
Abstract
Neurotropic viruses may cause meningitis, myelitis, encephalitis, or meningoencephalitis. These inflammatory conditions of the central nervous system (CNS) may have serious and devastating consequences if not treated adequately. In this review, we first summarize how neurotropic viruses can enter the CNS by (1) crossing the blood-brain barrier or blood-cerebrospinal fluid barrier; (2) invading the nose via the olfactory route; or (3) invading the peripheral nervous system. Neurotropic viruses may then enter the intracellular space of brain cells via endocytosis and/or membrane fusion. Antiviral drugs are currently used for different viral CNS infections, even though their use and dosing regimens within the CNS, with the exception of acyclovir, are minimally supported by clinical evidence. We therefore provide considerations to optimize drug treatment(s) for these neurotropic viruses. Antiviral drugs should cross the blood–brain barrier/blood cerebrospinal fluid barrier and pass the brain cellular membrane to inhibit these viruses inside the brain cells. Some antiviral drugs may also require intracellular conversion into their active metabolite(s). This illustrates the need to better understand these mechanisms because these processes dictate drug exposure within the CNS that ultimately determine the success of antiviral drugs for CNS infections. Finally, we discuss mathematical model-based approaches for optimizing antiviral treatments. Thereby emphasizing the potential of CNS physiologically based pharmacokinetic models because direct measurement of brain intracellular exposure in living humans faces ethical restrictions. Existing physiologically based pharmacokinetic models combined with in vitro pharmacokinetic/pharmacodynamic information can be used to predict drug exposure and evaluate efficacy of antiviral drugs within the CNS, to ultimately optimize the treatments of CNS viral infections.
Similar content being viewed by others

High mortality and severe health complications may result from viral infections of the central nervous system (CNS), indicating the need for better CNS antiviral treatment. |
As sampling of the human brain is rare, we propose mathematical model-based methodologies to inform and optimize CNS antiviral treatments. |
1 Introduction
Neurotropic viruses are viruses that tend to spread from initially infected organs to the central nervous system (CNS), where they can trigger severe inflammation of different CNS compartments. This includes the spinal cord (myelitis), the membranes that surround the brain and the spinal cord (meningitis), the brain itself (encephalitis), or brain and meninges (meningoencephalitis). Thus, the primary distinction between these diseases lies in the specific regions of the CNS that becomes inflamed following viral infection, as well as the associated mortality risk [1,2,3]. In general, viral meningitis is the most common CNS infection with an estimated incidence range from 0.26 to 17 cases per 100,000 people, while encephalitis and myelitis occur less frequently [4, 5]. However, in contrast to meningitis, encephalitis is a life-threatening disease that is linked to high mortality rates in patients and therefore requires adequate treatment. For example, encephalitis triggered by the herpes simplex virus is linked to 70% mortality in the absence of any treatment, which may be reduced but still is as high as 28% after antiviral treatment [6, 7]. This illustrates the need for finding new therapies and optimizing the use of existing antiviral drugs for the treatment of encephalitis.
After viruses enter the human body, they infect organs by binding to entry receptors on the surface of cellular membranes, and then reside within host cells [8]. The blood-brain barrier (BBB) and the blood-cerebrospinal fluid barrier (BCSFB) normally prevent toxins or pathogens from entering the brain; however, neurotropic viruses can cross these barriers, or bypass these barriers via the peripheral nervous system (PNS) and/or via the nasal route, and consequently invade the brain [9].
Antiviral drugs that can inhibit the entry and/or persistence of viral inflammation in the CNS are critical for the management of severe CNS infections and improving patient survival. Some antiviral drugs have proven to be effective to treat specific encephalitis. For instance, acyclovir and ganciclovir are currently used for encephalitis caused by herpesviruses, leading to significantly reduced mortality rates [10]. In addition, the combination antiretroviral therapy has markedly improved the human immunodeficiency virus (HIV)-related neurocognitive disorder [11]. However, the mortality/morbidity of encephalitis remains high after treatment [12,13,14]. Most neurotropic viruses (e.g., alphaviruses, bornaviruses, flaviviruses, rhabdoviruses) can be fatal (mortality rate range from 1 to 90%) because there is no effective drug treatment [15]. As such, preventing viral invasion of the CNS can be achieved solely through the use of appropriate vaccines [16], or by new drugs, or even by improvement of drug treatment modalities.
A key prerequisite for antiviral drugs to successfully treat CNS infections is their ability to effectively reach their target site, i.e., brain intracellular space, where the viruses reside. However, drug distribution into the CNS is limited by the BBB and the BCSFB [17]. The tight junctions between the cells of the BBB and the BCSFB limit paracellular permeability of drugs with high hydrophilicity and/or large molecular size. In addition, active influx and efflux transporters at the BBB and the BCSFB regulate the transport of their substrates, i.e., drugs, between blood and CNS. Apart from drug transport into the CNS, the intracellular exposure of drugs is also determined by the exchange of the antiviral drug between the extracellular and intracellular fluid (ECF and ICF). This is governed by passive diffusion and may as well be influenced by active brain cell membrane transport [18]. Only more lipophilic drugs with a low molecular weight can passively diffuse across the cell membrane. Hydrophilic drugs would need carriers. As brain cell membranes may have active transporters [18], the intracellular concentrations of antiviral drugs can be influenced if they are substrates of the transporters expressed on the brain cell membrane.
Furthermore, drug-metabolizing enzymes in the brain cells, such as some cytochrome P450 (CYP) enzymes and UDP-glucuronosyltransferase (UGT) [19, 20] may metabolize antiviral drugs. This may also affect the intracellular concentrations of antiviral drugs. Then, the antiviral drugs that are nucleoside/nucleotide analogs exert their function after undergoing the intracellular metabolism mediated by cellular and/or viral kinases. Therefore, the knowledge on intracellular profiles of antiviral drugs and their potentially active metabolites is important in relation to the antiviral effect.
Our knowledge of the intracellular pharmacokinetics of antiviral drugs in the human brain is currently very limited, while it is important for understanding optimal drug treatment of viral CNS infection. However, direct information on human brain pharmacokinetics is hampered by ethical limitations. Alternative approaches to study the antiviral drug distribution into the brain and the brain cells should therefore be considered.
Central nervous system physiologically-based pharmacokinetic (PBPK) modeling approaches are based on CNS physiological properties and drug physicochemical and biological properties, to predict PK profiles in different locations of the CNS. The PBPK model that distinguishes ECF and ICF spaces can be instrumental in predicting the distribution of antiviral drugs within the CNS and their reach to intracellular target sites during CNS viral infections. When combined with information on the efficacy of antiviral drugs, this model can be also applied for predicting drug effects, and then be further used to improve existing antiviral drug dosing regimens, or even to predict the effects of new antiviral drug candidates.
In this review, we first comprehensively summarize the mechanisms of viral invasion into the CNS as well as the current means of treating CNS viral infections. We then consider the potential factors that influence the effect of antiviral drugs. Finally, we discuss how we could utilize pharmacological modeling to improve antiviral treatment.
2 Mechanisms of Viral Invasion into the CNS
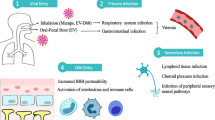
Viruses normally first infect peripheral tissues and may then distribute into the CNS. A list of neurotropic viruses and their abbreviations is given in Table 1. In general, there are three different routes for viruses to enter the CNS: (1) the virus can transmit through the peripheral nerves connecting to some part of the CNS; (2) the virus can bind to the olfactory receptor neuron located on the nasal epithelium, and (3) the virus can directly cross the blood-brain barriers (BBB/BCSFB) (Fig. 1).

Adapted from Koyuncu et al. [9]
Routes of viral invasion into the central nervous system (CNS). A Viruses infect the CNS through peripheral nerves or neuromuscular junctions (NMJ); B viruses spread into the CNS via olfactory nerves; C viruses enter brain across the blood-brain barrier (BBB).
2.1 PNS Route
2.1.1 Invasion Starting at the Sensory Neuron Terminal
Sensory neurons can receive external signals by stimulation of their nerve endings located in periphery tissues such as the skin or the viscera. Those signals are integrated in the cell body of sensory neurons and then conducted via axons that extend into the CNS. Similarly, viruses can bind to entry receptors on sensory nerve endings and then move along the peripheral nerves towards the PNS. As sensory neurons are in direct synaptic connection with CNS neurons, infection of PNS neurons provides a direct route of transmission into the CNS [21].
Alpha herpesviruses including herpes simplex virus 1 (HSV)-1, HSV-2, and varicella-zoster virus (VZV) normally enter the brain through the PNS route. For HSV, the Nectin cell adhesion molecule 1 (Nectin-1) serves as a primary entry receptor. For VZV, both myelin-associated glycoprotein and Nectin-1 are engaged in VZV entry into sensory neurons [22,23,24] (Fig. 1A). Severe acute respiratory syndrome coronavirus 2 (SARS‑CoV‑2) may also use the sensory neurons as a potential target to access the CNS as SARS‑CoV‑2 receptor angiotensin-converting enzyme 2 (ACE2) is broadly expressed on human sensory neurons [25].
