczies H, et al. Oral high-dose atorvastatin treatment in relapsing-remitting multiple sclerosis. PLoS ONE. 2008;3(4):e1928. https://doi.org/10.1371/journal.pone.0001928.
Lanzillo R, Orefice G, Quarantelli M, Rinaldi C, Prinster A, Ventrella G, et al. Atorvastatin combined to interferon to verify the efficacy (ACTIVE) in relapsing-remitting active multiple sclerosis patients: a longitudinal controlled trial of combination therapy. Mult Scler. 2010;16(4):450–4. https://doi.org/10.1177/1352458509358909.
Togha M, Karvigh SA, Nabavi M, Moghadam NB, Harirchian MH, Sahraian MA, et al. Simvastatin treatment in patients with relapsing-remitting multiple sclerosis receiving interferon beta 1a: a double-blind randomized controlled trial. Mult Scler. 2010;16(7):848–54. https://doi.org/10.1177/1352458510369147.
Lanzillo R, Quarantelli M, Pozzilli C, Trojano M, Amato MP, Marrosu MG, et al. No evidence for an effect on brain atrophy rate of atorvastatin add-on to interferon beta1b therapy in relapsing-remitting multiple sclerosis (the ARIANNA study). Mult Scler. 2016;22(9):1163–73. https://doi.org/10.1177/1352458515611222.
Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS). Neurology. 1983;33(11):1444–52. https://doi.org/10.1212/wnl.33.11.1444.
Amato MP, Portaccio E, Goretti B, Zipoli V, Ricchiuti L, De Caro MF, et al. The Rao’s Brief Repeatable Battery and Stroop Test: normative values with age, education and gender corrections in an Italian population. Mult Scler. 2006;12(6):787–93. https://doi.org/10.1177/1352458506070933.
Rudick RA, Pace A, Rani MR, Hyde R, Panzara M, Appachi S, et al. Effect of statins on clinical and molecular responses to intramuscular interferon beta-1a. Neurology. 2009;72(23):1989–93. https://doi.org/10.1212/WNL.0b013e3181a92b96.
Paz Soldan MM, Pittock SJ, Weigand SD, Yawn BP, Rodriguez M. Statin therapy and multiple sclerosis disability in a population-based cohort. Mult Scler. 2012;18(3):358–63. https://doi.org/10.1177/1352458511421920.
Ghasami K, Faraji F, Fazeli M, Ghazavi A, Mosayebi G. Interferon beta-1a and atorvastatin in the treatment of multiple sclerosis. Iran J Immunol. 2016;13(1):16–26 (Doi: IJIv13i1A3).
Kamm CP, El-Koussy M, Humpert S, Findling O, von Bredow F, Burren Y, et al. Atorvastatin added to interferon beta for relapsing multiple sclerosis: a randomized controlled trial. J Neurol. 2012;259(11):2401–13. https://doi.org/10.1007/s00415-012-6513-7.
Sorensen PS, Lycke J, Eralinna JP, Edland A, Wu X, Frederiksen JL, et al. Simvastatin as add-on therapy to interferon beta-1a for relapsing-remitting multiple sclerosis (SIMCOMBIN study): a placebo-controlled randomised phase 4 trial. Lancet Neurol. 2011;10(8):691–701. https://doi.org/10.1016/S1474-4422(11)70144-2.
Pihl-Jensen G, Tsakiri A, Frederiksen JL. Statin treatment in multiple sclerosis: a systematic review and meta-analysis. CNS Drugs. 2015;29(4):277–91. https://doi.org/10.1007/s40263-015-0239-x.
Wang J, Xiao Y, Luo M, Luo H. Statins for multiple sclerosis. Cochrane Database Syst Rev. 2011;12:CD008386. https://doi.org/10.1002/14651858.CD008386.pub3.
Salvatore E, Morra VB, Orefice G. Combining beta interferon and atorvastatin may increase disease activity in multiple sclerosis. Neurology. 2009;73(13):1078. https://doi.org/10.1212/WNL.0b013e3181ab6e08 (author reply-9).
Birnbaum G, Cree B, Altafullah I, Zinser M, Reder AT. Combining beta interferon and atorvastatin may increase disease activity in multiple sclerosis. Neurology. 2008;71(18):1390–5. https://doi.org/10.1212/01.wnl.0000319698.40024.1c.
Montero MT, Hernandez O, Suarez Y, Matilla J, Ferruelo AJ, Martinez-Botas J, et al. Hydroxymethylglutaryl-coenzyme A reductase inhibition stimulates caspase-1 activity and Th1-cytokine release in peripheral blood mononuclear cells. Atherosclerosis. 2000;153(2):303–13. https://doi.org/10.1016/s0021-9150(00)00417-2.
Miron VE, Rajasekharan S, Jarjour AA, Zamvil SS, Kennedy TE, Antel JP. Simvastatin regulates oligodendroglial process dynamics and survival. Glia. 2007;55(2):130–43. https://doi.org/10.1002/glia.20441.
Dhawan N, Reder A. Statins block interferon signaling in human immune cells: potential loss of the therapeutic effect of IFN-beta in multiple sclerosis. Neurology. 2007;68:A364.
Kieseier BC, Archelos JJ, Hartung HP. Different effects of simvastatin and interferon beta on the proteolytic activity of matrix metalloproteinases. Arch Neurol. 2004;61(6):929–32. https://doi.org/10.1001/archneur.61.6.929.
Kieseier BC. The mechanism of action of interferon-beta in relapsing multiple sclerosis. CNS Drugs. 2011;25(6):491–502. https://doi.org/10.2165/11591110-000000000-00000.
Chataway J, Schuerer N, Alsanousi A, Chan D, MacManus D, Hunter K, et al. Effect of high-dose simvastatin on brain atrophy and disability in secondary progressive multiple sclerosis (MS-STAT): a randomised, placebo-controlled, phase 2 trial. Lancet. 2014;383(9936):2213–21. https://doi.org/10.1016/S0140-6736(13)62242-4.
Multiple Sclerosis-Simvastatin Trial 2 (MS-STAT2). In: ClinicalTrials.gov Identifier: NCT03387670. Start: 2018. Accessed 3 Jun 2020.
Lanzillo R, Moccia M, Russo CV, Carotenuto A, Nozzolillo A, Petruzzo M, et al. Therapeutic lag in reducing disability progression in relapsing-remitting multiple sclerosis: 8-year follow-up of two randomized add-on trials with atorvastatin. Mult Scler Relat Disord. 2019;28:193–6. https://doi.org/10.1016/j.msard.2018.12.042.
Cakici N, Fakkel TM, van Neck JW, Verhagen AP, Coert JH. Systematic review of treatments for diabetic peripheral neuropathy. Diabet Med. 2016;33(11):1466–76. https://doi.org/10.1111/dme.13083.
Rochette L, Ghibu S, Richard C, Zeller M, Cottin Y, Vergely C. Direct and indirect antioxidant properties of alpha-lipoic acid and therapeutic potential. Mol Nutr Food Res. 2013;57(1):114–25. https://doi.org/10.1002/mnfr.201200608.
Seifar F, Khalili M, Khaledyan H, Amiri Moghadam S, Izadi A, Azimi A, et al. alpha-Lipoic acid, functional fatty acid, as a novel therapeutic alternative for central nervous system diseases: a review. Nutr Neurosci. 2019;22(5):306–16. https://doi.org/10.1080/1028415X.2017.1386755.
Ou P, Tritschler HJ, Wolff SP. Thioctic (lipoic) acid: a therapeutic metal-chelating antioxidant? Biochem Pharmacol. 1995;50(1):123–6. https://doi.org/10.1016/0006-2952(95)00116-h.
Schreibelt G, Musters RJ, Reijerkerk A, de Groot LR, van der Pol SM, Hendrikx EM, et al. Lipoic acid affects cellular migration into the central nervous system and stabilizes blood-brain barrier integrity. J Immunol. 2006;177(4):2630–7. https://doi.org/10.4049/jimmunol.177.4.2630.
Chaudhary P, Marracci GH, Bourdette DN. Lipoic acid inhibits expression of ICAM-1 and VCAM-1 by CNS endothelial cells and T cell migration into the spinal cord in experimental autoimmune encephalomyelitis. J Neuroimmunol. 2006;175(1–2):87–96. https://doi.org/10.1016/j.jneuroim.2006.03.007.
Wang KC, Tsai CP, Lee CL, Chen SY, Lin GJ, Yen MH, et al. alpha-Lipoic acid enhances endogenous peroxisome-proliferator-activated receptor-gamma to ameliorate experimental autoimmune encephalomyelitis in mice. Clin Sci (Lond). 2013;125(7):329–40. https://doi.org/10.1042/CS20120560.
Schillace RV, Pisenti N, Pattamanuch N, Galligan S, Marracci GH, Bourdette DN, et al. Lipoic acid stimulates cAMP production in T lymphocytes and NK cells. Biochem Biophys Res Commun. 2007;354(1):259–64. https://doi.org/10.1016/j.bbrc.2006.12.195.
Salinthone S, Yadav V, Schillace RV, Bourdette DN, Carr DW. Lipoic acid attenuates inflammation via cAMP and protein kinase A signaling. PLoS ONE. 2010. https://doi.org/10.1371/journal.pone.0013058.
Fiedler SE, Yadav V, Kerns AR, Tsang C, Markwardt S, Kim E, et al. Lipoic acid stimulates cAMP production in healthy control and secondary progressive MS subjects. Mol Neurobiol. 2018;55(7):6037–49. https://doi.org/10.1007/s12035-017-0813-y.
Yadav V, Marracci G, Lovera J, Woodward W, Bogardus K, Marquardt W, et al. Lipoic acid in multiple sclerosis: a pilot study. Mult Scler. 2005;11(2):159–65. https://doi.org/10.1191/1352458505ms1143oa.
Falardeau J, Fryman A, Wanchu R, Marracci GH, Mass M, Wooliscroft L, et al. Oral lipoic acid as a treatment for acute optic neuritis: a blinded, placebo controlled randomized trial. Mult Scler J Exp Transl Clin. 2019;5(2):2055217319850193. https://doi.org/10.1177/2055217319850193.
Chaudhary P, Marracci G, Yu X, Galipeau D, Morris B, Bourdette D. Lipoic acid decreases inflammation and confers neuroprotection in experimental autoimmune optic neuritis. J Neuroimmunol. 2011;233(1–2):90–6. https://doi.org/10.1016/j.jneuroim.2010.12.002.
Dietrich M, Helling N, Hilla A, Heskamp A, Issberner A, Hildebrandt T, et al. Early alpha-lipoic acid therapy protects from degeneration of the inner retinal layers and vision loss in an experimental autoimmune encephalomyelitis-optic neuritis model. J Neuroinflamm. 2018;15(1):71. https://doi.org/10.1186/s12974-018-1111-y.
