
Abstract
Introduction
OnabotulinumtoxinA is approved in the USA for the prevention of headache in adults with chronic migraine, a debilitating neurologic disease characterized by headaches occurring on ≥ 15 days per month for > 3 months and including migraine features on ≥ 8 days per month.
Objective
The COMPEL Study (NCT01516892), a 108-week, multi-center, open-label study, evaluated the long-term efficacy and safety of onabotulinumtoxinA in adults with chronic migraine. The objective of this subanalysis was to examine the safety and tolerability of onabotulinumtoxinA after each of nine treatment cycles.
Methods
OnabotulinumtoxinA 155 U was administered every 12 weeks. Safety and tolerability, overall and by treatment cycle, were assessed. Treatment-emergent adverse events reported between successive treatments were attributed to the preceding treatment. The safety population received one or more doses of onabotulinumtoxinA. The primary efficacy outcome was the reduction in headache days at week 108 compared with baseline.
Results
Of 716 patients enrolled, 373 patients (52.1%) completed the study and 343 (47.9%) withdrew; 481 patients (67.2%) received 60 weeks of treatment and 402 (56.1%) received 108 weeks of treatment. In total, 436 (60.9%) patients reported treatment-emergent adverse events; most were mild/moderate in severity. Thirty-two patients (4.5%) discontinued the study after experiencing treatment-emergent adverse events. The incidence of treatment-emergent adverse events typically decreased with repeated onabotulinumtoxinA treatment: first cycle, 24.2%; fourth cycle, 18.4%; ninth cycle, 12.2%. Neck pain (2.7%), eyelid ptosis (1.8%), musculoskeletal stiffness (1.4%), injection-site pain (1.3%), and headache (1.3%) were the most common treatment-emergent adverse events after the first cycle. Seventy-five patients (10.5%) reported serious treatment-emergent adverse events, 13 (1.8%) withdrew. Treatment-related adverse events were reported by 131 patients (18.3%), one was considered serious. OnabotulinumtoxinA significantly reduced headache day frequency by 10.7 (6.4) days per 28-day period (p < 0.0001) at week 108.
Conclusions
OnabotulinumtoxinA treatment was well tolerated over 108 weeks; no new safety signals were identified. The overall incidence of treatment-emergent adverse events and the most common individual events decreased with repeated onabotulinumtoxinA administration.
Clinical Trial Registration
ClinicalTrials.gov; NCT01516892.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Over 2 years of treatment, onabotulinumtoxinA was well tolerated and no new safety signals were identified. |
Cumulative tolerability issues with long-term onabotulinumtoxinA exposure do not occur with recommended treatment regimens. |
1 Introduction
Chronic migraine (CM) is a debilitating neurologic disease defined as headaches that occur on ≥ 15 days per month for > 3 months and have migraine features on ≥ 8 days per month [1]. Chronic migraine affects approximately 1.4–2.2% of adults worldwide [2, 3] and is associated with a substantial quality-of-life and economic burden [4,5,6,7,8,9]. Despite these adverse consequences, large epidemiologic studies have shown that many people with CM do not receive adequate treatment [10], with only approximately one-third of those with CM taking preventive treatments [11]. In a retrospective review of a US claims database including 75,870 patients with CM, of 8688 individuals prescribed an oral preventive treatment, only approximately 29% continued treatment after 6 months, with adherence rates dropping to 20% after 12 months [12]. Although reasons for not persisting with oral preventive treatments were not captured in the claims database, tolerability has been previously reported as one of the primary reasons for discontinuing oral preventive treatment [13].
OnabotulinumtoxinA offers a focal treatment for the management of multiple diseases. Its efficacy and safety have been established in more than 80 randomized placebo-controlled clinical trials over the past 30 years. It is currently approved in the USA for nine therapeutic and three aesthetic indications, including the prevention of headache in adults with CM [14]. Although the underlying mechanism of action of onabotulinumtoxinA in the treatment of CM is not fully characterized, it is deemed to act by interrupting the nociceptive and inflammatory pathways that cause peripheral nociceptor and central sensitization. At the molecular level, onabotulinumtoxinA cleaves synaptosomal-associated protein (SNAP-25) and inhibits the fusion of intracellular vesicles with the membrane of the nerve cell, thereby modulating neuropeptide release and down-regulating other mechanisms important in nociception. OnabotulinumtoxinA blocks the release of inflammatory neuropeptides, such as substance P and calcitonin gene-related peptide, from stimulated trigeminal sensory neurons [15]. Accordingly, people with CM who respond to onabotulinumtoxinA treatment demonstrate high interictal blood levels of calcitonin gene-related peptide and pentraxin 3 [16].