2.1.2 Invasion via Neuromuscular Junctions
Neuromuscular junctions (NMJs) are the synaptic connections between motor neurons and muscle fibers that are responsible for conversion of electrical impulses created by the motoneurons into action potentials in the target muscle fibers [26]. Neuromuscular junctions can provide an entrance for viruses from muscles into motor neurons, and subsequently facilitate invasion into the CNS as the somas of motor neurons are mostly located in the spinal cord, and therefore could connect to the motor centers in human brain via a synapse formation (Fig. 1A).
Poliovirus, rabies virus as well as SARS-CoV-2 might spread into the CNS through binding to the receptors on the axon terminals of motor neurons at the NMJs. The entry receptor of the poliovirus is a transmembrane glycoprotein called cluster of differentiation 155, whereas the entry of rabies virus is mediated by the synergistic action of receptors including nicotinic acetylcholine receptor and neural cell adhesion molecules [27, 28]. The nicotinic acetylcholine receptor, mainly presenting at NMJs, could concentrate viruses in front of the NMJs and improve the possibility of uptake by the neural cell adhesion molecules [29]. As the expression of ACE2 receptor is also found in skeletal muscle, SARS-CoV-2 has the potential to invade the CNS through the direct infection at NMJs [30].
2.2 ORN Route
Olfactory receptor neurons (ORNs), also called olfactory sensory neurons, are the sensory neurons of the olfactory system. The ORNs are located in the olfactory epithelium in the nasal cavity. The cell bodies of the ORNs are distributed among all three of the stratified layers of the olfactory epithelium in the nasal cavity. The ORNs have dendrites that face the external surface of the cribriform plate, and axons that pass through the cribriform foramina with terminal end at olfactory bulbs [31] (Fig. 1B). Some viruses including HSV-1, west Nile virus, SARS-CoV2, Borna disease virus, rabies virus, prions, influenza A virus, and parainfluenza viruses can propagate into theh brain along the olfactory nerves by infecting ORNs in the nasal epithelium [32, 33].
2.3 Brain Barrier (BBB and BCSFB) Routes
In addition to the routes above for viral infection of the CNS, viruses can also cross the brain barriers to enter the CNS. There are three viral pathways involved in crossing the brain barriers, including the trans/paracellular route and the “Trojan horse” route.
2.3.1 Trans- or Paracellular Route
Viruses present in the bloodstream may also infect and enter the brain microvascular endothelial cells, which make up the BBB. To that end, the viruses bind to specific membrane receptors depending on viral types (shown in Table 1). The viruses inside the cells may then traverse the abluminal membrane of the BBB to complete transcellular CNS penetration [34]. The viral infection of brain microvascular endothelial cells often causes the disruption of the tight junctions and impairs the integrity of the local BBB at the later stages of the CNS viral diseases [35,36,37]. The loss of tight junction proteins caused by viral infection might facilitate the further transmission of viruses via the paracellular route. Likewise, viruses may infect the choroid plexus epithelial cells and migrate to the CNS via those trans- or paracellular routes [38]. The BCSFB at the level of the blood vessels is more permeable than the BBB as the tight junctions between these endothelial cells are considered the “leaky type” [39].
The west Nile virus, John Cunningham virus, human cytomegalovirus (HCMV), and the hepatitis C virus are examples of viruses that can disrupt the BBB integrity and utilize both trans- and paracellular routes [9, 34] (Fig. 1C). The John Cunningham virus is involved in the viral infections at the BCSFB and may hire a direct transmigration to enter the CNS [38]. Furthermore, SARS-CoV-2 is also suspected to invade the CNS via brain microvascular endothelial cell/choroid plexus epithelial cell-mediated transmigration across the BBB/BCSFB [40]. This is mainly due to the extensive expression of the SARS-CoV2 receptor ACE2 on various types of cells including the endothelial cells of the BBB and the epithelial cells of the BCSFB [41]. Neurological symptoms were also observed in patients with coronavirus disease 2019, suggesting the CNS invasion of SARS-CoV2 [42].
2.3.2 Trojan Horse Route
Viruses may also use a Trojan horse strategy to cross the BBB or BCSFB (Fig. 1C), by first getting into white blood cells (leukocytes) that may enter the CNS and thereby carrying the viruses along into the CNS [43,44,45]. This process may be further boosted when viruses cause the impairment in the tight junction and BBB integrity [46, 47].
Human immunodeficiency virus may use this Trojan horse mechanism to get into the CNS. The process of viral entry into leukocytes is handled by the interaction between the HIV glycoproteins and the cell-surface receptor CD4 [48]. In addition to HIV, SARS-CoV2 possibly enters the CNS by a Trojan horse strategy as its ability to infect leukocytes has been reported [49].
3 Mechanisms of Viral Invasion into the Brain Cells
Once viruses have entered the CNS, they can invade specific cells in the brain through two routes, namely receptor-mediated endocytosis and receptor-mediated membrane fusion (Fig. 2).

Adapted from Doms [8]
General pathways of viral invasion into the cells. Viruses enter cells via receptor-mediated endocytosis or the membrane fusion pathway, wherein virion releases their genome by forming a pore or fusing with the endosome membrane after endocytosis or directly releasing DNA/RNA after fusion with the plasma membrane.
3.1 Receptor-Mediated Endocytosis
Cellular uptake of viruses can be achieved by receptor-mediated endocytosis [8]. For many enveloped viruses, they start with binding the cell-surface receptors, after which the receptor-virus complexes are transferred to the endosomes (Fig. 2). The low pH in late endosomes (~5.5) induces structural changes in viral spike glycoproteins via the protonation of acidic residues of the glycoproteins. This causes virus-endosome fusion through an interaction between the changed glycoproteins and the membrane lipids, which subsequently promotes the release of the viral genome into the host cells [50].
Nonenveloped viruses (i.e., a virus lacking a bilayer lipid membrane), such as polioviruses, may use a similar approach to invade cells (Fig. 2, mechanism 1B). However, after viral attachment to their entry receptors and subsequent transfer of the complexes to endosomes, the low pH environment could lead to the structural changes in viral capsids rather than that of the glycoprotein, and then expose the hydrophobic domains of viral capsids. Finally, the hydrophobic domains can insert into the endosome membrane and form a pore [51].
3.2 Receptor-Mediated Membrane Fusion
Viruses can enter the cell also through receptor-mediated membrane fusion [8]. This process, compared to receptor-mediated endocytosis, is characterized by the involvement of another cell surface receptor called a co-receptor to trigger the fusion of a viral membrane with a plasma membrane of a target cell.
For instance, HIV engages a non-endosomal dependent pathway to enter cells (Fig. 2). The process of membrane fusion by HIV requires the entry receptor CD4 and a co-receptor, such as the chemokine receptors CCR5 or CXCR4 [52]. Binding of HIV to CD4 induces the structural changes in the envelope glycoprotein (Env) enabling HIV to bind to the coreceptor. Coreceptor binding can further change the conformation of the Env, which triggers the fusion of the viral envelope with the plasma membrane. The virus-cell membrane fusion always occurs at the surface of the plasma membrane, without being affected by the acidic environment of endosomes.
Certain viruses can use both receptor-mediated endocytosis and receptor-mediated membrane fusion as entry mechanisms of the CNS cells, which depends on the expression of cell-surface receptors on host cells to be able to switch between entry mechanisms. For instance, HSV-1 could enter target cells via endocytosis by binding Nectin-1 while the overexpression of the viral co-receptor, paired immunoglobulin-like type 2 receptor alpha, could change the entry route to membrane fusion [53]. HIV-1 could also enter target cells via endocytosis [54,55,56,57], partly owing to the absence of co-receptors [58]. In the case of SARS-CoV-2, if there is transmembrane serine protease 2 expressed on the surface of target cells, SARS-CoV-2 binds to the receptor ACE2 and co-receptor transmembrane serine protease 2 in order, and then the conformational changes in viral glycoprotein is triggered to mediate SARS-CoV-2 entry through a membrane fusion pathway [59]. However, without the expression of transmembrane serine protease 2, ACE2 binding can directly lead to the endocytic entry. Thus, all the information on the viruses mentioned indicate these are able to invade cells through multiple routes and may therefore potentially be more neurotropic.
4 Brain Cell Preferences (Viral Tropism and Reservoir)
The brain parenchymal cells mainly consist of neurons and non-neuronal glial cells, such as astrocytes and microglia. At the cellular level, viral tropism is the specificity of a given virus to infect a particular type of cells within the brain/CNS to ultimately complete viral replication inside these cells. In general, viral tropism is mainly determined by two factors; namely the presence of specific receptors on the cell membrane that mediate viral entry and the subsequent intracellular events that are necessary for viral replication [60].
Once entering specific brain cells, virus can evade immune detection by remaining dormant, forming latent reservoirs rather than actively replicating [61]. By definition, a viral cell reservoir is a certain cell type where a replication-competent form of the virus accumulates and persists [61]. These cell reservoirs are significant as they remain viral genomes capable of producing infectious viruses and continuing to fuel the infection within the host organism. The persistence of viral genomes within these reservoirs poses challenges for treatment and eradication efforts.
4.1 Impact of Entry Receptors
For the majority of viruses, the determinant of viral tropism is the availability of entry receptors on the surface of the host cells (Table 2). In the CNS, viruses might selectively infect neurons, astrocytes and/or microglia owing to the presence of specific entry receptors on the different CNS cell types (Fig. 3).