Yadav V, Marracci GH, Munar MY, Cherala G, Stuber LE, Alvarez L, et al. Pharmacokinetic study of lipoic acid in multiple sclerosis: comparing mice and human pharmacokinetic parameters. Mult Scler. 2010;16(4):387–97. https://doi.org/10.1177/1352458509359722.
Bittner F, Murchison C, Koop D, Bourdette D, Spain R. Lipoic acid pharmacokinetics at baseline and 1 year in secondary progressive MS. Neurol Neuroimmunol Neuroinflamm. 2017;4(5):e380. https://doi.org/10.1212/NXI.0000000000000380.
Spain R, Powers K, Murchison C, Heriza E, Winges K, Yadav V, et al. Lipoic acid in secondary progressive MS: A randomized controlled pilot trial. Neurol Neuroimmunol Neuroinflamm. 2017;4(5):e374. https://doi.org/10.1212/NXI.0000000000000374.
Lipoic Acid for Progressive Multiple Sclerosis (MS) (LAPMS). In: ClinicalTrials.gov Identifier: NCT03161028. Start: 2018. Accessed 3 Jun 2020.
Dorr J, Wernecke KD, Wurfel J, Bellmann-Strobl J, Siffrin V, Sattler MB, et al. Disease modification in multiple sclerosis by flupirtine-results of a randomized placebo controlled phase II trial. Front Neurol. 2018;9:842. https://doi.org/10.3389/fneur.2018.00842.
Schwarz M, Nolden-Koch M, Purr J, Pergande G, Block F. Antiparkinsonian effect of flupirtine in monoamine-depleted rats. J Neural Transm (Vienna). 1996;103(5):581–90. https://doi.org/10.1007/BF01273155.
Muller WE, Romero FJ, Perovic S, Pergande G, Pialoglou P. Protection of flupirtine on beta-amyloid-induced apoptosis in neuronal cells in vitro: prevention of amyloid-induced glutathione depletion. J Neurochem. 1997;68(6):2371–7. https://doi.org/10.1046/j.1471-4159.1997.68062371.x.
Otto M, Cepek L, Ratzka P, Doehlinger S, Boekhoff I, Wiltfang J, et al. Efficacy of flupirtine on cognitive function in patients with CJD: A double-blind study. Neurology. 2004;62(5):714–8. https://doi.org/10.1212/01.wnl.0000113764.35026.ef.
Klinger F, Geier P, Dorostkar MM, Chandaka GK, Yousuf A, Salzer I, et al. Concomitant facilitation of GABAA receptors and KV7 channels by the non-opioid analgesic flupirtine. Br J Pharmacol. 2012;166(5):1631–42. https://doi.org/10.1111/j.1476-5381.2011.01821.x.
Jakob R, Krieglstein J. Influence of flupirtine on a G-protein coupled inwardly rectifying potassium current in hippocampal neurones. Br J Pharmacol. 1997;122(7):1333–8. https://doi.org/10.1038/sj.bjp.0701519.
Kornhuber J, Bleich S, Wiltfang J, Maler M, Parsons CG. Flupirtine shows functional NMDA receptor antagonism by enhancing Mg2+ block via activation of voltage independent potassium channels. Rapid communication. J Neural Transm (Vienna). 1999;106(9–10):857–67. https://doi.org/10.1007/s007020050206.
Sattler MB, Williams SK, Neusch C, Otto M, Pehlke JR, Bahr M, et al. Flupirtine as neuroprotective add-on therapy in autoimmune optic neuritis. Am J Pathol. 2008;173(5):1496–507. https://doi.org/10.2353/ajpath.2008.080491.
Puls F, Agne C, Klein F, Koch M, Rifai K, Manns MP, et al. Pathology of flupirtine-induced liver injury: a histological and clinical study of six cases. Virchows Arch. 2011;458(6):709–16. https://doi.org/10.1007/s00428-011-1087-9.
Nicoletti P, Werk AN, Sawle A, Shen Y, Urban TJ, Coulthard SA, et al. HLA-DRB1*16: 01-DQB1*05: 02 is a novel genetic risk factor for flupirtine-induced liver injury. Pharmacogenet Genomics. 2016;26(5):218–24. https://doi.org/10.1097/FPC.0000000000000209.
Konishi K, Fukami T, Ogiso T, Nakajima M. In vitro approach to elucidate the relevance of carboxylesterase 2 and N-acetyltransferase 2 to flupirtine-induced liver injury. Biochem Pharmacol. 2018;155:242–51. https://doi.org/10.1016/j.bcp.2018.07.019.
Munoz Garcia D, Midaglia L, Martinez Vilela J, Marin Sanchez M, Lopez Gonzalez FJ, Arias Gomez M, et al. Associated Inosine to interferon: results of a clinical trial in multiple sclerosis. Acta Neurol Scand. 2015;131(6):405–10. https://doi.org/10.1111/ane.12333.
von Geldern G, Mowry EM. The influence of nutritional factors on the prognosis of multiple sclerosis. Nat Rev Neurol. 2012;8(12):678–89. https://doi.org/10.1038/nrneurol.2012.194.
Spitsin S, Koprowski H. Role of uric acid in multiple sclerosis. Curr Top Microbiol Immunol. 2008;318:325–42. https://doi.org/10.1007/978-3-540-73677-6_13.
Touil T, Deloire-Grassin MS, Vital C, Petry KG, Brochet B. In vivo damage of CNS myelin and axons induced by peroxynitrite. NeuroReport. 2001;12(16):3637–44. https://doi.org/10.1097/00001756-200111160-00052.
Scott GS, Hooper DC. The role of uric acid in protection against peroxynitrite-mediated pathology. Med Hypotheses. 2001;56(1):95–100. https://doi.org/10.1054/mehy.2000.1118.
Cross AH, Manning PT, Keeling RM, Schmidt RE, Misko TP. Peroxynitrite formation within the central nervous system in active multiple sclerosis. J Neuroimmunol. 1998;88(1–2):45–56. https://doi.org/10.1016/s0165-5728(98)00078-2.
Hooper DC, Spitsin S, Kean RB, Champion JM, Dickson GM, Chaudhry I, et al. Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci USA. 1998;95(2):675–80. https://doi.org/10.1073/pnas.95.2.675.
Junqueira SC, Dos Santos CI, Lieberknecht V, Cunha MP, Calixto JB, Rodrigues ALS, et al. Inosine, an endogenous purine nucleoside, suppresses immune responses and protects mice from experimental autoimmune encephalomyelitis: a role for A2A adenosine receptor. Mol Neurobiol. 2017;54(5):3271–85. https://doi.org/10.1007/s12035-016-9893-3.
Liu B, Shen Y, Xiao K, Tang Y, Cen L, Wei J. Serum uric acid levels in patients with multiple sclerosis: a meta-analysis. Neurol Res. 2012;34(2):163–71. https://doi.org/10.1179/1743132811Y.0000000074.
Spitsin S, Hooper DC, Mikheeva T, Koprowski H. Uric acid levels in patients with multiple sclerosis: analysis in mono- and dizygotic twins. Mult Scler. 2001;7(3):165–6. https://doi.org/10.1177/135245850100700305.
Gonsette RE, Sindic C, D’Hooghe MB, De Deyn PP, Medaer R, Michotte A, et al. Boosting endogenous neuroprotection in multiple sclerosis: the ASsociation of Inosine and Interferon beta in relapsing- remitting Multiple Sclerosis (ASIIMS) trial. Mult Scler. 2010;16(4):455–62. https://doi.org/10.1177/1352458509360547.
Guerrero AL, Martin-Polo J, Laherran E, Gutierrez F, Iglesias F, Tejero MA, et al. Variation of serum uric acid levels in multiple sclerosis during relapses and immunomodulatory treatment. Eur J Neurol. 2008;15(4):394–7. https://doi.org/10.1111/j.1468-1331.2008.02087.x.
Kean RB, Spitsin SV, Mikheeva T, Scott GS, Hooper DC. The peroxynitrite scavenger uric acid prevents inflammatory cell invasion into the central nervous system in experimental allergic encephalomyelitis through maintenance of blood-central nervous system barrier integrity. J Immunol. 2000;165(11):6511–8. https://doi.org/10.4049/jimmunol.165.11.6511.
Kleber ME, Delgado G, Grammer TB, Silbernagel G, Huang J, Kramer BK, et al. Uric acid and cardiovascular events: a mendelian randomization study. J Am Soc Nephrol. 2015;26(11):2831–8. https://doi.org/10.1681/ASN.2014070660.
Low RK, Stoller ML. Uric acid-related nephrolithiasis. Urol Clin North Am. 1997;24(1):135–48. https://doi.org/10.1016/s0094-0143(05)70359-1.
Franklin RJ, Ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9(11):839–55. https://doi.org/10.1038/nrn2480.
Patrikios P, Stadelmann C, Kutzelnigg A, Rauschka H, Schmidbauer M, Laursen H, et al. Remyelination is extensive in a subset of multiple sclerosis patients. Brain. 2006;129(Pt 12):3165–72. https://doi.org/10.1093/brain/awl217.
Mi S, Miller RH, Lee X, Scott ML, Shulag-Morskaya S, Shao Z, et al. LINGO-1 negatively regulates myelination by oligodendrocytes. Nat Neurosci. 2005;8(6):745–51. https://doi.org/10.1038/nn1460.
Ruggieri S, Tortorella C, Gasperini C. Anti lingo 1 (opicinumab) a new monoclonal antibody tested in relapsing remitting multiple sclerosis. Expert Rev Neurother. 2017;17(11):1081–9. https://doi.org/10.1080/14737175.2017.1378098.
Mi S, Miller RH, Tang W, Lee X, Hu B, Wu W, et al. Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann Neurol. 2009;65(3):304–15. https://doi.org/10.1002/ana.21581.
Gresle MM, Liu Y, Kilpatrick TJ, Kemper D, Wu QZ, Hu B, et al. Blocking LINGO-1 in vivo reduces degeneration and enhances regeneration of the optic nerve. Mult Scler J Exp Transl Clin. 2016;2:2055217316641704. https://doi.org/10.1177/2055217316641704.
Mi S, Hu B, Hahm K, Luo Y, Kam Hui ES, Yuan Q, et al. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat Med. 2007;13(10):1228–33. https://doi.org/10.1038/nm1664.
Hanf KJM, Arndt JW, Liu Y, Gong BJ, Rushe M, Sopko R, et al. Functional activity of anti-LINGO-1 antibody opicinumab requires target engagement at a secondary binding site. MAbs. 2020;12(1):1713648. https://doi.org/10.1080/19420862.2020.1713648.
Tran JQ, Rana J, Barkhof F, Melamed I, Gevorkyan H, Wattjes MP, et al. Randomized phase I trials of the safety/tolerability of anti-LINGO-1 monoclonal antibody BIIB033. Neurol Neuroimmunol Neuroinflamm. 2014;1(2):e18. https://doi.org/10.1212/NXI.0000000000000018.
