
Abstract
Secukinumab (Cosentyx®), a first-in-class fully human monoclonal antibody against interleukin-17A, is approved in several countries, including the USA and those of the EU, for the treatment of ankylosing spondylitis (AS). Subcutaneous secukinumab significantly improved the clinical signs and symptoms of AS versus placebo in three of four phase III trials. The benefits of secukinumab were generally seen regardless of whether patients had or had not received previous tumour necrosis factor (TNF) inhibitor therapy, and were sustained during longer-term (up to 5 years) treatment. Secukinumab was also associated with improvements in spinal mobility, physical function, health-related quality of life and work productivity in some of the trials. In MEASURE 1, secukinumab reduced inflammation in the sacroiliac joint, and slowed radiographic progression. Secukinumab was generally well tolerated during up to 5 years’ treatment; the most commonly reported adverse event was nasopharyngitis. In the minority of patients who developed anti-drug antibodies (ADAs), ADAs did not decrease efficacy or increase adverse events. In conclusion, secukinumab is an effective therapy for TNF inhibitor-naive patients with active AS, and provides a useful treatment option for patients who have an inadequate response to or are intolerant of TNF inhibitors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Improves clinical signs and symptoms of AS, with benefits sustained during longer-term treatment |
Improves spinal mobility, physical function, health-related quality of life and work productivity in some trials |
Reduces inflammation in the sacroiliac joint, with a low rate of radiographic progression |
Generally well tolerated |
1 Introduction
Ankylosing spondylitis (AS) is a chronic, autoimmune inflammatory disease that primarily affects the axial skeleton [1]. Characteristic symptoms include chronic back pain, stiffness and progressive loss of spinal mobility [1, 2]. If not adequately treated, AS can lead to significant disability (including total fusion of the axial skeleton) and impaired quality of life (QOL) [1]. NSAIDs are the first-line recommended agents for the treatment of active AS [3, 4]. For patients whose disease remains active despite conventional treatment with NSAIDs [3, 4], the advent of tumour necrosis factor (TNF) inhibitors has revolutionized the treatment landscape [5]. However, some patients fail to respond adequately to TNF inhibitors or develop tolerability issues, and the efficacy of TNF inhibitors can wane over time. New treatment options for these patients are now available, including interleukin (IL)-17 inhibitors [5].
IL-17A, a member of the IL-17 family, is a cytokine involved in normal inflammatory and immune responses [6]. IL-17A has been shown to play an important role in the pathogenesis of AS [7]. Indeed, studies have demonstrated increased numbers of IL-17A-producing cells in the circulation and the subchondral bone marrow of joints in patients with AS [7]. Secukinumab (Cosentyx®) is the first IL-17A inhibitor approved for the treatment of AS. The pharmacological properties of secukinumab have been reviewed in detail previously [8] and are summarized in Table 1. This review focuses on the clinical use of secukinumab in adults with active AS [9, 10]. Secukinumab is also approved for the treatment of plaque psoriasis [11] and psoriatic arthritis [12]; discussion of these indications is beyond the scope of this review.
2 Therapeutic Efficacy of Secukinumab
The efficacy of subcutaneous secukinumab for the treatment of AS was primarily assessed in five multicentre, phase III trials, including four randomized, double-blind trials (MEASURE 1, a 2-year study with a 3-year extension [13]; MEASURE 2, a 5-year study [13]; MEASURE 3, a 3-year study [14]; and MEASURE 4, a 2-year-study [15]) and an open-label trial in Japanese patients (MEASURE 2-J) [16] (Sect. 2.1.4). The efficacy of secukinumab in the real-world setting is also briefly discussed (Sect. 2.2). Some data are available as abstracts [17,18,19,20,21,22,23,24,25,26,27].
2.1 MEASURE Trials
All trials included patients aged ≥ 18 years with active AS (according to the modified New York criteria), a Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) score of ≥ 4 and a spinal pain score of ≥ 4 cm on a 10 cm visual analogue scale (VAS), despite treatment with the maximum tolerated doses of NSAIDs [13,14,15,16]. Patients who had previously received no more than one TNF inhibitor were permitted if they had an inadequate response to an approved dose for ≥ 3 months or were intolerant to at least one dose (hereafter referred to as patients with an inadequate response to TNF inhibitors). Across all trials, the mean age of patients was 40–45 years and 61–78% of patients were TNF inhibitor-naive. The primary endpoint of all trials was the proportion of patients who met Assessment of SpondyloArthritis international Society 20 (ASAS20) response criteria at week 16 (Table 2) [13,14,15,16].
Patients in each of the MEASURE 1–4 trials were randomized to one of two secukinumab groups or a placebo group; randomization was stratified by baseline use of TNF inhibitors [13,14,15]. In MEASURE 1 and 3, secukinumab recipients received intravenous loading doses of secukinumab 10 mg/kg at weeks 0, 2 and 4, followed by subcutaneous secukinumab 75 or 150 mg (MEASURE 1) or 150 or 300 mg (MEASURE 3) every 4 weeks, starting at week 8 [13, 14]. In MEASURE 2, secukinumab recipients received subcutaneous injections of secukinumab 75 or 150 mg at weeks 0, 1, 2 and 3, then every 4 weeks, starting at week 4 [13]. In MEASURE 4, subcutaneous secukinumab was self-administered with a loading regimen (i.e. 150 mg at weeks 0, 1, 2 and 3, then every 4 weeks starting at week 4) or without a loading regimen (i.e. 150 mg every 4 weeks) [15]. In MEASURE 2-J, all patients self-administered subcutaneous secukinumab 150 mg at weeks 0, 1, 2, 3 and 4, then every 4 weeks [16]. In MEASURE 1, 2 and 3, placebo recipients were re-randomized to receive secukinumab 75 or 150 mg [13] or 150 or 300 mg [14] every 4 weeks from week 16 (MEASURE 2 and 3) [13, 14] or 24 (MEASURE 1) [13]. At week 16 in MEASURE 4, all placebo recipients were switched to open-label secukinumab 150 mg [15]. Discussion in this section focuses on the approved dose of secukinumab (150 mg), with data for the 75 and 300 mg doses presented in Table 2 for completeness.