Viral replication in different types of brain cells. CMV cytomegalovirus, HIV-1 human immunodeficiency virus type 1, HSV-1 herpes simplex virus 1, SARS-CoV-2 severe acute respiratory syndrome coronavirus 2 coronavirus, VZV varicella-zoster virus
HIV-1, for example, prefers to reside in microglia because these resident macrophages within the CNS primarily express the entry receptor CD4 antigen of HIV-1 [62]. Although HIV-1 could also enter human astrocytes using a CD4-independent pathway, i.e., human mannose receptor-mediated endocytosis [63], the CNS tropism of HIV-1 is specific for microglia where HIV-1 undergoes a productive infection after HIV-1 entry via utilizing CD4 as well as its co-receptor, while restricted replication in astrocytes was observed at different stages of the viral life cycle [64]. According to the definition of viral reservoirs, which involves productive viral replication, only microglia are acknowledged as the CNS reservoir for HIV-1.
Herpes simplex virus and VZV predominantly enter neurons as host cells through interactions with the receptor Nectin-1 that is highly expressed in neurons of the human CNS [65, 66]. The importance of Nectin for VZV tropism has been recently demonstrated in a human stem cell-based neuronal model in which both knock-down of endogenous Nectin-1 or introduction of soluble Nectin-1 as a decoy receptor markedly reduced VZV infection in neurons [24, 67]. Through functional studies in Nectin-1 knockout mice, Nectin-1 has been shown to play an important role in the CNS tropism of both HSV-1 and HSV-2 [68, 69]. Herpes simplex virus particles, like VZV, have a tendency to replicate within neurons [70, 71]. This propensity suggests that HSV might establish a latent reservoir in neurons within the CNS for ongoing virus production.
With respect to HCMV, there are numerous cell-surface receptors that can facilitate HCMV entry, which might explain the effective invasion of HCMV to various cell types [72]. Human cytomegalovirus productively infects CNS parenchymal cells, including astrocytes, microglia, and neurons, with susceptibility levels that range from high to low [73, 74]. Several post-mortem studies have shown that SARS-CoV-2 proteins were detected among neurons, astrocytes, and microglia [75,76,77]. Recently, Shen et al. found SARS-CoV-2 could invade the CNS cells (such as neurons, oligodendrocytes, and microglia) using both ACE2 and neuropilin-1 receptors [78]. They also revealed that SARS-CoV-2 not only infects mature neurons but also completes intracellular replication in these neurons, which is indicative of SARS-CoV-2 tropism for mature neurons and they may serve as a potential reservoir of SARS-CoV-2.
4.2 Intracellular Events in Restricting Viral Tropism
The specificity of viral tropism is also affected by other factors apart from the availability of entry receptors. Some viruses, such as poxvirus, can enter certain cells without being able to productively replicate in the cells they have invaded [79]. Interestingly, there are several downstream intracellular factors limiting poxvirus replication once they enter the cells, such as cell-cycle control, trans-regulatory elements, and innate cytokines [80]. Therefore, viral replication could be aborted by these intracellular restriction events occurring in the restricted cells and might result in specific infection of poxviruses to particular CNS cells.
Poliovirus is another example of whose tropism at a cellular level is determined by intracellular factors. Although human poliovirus receptor (or cluster of differentiation 155) proteins are generally expressed on the membrane of many cells of different organs including the intestine, lung, liver, heart, brain, and spinal cord, poliovirus replication is restricted to the skeletal muscle and the CNS, for the latter of which polioviruses prefer to infect neurons rather than glial cells [81]. However, it is still unclear why only neurons become primarily infected. As with poxvirus, some intracellular factors could limit viral replication and further affect cellular tropism of poliovirus.
4.2.1 IRES
Many studies have highlighted that the initiation process of viral translation mediated by the poliovirus internal ribosome entry site (IRES) could serve as an intracellular factor that limits viral replications [82,83,84,85]. As IRES is within a non-coding region of poliovirus genome, the mutation in this part of the viral genome or introducing new IRES from other viruses by recombinant DNA technology does not affect the utilization of host cell receptor by viruses, but influences the interaction between IRES and some downstream intracellular factors such as cell proteins and canonical translation initiation proteins that are involved in viral replication [86]. The neurovirulence or the replication of poliovirus is significantly reduced either by inducing the mutations of IRES or by incorporating new IRES from other viruses, which suggests that the IRES of poliovirus serves as a determinant of viral tropism [85, 87,88,89].
4.2.2 Interferon Production
In addition to the viral IRES, poliovirus tropism is also regulated by the secretion of alpha/beta interferons, which are self-protective cytokines that by binding to their cognate cellular receptors activate intracellular signaling events that can prevent viral infection [90]. In the IRES knockout mouse model, viral antigens were observed in non-target tissues that are not supposed to be infected by polioviruses, including the liver, spleen, and pancreas. Similar observations were also reported for other types of viruses and demonstrated altered cell and tissue tropism of viruses via ablation of interferon-α/β functions [79, 91, 92]. These results reinforce that interferon-induced responses are a common pathway against viral tropism that can prevent viral replication and the spread of different types of viruses.
In conclusion, viruses employ various pathways to enter the CNS and subsequently infect brain cells. Viral tropism signifies the preference of a virus for specific brain cell types, therefore influencing the extent of intracellular viral accumulation. As viral replication occurs within these cells, the intracellular concentrations of drugs are directly linked to their efficacy in inhibiting or eradicating the virus.
5 Current Treatments for CNS Viral Infections
Despite the presence of over 200 different virus species, the number of clinically approved antiviral drugs in the market is limited [93]. The ability of viruses to reside and replicate within (brain) cells for their survival, whereas other pathogenic micro-organisms such as bacteria remain in extracellular compartments, makes the development of antiviral drugs an additional challenge, as this requires that antiviral drugs can distribute into the cells that are infected.
Considering the persistence and latency of viruses, the best therapeutic strategy against CNS viral infections is to prevent the occurrence of viral infections by vaccines. Effective vaccines have been used to successfully control the outbreak of different viruses; however, life-threatening viral infections still happen and require antiviral drugs that can decrease the mortality [10]. For acute CNS viral infections, the immediate action by antiviral drugs should be emphasized to slow or stop, or even reverse the disease course. In such cases, patients would not get the obvious treatment benefit from virus vaccines, which require time to activate the immune system.
Viral meningitis is usually overcome by self-recovery, and drug treatment is therefore not needed [4]. There are also no recommended therapeutic agents for viral myelitis owing to the lack of potent drugs [94]. However, viral encephalitis is generally considered to be one of the most severe CNS infections and the mortality rate varies between virus types reaching up to 70% without antiviral therapies [7]. Currently there are only a few antiviral drugs commonly used for treating viral encephalitis (Table 3). Despite antiviral interventions, the treatment outcome is still poor, which could be explained partly by insufficient doses [95]. As specific antiviral medications are only available for viral encephalitis, this section mainly focuses on the current treatments for certain types of viral encephalitis in the CNS.
5.1 Antiviral Nucleoside Analogs
5.1.1 Pharmacological Properties
Nucleoside analogs, the mimics of endogenous nucleosides, are a common class of antiviral drugs used for viral encephalitis. The actions of antiviral nucleoside analogs are premised on intracellular metabolism to their active forms, which are triphosphorylated nucleosides [96]. In general, these nucleoside analogs undergo three steps of successive phosphorylations (Fig. 4). The first phosphorylation is typically mediated by viral kinases such as thymidine kinases (TK) of herpes viruses. For viruses such as HIV and hepatitis B virus that cannot produce virus-encoded TK, cellular kinases are required as the initial step to convert nucleoside analogs into monophosphorylated products [97]. The process mediated by viral kinases is considered more efficient than that of cellular kinases because virus-encoded TK has a higher affinity for their substrate. For the diphosphorylation and triphosphorylation steps, only nucleoside kinases from host cells are required to take charge of phosphorylation [98]. The active triphosphorylated derivatives thereby exert their functions in inhibiting viral replications by serving as a substrate for viral enzymes and incorporating their metabolites into viral DNA or RNA to induce the chain termination, as well as by acting as an inhibitor to suppress viral polymerases or ribonucleotide reductase [99]. As nucleoside analogs more readily interact with viral, but not human polymerase to block viral replication, most of these drugs are safe and well tolerated. Therefore, we mainly focus on drug efficacy instead of drug toxicity in the following discussion.

Process of intracellular phosphorylation of antiviral drugs in virus-infected cells. HBV hepatitis B virus, HCMV human cytomegalovirus, HIV human immunodeficiency virus type 1, HSV herpes simplex virus, HSV-TK herpes simplex virus-encoded thymidine kinase, pUL97 human cytomegalovirus UL97 protein, VZV varicella-zoster virus, VZV-TK varicella-zoster virus-encoded thymidine kinase
5.1.2 Clinical Efficacy: Herpesviruses
Herpesviruses are a common cause of encephalitis, most notably infections by HSV that account for 10–20% of all viral-mediated encephalitis cases [100]. In addition, VZV and CMV can also cause encephalitis where each of them comprises a relatively lower percentage (~3%) of the total patients with encephalitis [101]. The nucleoside analogs acyclovir and ganciclovir are the most commonly used treatments for viral encephalitis caused by herpesviruses [102].