Cadavid D, Balcer L, Galetta S, Aktas O, Ziemssen T, Vanopdenbosch L, et al. Safety and efficacy of opicinumab in acute optic neuritis (RENEW): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2017;16(3):189–99. https://doi.org/10.1016/S1474-4422(16)30377-5.
Klistorner A, Chai Y, Leocani L, Albrecht P, Aktas O, Butzkueven H, et al. Assessment of opicinumab in acute optic neuritis using multifocal visual evoked potential. CNS Drugs. 2018;32(12):1159–71. https://doi.org/10.1007/s40263-018-0575-8.
Aktas O, Ziemssen F, Ziemssen T, Comi G, Butzkueven H, Izquierdo G, et al. RENEWED: long-term electrophysiological and clinical outcomes in participants previously enrolled in the opicinumab phase 2 study RENEW (1599). Neurology. 2020;94(15 Supplement):1599.
Cadavid D, Mellion M, Hupperts R, Edwards KR, Calabresi PA, Drulovic J, et al. Safety and efficacy of opicinumab in patients with relapsing multiple sclerosis (SYNERGY): a randomised, placebo-controlled, phase 2 trial. Lancet Neurol. 2019;18(9):845–56. https://doi.org/10.1016/S1474-4422(19)30137-1.
Schwid SR, Goodman AD, McDermott MP, Bever CF, Cook SD. Quantitative functional measures in MS: what is a reliable change? Neurology. 2002;58(8):1294–6. https://doi.org/10.1212/wnl.58.8.1294.
Feys P, Lamers I, Francis G, Benedict R, Phillips G, LaRocca N, et al. The Nine-Hole Peg Test as a manual dexterity performance measure for multiple sclerosis. Mult Scler. 2017;23(5):711–20. https://doi.org/10.1177/1352458517690824.
Gronwall DM. Paced auditory serial-addition task: a measure of recovery from concussion. Percept Mot Skills. 1977;44(2):367–73. https://doi.org/10.2466/pms.1977.44.2.367.
Zhu B, Calabresi P, Giovannoni G, Kapoor R, Naismith R, Hartung H-P, et al. Phase 2 AFFINITY trial evaluates opicinumab in a targeted population of patients with relapsing multiple sclerosis: rationale, design and baseline characteristics (P32–072). Neurology. 2019;92(15 Supplement):P3.2-072.
Efficacy and Safety of BIIB033 (Opicinumab) as an Add-on Therapy to Disease-Modifying Therapies (DMTs) in Relapsing Multiple Sclerosis (MS) (AFFINITY). In: ClinicalTrials.gov Identifier: NCT03222973. Start: 2017. Accessed 3 Jun 2020.
MacCannell D, Nestorov I. Model-based identification of flat dosing regimen for opicinumab AFFINITY trial. ECTRIMS Online Library. 2019:P642.
Biogen. Biogen reports Q3 2020 Results. 2020.
Cadavid D, Balcer L, Galetta S, Aktas O, Ziemssen T, Vanopdenbosch LJ, et al. Predictors of response to opicinumab in acute optic neuritis. Ann Clin Transl Neurol. 2018;5(10):1154–62. https://doi.org/10.1002/acn3.620.
Neumann B, Segel M, Chalut KJ, Franklin RJ. Remyelination and ageing: reversing the ravages of time. Mult Scler. 2019;25(14):1835–41. https://doi.org/10.1177/1352458519884006.
Woodruff RH, Fruttiger M, Richardson WD, Franklin RJ. Platelet-derived growth factor regulates oligodendrocyte progenitor numbers in adult CNS and their response following CNS demyelination. Mol Cell Neurosci. 2004;25(2):252–62. https://doi.org/10.1016/j.mcn.2003.10.014.
Sheikh S, Calabresi P, Giovannoni G, Kapoor R, Arnold DL, Chai Y, et al. Predictors of an opicinumab treatment effect and identification of an efficacy subpopulation: a post hoc analysis of the SYNERGY Study (P3.408). Neurology. 2018;90(15 Supplement):P3.408.
Cerina M, Narayanan V, Gobel K, Bittner S, Ruck T, Meuth P, et al. The quality of cortical network function recovery depends on localization and degree of axonal demyelination. Brain Behav Immun. 2017;59:103–17. https://doi.org/10.1016/j.bbi.2016.08.014.
Larochelle C, Uphaus T, Prat A, Zipp F. Secondary progression in multiple sclerosis: neuronal exhaustion or distinct pathology? Trends Neurosci. 2016;39(5):325–39. https://doi.org/10.1016/j.tins.2016.02.001.
Correa MF, Dos Santos Fernandes JP. QSAR modeling of histamine H3R antagonists/inverse agonists as future drugs for neurodegenerative diseases. Curr Neuropharmacol. 2018;16(6):749–57. https://doi.org/10.2174/1570159X15666170818100644.
Gemkow MJ, Davenport AJ, Harich S, Ellenbroek BA, Cesura A, Hallett D. The histamine H3 receptor as a therapeutic drug target for CNS disorders. Drug Discov Today. 2009;14(9–10):509–15. https://doi.org/10.1016/j.drudis.2009.02.011.
Sadek B, Saad A, Sadeq A, Jalal F, Stark H. Histamine H3 receptor as a potential target for cognitive symptoms in neuropsychiatric diseases. Behav Brain Res. 2016;312:415–30. https://doi.org/10.1016/j.bbr.2016.06.051.
Chen Y, Zhen W, Guo T, Zhao Y, Liu A, Rubio JP, et al. Histamine Receptor 3 negatively regulates oligodendrocyte differentiation and remyelination. PLoS ONE. 2017;12(12):e0189380. https://doi.org/10.1371/journal.pone.0189380.
Merkestein M, Gispen WH, Adan RAH. Melanocortins: brain effects. In: Squire LR, editor. Encyclopedia of neuroscience. Oxford: Academic Press; 2009. p. 713–9.
Jarskog LF, Lowy MT, Grove RA, Keefe RS, Horrigan JP, Ball MP, et al. A Phase II study of a histamine H(3) receptor antagonist GSK239512 for cognitive impairment in stable schizophrenia subjects on antipsychotic therapy. Schizophr Res. 2015;164(1–3):136–42. https://doi.org/10.1016/j.schres.2015.01.041.
Grove RA, Harrington CM, Mahler A, Beresford I, Maruff P, Lowy MT, et al. A randomized, double-blind, placebo-controlled, 16-week study of the H3 receptor antagonist, GSK239512 as a monotherapy in subjects with mild-to-moderate Alzheimer’s disease. Curr Alzheimer Res. 2014;11(1):47–58. https://doi.org/10.2174/1567205010666131212110148.
Nathan PJ, Boardley R, Scott N, Berges A, Maruff P, Sivananthan T, et al. The safety, tolerability, pharmacokinetics and cognitive effects of GSK239512, a selective histamine H(3) receptor antagonist in patients with mild to moderate Alzheimer’s disease: a preliminary investigation. Curr Alzheimer Res. 2013;10(3):240–51. https://doi.org/10.2174/1567205011310030003.
Schwartzbach CJ, Grove RA, Brown R, Tompson D, Then Bergh F, Arnold DL. Lesion remyelinating activity of GSK239512 versus placebo in patients with relapsing-remitting multiple sclerosis: a randomised, single-blind, phase II study. J Neurol. 2017;264(2):304–15. https://doi.org/10.1007/s00415-016-8341-7.
Ashworth S, Berges A, Rabiner EA, Wilson AA, Comley RA, Lai RY, et al. Unexpectedly high affinity of a novel histamine H(3) receptor antagonist, GSK239512, in vivo in human brain, determined using PET. Br J Pharmacol. 2014;171(5):1241–9. https://doi.org/10.1111/bph.12505.
Maruff P, Thomas E, Cysique L, Brew B, Collie A, Snyder P, et al. Validity of the CogState brief battery: relationship to standardized tests and sensitivity to cognitive impairment in mild traumatic brain injury, schizophrenia, and AIDS dementia complex. Arch Clin Neuropsychol. 2009;24(2):165–78. https://doi.org/10.1093/arclin/acp010.
Teuscher C, Subramanian M, Noubade R, Gao JF, Offner H, Zachary JF, et al. Central histamine H3 receptor signaling negatively regulates susceptibility to autoimmune inflammatory disease of the CNS. Proc Natl Acad Sci USA. 2007;104(24):10146–51. https://doi.org/10.1073/pnas.0702291104.
Beghdadi W, Porcherie A, Schneider BS, Morisset S, Dubayle D, Peronet R, et al. Histamine H(3) receptor-mediated signaling protects mice from cerebral malaria. PLoS ONE. 2009;4(6):e6004. https://doi.org/10.1371/journal.pone.0006004.
Sharma HS, Vannemreddy P, Patnaik R, Patnaik S, Mohanty S. Histamine receptors influence blood-spinal cord barrier permeability, edema formation, and spinal cord blood flow following trauma to the rat spinal cord. Acta Neurochir Suppl. 2006;96:316–21. https://doi.org/10.1007/3-211-30714-1_67.
Shi Y, Li Z, Chen R, Zhang J, Hu X, He C, et al. Immethridine, histamine H3-receptor (H3R) agonist, alleviated experimental autoimmune encephalomyelitis via inhibiting the function of dendritic cells. Oncotarget. 2017;8(43):75038–49. https://doi.org/10.18632/oncotarget.20500.
Saligrama N, Noubade R, Case LK, del Rio R, Teuscher C. Combinatorial roles for histamine H1–H2 and H3–H4 receptors in autoimmune inflammatory disease of the central nervous system. Eur J Immunol. 2012;42(6):1536–46. https://doi.org/10.1002/eji.201141859.
Iida T, Yoshikawa T, Matsuzawa T, Naganuma F, Nakamura T, Miura Y, et al. Histamine H3 receptor in primary mouse microglia inhibits chemotaxis, phagocytosis, and cytokine secretion. Glia. 2015;63(7):1213–25. https://doi.org/10.1002/glia.22812.
Xu J, Zhang X, Qian Q, Wang Y, Dong H, Li N, et al. Histamine upregulates the expression of histamine receptors and increases the neuroprotective effect of astrocytes. J Neuroinflamm. 2018;15(1):41. https://doi.org/10.1186/s12974-018-1068-x.
Bittner S, Ruck T, Fernandez-Orth J, Meuth SG. TREK-king the blood-brain-barrier. J Neuroimmune Pharmacol. 2014;9(3):293–301. https://doi.org/10.1007/s11481-014-9530-8.
Bittner S, Ruck T, Schuhmann MK, Herrmann AM, Moha ou Maati H, Bobak N, et al. Endothelial TWIK-related potassium channel-1 (TREK1) regulates immune-cell trafficking into the CNS. Nat Med. 2013;19(9):1161–5. https://doi.org/10.1038/nm.3303.
Yamada K, Inagaki N. Neuroprotection by KATP channels. J Mol Cell Cardiol. 2005;38(6):945–9. https://doi.org/10.1016/j.yjmcc.2004.11.020.