2.1.1 Clinical Response
In MEASURE 1, 2 and 3, subcutaneous secukinumab 150 mg improved the signs and symptoms of AS, with significantly more secukinumab than placebo recipients achieving an ASAS20 response at week 16 (Table 2) [13, 14]. Secukinumab was also associated with significant improvements in all secondary endpoints, with the exception of ASAS partial remission rates in MEASURE 2 and 3 (Table 2) [13, 14]. In MEASURE 4, secukinumab (with or without a loading dose) was not superior to placebo at week 16 for the primary endpoint or for any of the secondary endpoints (Table 2) [15].
In prespecified subgroup analyses, the benefits of secukinumab 150 mg (although not always significant vs. placebo) were seen regardless of whether patients had or had not received previous TNF inhibitor therapy [14, 15, 23, 28]. For instance, ASAS20 response rates at week 16 were significantly higher with secukinumab than placebo among TNF inhibitor-naive patients in MEASURE 1 (66 vs. 33%; p < 0.0001 [23]), MEASURE 2 (68 vs. 31%; p < 0.001 [28]) and MEASURE 3 (63 vs. 39%; p < 0.05 [14]), but not MEASURE 4 (60% in loading-dose group and 62% in non-loading-dose group vs. 49% [15]) and among TNF inhibitor-experienced patients in MEASURE 1 (46 vs. 18%; p < 0.05) [23] and MEASURE 2 (50 vs. 24%; p < 0.05) [28], but not MEASURE 3 (41 vs. 29%) [14] or MEASURE 4 (58% in loading-dose group and 59% in non-loading-dose group vs. 41%) [15]. In MEASURE 1, 2 and 3, secukinumab was also associated with significant (p < 0.05) improvements in all secondary endpoints in TNF inhibitor-naive patients (with the exception of ASAS partial remission in MEASURE 2 and 3 and BASDAI in MEASURE 3), and for most secondary endpoints in TNF inhibitor-experienced patients [14, 23, 28]. In MEASURE 4, secukinumab was associated with numerically greater improvements than placebo in other efficacy endpoints regardless of prior TNF inhibitor use, although these differences were not statistically significant [15].
In a pooled subgroup analysis of MEASURE 1 and 2, ASAS20 response rates with secukinumab versus placebo at week 16 in Asian patients (n = 69; 70 vs. 26%; p < 0.01) were comparable with those in the overall study population [29]. In a post hoc analysis of pooled data from MEASURE 1 and 2, ASAS20 response rates at week 16 were significantly higher with secukinumab versus placebo in patients with normal C-reactive protein (CRP) levels (57 vs. 28%; p < 0.01) and in patients with elevated (≥ 5 mg/L) CRP levels (63 vs. 29%; p < 0.0001) [30].
In MEASURE 1–4, efficacy outcomes at week 16 were sustained through 52 weeks [13,14,15] and 104 weeks [15, 17, 31, 32] of treatment. For example, when missing values were imputed as non-responses, the ASAS20 response rate ranged from 58 to 72% at week 52 [13,14,15] and from 72 to 78% at week 104 [15, 17, 31, 32]. On the basis of observed data (where reported), the ASAS20 response rate ranged from 73 to 77% at week 52 [13,14,15] and from 75 to 82% at week 104 [15, 31].
Over the longer term, improvements in ASAS20 response rates and other major efficacy endpoints with secukinumab 150 mg were maintained for up to 260 weeks in MEASURE 1 [18, 33, 34], 208 weeks in MEASURE 2 [19, 35] and 156 weeks in MEASURE 3 [20]. For example, ASAS20 response rates (both as observed and with multiple imputations, where reported) remained high (68–80%) over 3–5 years of treatment [18,19,20, 33,34,35].
Patients who switched from placebo to secukinumab 150 mg at either week 16 or 24 had numerical improvements in ASAS20 response rates and other efficacy endpoints [13,14,15]; these were sustained over the longer term, up to week 104 in MEASURE 4 [15], week 156 in MEASURE 3 [20], week 208 in MEASURE 2 [19] and week 260 in MEASURE 1 [18].
2.1.2 Radiological Response
In MEASURE 1, secukinumab 150 mg reduced inflammation in the sacroiliac joint in patients with AS, as assessed by MRI [22, 34]. Among the subset of TNF inhibitor-naive patients (n = 105), secukinumab significantly (p < 0.01) improved Berlin sacroiliac joint total oedema scores at week 16 compared with placebo (baseline scores 2.22 vs. 2.40; mean improvement from baseline 59 vs. 7%) [22]. In addition, secukinumab was associated with mean reductions (indicating improvement) from baseline in MRI scores for spinal activity (ASspi-MRI-a) [– 1.13 vs. 2.70 at baseline] and the Berlin spine score (derived from ASspi-MRI-a) [– 1.08 vs. 2.23 at baseline], although these changes were not significant relative to those seen in the placebo group (– 0.66 vs. 5.73 at baseline and – 0.55 vs. 4.50 at baseline) [22]. The reductions in inflammation achieved with secukinumab at week 16 were maintained through 4 years of treatment [34]. The mean changes from baseline in Berlin sacroiliac joint total oedema and Berlin spine scores at week 208 were –1.6 and –0.6. MRI remission (Berlin spine score of ≤ 2) was achieved in 33% of patients [34].
Secukinumab 150 mg was associated with a low rate of radiographic progression during long-term therapy [34]. Of the 78 patients in MEASURE 1 who were originally randomized to secukinumab 150 mg and completed 4 years of treatment, 71 had evaluable x-rays at baseline and week 208. The mean change from baseline in modified Stoke Ankylosing Spondylitis Spinal Score (mSASSS) at week 208 was + 1.2 (mean baseline value 8.8). In 79% of patients, there was no definite radiographic progression (change from baseline in mSASSS < 2). Mean changes from baseline in mSASSS at week 208 were numerically higher in male versus female patients, those with versus without syndesmophytes at baseline and those with elevated versus normal CRP levels [34].