The Phase III REsearch Evaluating Migraine Prophylaxis Therapy (PREEMPT) clinical trials established the safety and efficacy of onabotulinumtoxinA for the treatment of CM [17,18,19]. OnabotulinumtoxinA was reported to reduce the frequency of headache days and the frequency of moderate or severe headache days at the end of the 24-week double-blind treatment period compared with placebo [18], with a further reduction in the frequencies of headache days and moderate or severe headache days reported at the end of the 32-week open-label phase [19]. Across the entire 56-week study period, adverse events (AEs) were mild to moderate in severity, with neck pain and muscular weakness the most commonly occurring treatment-related AEs [19]. Interestingly, in the PREEMPT studies, the incidence of treatment-related neck pain was reported to decline with repeated onabotulinumtoxinA treatment cycles [19]. Two post-marketing real-world studies have assessed the efficacy and tolerability of onabotulinumtoxinA in Europe and the UK; one assessed treatment utilization patterns and safety of onabotulinumtoxinA for up to 52 weeks [20], and the other assessed healthcare resource utilization and patient-reported outcomes, including patient-reported tolerability, over 24 months [21]. These real-world studies permitted more flexible dosing regimens than the PREEMPT studies, based on local standards of care, but similarly reported eyelid ptosis, neck pain, and musculoskeletal stiffness as the most frequently occurring treatment-related AEs, and an overall acceptable safety profile.
The Chronic migraine OnabotulinuMtoxinA Prolonged Efficacy open-Label (COMPEL) Study (NCT01516892) was designed to complement the current efficacy and safety data for onabotulinumtoxinA and evaluate the longer term efficacy and safety of onabotulinumtoxinA 155 U administered in a fixed-dose, fixed-site treatment paradigm for the prevention of headache in patients with CM in study centers in the USA and Asia/Pacific regions. Dosing and results reported in this study are specific to onabotulinumtoxinA. This formulation is not interchangeable with other botulinum toxin products, and units cannot be converted using a dose ratio. Therefore, the results of this study cannot be extrapolated to other formulations of botulinum toxins.
The primary objective of the COMPEL Study was to assess the change from baseline in the frequency of headache days per 28-day period at 108 weeks (i.e., after nine onabotulinumtoxinA treatment cycles) [22, 23]. The objective of this sub-analysis of the COMPEL Study was to evaluate the long-term safety and tolerability of onabotulinumtoxinA for the treatment of CM in adult patients over 108 weeks (nine treatment cycles). The safety and tolerability of onabotulinumtoxinA were also evaluated in the subset of patients with CM receiving concomitant oral preventive treatment. In addition, treatment-emergent AEs (TEAEs) were evaluated by treatment cycle to assess whether the incidence of TEAEs was consistent across multiple treatments.
2 Methods
2.1 Study Design
The COMPEL Study was a multicenter open-label postmarketing study in adult patients with CM at sites in the USA, Australia, and South Korea. Full study methodology has been previously published [22] and will be summarized briefly here for context. The duration of the COMPEL Study was 112 weeks, comprising a 4-week baseline period and a 108-week open-label treatment intervention phase. Data were collected at the screening and baseline visits and at follow-up visits every 3 months. Patients kept a record of headache frequency and duration, frequency of days with moderate or severe headache, and acute treatment use in a daily diary completed via an interactive voice response system during the 28-day period before visits 4, 7, 9, and 11 (after treatments 2, 5, 7, and 9, respectively).
OnabotulinumtoxinA (Botox®; Allergan plc, Dublin, Ireland) 155 U was administered every 12 weeks using a fixed-site, fixed-dose injection paradigm into 31 sites in seven muscle areas (5 U injected intramuscularly per site), as approved by the US Food and Drug Administration (FDA) [14]. Physician injectors were trained using insights gained from the PREEMPT studies and published recently [24].
Patients aged ≥ 18 years with a diagnosis of CM who had not previously received onabotulinumtoxinA were eligible for inclusion in the study. In addition to the study treatment, patients could take a single oral preventive treatment (defined as oral medication specifically prescribed for daily use for headache prophylaxis) concomitantly if the dose and regimen were stable for the 4 weeks before the first study treatment and remained unchanged until or after week 24. Acute headache treatment was permitted on an as-needed basis; patients were required to record the use of any acute headache treatment in their daily patient diary.
2.2 Assessment of Safety and Tolerability
Safety was evaluated by collecting data on the incidence and nature of TEAEs, including serious AEs (SAEs). Patients were screened for TEAEs at each follow-up visit through patient self-report, general non-directed questioning, and direct questioning via the Columbia-Suicide Severity Rating Scale [25] and the 9-item Patient Health Questionnaire (PHQ-9) [26]. The following definitions were applied to any TEAE detected: an AE was defined as any TEAE identified after the first treatment cycle through study completion (week 108), coded using nomenclature from the Medical Dictionary for Regulatory Activities®, Version 15.0 or later; a treatment-related AE was defined per protocol as any TEAE that was reasonably considered to be related to study treatment by the investigators; an SAE was defined as any TEAE occurring at any dose that resulted in death, a life-threatening AE, inpatient hospitalization or prolongation of an existing hospitalization, a persistent or significant disability/incapacity, a congenital anomaly/birth defect, any abortion (spontaneous or non-spontaneous), or any form of cancer [27].