Garlid KD, Paucek P, Yarov-Yarovoy V, Sun X, Schindler PA. The mitochondrial KATP channel as a receptor for potassium channel openers. J Biol Chem. 1996;271(15):8796–9. https://doi.org/10.1074/jbc.271.15.8796.
Shimizu K, Lacza Z, Rajapakse N, Horiguchi T, Snipes J, Busija DW. MitoK(ATP) opener, diazoxide, reduces neuronal damage after middle cerebral artery occlusion in the rat. Am J Physiol Heart Circ Physiol. 2002;283(3):H1005–11. https://doi.org/10.1152/ajpheart.00054.2002.
Yang Y, Liu X, Long Y, Wang F, Ding JH, Liu SY, et al. Activation of mitochondrial ATP-sensitive potassium channels improves rotenone-related motor and neurochemical alterations in rats. Int J Neuropsychopharmacol. 2006;9(1):51–61. https://doi.org/10.1017/S1461145705005547.
Liu D, Pitta M, Lee JH, Ray B, Lahiri DK, Furukawa K, et al. The KATP channel activator diazoxide ameliorates amyloid-beta and tau pathologies and improves memory in the 3xTgAD mouse model of Alzheimer’s disease. J Alzheimers Dis. 2010;22(2):443–57. https://doi.org/10.3233/JAD-2010-101017.
Virgili N, Mancera P, Wappenhans B, Sorrosal G, Biber K, Pugliese M, et al. K(ATP) channel opener diazoxide prevents neurodegeneration: a new mechanism of action via antioxidative pathway activation. PLoS ONE. 2013;8(9):e75189. https://doi.org/10.1371/journal.pone.0075189.
Zhou F, Yao HH, Wu JY, Ding JH, Sun T, Hu G. Opening of microglial K(ATP) channels inhibits rotenone-induced neuroinflammation. J Cell Mol Med. 2008;12(5A):1559–70. https://doi.org/10.1111/j.1582-4934.2007.00144.x.
Rodriguez MJ, Martinez-Moreno M, Ortega FJ, Mahy N. Targeting microglial K(ATP) channels to treat neurodegenerative diseases: a mitochondrial issue. Oxid Med Cell Longev. 2013;2013:194546. https://doi.org/10.1155/2013/194546.
Virgili N, Espinosa-Parrilla JF, Mancera P, Pasten-Zamorano A, Gimeno-Bayon J, Rodriguez MJ, et al. Oral administration of the KATP channel opener diazoxide ameliorates disease progression in a murine model of multiple sclerosis. J Neuroinflammation. 2011;8:149. https://doi.org/10.1186/1742-2094-8-149.
Virgili N, Mancera P, Chanvillard C, Wegner A, Wappenhans B, Rodriguez MJ, et al. Diazoxide attenuates autoimmune encephalomyelitis and modulates lymphocyte proliferation and dendritic cell functionality. J Neuroimmune Pharmacol. 2014;9(4):558–68. https://doi.org/10.1007/s11481-014-9551-3.
Villoslada P, Rovira A, Montalban X, Arroyo R, Paul F, Meca-Lallana V, et al. Effects of diazoxide in multiple sclerosis: a randomized, double-blind phase 2 clinical trial. Neurol Neuroimmunol Neuroinflamm. 2015;2(5):e147. https://doi.org/10.1212/NXI.0000000000000147.
Schafer G, Wegener C, Portenhauser R, Bojanovski D. Diazoxide, an inhibitor of succinate oxidation. Biochem Pharmacol. 1969;18(10):2678–81.
Nishino H, Shimano Y, Kumazaki M, Sakurai T. Chronically administered 3-nitropropionic acid induces striatal lesions attributed to dysfunction of the blood-brain barrier. Neurosci Lett. 1995;186(2–3):161–4. https://doi.org/10.1016/0304-3940(95)11311-j.
Fogal B, McClaskey C, Yan S, Yan H, Rivkees SA. Diazoxide promotes oligodendrocyte precursor cell proliferation and myelination. PLoS ONE. 2010;5(5):e10906. https://doi.org/10.1371/journal.pone.0010906.
Zhu Y, Wendler CC, Shi O, Rivkees SA. Diazoxide promotes oligodendrocyte differentiation in neonatal brain in normoxia and chronic sublethal hypoxia. Brain Res. 2014;1586:64–72. https://doi.org/10.1016/j.brainres.2014.08.046.
Ehling P, Bittner S, Budde T, Wiendl H, Meuth SG. Ion channels in autoimmune neurodegeneration. FEBS Lett. 2011;585(23):3836–42. https://doi.org/10.1016/j.febslet.2011.03.065.
Goodman AD, Brown TR, Schapiro RT, Klingler M, Cohen R, Blight AR. A pooled analysis of two phase 3 clinical trials of dalfampridine in patients with multiple sclerosis. Int J MS Care. 2014;16(3):153–60. https://doi.org/10.7224/1537-2073.2013-023.
Dietrich M, Koska V, Hecker C, Gottle P, Hilla AM, Heskamp A, et al. Protective effects of 4-aminopyridine in experimental optic neuritis and multiple sclerosis. Brain. 2020;143(4):1127–42. https://doi.org/10.1093/brain/awaa062.
Gobel K, Wedell JH, Herrmann AM, Wachsmuth L, Pankratz S, Bittner S, et al. 4-Aminopyridine ameliorates mobility but not disease course in an animal model of multiple sclerosis. Exp Neurol. 2013;248:62–71. https://doi.org/10.1016/j.expneurol.2013.05.016.
Moriguchi K, Miyamoto K, Fukumoto Y, Kusunoki S. 4-Aminopyridine ameliorates relapsing remitting experimental autoimmune encephalomyelitis in SJL/J mice. J Neuroimmunol. 2018;323:131–5. https://doi.org/10.1016/j.jneuroim.2018.08.007.
Cutter GR, Baier ML, Rudick RA, Cookfair DL, Fischer JS, Petkau J, et al. Development of a multiple sclerosis functional composite as a clinical trial outcome measure. Brain. 1999;122(Pt 5):871–82. https://doi.org/10.1093/brain/122.5.871.
Ruck T, Bittner S, Simon OJ, Gobel K, Wiendl H, Schilling M, et al. Long-term effects of dalfampridine in patients with multiple sclerosis. J Neurol Sci. 2014;337(1–2):18–24. https://doi.org/10.1016/j.jns.2013.11.011.
Goodman AD, Cohen JA, Cross A, Vollmer T, Rizzo M, Cohen R, et al. Fampridine-SR in multiple sclerosis: a randomized, double-blind, placebo-controlled, dose-ranging study. Mult Scler. 2007;13(3):357–68. https://doi.org/10.1177/1352458506069538.
Goodman AD, Brown TR, Cohen JA, Krupp LB, Schapiro R, Schwid SR, et al. Dose comparison trial of sustained-release fampridine in multiple sclerosis. Neurology. 2008;71(15):1134–41. https://doi.org/10.1212/01.wnl.0000326213.89576.0e.
Yrjanheikki J, Keinanen R, Pellikka M, Hokfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci USA. 1998;95(26):15769–74. https://doi.org/10.1073/pnas.95.26.15769.
Kim HS, Suh YH. Minocycline and neurodegenerative diseases. Behav Brain Res. 2009;196(2):168–79. https://doi.org/10.1016/j.bbr.2008.09.040.
Brundula V, Rewcastle NB, Metz LM, Bernard CC, Yong VW. Targeting leukocyte MMPs and transmigration: minocycline as a potential therapy for multiple sclerosis. Brain. 2002;125(Pt 6):1297–308. https://doi.org/10.1093/brain/awf133.
Giuliani F, Fu SA, Metz LM, Yong VW. Effective combination of minocycline and interferon-beta in a model of multiple sclerosis. J Neuroimmunol. 2005;165(1–2):83–91. https://doi.org/10.1016/j.jneuroim.2005.04.020.
Giuliani F, Metz LM, Wilson T, Fan Y, Bar-Or A, Yong VW. Additive effect of the combination of glatiramer acetate and minocycline in a model of MS. J Neuroimmunol. 2005;158(1–2):213–21. https://doi.org/10.1016/j.jneuroim.2004.09.006.
Giuliani F, Hader W, Yong VW. Minocycline attenuates T cell and microglia activity to impair cytokine production in T cell-microglia interaction. J Leukoc Biol. 2005;78(1):135–43. https://doi.org/10.1189/jlb.0804477.
Hou Y, Ryu CH, Park KY, Kim SM, Jeong CH, Jeun SS. Effective combination of human bone marrow mesenchymal stem cells and minocycline in experimental autoimmune encephalomyelitis mice. Stem Cell Res Ther. 2013;4(4):77. https://doi.org/10.1186/scrt228.
Niimi N, Kohyama K, Matsumoto Y. Minocycline suppresses experimental autoimmune encephalomyelitis by increasing tissue inhibitors of metalloproteinases. Neuropathology. 2013;33(6):612–20. https://doi.org/10.1111/neup.12039.
Tikka TM, Koistinaho JE. Minocycline provides neuroprotection against N-methyl-D-aspartate neurotoxicity by inhibiting microglia. J Immunol. 2001;166(12):7527–33. https://doi.org/10.4049/jimmunol.166.12.7527.
Faissner S, Mahjoub Y, Mishra M, Haupeltshofer S, Hahn JN, Gold R, et al. Unexpected additive effects of minocycline and hydroxychloroquine in models of multiple sclerosis: Prospective combination treatment for progressive disease? Mult Scler. 2018;24(12):1543–56. https://doi.org/10.1177/1352458517728811.
Metz LM, Li DKB, Traboulsee AL, Duquette P, Eliasziw M, Cerchiaro G, et al. Trial of minocycline in a clinically isolated syndrome of multiple sclerosis. N Engl J Med. 2017;376(22):2122–33. https://doi.org/10.1056/NEJMoa1608889.
Zhang Y, Metz LM, Yong VW, Bell RB, Yeung M, Patry DG, et al. Pilot study of minocycline in relapsing-remitting multiple sclerosis. Can J Neurol Sci. 2008;35(2):185–91. https://doi.org/10.1017/s0317167100008611.
Metz LM, Li D, Traboulsee A, Myles ML, Duquette P, Godin J, et al. Glatiramer acetate in combination with minocycline in patients with relapsing–remitting multiple sclerosis: results of a Canadian, multicenter, double-blind, placebo-controlled trial. Mult Scler. 2009;15(10):1183–94. https://doi.org/10.1177/1352458509106779.
Sorensen PS, Sellebjerg F, Lycke J, Farkkila M, Creange A, Lund CG, et al. Minocycline added to subcutaneous interferon beta-1a in multiple sclerosis: randomized RECYCLINE study. Eur J Neurol. 2016;23(5):861–70. https://doi.org/10.1111/ene.12953.