2.1.3 Patient-Reported Outcomes
Secukinumab improved spinal mobility and physical function in MEASURE 1, as indicated by the significantly greater mean changes from baseline to week 16 that were seen with the drug versus placebo in Bath Ankylosing Spondylitis Metrology Index (BASMI) [– 0.40 vs. – 0.12; p = 0.0114] [9] and Bath Ankylosing Spondylitis Functional Index (BASFI) [– 1.8 vs. – 0.4; p < 0.0001] [36] scores; these improvements were maintained through 5 years of treatment [18]. In MEASURE 2, the mean change from baseline in BASMI score at week 16 was numerically greater with secukinumab versus placebo (– 0.51 vs. – 0.22), although the difference was not statistically significant [9, 10].
Secukinumab improved health-related (HR)-QOL in MEASURE 1 and 2, as assessed by the Medical Outcomes Study 36-item Short-Form Health Survey (SF-36), the Ankylosing Spondylitis Quality of Life scale (ASQoL) and the Functional Assessment of Chronic Illness Therapy (FACIT) measurement system [13, 24, 36]. At week 16, secukinumab was associated with significantly (p < 0.05) greater improvements from baseline than placebo in SF-36 physical component summary (PCS) and ASQoL scores (Table 2) [13], as well as FACIT-Fatigue scores [24, 36]. At week 16, the proportion of secukinumab recipients with a fatigue response (defined as an increase/improvement from baseline in FACIT-Fatigue score of ≥ 4, based on the minimal clinically important difference) was 67% in MEASURE 1 and 78% in MEASURE 2 [24]. Improvements in HR-QOL seen with secukinumab at week 16 in MEASURE 1 and 2 were sustained for up to 260 weeks in MEASURE 1 [18, 21, 24, 36] and 156 weeks in MEASURE 2 [21, 24, 32, 35]. In MEASURE 4, the mean changes from baseline in SF-36 PCS and ASQoL scores were not significantly different between secukinumab (with or without a loading dose) and placebo at week 16 (Table 2), week 52 or week 104 [15].
Secukinumab improved several aspects of work productivity in MEASURE 1, as assessed by the Work Productivity and Activity Impairment-General Health (WPAI-GH) questionnaire [21, 36]. At week 16, absenteeism (i.e. work time missed due to health) decreased by 1% with secukinumab and increased by 2% with placebo [36]. Secukinumab was also associated with improvements from baseline in impairment while working due to health (– 20%), overall work impairment due to health (– 21%) and activity impairment due to health (– 19%), although these changes were not significant relative to those seen with placebo (– 13, – 10 and – 7%, respectively) [36]. Improvements in all WPAI-GH domains observed with secukinumab at week 16 were sustained through weeks 52 [21, 36] and 104 [21].
In MEASURE 3, all patients reported successful self-administration of secukinumab using the pre-filled syringe [14]. The only hazard experienced was ‘needle stick in a non-critical area’ (incidence of 4.6% across all treatment groups). Self-Injection Assessment Questionnaire scores for feeling about injections, self-confidence and satisfaction with self-injection improved over time [14].
2.1.4 In Japanese Patients
Results in Japanese patients were consistent with those seen in the MEASURE 1–4 trials. In MEASURE 2-J, the proportion of patients with an ASAS20 response at week 16 was 70% in the overall population (Table 2), 68% among TNF inhibitor-naive patients (n = 22) and 75% among TNF inhibitor-experienced patients (n = 8) [16]. Overall ASAS20 response rates at weeks 1, 4 and 24 were 23, 53 and 70%, respectively. Improvement was seen in all secondary endpoints at week 16 (Table 2), with these improvements sustained or further improved at week 24 [16].
With regard to exploratory endpoints, secukinumab was associated with improvements from baseline in disease severity [as assessed by Ankylosing Spondylitis Disease Activity Score (ASDAS)-CRP], patient’s global assessment of disease activity, total spinal pain and nocturnal back pain (as assessed by VAS scores), physical function (as assessed by BASFI scores) and spinal mobility (as assessed by BASMI scores) at week 16 [16]. The proportion of patients with a clinically important change (≥ 1.1), major improvement (≥ 2.0) or inactive disease (< 1.3) in ASDAS-CRP at week 16 was 63, 33 and 27%, respectively [16].
2.2 Real-World Clinical Practice
Real-world experience has confirmed the efficacy of secukinumab for the treatment of AS [25,26,27, 37]. In small (n < 50) observational studies conducted in Italy [26, 37] and Scotland [27], secukinumab 150 or 300 mg improved the signs and symptoms of AS. In a US web-based survey of 200 patients with AS, the majority of patients were very satisfied or mostly satisfied with secukinumab in terms of overall symptom improvement (90%), onset of symptom improvement (79%), patient support services (79%), frequency of administration (77%), ease of use (75%), side effects, if any (74%) and method of administration (70%) [25]. In addition, more than half of patients reported secukinumab to be better than their previous treatment with regard to overall symptom improvement (77%), patient support services (62%), ease of switching (58%), ease of use (57%) and onset of symptom improvement (55%) [25].
3 Tolerability of Secukinumab
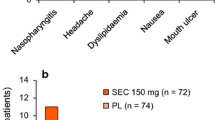
Subcutaneous secukinumab 150 mg was generally well tolerated in the MEASURE 1–4 trials [13,14,15]. The tolerability profile of secukinumab in patients with AS was consistent with that observed in patients with plaque psoriasis [9, 10]. During the 16-week placebo-controlled periods, adverse events (AEs) occurred in 46–70% of secukinumab recipients and 44–64% of placebo recipients, with serious AEs reported in 0–6% of secukinumab and 1–4% of placebo recipients [13,14,15]. During this period, the most commonly reported AE with secukinumab was nasopharyngitis [13,14,15].