In addition to screening for TEAEs at each visit, physical examinations were performed at screening, at week 48, and at the week 108 exit visit. Patients were withdrawn from the study for safety reasons and referred for follow-up medical care if they showed any signs of suicidal ideation or if they became pregnant. Suicidal ideation was identified through the PHQ-9 and the Columbia-Suicide Severity Rating Scale. If the patient had any change from baseline in their response to item 9 (“Thoughts that you would be better off dead, or hurting yourself in some way”) of the PHQ-9 or if they answered “yes” to questions 4 (active suicidal ideation with some intent to act but no specific plan) or 5 (active suicidal ideation with intent and a specific plan) on the Columbia-Suicide Severity Rating Scale they were deemed to have suicidal ideation. If a patient became pregnant, she was withdrawn from the study and followed up for 12 weeks after the last study treatment and for ≥ 4 weeks after the pregnancy was reported. These patients were further followed up via the Allergan Global Safety Database team. When a pregnancy was reported, Allergan made a minimum of three attempts to gather information on the pregnancy exposure and to obtain follow-up information on the pregnancy outcome [28]. With the patient’s consent, the patient’s physician was also contacted to obtain further information.
2.3 Efficacy Outcome Measure
The primary efficacy outcome measure was the mean change from baseline in the frequency of headache days per 28-day period at 108 weeks (after nine treatment cycles). A range of secondary efficacy outcome measures were also assessed, including the mean change in the frequency of headache days at week 60 (after five treatments) and the mean change from baseline in the 6-item Headache Impact Test scores over the 4-week periods preceding week 60 and week 108 [23].
3 Results
3.1 Patient Disposition and Demographics
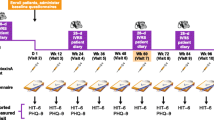
The safety population included all 716 enrolled patients who received one or more doses of onabotulinumtoxinA. The baseline demographics and disease characteristics of the population have previously been presented [23] and are briefly summarized here. Patients had a mean (SD) age of 43.0 (11.3) years and were typically female [n = 607 (84.8%)] and Caucasian [n = 582 (81.3%)]. The most common preventive headache medications used historically before study enrollment were anticonvulsants [n = 434 (60.6%)], antidepressants [n = 323 (45.1%)], and β-adrenergic antagonists [n = 211 (29.5%)]. A total of 343 patients (47.9%) discontinued treatment during the study. The primary reasons for discontinuation were withdrawal of consent [n = 92 (12.8%)], being lost to follow-up [n = 82 (11.5%)], or protocol violation [n = 60 (8.4%); Fig. 1] [23]. Treatment-emergent AEs were the primary reason for discontinuation for 25 patients (3.5%), and six patients withdrew from the study because of pregnancy. Of the 716 patients enrolled in the study, 34 (4.7%) had a significant protocol deviation where their treatment window exceeded ± 2 weeks within the allotted treatment visit schedule.

Patient disposition over the course of the study. Reproduced without modification from Blumenfeld et al. Long-term study of the efficacy and safety of OnabotulinumtoxinA for the prevention of chronic migraine: COMPEL study. J Headache Pain. 2018;19:13 [23] under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/)
3.2 Safety and Tolerability over the Duration of the Study
OnabotulinumtoxinA was well tolerated throughout the study, with no new safety signals identified (Table 1). A total of 436 patients (60.9%) reported one or more TEAEs; onabotulinumtoxinA treatment was discontinued in 32 patients (4.5%) who had experienced a TEAE. Treatment-related AEs were reported in 131 (18.3%) patients. The most frequently reported treatment-related AEs were neck pain [n = 29 (4.1%)], eyelid ptosis [n = 18 (2.5%)], musculoskeletal stiffness [n = 17 (2.4%)], and injection-site pain [n = 14 (2.0%); Table 1].
Serious AEs were reported in 75 patients (10.5%); migraine [n = 6 (0.8%)], suicidal ideation [n = 5 (0.7%)], headache, malignant melanoma, and non-cardiac chest pain [each n = 3 (0.4%)] were the most frequently reported (Table 2 and Electronic Supplementary Material [ESM] 1). The only SAE considered by the investigator to be treatment related was a generalized rash in one patient. The majority of SAEs resolved without sequelae. Treatment was discontinued in 13 patients (1.8%) who had an SAE, including the only SAE that was considered by the investigator to be treatment related (moderate generalized rash after the third treatment). Other SAEs reported in patients who discontinued the study included congestive cardiac failure and congestive cardiomyopathy (n = 1, occurred 26 days after the first treatment); malignant pulmonary neoplasm (n = 1, after the sixth treatment); bipolar disorder (n = 1, occurring after the ninth treatment); sinusitis, pyrexia, and headache (n = 1, after the seventh treatment); mantle cell lymphoma (n = 1, after the third treatment); breast cancer (n = 1, after the fifth treatment); suicidal ideation (n = 4, first reported after the first, second, fourth, and eighth treatments, respectively); Cushing syndrome (n = 1, after the second treatment); and depression (n = 1, after the sixth treatment). There were no deaths during the study.
No AEs that were identified were assessed as being due to the potential distant spread of toxins. The majority of findings on physical examinations were normal for all body systems. There were no clinically significant changes in vital signs during the study. Seven patients reported suicidal ideation during the study; all events were considered to be unrelated to onabotulinumtoxinA. Of these seven patients, all had medical histories that included depression (n = 5), or suicidal ideation (n = 1) or suicidal behavior (n = 1), and there were no completed suicides.