5.1.2.1 Acyclovir
Acyclovir is a US Food and Drug Administration-approved antiviral agent for the treatment of HSV encephalitis and is the sole drug for which a randomized controlled trial has been conducted to determine its efficacy for this specific type of encephalitis [12]. This study demonstrated that acyclovir can dramatically reduce mortality in adult patients with timely diagnosed HSV-1-mediated encephalitis. Despite the use of acyclovir, high mortality rates (up to 28%) are still seen with HSV-1-mediated encephalitis and survival can frequently be compromised by coexisting comorbidities. In neonates, encephalitis treated with intravenous acyclovir have been linked to lower mortality rates (6–19%) [103, 104], which may be explained by the not yet developed BBB in neonates that may facilitate the entry of acyclovir into the brain and therefore the treatment of encephalitis in these patients. However, the neurologic impairment remains very high (~70%) in newborns with HSV-mediated encephalitis after the use of acyclovir [104]. Within neonates, higher dosages of acyclovir (60 mg/kg/day) can further reduce the mortality to 6% [103], indicating that higher concentrations of acyclovir are beneficial/necessary for inhibiting the viral replication in the brain.
Intravenous acyclovir is also used to treat VZV encephalitis, with higher dosages proposed for these patients compared with those with HSV encephalitis. The reason underlying the higher dose is that acyclovir is less efficiently phosphorylated by the VZV-TK, which has lower affinity for acyclovir compared with HSV- TK [1]. The mortality rate in VZV encephalitis remains high, reaching up to 15% despite acyclovir treatment [105]. Furthermore, full recovery is observed in only up to 49% of patients after discharge [106].
Regarding viral meningitis, although there is no clinical controlled trial based on which the specific antiviral therapies could be recommended, acyclovir have been used to treat HSV-2 meningitis [107]. Acyclovir is also the principal treatment of HSV and VZV myelitis [108].
5.1.2.2 Valacyclovir
Valacyclovir is an l-valyl ester prodrug of acyclovir that can increase oral bioavailability of acyclovir, and is used as an alternative therapy for patients with HSV encephalitis if intravenous administration is not possible or because of the relatively high cost burden of acyclovir in resource-limited countries [109]. However, the use of valacyclovir within this patient population is first limited by the lack of clinical studies that have been able to thoroughly assess the clinical effectiveness of valacyclovir on the mortality that is associated with HSV encephalitis. Second, controlled trials have been suggesting that neurocognitive dysfunctions that develop following these types of infections may not improve following the use of valacyclovir [110]. Similar to acyclovir, valacyclovir have been used to treat HSV-2 meningitis [107], while clinical evidence shows the better outcomes were observed in patients treated with acyclovir [111].
5.1.2.3 Ganciclovir
Ganciclovir is a guanosine analog normally used in combination with foscarnet (a phosphonic acid derivative) to treat CMV encephalitis, which is the most common therapeutic regimen recommended by the experts [10, 13]. The two-drug combination regimen is further proved to be efficacious in CMV encephalitis/myelitis by a multi-center, non-randomized, uncontrolled single-arm trial [13]. The combination of ganciclovir plus foscarnet caused clinical improvements in 71% of the 17 patients with encephalitis who were enrolled, though within this small clinical study 24% of patients also died despite the use of the combination.
5.1.3 Clinical Efficacy: HIV
HIV-1 infection in the CNS may finally evolve into encephalitis and therefore damage the function of the human brain, and clinically result in the AIDS dementia complex [112]. Nucleoside reverse transcriptase inhibitors are the mainstay of antiretroviral therapeutic agents for HIV-associated CNS disease over the last 10 years [113, 114]. Antiretroviral drugs with good CNS penetration should be incorporated into the therapeutic strategies of HIV-1 encephalitis, as the inhibition of HIV-1 replication in the CNS seems to be an important factor for managing patients with neurological complications [115].
The revised 2010 version of the CNS Penetration Effectiveness (CPE) score is a tool that classifies the potential of drugs to penetrate the CNS. The penetration efficacy of different drugs is categorized into ranks (1–4) for which multiple factors are taken into consideration [116], including pharmacokinetic (PK) and pharmacodynamic (PD) data to assess whether a drug’s cerebrospinal fluid (CSF) concentration exceeds the half maximal inhibitory concentration (IC50), drug characteristics considering properties that may limit CNS penetration (e.g., large molecular weight), and clinical study outcomes evaluating improvements in patient cognition or reductions in the CSF viral load. With respect to antiviral nucleoside analogs, such as zidovudine (4, very good), emtricitabine (3, good), and abacavir (3, good), they have been allocated a higher CPE score, indicating a greater capability to penetrate the CNS. In contrast, lamivudine has modest penetration with a score of 2. Hence, theoretically, they are deemed more effective for patients experiencing HIV-associated neurological complications [117, 118]. However, relying solely on the CPE system to assess drug efficacy in the CNS is imprudent, as its application has not been uniformly validated. Certain studies suggest a link between higher CPE scores, cognitive enhancements, and a reduced viral load in the CSF, while other studies report no such correlation [118, 119].
In children with HIV encephalopathy, high CNS-penetrating antiretroviral regimens are recommended based on clinical evidence of reduced mortality of patients who were treated with the combination of highly permeable drugs, compared with those who never received any antiretroviral therapy (6% vs 24%) [120]. Despite effective antiretroviral therapy, more than 32% of patients with HIV-associated dementia have persistent neurocognitive impairment and 28% of them died according to one retrospective study [14]. With regard to HIV myelitis, it also arises in the early and late stages of HIV infection, while the optimal therapeutic regimens are unknown as antiretroviral drugs are often incapable of stopping the progression of related symptoms [108]. For the high CNS-penetrating antiviral drugs, still the question remains if adequate intracellular target site concentrations can be reached by these drugs.
5.2 Antiviral Nucleotide Analog Prodrug
5.2.1 Pharmacological Properties
In the case of mutations, viral kinases may fail to catalyze the initial phosphorylation of nucleoside analogs [121]. As nucleoside analogs have a similar structure to endogenous nucleosides, they can also hire cellular kinases to complete the first phosphorylation but this cell kinase-dependent reaction is typically slow and inefficient, and thus considered a rate-limiting step of the whole process of phosphorylation [122]. A nucleotide is the nucleoside with one to three phosphate groups and some nucleotide analogs that have only one phosphate attached (nucleoside monophosphate) have been developed to avoid the need for viral kinases and bypass the rate-limiting step in phosphorylation [121]. However, the cleavage of the P–O bond of a nucleoside monophosphate is prone to the catalysis of hydrolases before and after they enter viral-infected cells, which makes the chemical structure of phosphate analogs become less stable [123]. Hence, phosphonate nucleoside analogs, also a type of nucleotide analogs, are used to increase metabolic stability of these compounds by replacing the phosphonooxymethyl moiety (P–O–C) with the phosphonomethoxy (P–C–O) while remaining as substrates for a variety of cellular kinases so as to successfully achieve intracellular conversion into nucleoside triphosphate analogs [123].
Tenofovir and cidofovir are the representative nucleotide analogs with the P–C–O moiety. However, the presence of negative charges in the phosphonate group limits penetration of the drugs into the cell [124].
In order to mask negative charges for the further improvement of penetration, prodrugs of tenofovir, including tenofovir disoproxil fumarate (double ester prodrug) and tenofovir alafenamide (phosphoramidate prodrug), have been developed that contain modifications of the phosphonate moiety with either alkoxycarbonyl or with aryl and amino acid motifs [124]. These prodrugs can be rapidly converted into nucleoside phosphonate by removing the masking groups via intracellular enzymes [125]. Remdesivir is another example of a nucleotide analog prodrug, phosphoramidate, where the phosphate group instead of the phosphonate moiety is masked by modified groups.
5.2.2 Clinical Efficacy: HIV
Tenofovir disoproxil fumarate has proven effective in improving neurocognitive and neurological impairment by HIV in several clinical trials [126, 127]. Compared with tenofovir alafenamide, it might be more efficacious in HIV encephalitis because of a higher exposure to tenofovir in the CSF [128]. However, CSF concentrations may not reliably reflect brain tissue concentrations. This is evident from the notably lower levels of tenofovir observed in brain tissue compared with CSF in both human and animal models [129, 130]. Remdesivir is a Food and Drug Administration-approved drug for the treatment of SARS-CoV-2 [131]; however, in general, patients did obtain an obvious benefit from the treatment [132]. There is no relevant evidence to support the effect of remdesivir on reducing neurological symptoms, and remdesivir also needs more data from clinical trials to prove its own role in the fight against the pandemic of SARS-CoV-2 [133].
5.3 INSTIs
5.3.1 Pharmacological Properties
Integrase strand transfer inhibitors (INSTIs) stand as a pivotal class of antiretroviral drugs specifically used in the treatment of HIV. Integrase strand transfer inhibitors can interact with the active site of the integrase enzyme to effectively block its activity. As a result, the inactive integrase enzyme lacks the function to integrate viral DNA into the host cell DNA, hindering the viral replication. The first-generation INSTIs of raltegravir and elvitegravir have been approved for anti-HIV treatment over 10 years [134]. However, some HIV strains developed drug resistance mutations against first-generation INSTIs, limiting their effectiveness [135]. Second-generation INSTIs such as bictegravir and dolutegravir have been created to counteract the rapidly emerging resistance to first-generation INSTIs. These newer compounds demonstrate strengthened potency against viral replication and a reduced likelihood of resistance mutations, ensuring durable effectiveness compared with their predecessors [136].