In a pooled analysis of all secukinumab groups in MEASURE 1, 2 and 3 (n = 794), the tolerability profile of secukinumab during the entire safety period (156 weeks in MEASURE 1, 104 weeks in MEASURE 2 and 52 weeks in MEASURE 3) was generally consistent with that seen in previous reports (available as an abstract) [38]. In this analysis, the pooled exposure-adjusted incidence rate (EAIR) of AEs was 146.8 per 100 patient-years (PY) of exposure. The corresponding EAIR for serious AEs was 6.2 per 100 PY. The most frequent AEs were nasopharyngitis (12.3 per 100 PY), headache (6.0 per 100 PY), diarrhoea (5.7 per 100 PY) and upper respiratory tract infection (URTI; 5.1 per 100 PY) [38]. In MEASURE 4, the most commonly reported AEs during the entire treatment period (i.e. 104 weeks) when secukinumab was administered with a loading dose versus without a loading dose included nasopharyngitis (EAIR 19.2 vs. 16.7 per 100 PY), URTI (5.5 vs. 8.9 per 100 PY) and bronchitis (6.4 vs. 3.9 per 100 PY) [15].
The longer-term tolerability profile of secukinumab in the MEASURE 1–4 trials was consistent with that previously observed; no new safety signals or unexpected safety findings were observed [15, 17,18,19,20, 31,32,33,34,35].
3.1 Adverse Events of Special Interest
Treatment with secukinumab may increase the risk of infections [9, 10]. During the 16-week placebo-controlled period of MEASURE 1, infections or infestations occurred in 30% of secukinumab (any dose) and 12% of placebo recipients; corresponding rates in MEASURE 2 were 32 and 27% [13]. In the pooled analysis of MEASURE 1, 2 and 3, serious infections and infestations and Candida infections occurred at EAIRs of 1.1 and 0.7 per 100 PY of exposure to secukinumab (any dose) during the entire safety period [38]. In MEASURE 4, the EAIR of Candida infections with secukinumab was 1.9 per 100 PY when administered with a loading dose and 1.4 per 100 PY when administered without a loading dose [15]. Where reported, Candida infections resolved spontaneously or with standard antifungal therapy [13, 14]. Caution is advised when considering the use of secukinumab in patients with a chronic infection or a history of recurrent infection [9, 10]. No increased susceptibility to tuberculosis was reported in clinical trials [9]. However, patients should be evaluated for tuberculosis infection prior to initiating secukinumab treatment [10] and secukinumab should not be given to patients with active tuberculosis [9, 10].
The pooled EAIR of grade 3 or 4 neutropenia with secukinumab (any dose) over the entire safety period in MEASURE 1 and 2 was 0.7 per 100 PY; only one case of neutropenia was associated with infection (a non-serious URTI) [13]. In MEASURE 3, the EAIR of grade 3/4 neutropenia with secukinumab 150 mg during the 52-week safety-data period was 0.9/1.8 per 100 PY [14]. In MEASURE 4, the EAIR of neutropenia with secukinumab 150 mg was 0.5 per 100 PY when administered with a loading dose and 0.0 per 100 PY when administered without a loading dose [15]. In all studies, neutropenia resolved without the need for discontinuation of treatment [13,14,15].
There have been reports of new-onset or exacerbations of inflammatory bowel disease (IBD), including some serious cases, in patients receiving secukinumab [9, 10]. Across MEASURE 1, 2 and 3 (1943.1 PY of exposure to secukinumab), the pooled EAIRs of Crohn’s disease, ulcerative colitis and IBD not otherwise specified with secukinumab over the entire treatment period were 0.4, 0.2 and 0.1 per 100 PY, respectively [39]. In MEASURE 4, the EAIR of Crohn’s disease with secukinumab was 0.0 per 100 PY when administered with a loading dose and 0.5 per 100 PY when administered without a loading dose [15]. Patients receiving secukinumab should be closely monitored for signs and symptoms of IBD, and caution is advised when prescribing secukinumab to patients with IBD, including Crohn’s disease and ulcerative colitis [9, 10].
In the pooled analysis of MEASURE 1, 2 and 3, the EAIR of major adverse cardiac events (MACE) with secukinumab (any dose) during the entire safety period was 0.7 per 100 PY [38]. In MEASURE 4, the EAIR of MACE was 0.9 per 100 PY when administered with a loading dose and 0.0 per 100 PY when administered without a loading dose [15].
Across MEASURE 1, 2 and 3, there were 26 cases of uveitis, including 12 new-onset cases [40]. The pooled EAIR of uveitis with secukinumab over the entire treatment period was 1.4 per 100 PY [40]. In MEASURE 4, uveitis occurred at an EAIR of 0.9 per 100 PY of exposure to secukinumab [15].
As a monoclonal antibody, secukinumab has the potential for immunogenicity [41]. Among secukinumab-treated patients with evaluable samples in MEASURE 1–4 (n = 1163), treatment-emergent anti-drug antibodies (ADAs) were detected in eight (0.7%) patients, all of which were non-neutralizing. The emergence of antibodies was not associated with abnormal pharmacokinetics, immunogenicity-related AEs or a loss of secukinumab efficacy over 52 weeks [41].
In a post-marketing safety study based on cumulative secukinumab exposure of > 96 000 PY across all approved indications (psoriasis, psoriatic arthritis and AS), the overall safety profile of secukinumab was consistent with that reported in clinical trials [42]. Across five successive periodic safety update reporting periods between December 2014 and June 2017, the exposure-adjusted reporting rates of infections and infestations, neutropenia, hypersensitivity, malignant or unspecified tumours, IBD and MACE with secukinumab were 4.7, 0.07, 2.4, 0.2, 0.2 and 0.2 per 100 PY, respectively. There was one case of immunogenicity [42].
4 Dosage and Administration of Secukinumab
Subcutaneous secukinumab is indicated for the treatment of adults with active AS [9, 10] who have responded inadequately to conventional therapy [10]. Secukinumab is available as a lyophilized powder (150 mg) in a vial for reconstitution, or as a 150 mg/mL solution for injection in a pre-filled pen or syringe [9, 10]. The lyophilized powder for reconstitution is to be administered by healthcare professionals only, whereas secukinumab in a pre-filled pen or syringe may be self-administered following proper training in subcutaneous injection technique [10].