3.3 Safety and Tolerability by Treatment Cycle
The incidence of TEAEs typically decreased with repeated onabotulinumtoxinA treatment cycles, from 24.2% after the first treatment cycle to 16.3% after the fifth treatment cycle and 12.2% after the ninth (last) treatment cycle (Fig. 2a). Similarly, the incidence of most individual TEAEs generally decreased with repeated onabotulinumtoxinA treatment (Table 3). For example, neck pain decreased from 2.7% after the first treatment to 0.2% after the last treatment. Correspondingly, eyelid ptosis decreased from 1.8 to 0.0%, musculoskeletal stiffness decreased from 1.4 to 0.2%, headache decreased from 1.3 to 0.5%, and injection-site pain decreased from 1.3 to 0.0%. The percentage of patients withdrawing from the study and citing a TEAE as the reason also decreased with repeated onabotulinumtoxinA treatment (Fig. 2b).

Percentage of patients at each treatment cycle (a) with treatment-emergent adverse events (TEAEs) and (b) withdrawing from the study citing adverse events as the reason
At the system organ class level, TEAEs generally decreased with repeated treatment (Table 4). However, the incidence of specific TEAEs related to nervous system disorders (Fig. 3a), musculoskeletal and connective tissue disorders (Fig. 3b), and infections (Fig. 3c) varied throughout the treatment cycles, showing no clear trend to decrease over repeated treatments. Treatment-emergent AEs related to other system organ classes showed a clearer reduction in incidence with repeated treatment (Fig. 3d).

Percentage of patients with treatment-emergent adverse events (TEAEs) by treatment cycle related to (a) nervous system disorders, (b) musculoskeletal and connective disorders, (c) infections, and (d) other system organ classes. Classes of adverse events based on Medical Dictionary for Regulatory Activities®, Version 15.1 terminology
3.4 Safety and Tolerability in Patients Receiving Oral Preventive Treatment
A total of 89 patients (12.4%) received one or more stable oral preventive treatments during the study. This included 44 patients (6.1%) who had oral preventive treatments added to their treatment regimen on or after week 24; of these, the most commonly used were topiramate [n = 18 (20.2%)], amitriptyline [n = 6 (6.7%)], valproate [n = 4 (4.5%)], gabapentin [n = 3 (3.4%)], non-selective β-adrenergic antagonists [n = 3 (3.4%)], selective β-adrenergic antagonists [n = 2 (2.2%)], zonisamide [n = 2 (2.2%)], fiorinal-C [n = 1 (1.1%)], lamotrigine [n = 1 (1.1%)], and lisinopril [n = 1 (1.1%)]. Baseline demographics of those receiving concomitant oral preventive treatment vs. those not receiving any oral preventive treatment were similar, except those receiving oral preventive treatments were more likely to be male (21.3% vs. 14.4%), less likely to have a family history of migraine (57.3% vs. 63.5%), and more likely to have been taking oral preventive treatment at baseline (89.9% vs. 79.6%) compared with those who did not receive oral preventive treatment (ESM 2).
Treatment-emergent AEs (71.9% vs. 59.3%) and treatment-related AEs (25.8% vs. 17.2%) were more common among patients receiving vs. not receiving oral preventive treatment. However, TEAEs led to study discontinuation in a lower percentage of patients taking oral preventive treatment (1.1% vs. 4.9%). The incidence of SAEs was similar in both subgroups (9.0% vs. 10.7%, respectively), and no patient receiving oral preventive treatment presented with a treatment-related SAE. The nature of treatment-related AEs was different among patients receiving vs. not receiving oral preventive treatment, with a lower incidence of neck pain (1.1% vs. 4.5%) and a higher incidence of musculoskeletal stiffness (6.7% vs. 1.8%) and injection-site pain (4.5% vs. 1.6%; Table 5). The incidence of TEAEs reduced from 23.6% after treatment cycle 1 to 14.3% after treatment cycle 9 in patients receiving oral preventive treatment; the most common TEAEs associated with onabotulinumtoxinA also tended to decrease over repeated treatment cycles (ESM 3).
3.5 Pregnancy
Six women became pregnant after enrollment. Once their pregnancy was confirmed, the women received no further onabotulinumtoxinA treatment and were followed up for 12 weeks before being withdrawn from the study; follow-up information was captured in the Allergan Global Safety Database. One patient, who was reported to have become pregnant on the same day as the last onabotulinumtoxinA treatment, gave birth to a normal healthy female by caesarean section. One pregnancy, reported 5 days after the first study treatment, resulted in a normal healthy baby at term without complications. One pregnancy in a patient who had received a single treatment with onabotulinumtoxinA ended as a spontaneous abortion > 12 weeks after the treatment with onabotulinumtoxinA; this event occurred outside of the study period (i.e., > 12 weeks after the last study treatment and > 4 weeks after the pregnancy was reported) and was considered to be unrelated to onabotulinumtoxinA. The three remaining cases were lost to follow-up, and pregnancy outcomes are unknown.