5.3.2 Clinical Efficacy: HIV
Bictegravir and dolutegravir are currently the first-line drugs for HIV treatment, along with the use of a nucleoside analog such as lamivudine and emtricitabine and the nucleotide analog tenofovir [137]. As the concentrations of bictegravir and dolutegravir in CSF are much higher above their respective IC50 values, they might exert a sufficient inhibitive effect on viral replication in the CNS regions, despite modest penetration to the CNS (total CSF-to-plasma ratio less than 1%) [138,139,140]. Nevertheless, increased exposure of dolutegravir in the CNS has shown an association with an increased risk of CNS disorders, including dizziness, headache, and anxiety [141]. Some preclinical studies also suggested that both dolutegravir and bictegravir could disrupt the BBB integrity and enhance its permeability [142, 143].
Currently, there are no specific recommendations for INSTIs concerning HIV-related CNS infections. Further clinical trials are essential to ascertain the efficacy of bictegravir and dolutegravir in managing HIV-associated CNS infections.
Taken together, different nucleoside and nucleotide analogs are available for the treatment of viral encephalitis and may drastically lower mortality. Nevertheless, patients with viral encephalitis remain at a high risk for mortality (>10%) and long-term complications, indicating the need for new drugs and/or improvements in the dosing of existing drugs. With respect to these classes of drugs, it is important to note that the desired inhibitory activity on viral replications takes place inside the neuron, microglia, or astrocyte in which further metabolic conversion/activation of the different antiviral drugs may be required. Thus, knowledge of the intracellular concentrations of antiviral drugs within brain cells is most relevant for future strategies that aim to optimize dosing regimens of antiviral drugs.
6 Challenges in CNS Antiviral Treatment
As antiviral drugs should reach the target cells and maintain a sufficient concentration to completely suppress viral replication, insufficient intracellular concentrations might be one reason for treatment failure. To better assess the drug exposure–response relationship, the processes determining to which extent antiviral drugs remain inside the cells should be taken into account. These processes can influence drug intracellular concentrations by two different ways. More specifically, passive passage and active influx transport across the brain cell membrane determine how much antiviral drugs can enter brain cells, while intracellular metabolism and active efflux transport across the brain cell membrane can reduce the amount of drugs inside the cells.
Drug resistance caused by viral gene mutation is another challenge for antiviral treatment. Despite a low prevalence in immunocompetent individuals, antiviral drug resistance among a specific population (HIV or organ transplant patients) is of concern given the observed high incidence (up to 30%) of drug resistance [144, 145].
6.1 Passive Diffusion Across the Brain Cell Membrane
Antiviral drugs can pass through the lipid bilayer into the intracellular space by means of transmembrane passive diffusion, which is favored by small molecules. Many antiviral drugs cross the cell membrane through this route. This non-saturable process is facilitated by a drug concentration gradient from high to low [146]. In addition, the balance between lipophilicity and hydrophilicity is very important as compounds with a real high lipid solubility can readily penetrate the lipid membrane, but tend to accumulate there (i.e., non-specific binding) [147]. However, low lipophilicity can be a problem. As an example, for the hydrophilic drug acyclovir, limited penetration has been observed with the values of 50% for intravenous administration and 20% for oral administration [148, 149], which also explains the limited permeability of acyclovir across the brain cell membrane into the brain ICF. Tenofovir is another hydrophilic drug with much lower concentrations in the CSF compared to plasma concentrations [150]. It has also been shown that only a small amount of tenofovir can cross the cell membrane of microglia [151]. The hydrophilicity of both drugs might constrain their ability to cross the BBB/BCSFB barriers as well as the brain cell membrane, potentially leading to lower intracellular concentrations of these two prodrugs and, consequently, their active metabolites—acyclovir-triphosphate and tenofovir-diphosphate.
6.2 Active Drug Transporters on Brain Cell Membranes
Apart from the BBB and the BCSFB, drug efflux and influx transporters have been also identified on the brain cellular membranes [18, 152]. However, the presence and functional consequences of active transporters are primarily understood at the level of the BBB/BCSFB, whereas its impact on the membranes of different cell types within the brain remains largely unclear. Based on recent findings (see references in the text below), we summarized the expressions of active transporters that can transport antiviral drugs across the brain cell membrane (Fig. 5).

Main active transporters expressed on human and rat brain parenchymal cells for transferring antiviral drugs. MRP multidrug resistance associated protein, OAT organic anion transporter, OCT organic cation transporter, P-gp P-glycoprotein
6.2.1 Influx Transporters
The messenger RNA (mRNA) and protein expression of different influx transporters have been identified in brain parenchymal cells. Most of them belong to two families, namely the organic anion transporters (OATs) and the organic cation transporters (OCTs) [153,154,155,156], which may act as cellular influx transporters.
The antiviral drugs that are used for the treatment of encephalitis have been confirmed to be substrates of these influx transporters, as the intracellular drug concentrations decreased after blocking the function of corresponding transporters within cell models in which the respective human influx transporter gene was transfected [157, 158]. Acyclovir is eliminated by renal excretion involving interactions with human OAT1 (SLC22A6) and OAT3 (SLC22A8), both of which contribute to the active uptake of drugs from the blood to kidney cells [159]. It is however unclear if acyclovir can interact with hOAT1/3 in brain parenchyma because of the lack of knowledge of hOAT distribution in brain parenchymal cells. However, OAT1/OAT3 has been found to be expressed on mouse and rat neurons [160, 161], suggesting that the intracellular concentration of acyclovir in neurons might be increased by these two influx transporters at least in rodent animal models [162, 163]. Some studies also showed that tenofovir and ganciclovir are substrates of OAT1 or OAT3 [164, 165], and the intracellular concentrations of these drugs might vary in different types of brain cells because only neurons express OAT1/3 that take their substrates into brain cells. The expression of OCTs have been also observed at neurons and astrocytes [152, 166,167,168]. Hence, OCTs may be involved in the intracellular distribution of the antiretroviral drugs abacavir and lamivudine, which are high-affinity substrates for OCTs [157], and widen the concentration gaps in OCT-expressing and non-expressing brain cells.
6.2.2 Efflux Transporters
The best-known efflux transporters are the ATP-binding cassette (ABC) transporters, including P-glycoprotein (P-gp/ABCB1), multidrug resistance associated protein 1 (MRP1/ABCC1), and breast cancer resistance protein (BCRP/ABCG2). Within the brain, P-gp and MRP1 are not only expressed at the BBB but also on the brain cells [169], whereas BCRP is primarily expressed on the endothelial cells that form the BBB [170,171,172,173].
In this section, we do not go further to discuss BCRP expression at the BBB as our main focus is on the active transport on the brain cells. A species difference in MRP1 expression in brain cells has been observed between humans and rats, where the gene expression was detected in the three main types of brain cells in rats [174,175,176] but not in humans [177]. P-glycoprotein is mainly identified at glial cells [175, 178], but pathological conditions (such as seizures) and/or aging can increase the expression of P-gp also at neurons [179, 180]. The efflux transporters present on the brain cell membrane probably have an important impact on the distribution of antiretroviral drugs into the brain cells (such as abacavir and lamivudine), as antiretroviral drugs are substrates of these transporters [164, 181, 182]. By using different CNS cell lines, Patel et al. tested three antiretroviral drugs and revealed cell-type specific differences in intracellular drug concentrations (up to three-fold) between microglia and astrocytes, although the role of certain active transporters still remains to be further elucidated [183].
6.3 Drug-Metabolizing Enzymes Inside Brain Cells
Cytochrome P450 families and UGTs are the most predominant metabolic enzymes involved in phase I and II biotransformation reactions in the liver that are required for drug elimination [184,185,186]. Even though drug-metabolizing enzymes are less expressed in the brain compared with the liver, some of these drug-metabolizing enzymes may also exhibit functional activity in the brain. [19, 20]. Different methodological approaches have been used to demonstrate the presence and functional consequences of the different CYP and UGT isoforms on the distinct cell types within the brains of rodents and humans [20, 187]. Through the use of rat and human brain microsomes, Voirol et al. demonstrated functional metabolic activities of two essential CYP enzymes, CYP3A4 and CYP2D6 [188]. Further studies have confirmed mRNA expression of CYP2D6 and the protein presence of CYP3A4 in human neurons [189, 190]. In the brain, limited mRNA and/or protein expression of different UGT isoforms was also detected in both neuronal and non-neuronal cells [187]. Noteworthy, studies in human brain microsomes demonstrated UGT-dependent glucuronidation of morphine, which provides evidence for functional enzymatic activity of UGTs within the brain [191].