The recommended dosage of secukinumab is 150 mg administered at weeks 0, 1, 2, 3 and 4 followed by 4-weekly (USA) or monthly (EU) dosing commencing at week 4 [9, 10] or, alternatively, in the USA [10], commencing on a 4-weekly schedule with the omission of the loading doses. Each injection should be administered at a different site (upper arm, abdomen or thigh) than the previous injection [10]. Discontinuation of secukinumab should be considered if no response is seen after 16 weeks of treatment; patients with an initial partial response may subsequently improve with continued treatment beyond 16 weeks [9].
The efficacy and tolerability of secukinumab in paediatric patients has not been established [9, 10]. There are limited data regarding the use of secukinumab in pregnant women [9, 10] and it is preferable to avoid its use during pregnancy as a precaution [9]. It is unknown whether secukinumab is excreted in human milk [9, 10]. The benefits of breastfeeding for the infant and the clinical benefits of secukinumab therapy for the mother should be considered [9, 10]. Local prescribing information should be consulted for full details regarding the administration of secukinumab, including further information on warnings and precautions, contraindications and use in special populations.
5 Place of Secukinumab in the Management of Ankylosing Spondylitis
Secukinumab is a first-in-class IL-17A inhibitor approved for the treatment of active AS (Sect. 4). American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network (ACR/SAA/SPARTAN) guidelines published prior to the US approval of secukinumab strongly recommend TNF inhibitors for AS patients whose disease remains active despite first-line treatment with NSAIDs [4]. However, updated recommendations are currently being developed to incorporate new treatment options, including secukinumab, the JAK inhibitor tofacitinib, and TNF inhibitor biosimilars [43]. The recently updated ASAS-European League Against Rheumatism (EULAR) guidelines recommend that biologic disease-modifying antirheumatic drugs (i.e. TNF inhibitors and IL-17 inhibitors) should be considered in patients with persistently high disease activity despite conventional treatments (including NSAIDs) [3]. The UK National Institute for Health and Care Excellence (NICE) recommends secukinumab as an option for treating active AS in adults whose disease has responded inadequately to conventional NSAID or TNF inhibitor therapy [44].
In several well-designed phase III trials (MEASURE 1, 2 and 3) in adults with active AS, secukinumab 150 mg improved the signs and symptoms of disease (Sect. 2.1.1). The clinical efficacy of secukinumab observed during the first year of treatment was sustained over the longer term (through 3 years in MEASURE 1, 2 and 3, 4 years in MEASURE 1 and 2, and 5 years in MEASURE 1). Final results of the ongoing MEASURE 2 trial will provide additional long-term data through 5 years of secukinumab treatment. Another phase III trial (MEASURE 4) did not demonstrate significant efficacy with secukinumab (with or without a loading dose) versus placebo for primary or key secondary endpoints (Sect. 2.1.1); this finding may have been the result of higher than expected placebo response rates [15].
The benefits of secukinumab were generally seen regardless of previous TNF inhibitor use (Sect. 2.1.1), suggesting that secukinumab may be an effective treatment in both TNF inhibitor-naive and -experienced patients. The significance of this finding is evident in the ASAS-EULAR guidelines, which recommend switching to either another TNF inhibitor or an IL-17 inhibitor after failure of the first TNF inhibitor, stating that it may be more rational to switch to an IL-17 inhibitor [3]. Current ACR/SAA/SPARTAN guidelines conditionally recommend switching to a different TNF inhibitor after failure of the first TNF inhibitor [4].
To date, no randomized controlled trials have directly compared secukinumab with other pharmacological agents in patients with AS. Matching-adjusted indirect comparisons have demonstrated no apparent differences in efficacy between secukinumab and adalimumab [45] or between secukinumab and golimumab [46] in placebo-anchored ASAS20/40 comparisons. In non-placebo-anchored analyses, secukinumab demonstrated higher ASAS20/40 response rates than adalimumab after week 12 [45] and golimumab at week 24 [46]. However, given the limitations of indirect comparisons, these results should be interpreted with caution. Clinical trials comparing secukinumab with other agents (particularly TNF inhibitors) would help to definitively place secukinumab in the management of AS. The randomized, phase IIIb SURPASS trial is currently underway comparing the effects of secukinumab and GP2017, an adalimumab biosimilar, on radiographic progression in biologic-naive patients with AS [47]. Results from this trial are awaited with interest.
Given the chronic and often disabling nature of AS, it is important to minimize the impact of the disease on patients in terms of pain, stiffness, fatigue, interference with activities of daily living and work disability [48]. In some trials, secukinumab provided clinically meaningful and sustained improvements in patient-reported spinal mobility, physical function, HR-QOL, fatigue levels and work productivity (Sect. 2.1.3).
The prevention of structural damage is another important long-term goal in the management of AS [31]. Secukinumab was associated with reductions in inflammatory biomarkers in patients with AS (Tables 1, 2). In addition, data from MEASURE 1 demonstrated that secukinumab reduced inflammation in the sacroiliac joint, and slowed radiographic progression (Sect. 2.1.2). Further long-term controlled studies are needed to clearly establish the efficacy of secukinumab in inhibiting radiographic progression in patients with AS.
Safety and tolerability may influence the selection of therapy for AS [1, 5]. Secukinumab was generally well tolerated in clinical trials, with a tolerability profile consistent with that observed in patients with plaque psoriasis (Sect. 3). Nasopharyngitis was the most commonly reported AE. Secukinumab remained generally well tolerated over the longer term (up to 5 years), with no new safety signals or unexpected safety findings (Sect. 3). ADAs developed in a minority of secukinumab recipients but were not associated with AEs or decreased efficacy (Sect. 3.1).