3.6 Efficacy Outcomes
The efficacy results of the COMPEL Study have been reported in full elsewhere [23] and will be briefly summarized here. OnabotulinumtoxinA treatment achieved statistically significant changes from baseline for all of the primary and secondary efficacy endpoints. At baseline, patients reported a mean (SD) of 22.0 (4.8) headache days per 28-day period. OnabotulinumtoxinA significantly reduced the number of headache days per 28-day period at all time points (p < 0.0001), including week 60 (secondary endpoint; mean [SD] headache frequency reduced to 12.7 [7.6] days) and week 108 (primary endpoint; mean [SD] headache frequency reduced to 11.3 [7.4] days). OnabotulinumtoxinA also significantly reduced 6-item Headache Impact Test total scores at all time points (p < 0.0001) from a mean (SD) total score of 64.7 (4.8) at baseline, including at week 60 [reduced to 58.0 (7.0)] and week 108 [reduced to 57.7 (7.8)]. The frequency of moderate-to-severe headache days was also reduced from a mean (SD) at baseline of 18.0 (5.7) days per 28-day period to 8.5 (6.4) days at week 108.
4 Discussion
This analysis of the COMPEL Study provides additional data on the safety and tolerability of onabotulinumtoxinA to complement data previously reported [23]. Treatment-emergent AEs were evaluated by treatment cycle, demonstrating that the incidence of TEAEs diminished with repeated administration of onabotulinumtoxinA, as did the percentage of patients withdrawing from the study after experiencing an AE. Data are also presented on the incidence and nature of TEAEs in patients receiving concomitant oral preventive treatment, demonstrating that onabotulinumtoxinA remains well tolerated in this subgroup. Data on the outcomes of pregnancy in patients receiving onabotulinumtoxinA treatment are also reported. Consistent with the findings of a 24-year retrospective review of women exposed to onabotulinumtoxinA before or during pregnancy [28], no new safety concerns were identified. Three patients were lost to follow-up, despite attempts to obtain detailed information regarding pregnancy outcomes.
The incidence of TEAEs in this study (60.9%) was similar to the incidence reported in the PREEMPT study (62.4%) [19] and higher than that reported in a real-world European Observational Study (41.2%) [20]. The incidence of treatment-related AEs (18.3%) was lower than that reported in the PREEMPT study (29.4%) and lower than or similar to that reported in real-world observational studies (European Observational Study, 25.1% [20]; REPOSE Study, 18.3% [21]), further supporting the long-term safety and tolerability of onabotulinumtoxinA. The injection experience gained from the PREEMPT study and real-world practice with regard to proper identification of injection sites was shared with the injectors in the current study and may have contributed to the lower incidence of AEs [24]. Neck pain (4.6%), muscular weakness (3.9%), eyelid ptosis (2.5%), muscle tightness (2.2%), and injection-site pain (2.0%) were the most commonly reported treatment-related AEs over the 32-week open-label phase in PREEMPT [19], with an overall incidence rate similar to the most commonly reported treatment-related AEs in this study (neck pain, 4.1%; musculoskeletal stiffness, 2.4%; eyelid ptosis, 2.5%; and injection-site pain, 2.0%). Despite the more flexible injection paradigms in the real-world observational studies, the overall incidences of the most common treatment-related AEs were similar (European Observational Study: neck pain, 4.4%; eyelid ptosis, 4.1%; muscular weakness, 2.7%; headache, 2.2%; and musculoskeletal stiffness, 2.0% [20]; REPOSE: eyelid ptosis, 5.4%; neck pain, 3.0%; musculoskeletal stiffness, 2.7% [21]). Interestingly, eyelid ptosis was a more frequent treatment-related AE in REPOSE, potentially owing to the diverse mix of physician specialists and patients in this real-world observational study. Suicidal ideation was reported in approximately 1% of patients; however, this should be considered in the context of previous reports stating a strong association between migraine and suicidal ideation [29], with a lifetime incidence of suicidal ideation of 3.8% in elderly patients (aged > 65 years) with migraine or non-migraine headaches [30].
The incidence of overall TEAEs and many individual TEAEs decreased with repeated administration of onabotulinumtoxinA from 24.2% after the first treatment cycle to 16.3% after the fifth treatment cycle and 12.2% after the ninth treatment cycle. Similarly, in the real-world observational study in the UK and Europe, the percentage of patients reporting a TEAE declined from 26.8% after the first treatment cycle to 18.6% after the fifth treatment cycle [20]. Additionally, this is consistent with the TEAE profile reported in a separate long-term study of onabotulinumtoxinA in patients with CM and medication overuse reported by Negro et al [31], in which two of the most frequently reported events (i.e., neck pain and musculoskeletal weakness) were most prominent in the first three injection cycles. However, Guerzoni et al. [32] did not find a correlation between AE frequency and injection cycle. Taken together, these results would suggest that there is no evidence of cumulative tolerability issues with long-term onabotulinumtoxinA exposure and that injectors gain insights with experience that enables adverse effects to be mitigated to a degree over time.