Antiviral agents used for treating viral encephalitis could be the substrates of some isoforms of these enzyme families. For instance, dolutegravir is primarily metabolized by UGT1A1, with a small portion metabolized by CYP3A4 [192]. Expression of UGT1A1 mRNA and CYP3A4 protein were found in rat astrocytes and human neurons, respectively [188, 193]. In addition, abacavir is metabolized in the liver by alcohol dehydrogenase (ADH1A) and UGT1A1 at a ratio of 1:1 [194]. This isoform of ADH1 also exists in the rat CNS [195] and significant ADH activity was found in the rat brain, although it is still lower than its counterpart in the rat liver [196].
With the information provided above, intracellular drug concentrations might be also affected by these enzymes, which are differentially expressed in each type of brain cell.
6.4 Virus Resistance to Antiviral Treatment
Mutations at the genetic structure of virus-encoded enzymes are thought to be the main reason for the resistance to antiviral drugs [197]. These mutations are often related to different modes of action of the antiviral compounds. For instance, the existence of viral kinase mutants could largely explain the viral resistance to acyclovir or ganciclovir in the treatment of HSV or HCMV as the mutants cannot activate the initial phosphorylation of these two compounds [197]. Although less frequent, the mutations in the gene of viral DNA polymerase are the primary reason for the development of HSV/HCMV resistance to foscarnet and cidofovir because these two drugs do not require the involvement of viral kinases to inhibit viral replications [197]. The mechanism of HIV drug resistance is complex and drug resistance can be driven by different point mutations in the gene of HIV-encoded enzymes, which does not impair HIV replication but blocks the action of antiviral drugs [198].
The occurrence of acyclovir resistance is rare (~0.3%) in immunocompetent patients with HSV. However, in immunocompromised hosts, the prevalence of viral resistance to acyclovir is relatively higher, typically between 4 and 7% [199]. Ganciclovir-resistant HCMV infection often occurs in organ transplant recipients with an incidence rate of 5–10% and in immunodeficient patients with an incidence rate of 5% [200]. Foscarnet and cidofovir are the options when first-line therapies fail in HCMV/HSV infection because of drug resistance, as their actions do not require prior activation by viral kinases, although resistance to foscarnet and cidofovir can occur as well [201,202,203]. The prevalence of HIV resistant to antiretroviral therapy ranges from 3.5 to 25.8% across different countries [204]. Some recent studies also suggest the rising levels of HIV drug resistance in low-income countries due to poor antiretroviral therapy and surveillance of resistance-related mutations [205, 206].
It seems that for both immunocompetent and immunocompromised patients drug resistance in CNS viral infection is very rare, with limited information from a few case reports [207,208,209,210]. The diagnosis of drug resistance is normally conducted by genotype sequencing of both virus-encoded TK and DNA polymerase in CSF samples, based on these case reports. Furthermore, once drug resistance is confirmed, current therapeutic agents need to be substituted by or combined with other drugs having a distinct mechanism of action to overcome the resistance. For instance, among patients who initially received acyclovir or ganciclovir treatment, the immediate involvement of foscarnet should be considered after the occurrence of drug resistance [207, 208]. Insufficient CNS exposure is often associated with inadequate viral suppression, allowing viruses ample time to develop resistance-related gene mutations [211]. Increasing the drug dose or using drugs with better CNS penetration therefore is a potential strategy to prevent antiviral drug resistance [209, 210].
In summary, it seems that insufficient intracellular concentrations and antiviral drug resistance are important reasons for drug treatment failures. The former might also facilitate the evolution of resistance to antiviral drugs owing to incomplete suppression of viral replication. For a specific population such as immunocompromised patients, they have a higher risk of developing drug resistance during the treatment course, and thus optimization of dosing regimens as well as gene mutation surveillance are warranted for them.
It is important to note that there are no available intracellular PK data from clinical studies on viral CNS infections that can be used for investigating the direct relationship of (in)adequate intracellular exposure and drug treatment success/failure. As sampling from the human brain, including brain cells, is highly restricted, indirect approaches should be developed as an alternative to obtain the intracellular PK information that can be used to assess whether antiviral drugs fail to effectively inhibit viral replication. In many current studies involved with CNS viral infection, PK data are commonly generated from CSF, which typically do not represent brain ECF [212], let alone brain ICF concentrations at the site where viruses reside.
7 Mathematical Modeling Approaches for Optimizing Antiviral Treatment
Although it is crucial to know whether antiviral drugs can cross cellular membranes of brain cells in a sufficient manner, there remains a knowledge gap in brain intracellular pharmacokinetics because direct sampling from the human brain to obtain PK data on intracellular drug concentrations is highly restricted. Mathematical modeling approaches are an alternative way to predict drug concentrations, wherein population pharmacokinetic (POP-PK) models take up a relatively large proportion of the whole methods in antiviral treatment [213, 214].
7.1 POP-PK Model
Population-pharmacokinetic modeling have been successfully applied for HIV drugs [215,216,217]. A key advantage of these models is that they link plasma pharmacokinetics to the peripheral blood mononuclear cell compartment that could be a biomarker of intracellular target site concentrations, and related to intracellular drug action. Letendre et al. developed a POP-PK model using data from both plasma and CSF to investigate the CNS penetration of indinavir [218]. The model predicted that CSF drug concentrations exceed a 95% inhibitory concentration range for clinical isolates, which suggests that the current dosing regimen may be sufficient. As indicated earlier, CSF concentrations cannot generally be taken as a good indication of brain ECF/ICF concentrations [212]. Population-pharmacokinetic models, being data driven, are heavily based on actual measurements of drug concentrations in either plasma or CSF, which cannot address intracellular concentrations of brain cells.
7.2 PBPK Model
Here, we discuss the use of in silico PBPK models for enhancing our comprehension of drug treatment by predicting the intracellular drug concentrations. Compared to the POP-PK models that fit the model with existing data, the physiologically based models need a good understanding of mechanisms and integration of the parameters on both human brain physiology and drug properties into a mathematical model to predict intracellular drug concentrations, i.e., a knowledge-driven or bottom-up approach. Then PK-PD relationships can be interpreted by comparing the predicted intracellular concentrations to the in vitro concentrations based on drug effects (IC50 or half maximal effective concentration).
The PBPK models are “bottom up”, which require a deep understanding of the physiological properties of organs, tissues as well as tissue-related cells. Compared to empirical models, one of the advantages of PBPK models is that target site concentrations can be predicted based on tissue-specific and cell-specific data. Thus, both drug-specific and system-specific information can be used as inputs to help construct these models. Moreover, cross-species PBPK modeling for drug development could be achieved by replacing the physiological values of one species by another.
7.2.1 Semi-PBPK Modeling for CNS Regions
Some PBPK models have been applied for evaluating the toxicity or effectiveness of antiviral drugs based on the predicted data of intracellular drug concentrations [219, 220]. These models are full PBPK models that represent the principal organs in the human body but miss the depth of specific mechanisms in the CNS. Of note, the intracellular concentrations in the brain were not simulated as these models serve for improving the treatments of patients with infections in locations other than the CNS.
To investigate exposure-related effects of antiviral drugs in the brain, Ito et al. constructed a CNS PBPK model including three compartments, i.e., the blood, the BBB, and the whole brain region [221]. Active transport at the BBB was also taken into account, by utilizing the information on the functional characteristics of drug transporters in mice studies. Given that this model employs a single general compartment to represent the entire brain, the human simulation focused on generating the area under the unbound brain drug concentration–time curve to reflect CNS exposure. However, it is important to note that this simplified representation may not fully capture the complex distribution patterns within the brain. The use of a single general compartment for the entire brain overlooks regional variations in drug distribution and may lead to an oversimplification of the drug PK profile. Despite these limitations, simulating the area under the unbound brain drug concentration–time curve provides a practical approximation of drug CNS exposure. According to the analysis from Ito et al., high brain exposure (seven-fold higher area under the concentration–time curve) to oseltamivir occurs among the population with rare genetic variants in active transporters which may explain why only a few patients experienced neuropsychiatric adverse reactions.
7.2.2 PBPK Modeling of CNS Regions into Physiological Compartments
Several PBPK models [222,223,224,225] have been developed to enhance the structural accuracy and mechanistic representation of the anatomy and physiology in CNS regions. These models divided the CNS regions into multiple physiological compartments representing CSF and brain parenchyma, interlinking these physiological compartments through fluid bulk flows. Despite validation of the models in animal and human data, their application is limited to predicting either brain extracellular or total concentrations, owing to the simplification of the brain structure within these models.
However, we need a model that explicitly distinguishes the extracellular and intracellular space, as different mechanisms are responsible for the pharmacokinetics in these physiological CNS compartments. To achieve this, more sophisticated CNS models were built to better tell apart the brain parenchyma and its surrounding environment.