Following proper training, secukinumab may be self-administered by subcutaneous injection using a pre-filled pen or syringe (Sect. 4). In MEASURE 3, the secukinumab pre-filled syringe was associated with a high level of usability, acceptability and satisfaction (Sect. 2.1.3). Similarly, in the real-world setting, most patients were satisfied with secukinumab and more than half of patients reported secukinumab to be better than their previous treatment, including in terms of method of administration and ease of use (Sect. 2.2). An additional phase III trial investigating the efficacy and tolerability of subcutaneous secukinumab 150 mg in pre-filled syringes in 459 patients with active AS is currently underway (MEASURE 5; NCT02896127).
Chronic, immune-mediated, inflammatory diseases such as psoriasis, psoriatic arthritis and AS are associated with substantial healthcare costs [49]. In the NICE guidance, secukinumab was reported to be a cost-effective use of health system resources for AS, regardless of previous TNF inhibitor use [44]. Indeed, a modelled pharmacoeconomic study from a UK National Health Service perspective suggests that secukinumab is a cost-effective treatment option for biologic-naive and -experienced patients with active AS [50]. Model-based budget impact analyses in Italy [49], Finland [51], Greece [52] and Brazil [53] also demonstrated that secukinumab is a cost-saving option for the treatment of AS. In cost-effectiveness analyses in Portugal [54], Tunisia [55] and Korea [56], the costs per responder were lower with secukinumab than with adalimumab in patients with AS.
In conclusion, secukinumab is effective and generally well tolerated for the treatment of adults with active AS, with efficacy and tolerability sustained over the longer term. Secukinumab is an effective therapy for TNF inhibitor-naive patients with active AS, and provides a useful treatment option for patients who have an inadequate response to or are intolerant of TNF inhibitors.
Data Selection Secukinumab: 343 records identified
Duplicates removed | 136 |
Excluded during initial screening (e.g. press releases; news reports; not relevant drug/indication; preclinical study; reviews; case reports; not randomized trial) | 135 |
Excluded during writing (e.g. reviews; duplicate data; small patient number; nonrandomized/phase I/II trials) | 16 |
Cited efficacy/tolerability articles | 32 |
Cited articles not efficacy/tolerability | 24 |
Search Strategy: EMBASE, MEDLINE and PubMed from 2016 to present. Previous Adis Drug Evaluation published in 2016 was hand-searched for relevant data. Clinical trial registries/databases and websites were also searched for relevant data. Key words were secukinumab, Cosentyx, AIN-457, ankylosing spondylitis, spondyloarthritis. Records were limited to those in English language. Searches last updated 11 February 2019 | |
Change history
11 March 2019
The article Secukinumab: A Review in Ankylosing Spondylitis, written by Hannah A. Blair, was originally published Online First without open access. After online publication Novartis Pharma AG requested that the article be Open Choice to make the article an open access publication.
References
Mahmood F, Helliwell P. Ankylosing spondylitis: a review. Eur Med J. 2017;2(4):134–9.
Braun J, Sieper J. Ankylosing spondylitis. Lancet. 2007;369(9570):1379–90.
van der Heijde D, Ramiro S, Landewe R, et al. 2016 update of the ASAS-EULAR management recommendations for axial spondyloarthritis. Ann Rheum Dis. 2017;76(6):978–91.
Ward MM, Deodhar A, Akl EA, et al. American College of Rheumatology/Spondylitis Association of America/Spondyloarthritis Research and Treatment Network 2015 recommendations for the treatment of ankylosing spondylitis and nonradiographic axial spondyloarthritis. Arthritis Rheumatol. 2016;68(2):282–98.
Antonelli M, Khan MA, Magrey MN. Differential adverse events between TNF-α inhibitors and IL-17 axis inhibitors for the treatment of spondyloarthritis. Curr Treatm Opt Rheumatol. 2015;1(2):239–54.
Kuwabara T, Ishikawa F, Kondo M, et al. The role of IL-17 and related cytokines in inflammatory autoimmune diseases. Mediators Inflamm. 2017. https://doi.org/10.1155/2017/3908061.
Chyuan IT, Chen JY. Role of interleukin- (IL-) 17 in the pathogenesis and targeted therapies in spondyloarthropathies. Mediators Inflamm. 2018. https://doi.org/10.1155/2018/2403935.
Blair HA, Dhillon S. Secukinumab: a review in ankylosing spondylitis. Drugs. 2016;76(10):1023–30.
European Medicines Agency. Cosentyx: EU summary of product characteristics. 2018. http://www.ema.europa.eu. Accessed 11 Feb 2019.
Novartis. Cosentyx® (secukinumab): US prescribing information. 2018. http://www.pharma.us.novartis.com. Accessed 11 Feb 2019.
Garnock-Jones KP. Secukinumab: a review in moderate to severe plaque psoriasis. Am J Clin Dermatol. 2015;16(4):323–30.
Shirley M, Scott LJ. Secukinumab: a review in psoriatic arthritis. Drugs. 2016;76(11):1135–45.
Baeten D, Sieper J, Braun J, et al. Secukinumab, an interleukin-17A inhibitor, in ankylosing spondylitis. N Engl J Med. 2015;373(26):2534–48.
Pavelka K, Kivitz A, Dokoupilova E, et al. Efficacy, safety, and tolerability of secukinumab in patients with active ankylosing spondylitis: a randomized, double-blind phase 3 study, MEASURE 3. Arthritis Res Ther. 2017;19(285):1–10.
Kivitz AJ, Wagner U, Dokoupilova E, et al. Efficacy and safety of secukinumab 150 mg with and without loading regimen in ankylosing spondylitis: 104-week results from MEASURE 4 study. Rheumatol Ther. 2018;5(2):447–62.
Kishimoto M, Taniguchi A, Fujishige A, et al. Efficacy and safety of secukinumab in Japanese patients with active ankylosing spondylitis: 24-week results from an open-label phase 3 study (MEASURE 2-J). Mod Rheumatol. 2018. https://doi.org/10.1080/14397595.2018.1538004.
Pavelka K, Kivitz AJ, Dokoupilova E, et al. Secukinumab provides sustained improvements in the signs and symptoms of active ankylosing spondylitis: 2-year results from a phase 3 study [abstract no. 1828]. In: American College of Rheumatology/Association of Rheumatology Health Professionals Annual Scientific Meeting. 2017.