By allowing the use of stable concomitant oral preventive treatments, the COMPEL Study data also provide reassurance of the safety and tolerability of onabotulinumtoxinA in clinical practice. There was no evidence in this study, in which 89 patients (12.4%) received oral preventive treatment during the study, that the concomitant use of oral preventive treatment had any detrimental effect on the overall safety profile of onabotulinumtoxinA. Given the tolerability issues previously reported with oral preventive treatment [13], it is not surprising that the incidence of TEAEs and treatment-related AEs was slightly higher among those receiving concomitant oral preventive treatment. However, the incidences of SAEs and the TEAEs leading to study discontinuation were similar in both groups, suggesting that onabotulinumtoxinA remained well tolerated in this subgroup of patients. This is consistent with the experience from the REPOSE study, in which 19.3% of patients receiving concomitant headache treatments at baseline and throughout the study experienced treatment-related AEs (Allergan plc, data on file).
OnabotulinumtoxinA has previously been classified as “category C” owing to the developmental toxicity observed in mice, rats, and rabbits when administered during organogenesis at doses equivalent to less than the maximum recommended human dose of 400 U (on a body weight basis) [14]. Further, because of the lack of adequate and well-controlled studies in pregnant women, onabotulinumtoxinA should only be used in pregnancy if the potential benefit justifies the potential risk to the fetus [14]. Allergan monitors and evaluates safety if exposure occurs during pregnancy. The numbers of pregnancy exposures were low, and pregnancy outcomes were only available for half of the pregnancies; therefore, the results of this study can only augment the current data for onabotulinumtoxinA exposure in women who became pregnant.
Pregnancy outcomes after exposure to onabotulinumtoxinA over 24 years (1990–2013) have been previously reported [28]. During this period, 574 pregnancies were reported after maternal exposure to onabotulinumtoxinA, and the outcomes of 232 (137 prospective cases) of those pregnancies were known. Approximately 50.5% of the patients exposed were receiving onabotulinumtoxinA for cosmetic purposes, and 14.2% for pain disorders, such as migraine, with 45.3% of known doses ≥ 100 U (26.3% of doses were ≥ 150 U). The prevalence rates for fetal loss (20.9% [95% confidence interval 14.0–30.0]) and defects (2.7% [95% confidence interval 0.6–8.0]), calculated based on prospective cases as recommended by the Food and Drug Administration [33], were lower than underlying population estimates for fetal loss (35.4%) [34] and similar to population estimates for birth defects (2.8%) [35]. Given the relatively low doses of onabotulinumtoxinA in this population, these results should not be extrapolated to the general use of onabotulinumtoxinA treatment during pregnancy [36]. A preliminary report [37] of pregnancy outcomes in 34 patients with CM at a single center who were exposed to onabotulinumtoxinA included 13 (38.2%) who discontinued treatment and 21 (65.6%) who wished to continue at 3-month treatment intervals. All pregnancies but one (miscarriage) resulted in normal vaginal delivery with live births and no fetal malformations. Overall, it remains important to continue to balance the benefits and risks of onabotulinumtoxinA treatment in women of childbearing age and to report and monitor pregnancies in those exposed to onabotulinumtoxinA.
This study has a number of strengths that make it an important contribution to the understanding of the safety and tolerability of onabotulinumtoxinA in clinical practice. A relatively large population was followed up for more than 2 years, with TEAE data collected systematically over nine treatment cycles. The evaluation of TEAE data over repeated treatments enables an increased understanding of the long-term safety and tolerability of onabotulinumtoxinA after repeated administration. Subgroup analysis of those receiving concomitant oral preventive treatment provides additional support for the safety and tolerability of onabotulinumtoxinA in clinical practice.
As a non-randomized open-label study, our study is subject to inherent limitations. Any open-label study with a long-term follow-up can be subject to unintentional bias, low persistency rates, and concomitant medication changes. The relatively low persistency rates observed in this study may understate the short-term safety and tolerability because patients who experienced TEAEs may not have persisted with treatment over the 108-week follow-up period, leading to a lower prevalence of TEAEs as the study progressed. Furthermore, the exclusion of patients with severe depression and suicidal ideation could have meant that safety and tolerability data from those patients who are potentially the most challenged by CM would not have been captured.
5 Conclusions
The results of this long-term non-randomized open-label study support the efficacy and safety of onabotulinumtoxinA for headache prevention in adults with CM for up to nine treatment cycles (108 weeks). OnabotulinumtoxinA was well tolerated over nine cycles of treatment, and no new safety concerns were identified. Importantly, the overall incidence of AEs considered to be related to treatment was low.
In this first study to assess AEs over nine treatment cycles, the overall incidence of TEAEs and rates of many individual TEAEs decreased with administration of onabotulinumtoxinA, suggesting that cumulative tolerability issues with long-term onabotulinumtoxinA exposure do not occur with recommended treatment regimens.
References
Headache Classification Committee of the International Headache Society (IHS). The international classification of headache disorders, 3rd edition. Cephalalgia. 2018;38:1–211.
Manack AN, Buse DC, Lipton RB. Chronic migraine: epidemiology and disease burden. Curr Pain Headache Rep. 2011;15:70–8.