An example of such a model is the Leiden CNS PBPK model (LeiCNS-PK3.0), which is capable of distinguishing extracellular and intracellular concentrations. This model is constructed by utilizing the data on CNS physiological characteristics, drug properties, and plasma pharmacokinetics. With such information available, it can adequately predict drug concentrations in brain ECF, brain ICF, subcellular lysosomes, and CSF in lateral ventricles, the third and fourth ventricle, cisterna magma, and subarachnoid space that includes the lumbar CSF [226]. In this model, drug transport across the brain barriers and brain cell membrane is mechanistically divided into passive diffusion and active transport, and includes the pH partition theory on the basis of actual pH in the different physiological compartments, and non-specific binding to brain cell lipid bilayers. Using brain ICF pharmacokinetic predictions, and information on metabolic conversion where needed, active metabolite concentrations can be predicted and then related to PD indicators (IC50, half maximal effective concentration, or Km) for the evaluation of antiviral efficacy. In a recent application of the LeiCNS-PK3.0 model [227], CNS antiviral efficacy was evaluated for coronavirus disease 2019 drug candidates including nirmatrelvir, molnupiravir, and remdesivir. By incorporating the information on the conversion of parent drug to metabolites in in vitro studies, the model further predicted the intracellular concentrations of active metabolites. The ICF PK predictions were subsequently used to assess antiviral drug activity by comparing them to reported EC90 values of nirmatrelvir, molnupiravir, and remdesivir against coronavirus variants. Based on these LeiCNS-PK3.0 predictions, it was shown that only nirmatrelvir could reach an effective PK exposure based on the current dosing regimen (above the variants’ EC90).
Heitman et al. [228] used the LeiCNS PBPK model structure and divided the brain tissue over different regions—the thalamus, cortex, basal ganglia, and the rest of the brain—and added different lipid compositions for these regions to predict how drugs distribute intracellularly among various brain locations. It holds the potential to assess variations in the efficacy of antiviral drugs based on their exposure within different brain regions. Such predictive capabilities enable a more nuanced understanding of drug performance within specific anatomical zones of the CNS, offering insights crucial for optimizing therapeutic regimens targeting CNS viral infections.
7.2.3 Application of CNS PBPK Model with the Consideration of a Sex Difference
A sex difference may play a role in both the pharmacokinetics (drug distribution) and pharmacodynamics (severity of CNS viral infection) of antiviral agents. Regarding PK differences between men and women, several studies [229,230,231] suggest that these variations might arise from physiological factors such as the plasma protein level, renal clearance, and hepatic metabolism. Zooming in on the CNS, sex-specific differences have been observed in human physiological parameters such as the cerebral blood flow rate and the total brain volume [232], and the CYP enzyme expression at least in rat models [233].
Concerning sex-related pharmacodynamics, differences in the severity and incidence of CNS viral infections have been reported [234,235,236]. The differential outcomes and pathogenesis of viral infections between sexes are associated with an immunity to viruses [237]. Multiple factors contribute to this, including variations in the expression of sex chromosome-encoded viral-sensing receptors, virus entry receptors, as well as the quantity and quality of immune cells. However, the variation in immunity due to sex mainly affects viral dynamics.
To address these PK/PD differences in the whole population, the first step is to identify the factors that potentially affect the antiviral drug pharmacokinetics at the CNS cellular target site. After incorporating such information into the PBPK model, the subsequent step should be the development of a mechanistic model linking viral dynamics to pharmacokinetics (PK-PD model). For this, information on the impact of sex on immunity is crucial. However, constructing an accurate PD model faces challenges because of the unclear underlying mechanisms governing the interaction between the virus and cell receptors, and the scarcity of quantitative data. Hence, experimental data are required.
In summary, CNS PBPK models stand out for their effectiveness in predicting brain ICF drug concentrations. To account for the distinctive PK profiles between ECF and ICF, it is imperative to mechanistically divide the PBPK model into two segments corresponding to ECF and ICF, for the sake of enhancing the precision of predicting drug intracellular distribution. For those interested in exploring the brain’s regional distribution of antiviral drugs, it is advisable to include regions of interest within the model. To explore even further, it becomes intriguing to distinct drug intracellular distributions among neurons, microglia, and astrocytes based on the PBPK model approach. Understanding cellular membrane transporter expression and functionality also adds valuable information, which can be integrated into the PBPK model to account for active transport mechanisms on the brain cell membrane. With the consideration of PK/PD differences related to sex, the model can be used to optimize the existing and novel therapies for the entire population.
8 Concluding Remarks
Neurotropic viruses have multiple pathways to enter the CNS and then target the interior of the brain cells where they reside, replicate, and cause disease conditions. Although the use of vaccines has effectively reduced the occurrence of CNS viral infection, people still suffer or even die from viral infections of the CNS. While for certain types of viruses such as HIV or HSV, efficacious vaccines are under development, adequate drug treatments are still necessary and important if vaccines do not work properly or are not taken. One reason for why antiviral drugs may lack efficacy for the treatment of CNS infections is that they may reach inadequate intracellular concentrations in the CNS to block viral replication. Intracellular exposure-response relationships should be further investigated to understand which dosing regimens can provide the desired intracellular concentrations that are necessary to block viral replication. Such information can be difficult to obtain from humans and requires therefore alternative strategies. The use of comprehensive CNS PBPK models, that incorporate predictions of the brain ICF, can therefore be an important approach to explore intracellular drug PK profiles and its relationship to antiviral drug effects in the CNS.
References
Bookstaver PB, Mohorn PL, Shah A, et al. Management of viral central nervous system infections: a primer for clinicians. J Central Nerv Syst Dis. 2017;9:1179573517703342.
Richie MB, Josephson SA. A practical approach to meningitis and encephalitis. Semin Neurol. 2015;35(6):611–20.
Swanson PA, McGavern DB. Viral diseases of the central nervous system. Curr Opin Virol. 2015;11:44–54.
Cantu RM, Das JM. Viral meningitis. Treasure Island: StatPearls Publishing LLC; 2022.
Ben Abid F, Abukhattab M, Ghazouani H, et al. Epidemiology and clinical outcomes of viral central nervous system infections. Int J Infect Dis. 2018;73:85–90.
Sköldenberg B, Forsgren M, Alestig K, et al. Acyclovir versus vidarabine in herpes simplex encephalitis: randomised multicentre study in consecutive Swedish patients. Lancet. 1984;2(8405):707–11.
Whitley RJ, Soong SJ, Dolin R, et al. Adenine arabinoside therapy of biopsy-proved herpes simplex encephalitis. National Institute of Allergy and Infectious Diseases collaborative antiviral study. N Engl J Med. 1977;297(6):289–94.
Doms RW. Basic concepts: a step-by-step guide to viral infection. In: Viral pathogenesis. Elsevier; 2016. p. 29–40.
Koyuncu OO, Hogue IB, Enquist LW. Virus infections in the nervous system. Cell Host Microbe. 2013;13(4):379–93.
Tunkel AR, Glaser CA, Bloch KC, et al. The management of encephalitis: clinical practice guidelines by the Infectious Diseases Society of America. Clin Infect Dis. 2008;47(3):303–27.
Palella FJ Jr, Delaney KM, Moorman AC, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338(13):853–60.
Whitley RJ, Alford CA, Hirsch MS, et al. Vidarabine versus acyclovir therapy in herpes simplex encephalitis. N Engl J Med. 1986;314(3):144–9.
Anduze-Faris BM, Fillet AM, Gozlan J, et al. Induction and maintenance therapy of cytomegalovirus central nervous system infection in HIV-infected patients. AIDS. 2000;14(5):517–24.
Cutler R, Trentman S, Jandarov R, et al. Severe neurologic impairment persists despite potent ART in HIV encephalopathy. Open Forum Infect Dis. 2019;6(Suppl. 2):S192. https://doi.org/10.1093/ofid/ofz360.439.
Ludlow M, Kortekaas J, Herden C, et al. Neurotropic virus infections as the cause of immediate and delayed neuropathology. Acta Neuropathol. 2016;131(2):159–84.
Tyler KL. Acute viral encephalitis. N Engl J Med. 2018;379(6):557–66.
Graff CL, Pollack GM. Drug transport at the blood-brain barrier and the choroid plexus. Curr Drug Metab. 2004;5(1):95–108.
Lee G, Dallas S, Hong M, et al. Drug transporters in the central nervous system: brain barriers and brain parenchyma considerations. Pharmacol Rev. 2001;53(4):569–96.
Minn A, Ghersi-Egea JF, Perrin R, et al. Drug metabolizing enzymes in the brain and cerebral microvessels. Brain Res Brain Rev. 1991;16(1):65–82.
Meyer RP, Gehlhaus M, Knoth R, et al. Expression and function of cytochrome p450 in brain drug metabolism. Curr Drug Metab. 2007;8(4):297–306.
Tirabassi RS, Townley RA, Eldridge MG, et al. Molecular mechanisms of neurotropic herpesvirus invasion and spread in the CNS. Neurosci Biobehav Rev. 1998;22(6):709–20.
Suenaga T, Satoh T, Somboonthum P, et al. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc Natl Acad Sci U S A. 2010;107(2):866–71.
Smith G. Herpesvirus transport to the nervous system and back again. Ann Rev Microbiol. 2012;66:153–76.
Rajbhandari L, Shukla P, Jagdish B, et al. Nectin-1 is an entry mediator for varicella-zoster virus infection of human neurons. J Virol. 2021;95(22):e0122721.
Shiers S, Ray PR, Wangzhou A, et al. ACE2 and SCARF expression in human dorsal root ganglion nociceptors: implications for SARS-CoV-2 virus neurological effects. Pain. 2020;161(11):2494–501.
Rodríguez Cruz PM, Cossins J, Beeson D, et al. The neuromuscular junction in health and disease: molecular mechanisms governing synaptic formation and homeostasis. Front Mol Neurosci. 2020;13:610964.