Baer AS, Syed YA, Kang SU, Mitteregger D, Vig R, Ffrench-Constant C, et al. Myelin-mediated inhibition of oligodendrocyte precursor differentiation can be overcome by pharmacological modulation of Fyn-RhoA and protein kinase C signalling. Brain. 2009;132(Pt 2):465–81. https://doi.org/10.1093/brain/awn334.
Miron VE. Microglia-driven regulation of oligodendrocyte lineage cells, myelination, and remyelination. J Leukoc Biol. 2017;101(5):1103–8. https://doi.org/10.1189/jlb.3RI1116-494R.
Li J, Zhang L, Chu Y, Namaka M, Deng B, Kong J, et al. Astrocytes in oligodendrocyte lineage development and white matter pathology. Front Cell Neurosci. 2016;10:119. https://doi.org/10.3389/fncel.2016.00119.
Larsen PH, Wells JE, Stallcup WB, Opdenakker G, Yong VW. Matrix metalloproteinase-9 facilitates remyelination in part by processing the inhibitory NG2 proteoglycan. J Neurosci. 2003;23(35):11127–35.
Kozovska ME, Hong J, Zang YC, Li S, Rivera VM, Killian JM, et al. Interferon beta induces T-helper 2 immune deviation in MS. Neurology. 1999;53(8):1692–7. https://doi.org/10.1212/wnl.53.8.1692.
Waldmann R, Champigny G, Bassilana F, Heurteaux C, Lazdunski M. A proton-gated cation channel involved in acid-sensing. Nature. 1997;386(6621):173–7. https://doi.org/10.1038/386173a0.
Arias RL, Sung ML, Vasylyev D, Zhang MY, Albinson K, Kubek K, et al. Amiloride is neuroprotective in an MPTP model of Parkinson’s disease. Neurobiol Dis. 2008;31(3):334–41. https://doi.org/10.1016/j.nbd.2008.05.008.
Xiong ZG, Zhu XM, Chu XP, Minami M, Hey J, Wei WL, et al. Neuroprotection in ischemia: blocking calcium-permeable acid-sensing ion channels. Cell. 2004;118(6):687–98. https://doi.org/10.1016/j.cell.2004.08.026.
Wong HK, Bauer PO, Kurosawa M, Goswami A, Washizu C, Machida Y, et al. Blocking acid-sensing ion channel 1 alleviates Huntington’s disease pathology via an ubiquitin-proteasome system-dependent mechanism. Hum Mol Genet. 2008;17(20):3223–35. https://doi.org/10.1093/hmg/ddn218.
Vergo S, Craner MJ, Etzensperger R, Attfield K, Friese MA, Newcombe J, et al. Acid-sensing ion channel 1 is involved in both axonal injury and demyelination in multiple sclerosis and its animal model. Brain. 2011;134(Pt 2):571–84. https://doi.org/10.1093/brain/awq337.
Friese MA, Craner MJ, Etzensperger R, Vergo S, Wemmie JA, Welsh MJ, et al. Acid-sensing ion channel-1 contributes to axonal degeneration in autoimmune inflammation of the central nervous system. Nat Med. 2007;13(12):1483–9. https://doi.org/10.1038/nm1668.
Bernardinelli L, Murgia SB, Bitti PP, Foco L, Ferrai R, Musu L, et al. Association between the ACCN1 gene and multiple sclerosis in Central East Sardinia. PLoS ONE. 2007;2(5):e480. https://doi.org/10.1371/journal.pone.0000480.
Arun T, Tomassini V, Sbardella E, de Ruiter MB, Matthews L, Leite MI, et al. Targeting ASIC1 in primary progressive multiple sclerosis: evidence of neuroprotection with amiloride. Brain. 2013;136(Pt 1):106–15. https://doi.org/10.1093/brain/aws325.
McKee JB, Cottriall CL, Elston J, Epps S, Evangelou N, Gerry S, et al. Amiloride does not protect retinal nerve fibre layer thickness in optic neuritis in a phase 2 randomised controlled trial. Mult Scler. 2019;25(2):246–55. https://doi.org/10.1177/1352458517742979.
Chataway J, De Angelis F, Connick P, Parker RA, Plantone D, Doshi A, et al. Efficacy of three neuroprotective drugs in secondary progressive multiple sclerosis (MS-SMART): a phase 2b, multiarm, double-blind, randomised placebo-controlled trial. Lancet Neurol. 2020;19(3):214–25. https://doi.org/10.1016/S1474-4422(19)30485-5.
Pasternak B, Svanstrom H, Nielsen NM, Melbye M, Hviid A. Use of amiloride and multiple sclerosis: registry-based cohort studies. Pharmacoepidemiol Drug Saf. 2012;21(8):890–5. https://doi.org/10.1002/pds.3269.
Toosy AT, Mason DF, Miller DH. Optic neuritis. Lancet Neurol. 2014;13(1):83–99. https://doi.org/10.1016/S1474-4422(13)70259-X.
Beck RW, Cleary PA, Anderson MM Jr, Keltner JL, Shults WT, Kaufman DI, et al. A randomized, controlled trial of corticosteroids in the treatment of acute optic neuritis. The Optic Neuritis Study Group. N Engl J Med. 1992;326(9):581–8. https://doi.org/10.1056/NEJM199202273260901.
Bitsch A, Bruhn H, Vougioukas V, Stringaris A, Lassmann H, Frahm J, et al. Inflammatory CNS demyelination: histopathologic correlation with in vivo quantitative proton MR spectroscopy. AJNR Am J Neuroradiol. 1999;20(9):1619–27.
pH-weighted MRI in Multiple Sclerosis: A Surrogate Marker of Tissue Metabolic Stress. In: ClinicalTrials.gov Identifier: NCT03028675. Start: 2017. Accessed 3 Jun 2020.
Wong DT, Perry KW, Bymaster FP. Case history: the discovery of fluoxetine hydrochloride (Prozac). Nat Rev Drug Discov. 2005;4(9):764–74. https://doi.org/10.1038/nrd1821.
Flax JW, Gray J, Herbert J. Effect of fluoxetine on patients with multiple sclerosis. Am J Psychiatry. 1991;148(11):1603. https://doi.org/10.1176/ajp.148.11.1603a.
Yuan XQ, Qiu G, Liu XJ, Liu S, Wu Y, Wang X, et al. Fluoxetine promotes remission in acute experimental autoimmune encephalomyelitis in rats. NeuroImmunoModulation. 2012;19(4):201–8. https://doi.org/10.1159/000334095.
Bhat R, Mahapatra S, Axtell RC, Steinman L. Amelioration of ongoing experimental autoimmune encephalomyelitis with fluoxetine. J Neuroimmunol. 2017;313:77–81. https://doi.org/10.1016/j.jneuroim.2017.10.012.
Mostert JP, Sijens PE, Oudkerk M, De Keyser J. Fluoxetine increases cerebral white matter NAA/Cr ratio in patients with multiple sclerosis. Neurosci Lett. 2006;402(1–2):22–4. https://doi.org/10.1016/j.neulet.2006.03.042.
Kong EK, Peng L, Chen Y, Yu AC, Hertz L. Up-regulation of 5-HT2B receptor density and receptor-mediated glycogenolysis in mouse astrocytes by long-term fluoxetine administration. Neurochem Res. 2002;27(1–2):113–20. https://doi.org/10.1023/a:1014862808126.
Allaman I, Fiumelli H, Magistretti PJ, Martin JL. Fluoxetine regulates the expression of neurotrophic/growth factors and glucose metabolism in astrocytes. Psychopharmacology. 2011;216(1):75–84. https://doi.org/10.1007/s00213-011-2190-y.
Deak F, Lasztoczi B, Pacher P, Petheo GL, Valeria K, Spat A. Inhibition of voltage-gated calcium channels by fluoxetine in rat hippocampal pyramidal cells. Neuropharmacology. 2000;39(6):1029–36. https://doi.org/10.1016/s0028-3908(99)00206-3.
Pancrazio JJ, Kamatchi GL, Roscoe AK, Lynch C 3rd. Inhibition of neuronal Na+ channels by antidepressant drugs. J Pharmacol Exp Ther. 1998;284(1):208–14.
Lee JY, Kim HS, Choi HY, Oh TH, Yune TY. Fluoxetine inhibits matrix metalloprotease activation and prevents disruption of blood-spinal cord barrier after spinal cord injury. Brain. 2012;135(Pt 8):2375–89. https://doi.org/10.1093/brain/aws171.
Lee JY, Lee HE, Kang SR, Choi HY, Ryu JH, Yune TY. Fluoxetine inhibits transient global ischemia-induced hippocampal neuronal death and memory impairment by preventing blood-brain barrier disruption. Neuropharmacology. 2014;79:161–71. https://doi.org/10.1016/j.neuropharm.2013.11.011.
Branco-de-Almeida LS, Kajiya M, Cardoso CR, Silva MJ, Ohta K, Rosalen PL, et al. Selective serotonin reuptake inhibitors attenuate the antigen presentation from dendritic cells to effector T lymphocytes. FEMS Immunol Med Microbiol. 2011;62(3):283–94. https://doi.org/10.1111/j.1574-695X.2011.00816.x.
Diamond M, Kelly JP, Connor TJ. Antidepressants suppress production of the Th1 cytokine interferon-gamma, independent of monoamine transporter blockade. Eur Neuropsychopharmacol. 2006;16(7):481–90. https://doi.org/10.1016/j.euroneuro.2005.11.011.
Cambron M, Mostert J, D’Hooghe M, Nagels G, Willekens B, Debruyne J, et al. Fluoxetine in progressive multiple sclerosis: the FLUOX-PMS trial. Mult Scler. 2019;25(13):1728–35. https://doi.org/10.1177/1352458519843051.
Mostert J, Heersema T, Mahajan M, Van Der Grond J, Van Buchem MA, De Keyser J. The effect of fluoxetine on progression in progressive multiple sclerosis: a double-blind, randomized, placebo-controlled trial. ISRN Neurol. 2013;2013:370943. https://doi.org/10.1155/2013/370943.
Kenis G, Maes M. Effects of antidepressants on the production of cytokines. Int J Neuropsychopharmacol. 2002;5(4):401–12. https://doi.org/10.1017/S1461145702003164.
Mitsonis CI, Zervas IM, Potagas CM, Mitropoulos PA, Dimopoulos NP, Sfagos CA, et al. Effects of escitalopram on stress-related relapses in women with multiple sclerosis: an open-label, randomized, controlled, one-year follow-up study. Eur Neuropsychopharmacol. 2010;20(2):123–31. https://doi.org/10.1016/j.euroneuro.2009.10.004.
Mostert JP, Admiraal-Behloul F, Hoogduin JM, Luyendijk J, Heersema DJ, van Buchem MA, et al. Effects of fluoxetine on disease activity in relapsing multiple sclerosis: a double-blind, placebo-controlled, exploratory study. J Neurol Neurosurg Psychiatry. 2008;79(9):1027–31. https://doi.org/10.1136/jnnp.2007.139345.