Baraliakos X, Braun J, Deodhar A, et al. Long-term evaluation of secukinumab in ankylosing spondylitis: 5 year efficacy and safety results from a phase 3 trial [abstract no. L13]. In: American College of Rheumatology/Association of Rheumatology Health Professionals Annual Scientific Meeting. 2018.
Marzo-Ortega H, Sieper J, Kivitz A, et al. Secukinumab 150 mg provides sustained improvements in the signs and symptoms of active ankylosing spondylitis with high retention rate: 4-year results from the phase III trial, MEASURE 2 [abstract no. SAT0283]. Ann Rheum Dis. 2018;77 (Suppl 2):1005.
Kivitz A, Pavelka K, Dokoupilova E, et al. Sustained improvements in signs and symptoms of active ankylosing spondylitis and reassuring safety with secukinumab 300 mg: 3-year results from a phase 3 study [abstract no. 1869]. In: American College of Rheumatology/Association of Rheumatology Health Professionals Annual Scientific Meeting. 2018.
Deodhar A, Conaghan PG, Strand V, et al. Secukinumab provides sustained improvements in work productivity and health related quality of life in patients with ankylosing spondylitis: long-term results from MEASURE 1 and MEASURE 2 [abstract no. AB0685]. Ann Rheum Dis. 2017;76(Suppl 2):1293.
Baraliakos X, Braun J, Sieper J, et al. Secukinumab reduces sacroiliac joint and spinal inflammation in patients with ankylosing spondylitis: MRI data from a phase 3 randomized, double-blind, placebo-controlled study (MEASURE 1) [abstract no. THU0233]. Ann Rheum Dis. 2015;74:281.
Baeten D, Blanco R, Geusens P, et al. Secukinumab provides sustained improvements in the signs and symptoms of active ankylosing spondylitis in anti-TNF-naive patients and those previously exposed to anti-TNF therapy: 52-week results from two randomized, double-blind, placebo-controlled phase 3 trials [abstract no. 2890]. Arthritis Rheumatol. 2015;67(Suppl 10):3482–4.
Marzo-Ortega H, Kvien TK, Deodhar AA, et al. Secukinumab provides sustained reduction in fatigue in patients with ankylosing spondylitis through three years: long-term results of two randomised double-blind placebo-controlled phase III studies [abstract no. 176]. Rheumatology (Oxford). 2018;57(Suppl 3):iii112–iii113.
Magrey M, Bozyczko M, Wolin D, et al. Treatment experience and satisfaction in ankylosing spondylitis patients treated with secukinumab: results from a US web-based survey [abstract no. SAT0300]. Ann Rheum Dis. 2018;77(Suppl 2):1014.
Martinis F, Caimmi C, Carletto A. Real-world efficacy and safety of secukinumab: data from Verona’s cohort [abstract no. AB0833]. Ann Rheum Dis. 2018;77 (Suppl 2):1544–5.
Robertson J, Tindell A, Crosbie D, et al. Real-world experience of secukinumab for psoriatic arthritis and axial spondyloarthritis [abstract no. 190]. Rheumatology (United Kingdom). 2018;57(Suppl 3):iii120.
Sieper J, Deodhar A, Marzo-Ortega H, et al. Secukinumab efficacy in anti-TNF-naive and anti-TNF-experienced subjects with active ankylosing spondylitis: results from the MEASURE 2 study. Ann Rheum Dis. 2017;76(3):571–92.
Wei JC, Baeten D, Sieper J, et al. Efficacy and safety of secukinumab in Asian patients with active ankylosing spondylitis: 52-week pooled results from two phase 3 studies. Int J Rheum Dis. 2017;20(5):589–96.
Braun J, Deodhar A, Landewe R, et al. Impact of baseline C-reactive protein levels on the response to secukinumab in ankylosing spondylitis: 3-year pooled data from two phase III studies. RMD Open. 2018;4(2):e000749.
Braun J, Baraliakos X, Deodhar A, et al. Effect of secukinumab on clinical and radiographic outcomes in ankylosing spondylitis: 2-year results from the randomised phase III MEASURE 1 study. Ann Rheum Dis. 2017;76(6):1070–7.
Marzo-Ortega H, Sieper J, Kivitz A, et al. Secukinumab and sustained improvement in signs and symptoms of patients with active ankylosing spondylitis through two years: results from a phase III study. Arthritis Care Res. 2017;69(7):1020–9.
Baraliakos X, Kivitz AJ, Deodhar AA, et al. Long-term effects of interleukin-17A inhibition with secukinumab in active ankylosing spondylitis: 3-year efficacy and safety results from an extension of the phase 3 MEASURE 1 trial. Clin Exp Rheumatol. 2018;36(1):50–5.
Braun J, Baraliakos X, Deodhar A, et al. Secukinumab shows sustained efficacy and low structural progression in ankylosing spondylitis: 4-year results from the MEASURE 1 study. Rheumatology (Oxford). 2018. https://doi.org/10.1093/rheumatology/key375.
Marzo-Ortega H, Sieper J, Kivitz A, et al. Secukinumab provides sustained improvements in the signs and symptoms of active ankylosing spondylitis with high retention rate: 3-year results from the phase III trial, MEASURE 2. RMD Open. 2017;3(2):e000592.
Deodhar AA, Dougados M, Baeten DL, et al. Effect of secukinumab on patient-reported outcomes in patients with active ankylosing spondylitis: a phase III randomized trial (MEASURE 1). Arthritis Rheumatol. 2016;68(12):2901–10.
Gentileschi S, Vitale A, Rigante D, et al. Prompt clinical response to secukinumab in patients with axial spondyloarthritis: real life observational data from three Italian referral centers. Isr Med Assoc J. 2018;20(7):438–41.
Deodhar AA, Baraliakos X, Marzo-Ortega H, et al. Secukinumab demonstrates consistent safety over long-term exposure (up to 3 years) in patients with active ankylosing spondylitis: pooled analysis of three phase 3 trials [abstract no. 1529]. Arthritis Rheumatol. 2017;69(Suppl 10).