Natoli JL, Manack A, Dean B, Butler Q, Turkel CC, Stovner L, et al. Global prevalence of chronic migraine: a systematic review. Cephalalgia. 2010;30:599–609.
Blumenfeld AM, Varon SF, Wilcox TK, Buse DC, Kawata AK, Manack A, et al. Disability, HRQoL and resource use among chronic and episodic migraineurs: results from the International Burden of Migraine Study (IBMS). Cephalalgia. 2011;31:301–15.
Buse DC, Manack AN, Serrano D, Reed ML, Varon S, Turkel CC, et al. Headache impact of chronic and episodic migraine: results from the American migraine prevalence and prevention study. Headache. 2012;52:3–17.
Buse DC, Manack A, Serrano D, Turkel C, Lipton RB. Sociodemographic and comorbidity profiles of chronic migraine and episodic migraine sufferers. J Neurol Neurosurg Psychiatry. 2010;81:428–32.
Chen YC, Tang CH, Ng K, Wang SJ. Comorbidity profiles of chronic migraine sufferers in a national database in Taiwan. J Headache Pain. 2012;13:311–9.
Lipton RB, Varon SF, Grosberg B, McAllister PJ, Freitag F, Aurora SK, et al. OnabotulinumtoxinA improves quality of life and reduces impact of chronic migraine. Neurology. 2011;77:1465–72.
Buse DC, Scher AI, Dodick DW, Reed ML, Fanning KM, Manack Adams A, et al. Impact of migraine on the family: perspectives of people with migraine and their spouse/domestic partner in the CaMEO Study. Mayo Clin Proc. 2016;91:596–611.
Dodick DW, Loder EW, Manack Adams A, Buse DC, Fanning KM, Reed ML, et al. Assessing barriers to chronic migraine consultation, diagnosis, and treatment: results from the Chronic Migraine Epidemiology and Outcomes (CaMEO) study. Headache. 2016;56:821–34.
Bigal ME, Serrano D, Reed M, Lipton RB. Chronic migraine in the population: burden, diagnosis, and satisfaction with treatment. Neurology. 2008;71:559–66.
Hepp Z, Dodick DW, Varon SF, Gillard P, Hansen RN, Devine EB. Adherence to oral migraine-preventive medications among patients with chronic migraine. Cephalalgia. 2015;35:478–88.
Hepp Z, Bloudek LM, Varon SF. Systematic review of migraine prophylaxis adherence and persistence. J Manag Care Pharm. 2014;20:22–33.
Botox [package insert]. Irvine: Allergan plc; 2018.
Aurora SK, Brin MF. Chronic migraine: an update on physiology, imaging, and the mechanism of action of two available pharmacologic therapies. Headache. 2017;57:109–25.
Dominguez C, Vieites-Prado A, Perez-Mato M, Sobrino T, Rodriguez-Osorio X, Lopez A, et al. CGRP and PTX3 as predictors of efficacy of onabotulinumtoxin type A in chronic migraine: an observational study. Headache. 2018;58:78–87.
Diener HC, Dodick DW, Aurora SK, Turkel CC, DeGryse RE, Lipton RB, et al. OnabotulinumtoxinA for treatment of chronic migraine: results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 2 trial. Cephalalgia. 2010;30:804–14.
Aurora SK, Dodick DW, Turkel CC, DeGryse RE, Silberstein SD, Lipton RB, et al. OnabotulinumtoxinA for treatment of chronic migraine: results from the double-blind, randomized, placebo-controlled phase of the PREEMPT 1 trial. Cephalalgia. 2010;30:793–803.
Aurora SK, Winner P, Freeman MC, Spierings EL, Heiring JO, DeGryse RE, et al. OnabotulinumtoxinA for treatment of chronic migraine: pooled analyses of the 56-week PREEMPT clinical program. Headache. 2011;51:1358–73.
Matharu M, Pascual J, Nilsson Remahl I, Straube A, Lum A, Davar G, et al. Utilization and safety of onabotulinumtoxinA for the prophylactic treatment of chronic migraine from an observational study in Europe. Cephalalgia. 2017;37:1384–97.
Ahmed F, Gaul C, Martelletti P, Garcia-Monco JC, Manack Adams A, editors. Real-life use of onabotulinumtoxinA for the symptomatic treatment of chronic migraine: the REPOSE study. International Headache Congress; 7–10 Sept 2017; Vancouver.
Blumenfeld AM, Aurora SK, Laranjo K, Papapetropoulos S. Unmet clinical needs in chronic migraine: rationale for study and design of COMPEL, an open-label, multicenter study of the long-term efficacy, safety, and tolerability of onabotulinumtoxinA for headache prophylaxis in adults with chronic migraine. BMC Neurol. 2015;15:100.
Blumenfeld AM, Stark RJ, Freeman MC, Orejudos A, Manack Adams A. Long-term study of the efficacy and safety of onabotulinumtoxinA for the prevention of chronic migraine: COMPEL study. J Headache Pain. 2018;19:13.
Blumenfeld AM, Silberstein SD, Dodick DW, Aurora SK, Brin MF, Binder WJ. Insights into the functional anatomy behind the PREEMPT injection paradigm: guidance on achieving optimal outcomes. Headache. 2017;57:766–77.