Macrez R, Stys PK, Vivien D, Lipton SA, Docagne F. Mechanisms of glutamate toxicity in multiple sclerosis: biomarker and therapeutic opportunities. Lancet Neurol. 2016;15(10):1089–102. https://doi.org/10.1016/S1474-4422(16)30165-X.
Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6(1):67–70. https://doi.org/10.1038/71555.
Gilgun-Sherki Y, Panet H, Melamed E, Offen D. Riluzole suppresses experimental autoimmune encephalomyelitis: implications for the treatment of multiple sclerosis. Brain Res. 2003;989(2):196–204. https://doi.org/10.1016/s0006-8993(03)03343-2.
Bellingham MC. A review of the neural mechanisms of action and clinical efficiency of riluzole in treating amyotrophic lateral sclerosis: what have we learned in the last decade? CNS Neurosci Ther. 2011;17(1):4–31. https://doi.org/10.1111/j.1755-5949.2009.00116.x.
Killestein J, Kalkers NF, Polman CH. Glutamate inhibition in MS: the neuroprotective properties of riluzole. J Neurol Sci. 2005;233(1–2):113–5. https://doi.org/10.1016/j.jns.2005.03.011.
Waubant E, Maghzi AH, Revirajan N, Spain R, Julian L, Mowry EM, et al. A randomized controlled phase II trial of riluzole in early multiple sclerosis. Ann Clin Transl Neurol. 2014;1(5):340–7. https://doi.org/10.1002/acn3.60.
Lovera JF, Frohman E, Brown TR, Bandari D, Nguyen L, Yadav V, et al. Memantine for cognitive impairment in multiple sclerosis: a randomized placebo-controlled trial. Mult Scler. 2010;16(6):715–23. https://doi.org/10.1177/1352458510367662.
Jaber S, Polster BM. Idebenone and neuroprotection: antioxidant, pro-oxidant, or electron carrier? J Bioenerg Biomembr. 2015;47(1–2):111–8. https://doi.org/10.1007/s10863-014-9571-y.
Suno M, Nagaoka A. Inhibition of lipid peroxidation by a novel compound, idebenone (CV-2619). Jpn J Pharmacol. 1984;35(2):196–8. https://doi.org/10.1254/jjp.35.196.
Shirani A, Okuda DT, Stuve O. Therapeutic advances and future prospects in progressive forms of multiple sclerosis. Neurotherapeutics. 2016;13(1):58–69. https://doi.org/10.1007/s13311-015-0409-z.
Mordente A, Martorana GE, Minotti G, Giardina B. Antioxidant properties of 2,3-dimethoxy-5-methyl-6-(10-hydroxydecyl)-1,4-benzoquinone (idebenone). Chem Res Toxicol. 1998;11(1):54–63. https://doi.org/10.1021/tx970136j.
Erb M, Hoffmann-Enger B, Deppe H, Soeberdt M, Haefeli RH, Rummey C, et al. Features of idebenone and related short-chain quinones that rescue ATP levels under conditions of impaired mitochondrial complex I. PLoS ONE. 2012;7(4):e36153. https://doi.org/10.1371/journal.pone.0036153.
Haefeli RH, Erb M, Gemperli AC, Robay D, Courdier Fruh I, Anklin C, et al. NQO1-dependent redox cycling of idebenone: effects on cellular redox potential and energy levels. PLoS ONE. 2011;6(3):e17963. https://doi.org/10.1371/journal.pone.0017963.
Yan A, Liu Z, Song L, Wang X, Zhang Y, Wu N, et al. Idebenone alleviates neuroinflammation and modulates microglial polarization in LPS-stimulated BV2 cells and MPTP-induced Parkinson’s disease mice. Front Cell Neurosci. 2018;12:529. https://doi.org/10.3389/fncel.2018.00529.
Kosa P, Wu T, Phillips J, Leinonen M, Masvekar R, Komori M, et al. Idebenone does not inhibit disability progression in primary progressive MS. Mult Scler Relat Disord. 2020;45:102434. https://doi.org/10.1016/j.msard.2020.102434.
Weideman AM, Barbour C, Tapia-Maltos MA, Tran T, Jackson K, Kosa P, et al. New multiple sclerosis disease severity scale predicts future accumulation of disability. Front Neurol. 2017;8:598. https://doi.org/10.3389/fneur.2017.00598.
Idebenone for Primary Progressive Multiple Sclerosis. In: ClinicalTrials.gov Identifier: NCT01854359. Start: 2013. Accessed 3 Jun 2020.
Fiebiger SM, Bros H, Grobosch T, Janssen A, Chanvillard C, Paul F, et al. The antioxidant idebenone fails to prevent or attenuate chronic experimental autoimmune encephalomyelitis in the mouse. J Neuroimmunol. 2013;262(1–2):66–71. https://doi.org/10.1016/j.jneuroim.2013.07.002.
King MS, Sharpley MS, Hirst J. Reduction of hydrophilic ubiquinones by the flavin in mitochondrial NADH:ubiquinone oxidoreductase (Complex I) and production of reactive oxygen species. Biochemistry. 2009;48(9):2053–62. https://doi.org/10.1021/bi802282h.
Esposti MD, Ngo A, Ghelli A, Benelli B, Carelli V, McLennan H, et al. The interaction of Q analogs, particularly hydroxydecyl benzoquinone (idebenone), with the respiratory complexes of heart mitochondria. Arch Biochem Biophys. 1996;330(2):395–400. https://doi.org/10.1006/abbi.1996.0267.
Stringer JL, Gaikwad A, Gonzales BN, Long DJ Jr, Marks LM, Jaiswal AK. Presence and induction of the enzyme NAD(P)H: quinone oxidoreductase 1 in the central nervous system. J Comp Neurol. 2004;471(3):289–97. https://doi.org/10.1002/cne.20048.
Jaber SM, Ge SX, Milstein JL, VanRyzin JW, Waddell J, Polster BM. Idebenone has distinct effects on mitochondrial respiration in cortical astrocytes as compared to cortical neurons due to differential NQO1 activity. J Neurosci. 2020. https://doi.org/10.1523/JNEUROSCI.1632-17.2020.
Sugawa M, Sakurai Y, Ishikawa-Ieda Y, Suzuki H, Asou H. Effects of erythropoietin on glial cell development; oligodendrocyte maturation and astrocyte proliferation. Neurosci Res. 2002;44(4):391–403. https://doi.org/10.1016/s0168-0102(02)00161-x.
Bernaudin M, Bellail A, Marti HH, Yvon A, Vivien D, Duchatelle I, et al. Neurons and astrocytes express EPO mRNA: oxygen-sensing mechanisms that involve the redox-state of the brain. Glia. 2000;30(3):271–8.
Hasselblatt M, Ehrenreich H, Siren AL. The brain erythropoietin system and its potential for therapeutic exploitation in brain disease. J Neurosurg Anesthesiol. 2006;18(2):132–8. https://doi.org/10.1097/00008506-200604000-00007.
Brines M, Cerami A. Emerging biological roles for erythropoietin in the nervous system. Nat Rev Neurosci. 2005;6(6):484–94. https://doi.org/10.1038/nrn1687.
Kang SY, Kang JH, Choi JC, Lee JS, Lee CS, Shin T. Expression of erythropoietin in the spinal cord of lewis rats with experimental autoimmune encephalomyelitis. J Clin Neurol. 2009;5(1):39–45. https://doi.org/10.3988/jcn.2009.5.1.39.
Savino C, Pedotti R, Baggi F, Ubiali F, Gallo B, Nava S, et al. Delayed administration of erythropoietin and its non-erythropoietic derivatives ameliorates chronic murine autoimmune encephalomyelitis. J Neuroimmunol. 2006;172(1–2):27–37. https://doi.org/10.1016/j.jneuroim.2005.10.016.
Yuan R, Maeda Y, Li W, Lu W, Cook S, Dowling P. Erythropoietin: a potent inducer of peripheral immuno/inflammatory modulation in autoimmune EAE. PLoS ONE. 2008;3(4):e1924. https://doi.org/10.1371/journal.pone.0001924.
Diem R, Sattler MB, Merkler D, Demmer I, Maier K, Stadelmann C, et al. Combined therapy with methylprednisolone and erythropoietin in a model of multiple sclerosis. Brain. 2005;128(Pt 2):375–85. https://doi.org/10.1093/brain/awh365.
Cho YK, Kim G, Park S, Sim JH, Won YJ, Hwang CH, et al. Erythropoietin promotes oligodendrogenesis and myelin repair following lysolecithin-induced injury in spinal cord slice culture. Biochem Biophys Res Commun. 2012;417(2):753–9. https://doi.org/10.1016/j.bbrc.2011.12.029.
Mirzaie J, Raoofi A, Jamalpoor Z, Nezhadi A, Golmohammadi R. Protective impacts of erythropoietin on myelinization of oligodendrocytes and schwann cells in CNS and PNS following cuprizone-induced multiple sclerosis- histology, molecular, and functional studies. J Chem Neuroanat. 2020;104:101750. https://doi.org/10.1016/j.jchemneu.2020.101750.
Zhang J, Li Y, Cui Y, Chen J, Lu M, Elias SB, et al. Erythropoietin treatment improves neurological functional recovery in EAE mice. Brain Res. 2005;1034(1–2):34–9. https://doi.org/10.1016/j.brainres.2004.11.036.
Campana WM, Myers RR. Erythropoietin and erythropoietin receptors in the peripheral nervous system: changes after nerve injury. FASEB J. 2001;15(10):1804–6. https://doi.org/10.1096/fj.00-0857fje.
Chattopadhyay A, Choudhury TD, Bandyopadhyay D, Datta AG. Protective effect of erythropoietin on the oxidative damage of erythrocyte membrane by hydroxyl radical. Biochem Pharmacol. 2000;59(4):419–25. https://doi.org/10.1016/s0006-2952(99)00277-4.
Siren AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P, et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proc Natl Acad Sci USA. 2001;98(7):4044–9. https://doi.org/10.1073/pnas.051606598.
Ehrenreich H, Fischer B, Norra C, Schellenberger F, Stender N, Stiefel M, et al. Exploring recombinant human erythropoietin in chronic progressive multiple sclerosis. Brain. 2007;130(Pt 10):2577–88. https://doi.org/10.1093/brain/awm203.
Creange A, Lefaucheur JP, Balleyguier MO, Galacteros F. Iron depletion induced by bloodletting and followed by rhEPO administration as a therapeutic strategy in progressive multiple sclerosis: a pilot, open-label study with neurophysiological measurements. Neurophysiol Clin. 2013;43(5–6):303–12. https://doi.org/10.1016/j.neucli.2013.09.004.
Schreiber K, Magyari M, Sellebjerg F, Iversen P, Garde E, Madsen CG, et al. High-dose erythropoietin in patients with progressive multiple sclerosis: a randomized, placebo-controlled, phase 2 trial. Mult Scler. 2017;23(5):675–85. https://doi.org/10.1177/1352458516661048.