Schreiber S, Colombel JF, Feagan BG, et al. Incidence rates of inflammatory bowel disease in patients with psoriasis, psoriatic arthritis and ankylosing spondylitis treated with secukinumab: a retrospective analysis of pooled data from 21 clinical trials. Ann Rheum Dis. 2019. https://doi.org/10.1136/annrheumdis-2018-214273.
Deodhar A, Miceli-Richard C, Baraliakos X, et al. Low incidence of both new-onset and flares of uveitis in secukinumab-treated patients with ankylosing spondylitis: clinical trial and post-marketing safety analysis [abstract no. SAT0270]. Ann Rheum Dis. 2018;77(Suppl 2):999.
Deodhar A, Gladman D, McInnes I, et al. Secukinumab, a fully human anti-interleukin-17A monoclonal antibody, exhibits low immunogenicity in patients with psoriatic arthritis and ankylosing spondylitis during a 52-week treatment period [abstract no. SAT0271]. Ann Rheum Dis. 2018;77(Suppl 2):999–1000.
Deodhar AA, Gladman DD, McInnes I, et al. Post-marketing safety study of secukinumab in adult patients with psoriasis, psoriatic arthritis and ankylosing spondylitis: cumulative analysis across > 96,000 patient-treatment years exposure [abstract no. 2582]. In: American College of Rheumatology/Association of Rheumatology Health Professionals Annual Scientific Meeting. 2018.
2018 ACR/SAA/SPARTAN updated recommendations for the management of axial spondyloarthritis. 2017. http://www.rheumatology.org/Portals/0/Files/Axial-SpA-Guideline-Project-Plan-2017.pdf. Accessed 11 Feb 2019.
National Institute for Health and Care Excellence. Secukinumab for ankylosing spondylitis after treatment with non-steroidal anti-inflammatory drugs or TNF-alpha inhibitors. 2016. http://www.nice.org.uk. Accessed 11 Feb 2019.
Maksymowych WP, Strand V, Nash P, et al. Comparative effectiveness of secukinumab and adalimumab in ankylosing spondylitis as assessed by matching-adjusted indirect comparison. Eur J Rheumatol. 2018;5(4):216–23.
Tahir H, Maksymowych W, Choy E, et al. Comparative effectiveness of secukinumab and golimumab in ankylosing spondylitis assessed by matching-adjusted indirect comparison using pivotal phase III clinical trial data [abstract no. 173]. Rheumatology (United Kingdom). 2018;57(Suppl 3):iii110-iii111.
Baraliakos X, Ostergaard M, Gensler LS, et al. Comparison of secukinumab and adalimumab biosimilar on radiographic progression in ankylosing spondylitis patients: design of a randomized, phase-IIIb study (SURPASS) [abstract no. SU147]. Int J Rheum Dis. 2018;21(Suppl 1):121–2.
Hamilton-West K. Managing the impact of ankylosing spondylitis on the patient and society. Int J Clin Rheumatol. 2010;5(5):537–46.
Colombo GL, Di Matteo S, Martinotti C, et al. Budget impact model of secukinumab for the treatment of moderate-to-severe psoriasis, psoriatic arthritis, and ankylosing spondylitis in Italy: a cross-indication initiative. Clinicoecon Outcomes Res. 2018;10:477–91.
Emery P, Van Keep M, Beard S, et al. Cost effectiveness of secukinumab for the treatment of active ankylosing spondylitis in the UK. Pharmacoeconomics. 2018;36(8):1015–27.
Purmonen T, Tormalehto S, Wahlman H, et al. Budget impact analysis of secukinumab versus adalimumab in the treatment of ankylosing spondylitis. J Med Econ. 2018;22(2):151–7.
Kalogeropoulou M, Kushwaha S, Jain M, et al. A cross indication budget impact analysis of secukinumab: a Greek perspective [abstract no. PMU28]. Value Health. 2018;21:S312–3.
Tozato C, Huerta C, Suzuki C, et al. Budget impact of introducing secukinumab in the treatment of ankylosing spondylitis from the private payer perspective in Brazil [abstract no. PMS20]. Value Health. 2018;21:S291.
Laires PA, Esparteiro J, Fonseca A, et al. Secukinumab versus adalimumab for the treatment of ankylosing spondylitis: a 52-week cost per responder analysis from the Portuguese payer perspective [abstract no. PMS42]. Value Health. 2018;21:S294–5.
Yahiaoui S, Ramakrishna G, Toumi J, et al. Secukinumab versus adalimumab in the treatment of ankylosing spondylitis: a cost per responder analysis among 52 weeks from a Tunisian perspective [abstract no. PMS29]. Value Health. 2018;21:S292.
Kim D, Kim H, Cho S, et al. Secukinumab as a more recent efficient alternative for ankylosing spondylitis treatment: a cost per responder analysis from Korean perspective [abstract no. PMS31]. Value Health. 2018;21:S292–3.
Acknowledgements
During the peer review process, the manufacturer of secukinumab was also offered an opportunity to review this article. Changes resulting from comments received were made on the basis of scientific and editorial merit.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
The preparation of this review was not supported by any external funding.
Conflict of interest
Hannah Blair is a salaried employee of Adis/Springer, is responsible for the article content and declares no relevant conflicts of interest.
Additional information
The manuscript was reviewed by: K. Pavelka, Institute of Rheumatology, Prague, Czech Republic; M. J. Puszczewicz, Department of Rheumatology and Internal Medicine, Poznan University of Medical Sciences, Poznan, Poland; D. Wendling, Department of Rheumatology, University Teaching Hospital, CHRU de Besancon, Besancon, France.
The original version of this article was revised due to a retrospective Open Access request.
Rights and permissions
OpenAccess This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Blair, H.A. Secukinumab: A Review in Ankylosing Spondylitis. Drugs 79, 433–443 (2019). https://doi.org/10.1007/s40265-019-01075-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-019-01075-3