Posner K, Brown GK, Stanley B, Brent DA, Yershova KV, Oquendo MA, et al. The Columbia-Suicide Severity Rating Scale: initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry. 2011;168:1266–77.
Seo JG, Park SP. Validation of the Patient Health Questionnaire-9 (PHQ-9) and PHQ-2 in patients with migraine. J Headache Pain. 2015;16:65.
Brin MF, Boodhoo TI, Pogoda JM, James LM, Demos G, Terashima Y, et al. Safety and tolerability of onabotulinumtoxinA in the treatment of facial lines: a meta-analysis of individual patient data from global clinical registration studies in 1678 participants. J Am Acad Dermatol. 2009;61:961–70.e1–11.
Brin MF, Kirby RS, Slavotinek A, Miller-Messana MA, Parker L, Yushmanova I, et al. Pregnancy outcomes following exposure to onabotulinumtoxinA. Pharmacoepidemiol Drug Saf. 2016;25:179–87.
Novic A, Kolves K, O’Dwyer S, De Leo D. Migraine and suicidal behaviors: a systematic literature review. Clin J Pain. 2016;32:351–64.
Calati R, Courtet P, Norton J, Ritchie K, Artero S. Association between lifetime headache and history of suicide attempts in the elderly. Eur Psychiatry. 2017;41:132–9.
Negro A, Curto M, Lionetto L, Crialesi D, Martelletti P. OnabotulinumtoxinA 155 U in medication overuse headache: a two years prospective study. Springerplus. 2015;4:826.
Guerzoni S, Pellesi L, Baraldi C, Cainazzo MM, Negro A, Martelletti P, et al. Long-term treatment benefits and prolonged efficacy of onabotulinumtoxinA in patients affected by chronic migraine and medication overuse headache over 3 years of therapy. Front Neurol. 2017;8:586.
US Department of Health and Human Services, Food and Drug Administration. Guidance for industry: establishing pregnancy exposure registries (2002). https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm071639.pdf. Accessed 23 Apr 2019.
Ventura SJ, Curtin SC, Abma JC, Henshaw SK. Estimated pregnancy rates and rates of pregnancy outcomes for the United States, 1990–2008. Natl Vital Stat Rep. 2012;60:1–21.
Centers for Disease Control and Prevention. Update on overall prevalence of major birth defects: Atlanta, Georgia, 1978–2005. MMWR Morb Mortal Wkly Rep. 2008;57:1–5.
Polo JM, Martin J, Berciano J. Botulism and pregnancy. Lancet. 1996;348:195.
Ahmed F, Khalil M, Tanvir T, Buture A, editors. OnabotulinumtoxinA for chronic migraine during pregnancy; experience from Hull migraine clinic, United Kingdom [abstract MTIS2018-104]. Biennial Migraine Trust International Symposium; 6–9 Sep 2018; London.
Acknowledgments
Editorial support for development of this manuscript was provided by Lee B. Hohaia, PharmD, at Complete Healthcare Communications, LLC (North Wales, PA), a CHC Group company, and funded by Allergan plc (Dublin, Ireland).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by Allergan plc (Dublin, Ireland). The authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Conflict of interest
Paul K. Winner has received consulting fees/honoraria from Allergan, Amgen, and Supernus; has served on the speakers’ bureau for Allergan, Amgen, Avanir, and Supernus; and has received research grants from Allergan, Amgen, NuPathe, AstraZeneca, Avanir, Eli Lilly, and Novartis. Andrew M. Blumenfeld has served on advisory boards for Allergan, Amgen, Alder, Teva, Supernus, Lilly, Promius, and Eaglet, has received speaking fees from Allergan, Amgen, Pernix, Supernus, Depomed, Avanir and Promius, and holds patents on onabotulinumtoxinA in migraine that Allergan owns. Eric J. Eross has received grant/research support from Allergan; has served as an advisory board member/consultant for Amgen, Promius, and Supernus; has served on the speakers’ bureau for Allergan, Amgen, Avanir, Depomed, Pernix, Supernus, and Teva; and is the owner and president of Glia Sciences. Amelia Orejudos is an employee of Allergan plc. Debbie L. Mirjah, Aubrey Manack Adams, and Mitchell F. Brin are current or former employees of Allergan plc and own stock in the company.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. This study received ethical approval from the institutional review board or independent ethics committee at each site.
Consent to participate
Written informed consent was obtained from each patient prior to enrollment in the study.
Data availability
Data reported in this manuscript are available within the article and online resources. Additional data from the COMPEL Study (Clinical Trial Registration: ClinicalTrials.gov; NCT01516892) may be requested at http://www.allerganclinicaltrials.com/PatientDataRequest.htm.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Winner, P.K., Blumenfeld, A.M., Eross, E.J. et al. Long-Term Safety and Tolerability of OnabotulinumtoxinA Treatment in Patients with Chronic Migraine: Results of the COMPEL Study. Drug Saf 42, 1013–1024 (2019). https://doi.org/10.1007/s40264-019-00824-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40264-019-00824-3