Freeman JA, Hobart JC, Langdon DW, Thompson AJ. Clinical appropriateness: a key factor in outcome measure selection: the 36 item short form health survey in multiple sclerosis. J Neurol Neurosurg Psychiatry. 2000;68(2):150–6. https://doi.org/10.1136/jnnp.68.2.150.
Najmi Varzaneh F, Najmi Varzaneh F, Azimi AR, Rezaei N, Sahraian MA. Efficacy of combination therapy with erythropoietin and methylprednisolone in clinical recovery of severe relapse in multiple sclerosis. Acta Neurol Belg. 2014;114(4):273–8. https://doi.org/10.1007/s13760-014-0286-y.
Suhs KW, Hein K, Sattler MB, Gorlitz A, Ciupka C, Scholz K, et al. A randomized, double-blind, phase 2 study of erythropoietin in optic neuritis. Ann Neurol. 2012;72(2):199–210. https://doi.org/10.1002/ana.23573.
Diem R, Molnar F, Beisse F, Gross N, Druschler K, Heinrich SP, et al. Treatment of optic neuritis with erythropoietin (TONE): a randomised, double-blind, placebo-controlled trial-study protocol. BMJ Open. 2016;6(3):e010956. https://doi.org/10.1136/bmjopen-2015-010956.
Pollock C, Johnson DW, Horl WH, Rossert J, Casadevall N, Schellekens H, et al. Pure red cell aplasia induced by erythropoiesis-stimulating agents. Clin J Am Soc Nephrol. 2008;3(1):193–9. https://doi.org/10.2215/CJN.02440607.
Yasuda Y, Fujita Y, Matsuo T, Koinuma S, Hara S, Tazaki A, et al. Erythropoietin regulates tumour growth of human malignancies. Carcinogenesis. 2003;24(6):1021–9. https://doi.org/10.1093/carcin/bgg060.
Henke M, Laszig R, Rube C, Schafer U, Haase KD, Schilcher B, et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet. 2003;362(9392):1255–60. https://doi.org/10.1016/S0140-6736(03)14567-9.
Pajonk F, Weil A, Sommer A, Suwinski R, Henke M. The erythropoietin-receptor pathway modulates survival of cancer cells. Oncogene. 2004;23(55):8987–91. https://doi.org/10.1038/sj.onc.1208140.
Brines M, Grasso G, Fiordaliso F, Sfacteria A, Ghezzi P, Fratelli M, et al. Erythropoietin mediates tissue protection through an erythropoietin and common beta-subunit heteroreceptor. Proc Natl Acad Sci USA. 2004;101(41):14907–12. https://doi.org/10.1073/pnas.0406491101.
Yuan R, Wang B, Lu W, Maeda Y, Dowling P. A Distinct Region in erythropoietin that induces immuno/inflammatory modulation and tissue protection. Neurotherapeutics. 2015;12(4):850–61. https://doi.org/10.1007/s13311-015-0379-1.
Magnetic Resonance Imaging Study of JM-4 in Multiple Sclerosis/Clinically Patients. In: ClinicalTrials.gov Identifier: NCT03887065. Start: 2019. Accessed 3 Jun 2020.
Paul PSL, Debruyne D, Bernard D, Mock DM, Defer GL. Pharmacokinetics and pharmacodynamics of MD1003 (high-dose biotin) in the treatment of progressive multiple sclerosis. Expert Opin Drug Metab Toxicol. 2016;12(3):327–44. https://doi.org/10.1517/17425255.2016.1136288.
Mock DM. Biotin: from nutrition to therapeutics. J Nutr. 2017;147(8):1487–92. https://doi.org/10.3945/jn.116.238956.
Anagnostouli M, Livaniou E, Nyalala JO, Evangelatos G, Zournas C, Ithakissios DS, et al. Cerebrospinal fluid levels of biotin in various neurological disorders. Acta Neurol Scand. 1999;99(6):387–92. https://doi.org/10.1111/j.1600-0404.1999.tb07369.x.
Wolf B, Grier RE, Allen RJ, Goodman SI, Kien CL. Biotinidase deficiency: the enzymatic defect in late-onset multiple carboxylase deficiency. Clin Chim Acta. 1983;131(3):273–81. https://doi.org/10.1016/0009-8981(83)90096-7.
Wolf B. Biotinidase deficiency should be considered in individuals thought to have multiple sclerosis and related disorders. Mult Scler Relat Disord. 2019;28:26–30. https://doi.org/10.1016/j.msard.2018.11.030.
Tong L. Structure and function of biotin-dependent carboxylases. Cell Mol Life Sci. 2013;70(5):863–91. https://doi.org/10.1007/s00018-012-1096-0.
Sedel F, Bernard D, Mock DM, Tourbah A. Targeting demyelination and virtual hypoxia with high-dose biotin as a treatment for progressive multiple sclerosis. Neuropharmacology. 2016;110(Pt B):644–53. https://doi.org/10.1016/j.neuropharm.2015.08.028.
Rasband MN, Macklin WB. Chapter 10—myelin structure and biochemistry. In: Brady ST, Siegel GJ, Albers RW, Price DL, editors. Basic neurochemistry. 8th ed. New York: Academic Press; 2012. p. 180–99.
Sedel F, Papeix C, Bellanger A, Touitou V, Lebrun-Frenay C, Galanaud D, et al. High doses of biotin in chronic progressive multiple sclerosis: a pilot study. Mult Scler Relat Disord. 2015;4(2):159–69. https://doi.org/10.1016/j.msard.2015.01.005.
Tourbah A, Lebrun-Frenay C, Edan G, Clanet M, Papeix C, Vukusic S, et al. MD1003 (high-dose biotin) for the treatment of progressive multiple sclerosis: a randomised, double-blind, placebo-controlled study. Mult Scler. 2016;22(13):1719–31. https://doi.org/10.1177/1352458516667568.
Cree BAC, Cutter G, Wolinsky JS, Freedman MS, Comi G, Giovannoni G, et al. Safety and efficacy of MD1003 (high-dose biotin) in patients with progressive multiple sclerosis (SPI2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 2020;19(12):988–97. https://doi.org/10.1016/S1474-4422(20)30347-1.
Birnbaum G, Stulc J. High dose biotin as treatment for progressive multiple sclerosis. Mult Scler Relat Disord. 2017;18:141–3. https://doi.org/10.1016/j.msard.2017.09.030.
Couloume L, Barbin L, Leray E, Wiertlewski S, Le Page E, Kerbrat A, et al. High-dose biotin in progressive multiple sclerosis: a prospective study of 178 patients in routine clinical practice. Mult Scler. 2019. https://doi.org/10.1177/1352458519894713.
Buonvicino D, Ranieri G, Pratesi S, Guasti D, Chiarugi A. Neuroimmunological characterization of a mouse model of primary progressive experimental autoimmune encephalomyelitis and effects of immunosuppressive or neuroprotective strategies on disease evolution. Exp Neurol. 2019;322:113065. https://doi.org/10.1016/j.expneurol.2019.113065.
Tourbah A, Gout O, Vighetto A, Deburghgraeve V, Pelletier J, Papeix C, et al. MD1003 (High-Dose Pharmaceutical-Grade Biotin) for the treatment of chronic visual loss related to optic neuritis in multiple sclerosis: a randomized, double-blind, Placebo-Controlled Study. CNS Drugs. 2018;32(7):661–72. https://doi.org/10.1007/s40263-018-0528-2.
Lebrun C, Cohen M, Mondot L, Ayrignac X, Labauge P. A case report of solitary sclerosis: this is really multiple sclerosis. Neurol Ther. 2017;6(2):259–63. https://doi.org/10.1007/s40120-017-0082-8.
Demas A, Cochin JP, Hardy C, Vaschalde Y, Bourre B, Labauge P. Tardive reactivation of progressive multiple sclerosis during treatment with biotin. Neurol Ther. 2020;9(1):181–5. https://doi.org/10.1007/s40120-019-00175-2.
Granella F, Curti E, Tsantes E, Siena E. Breakthrough disease under high-dose biotin treatment in progressive multiple sclerosis. ECTRIMS Online Libr. 2017;200405:P750.
Branger P, Parienti JJ, Derache N, Kassis N, Assouad R, Maillart E, et al. Relapses during high-dose biotin treatment in progressive multiple sclerosis: a case-crossover and propensity score-adjusted prospective cohort. Neurotherapeutics. 2020. https://doi.org/10.1007/s13311-020-00880-z.
Pignolet B, Ciron J, Bucciarelli F, Scandella L, Biotti D, Lerebours F, et al. Immunomodulation associated with clinical and MRI worsening in patients with progressive MS treated with MD1003 (high dose pharmaceutical grade biotin). ECTRIMS Online Libr. 2018;228721:P878.
Moisset X, Mathais S, Pereira B, Taithe F, Ciron J, Labauge P, et al. Incidence of relapses in patients with purified high dose biotin-treated progressive multiple sclerosis (IPBio-SeP study): a second intermediate analysis of a French, multicenter study. ECTRIMS Online Library. 2019;279034:P674.
Young W. Review of lithium effects on brain and blood. Cell Transplant. 2009;18(9):951–75. https://doi.org/10.3727/096368909X471251.
De Sarno P, Axtell RC, Raman C, Roth KA, Alessi DR, Jope RS. Lithium prevents and ameliorates experimental autoimmune encephalomyelitis. J Immunol. 2008;181(1):338–45. https://doi.org/10.4049/jimmunol.181.1.338.
Ahn M, Kim J, Park C, Cho J, Jee Y, Jung K, et al. Potential involvement of glycogen synthase kinase (GSK)-3beta in a rat model of multiple sclerosis: evidenced by lithium treatment. Anat Cell Biol. 2017;50(1):48–59. https://doi.org/10.5115/acb.2017.50.1.48.
Rowse AL, Naves R, Cashman KS, McGuire DJ, Mbana T, Raman C, et al. Lithium controls central nervous system autoimmunity through modulation of IFN-gamma signaling. PLoS ONE. 2012;7(12):e52658. https://doi.org/10.1371/journal.pone.0052658.
Beurel E, Yeh WI, Michalek SM, Harrington LE, Jope RS. Glycogen synthase kinase-3 is an early determinant in the differentiation of pathogenic Th17 cells. J Immunol. 2011;186(3):1391–8. https://doi.org/10.4049/jimmunol.1003511.
Li W, Li R, Zhao S, Jiang C, Liu Z, Tang X. Lithium posttreatment alleviates blood-brain barrier injury after intracerebral hemorrhage in rats. Neuroscience. 2018;383:129–37. https://doi.org/10.1016/j.neuroscience.2018.05.001.
Karimi A, Bahrampour K, Momeni Moghaddam MA, Asadikaram G, Ebrahimi G, Torkzadeh-Mahani M, et al. Evaluation of lithium serum level in multiple sclerosis patients: a neuroprotective element. Mult Scler Relat Disord. 2017;17:244–8. https://doi.org/10.1016/j.msard.2017.08.019.
Booth DR, Arthur AT, Teutsch SM, Bye C, Rubio J, Armati PJ, et al. Gene expression and genotyping studies implicate the interleukin 7 receptor in the pathogenesis of primary progressive multiple sclerosis. J Mol Med (Berl). 2005;83(10):822–30. https://doi.org/10.1007/s00109-005-0684-y.
Rinker JR 2nd, Meador WR, King P. Randomized feasibility trial to assess tolerance and clinical effects of lithium in progressive multiple sclerosis. Heliyon. 2020;6(7):e04528. https://doi.org/10.1016/j.heliyon.2020.e04528.
Vickrey BG, Hays RD, Harooni R, Myers LW, Ellison GW. A health-related quality of life measure for multiple sclerosis. Qual Life Res. 1995;4(3):187–206. https://doi.org/10.1007/BF02260859.
Fancy SP, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, et al. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009;23(13):1571–85. https://doi.org/10.1101/gad.1806309.
Ouyang S, Zeng Q, Tang N, Guo H, Tang R, Yin W, et al. Akt-1 and Akt-2 differentially regulate the development of experimental autoimmune encephalomyelitis by controlling proliferation of thymus-derived regulatory T cells. J Immunol. 2019;202(5):1441–52. https://doi.org/10.4049/jimmunol.1701204.
Rinker JR 2nd, Cossey TC, Cutter GR, Culpepper WJ. A retrospective review of lithium usage in veterans with multiple sclerosis. Mult Scler Relat Disord. 2013;2(4):327–33. https://doi.org/10.1016/j.msard.2013.03.004.
Wiendl H, Hohlfeld R. Therapeutic approaches in multiple sclerosis: lessons from failed and interrupted treatment trials. BioDrugs. 2002;16(3):183–200. https://doi.org/10.2165/00063030-200216030-00003.
Meuth SG, Bittner S, Ulzheimer JC, Kleinschnitz C, Kieseier BC, Wiendl H. Therapeutic approaches to multiple sclerosis: an update on failed, interrupted, or inconclusive trials of neuroprotective and alternative treatment strategies. BioDrugs. 2010;24(5):317–30. https://doi.org/10.2165/11537190-000000000-00000.
Ulzheimer JC, Meuth SG, Bittner S, Kleinschnitz C, Kieseier BC, Wiendl H. Therapeutic approaches to multiple sclerosis: an update on failed, interrupted, or inconclusive trials of immunomodulatory treatment strategies. BioDrugs. 2010;24(4):249–74. https://doi.org/10.2165/11537160-000000000-00000.
Pfeuffer S, Ruck T, Kleinschnitz C, Wiendl H, Meuth SG. Failed, interrupted and inconclusive trials on relapsing multiple sclerosis treatment: update 2010–2015. Expert Rev Neurother. 2016;16(6):689–700. https://doi.org/10.1080/14737175.2016.1176531.
Wooliscroft L, Silbermann E, Cameron M, Bourdette D. Approaches to remyelination therapies in multiple sclerosis. Curr Treat Options Neurol. 2019;21(7):34. https://doi.org/10.1007/s11940-019-0574-1.
Jensen SK, Michaels NJ, Ilyntskyy S, Keough MB, Kovalchuk O, Yong VW. Multimodal enhancement of remyelination by exercise with a pivotal role for oligodendroglial PGC1alpha. Cell Rep. 2018;24(12):3167–79. https://doi.org/10.1016/j.celrep.2018.08.060.
Yednock TA, Cannon C, Fritz LC, Sanchez-Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature. 1992;356(6364):63–6. https://doi.org/10.1038/356063a0.
Polman CH, O’Connor PW, Havrdova E, Hutchinson M, Kappos L, Miller DH, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med. 2006;354(9):899–910. https://doi.org/10.1056/NEJMoa044397.
Hall SM. The effect of injections of lysophosphatidyl choline into white matter of the adult mouse spinal cord. J Cell Sci. 1972;10(2):535–46.
Plemel JR, Liu WQ, Yong VW. Remyelination therapies: a new direction and challenge in multiple sclerosis. Nat Rev Drug Discov. 2017;16(9):617–34. https://doi.org/10.1038/nrd.2017.115.
Basso AS, Frenkel D, Quintana FJ, Costa-Pinto FA, Petrovic-Stojkovic S, Puckett L, et al. Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J Clin Invest. 2008;118(4):1532–43. https://doi.org/10.1172/JCI33464.
Constantinescu CS, Farooqi N, O’Brien K, Gran B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br J Pharmacol. 2011;164(4):1079–106. https://doi.org/10.1111/j.1476-5381.2011.01302.x.
Llovera G, Liesz A. The next step in translational research: lessons learned from the first preclinical randomized controlled trial. J Neurochem. 2016;139(Suppl 2):271–9. https://doi.org/10.1111/jnc.13516.
Schmierer K, Scaravilli F, Altmann DR, Barker GJ, Miller DH. Magnetization transfer ratio and myelin in postmortem multiple sclerosis brain. Ann Neurol. 2004;56(3):407–15. https://doi.org/10.1002/ana.20202.
Cerina M, Muthuraman M, Gallus M, Koirala N, Dik A, Wachsmuth L, et al. Myelination- and immune-mediated MR-based brain network correlates. J Neuroinflamm. 2020;17(1):186. https://doi.org/10.1186/s12974-020-01827-z.
Bodini B, Veronese M, Garcia-Lorenzo D, Battaglini M, Poirion E, Chardain A, et al. Dynamic imaging of individual remyelination profiles in multiple sclerosis. Ann Neurol. 2016;79(5):726–38. https://doi.org/10.1002/ana.24620.
Gingele S, Stangel M. Emerging myelin repair agents in preclinical and early clinical development for the treatment of multiple sclerosis. Expert Opin Investig Drugs. 2020. https://doi.org/10.1080/13543784.2020.1762567.
Klistorner A, Fraser C, Garrick R, Graham S, Arvind H. Correlation between full-field and multifocal VEPs in optic neuritis. Doc Ophthalmol. 2008;116(1):19–27. https://doi.org/10.1007/s10633-007-9072-y.
Varhaug KN, Torkildsen O, Myhr KM, Vedeler CA. Neurofilament light chain as a biomarker in multiple sclerosis. Front Neurol. 2019;10:338. https://doi.org/10.3389/fneur.2019.00338.
Thebault S, Abdoli M, Fereshtehnejad SM, Tessier D, Tabard-Cossa V, Freedman MS. Serum neurofilament light chain predicts long term clinical outcomes in multiple sclerosis. Sci Rep. 2020;10(1):10381. https://doi.org/10.1038/s41598-020-67504-6.
Bittner S, Steffen F, Uphaus T, Muthuraman M, Fleischer V, Salmen A, et al. Clinical implications of serum neurofilament in newly diagnosed MS patients: a longitudinal multicentre cohort study. EBioMedicine. 2020;56:102807. https://doi.org/10.1016/j.ebiom.2020.102807.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL.
Conflicts of interest
Niklas Huntemann: declares no conflicts of interest. Leoni Rolfes: declares no conflicts of interest. Marc Pawlitzki: declares no conflicts of interest. Tobias Ruck: received personal fees from Alexion, Biogen, Merck Serono, Sanofi-Genzyme, Roche, and Teva, grants from Alexion and Sanofi-Genzyme, and nonfinancial support from Merck Serono. Steffen Pfeuffer: received travel reimbursements from Sanofi Genzyme and Merck Serono, honoraria for lecturing from Sanofi Genzyme, Biogen, and Mylan Healthcare, and research support from Merck Serono, Diamed, and the German Multiple Sclerosis Society North Rhine-Westphalia. Heinz Wiendl: received grants from German Ministry for Education and Research (BMBF), Deutsche Forschungsgesellschaft (DFG), Else Kröner Fresenius Foundation, Fresenius Foundation, the European Union, Hertie Foundation, NRW Ministry of Education and Research, Interdisciplinary Center for Clinical Studies (IZKF) Muenster, Biogen, GlaxoSmithKline GmbH, Roche, and Sanofi-Genzyme; consulting fees from AbbVie, Actelion, Argenx, Biogen, EMD Serono, Idorsia, IGES, Immunic, Merck, Novartis, Roche, Sanofi-Aventis, the Swiss Multiple Sclerosis Society, and UCB; support for travel to meetings for other purposes from Alexion, Biogen, Cognomed, F. Hoffmann-La Roche Ltd., Hertie Foundation, Merck Serono, Novartis, Roche, Genzyme, Teva, and WebMD Global; fees for participation in review activities such as data monitoring boards from Polpharma Biologics; payment for lectures from Alexion, Biogen, Cognomed, F. Hoffmann-La Roche Ltd., Hertie Foundation, Merck Serono, Novartis, Roche, Genzyme, Teva, and WebMD Global; honorarium for expert testimony from the Drug Commission of the German Medical Association. Sven G. Meuth: received honoraria for lecturing and travel reimbursement for attending meetings from Almirall, Amicus Therapeutics Germany, Bayer Health Care, Biogen, Celgene, Diamed, Genzyme, MedDay Pharmaceuticals, Merck Serono, Novartis, Novo Nordisk, ONO Pharma, Roche, Sanofi-Aventis, Chugai Pharma, QuintilesIMS, and Teva. His research is funded by the German Ministry for Education and Research (BMBF), Bundesinstitut für Risikobewertung (BfR), Deutsche Forschungsgemeinschaft (DFG), Else Kröner Fresenius Foundation, Gemeinsamer Bundesausschuss (G-BA), German Academic Exchange Service, Hertie Foundation, Interdisciplinary Center for Clinical Studies (IZKF) Muenster, German Foundation Neurology and Alexion, Almirall, Amicus Therapeutics Germany, Biogen, Diamed, Fresenius Medical Care, Genzyme, HERZ Burgdorf, Merck Serono, Novartis, ONO Pharma, Roche, and Teva.
Availability of data and material
Not applicable.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Code availability
Not applicable.
Author contributions
NH: study concept and design, acquisition of data, analysis and interpretation of data, writing of the manuscript. LR: study concept and design, acquisition of data, writing of the manuscript. MP: critical revision of manuscript for intellectual content. TR: critical revision of manuscript for intellectual content. SP: critical revision of manuscript for intellectual content. HW: critical revision of manuscript for intellectual content. SGM: study concept and design, critical revision of manuscript for intellectual content.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Huntemann, N., Rolfes, L., Pawlitzki, M. et al. Failed, Interrupted, or Inconclusive Trials on Neuroprotective and Neuroregenerative Treatment Strategies in Multiple Sclerosis: Update 2015–2020. Drugs 81, 1031–1063 (2021). https://doi.org/10.1007/s40265-021-01526-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-021-01526-w