Ren R, Racaniello VR. Human poliovirus receptor gene expression and poliovirus tissue tropism in transgenic mice. J Virol. 1992;66(1):296–304.
Ugolini G. Rabies virus as a transneuronal tracer of neuronal connections. Adv Virus Res. 2011;79:165–202.
Lafon M. Rabies virus receptors. J Neurovirol. 2005;11(1):82–7.
Suh J, Amato AA. Neuromuscular complications of coronavirus disease-19. Curr Opin Neurol. 2021;34(5):669–74.
Wilson RI, Mainen ZF. Early events in olfactory processing. Ann Rev Neurosci. 2006;29:163–201.
Mori I, Nishiyama Y, Yokochi T, et al. Olfactory transmission of neurotropic viruses. J Neurovirol. 2005;11(2):129–37.
Bilinska K, Jakubowska P, Von Bartheld CS, et al. Expression of the SARS-CoV-2 entry proteins, ACE2 and TMPRSS2, in cells of the olfactory epithelium: identification of cell types and trends with age. ACS Chem Neurosci. 2020;11(11):1555–62.
Daniels BP, Klein RS. Viral sensing at the blood-brain barrier: new roles for innate immunity at the CNS vasculature. Clin Pharmacol Ther. 2015;97(4):372–9.
Růžek D, Salát J, Singh SK, et al. Breakdown of the blood–brain barrier during tick-borne encephalitis in mice is not dependent on CD8+ T cells. PLoS ONE. 2011;6(5):e20472.
Bonney S, Seitz S, Ryan CA, et al. Gamma interferon alters junctional integrity via Rho kinase, resulting in blood-brain barrier leakage in experimental viral encephalitis. MBio. 2019;10(4):10–1128.
Wang P, Dai J, Bai F, et al. Matrix metalloproteinase 9 facilitates West Nile virus entry into the brain. J Virol. 2008;82(18):8978–85.
Thompson D, Brissette CA, Watt JA. The choroid plexus and its role in the pathogenesis of neurological infections. Fluids Barriers CNS. 2022;19(1):75.
Johanson CE. Chapter 11: Choroid plexus-cerebrospinal fluid transport dynamics: support of brain health and a role in neurotherapeutic. In: Conn PM, editor. Conn’s translational neuroscience. Academic Press; 2017. p. 233–61.
Jakhmola S, Indari O, Chatterjee S, et al. SARS-CoV-2, an underestimated pathogen of the nervous system. SN Compr Clin Med. 2020;2:1–10.
Hamming I, Timens W, Bulthuis MLC, et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus: a first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631–7.
Harapan BN, Yoo HJ. Neurological symptoms, manifestations, and complications associated with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease 19 (COVID-19). J Neurol. 2021;268(9):3059–71.
Man S, Ubogu EE, Ransohoff RM. Inflammatory cell migration into the central nervous system: a few new twists on an old tale. Brain Pathol. 2007;17(2):243–50.
Engelhardt B, Ransohoff RM. Capture, crawl, cross: the T cell code to breach the blood-brain barriers. Trends Immunol. 2012;33(12):579–89.
Solár P, Zamani A, Kubíčková L, et al. Choroid plexus and the blood-cerebrospinal fluid barrier in disease. Fluids Barriers CNS. 2020;17(1):35.
Xu Z, Waeckerlin R, Urbanowski MD, et al. West Nile virus infection causes endocytosis of a specific subset of tight junction membrane proteins. PLoS ONE. 2012;7(5):e37886.
Casiraghi C, Dorovini-Zis K, Horwitz MS. Epstein-Barr virus infection of human brain microvessel endothelial cells: a novel role in multiple sclerosis. J Neuroimmunol. 2011;230(1–2):173–7.
Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410(6831):988–94.
Huang J, Zheng M, Tang X, et al. Potential of SARS-CoV-2 to cause CNS infection: biologic fundamental and clinical experience. Front Neurol. 2020;11:659.
Helenius A, Kartenbeck J, Simons K, et al. On the entry of Semliki forest virus into BHK-21 cells. J Cell Biol. 1980;84(2):404–20.
Dimitrov DS. Virus entry: molecular mechanisms and biomedical applications. Nat Rev Microbiol. 2004;2(2):109–22.
Wilen CB, Tilton JC, Doms RW. HIV: cell binding and entry. Cold Spring Harbor Perspect Med. 2012;2(8):a006866.
Hilterbrand AT, Heldwein EE. Go go gadget glycoprotein!: HSV-1 draws on its sizeable glycoprotein tool kit to customize its diverse entry routes. PLoS Pathog. 2019;15(5):e1007660.
Pauza CD, Price TM. Human immunodeficiency virus infection of T cells and monocytes proceeds via receptor-mediated endocytosis. J Cell Biol. 1988;107(3):959–68.
Schaeffer E, Geleziunas R, Greene WC. Human immunodeficiency virus type 1 Nef functions at the level of virus entry by enhancing cytoplasmic delivery of virions. J Virol. 2001;75(6):2993–3000.
Stein BS, Gowda SD, Lifson JD, et al. pH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell. 1987;49(5):659–68.
Permanyer M, Ballana E, Esté JA. Endocytosis of HIV: anything goes. Trends Microbiol. 2010;18(12):543–51.
Schaeffer E, Soros VB, Greene WC. Compensatory link between fusion and endocytosis of human immunodeficiency virus type 1 in human CD4 T lymphocytes. J Virol. 2004;78(3):1375–83.
Duan L, Zheng Q, Zhang H, et al. The SARS-CoV-2 spike glycoprotein biosynthesis, structure, function, and antigenicity: implications for the design of spike-based vaccineiImmunogens. Front Immunol. 2020;11:576622.
Nomaguchi M, Fujita M, Miyazaki Y, et al. Viral tropism. Front Microbiol. 2012;3:281.
Blankson JN, Persaud D, Siliciano RF. The challenge of viral reservoirs in HIV-1 infection. Annu Rev Med. 2002;53:557–93.
Jordan CA, Watkins BA, Kufta C, et al. Infection of brain microglial cells by human immunodeficiency virus type 1 is CD4 dependent. J Virol. 1991;65(2):736–42.
Liu Y, Liu H, Kim BO, et al. CD4-independent infection of astrocytes by human immunodeficiency virus type 1: requirement for the human mannose receptor. J Virol. 2004;78(8):4120–33.
Wallet C, De Rovere M, Van Assche J, et al. Microglial cells: the main HIV-1 reservoir in the brain. Front Cell Infect Microbiol. 2019;9:362.
Guzman G, Oh S, Shukla D, et al. Expression of entry receptor nectin-1 of herpes simplex virus 1 and/or herpes simplex virus 2 in normal and neoplastic human nervous system tissues. Acta Virol. 2006;50(1):59–66.
Castellanos KJ, Gagyi E, Kormos B, et al. Increased axonal expression of nectin-1 in multiple sclerosis plaques. Neurol Sci. 2013;34(4):465–9.
Pugazhenthi S, Nair S, Velmurugan K, et al. Varicella-zoster virus infection of differentiated human neural stem cells. J Virol. 2011;85(13):6678–86.
Kopp SJ, Ranaivo HR, Wilcox DR, et al. Herpes simplex virus serotype and entry receptor availability alter CNS disease in a mouse model of neonatal HSV. Pediatr Res. 2014;76(6):528–34.
Kopp SJ, Banisadr G, Glajch K, et al. Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc Natl Acad Sci U S A. 2009;106(42):17916–20.
Shives KD, Tyler KL, Beckham JD. Molecular mechanisms of neuroinflammation and injury during acute viral encephalitis. J Neuroimmunol. 2017;308:102–11.
D'Aiuto L, Bloom DC, Naciri JN, et al. Modeling herpes simplex virus 1 infections in human central nervous system neuronal cells using two- and three-dimensional cultures derived from induced pluripotent stem cells. J Virol. 2019;93(9):00111.
Connolly SA, Jardetzky TS, Longnecker R. The structural basis of herpesvirus entry. Nat Rev Microbiol. 2021;19(2):110–21.
Teissier N, Fallet-Bianco C, Delezoide A-L, et al. Cytomegalovirus-induced brain malformations in fetuses. J Neuropathol Exp Neurol. 2014;73(2):143–58.
Krstanović F, Britt WJ, Jonjić S, et al. Cytomegalovirus infection and inflammation in developing brain. Viruses. 2021;13(6):1078.
Cama VF, Marín-Prida J, Acosta-Rivero N, et al. The microglial NLRP3 inflammasome is involved in human SARS-CoV-2 cerebral pathogenicity: a report of three post-mortem cases. J Neuroimmunol. 2021;361:577728.
Matschke J, Lütgehetmann M, Hagel C, et al. Neuropathology of patients with COVID-19 in Germany: a post-mortem case series. Lancet Neurol. 2020;19(11):919–29.
Song E, Zhang C, Israelow B, et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J Exp Med. 2021;218(3):e20202135.
Shen W-B, Logue J, Yang P, et al. SARS-CoV-2 invades cognitive centers of the brain and induces Alzheimer’s-like neuropathology. BioRxiv. 2022;8:416.
McFadden G, Mohamed MR, Rahman MM, et al. Cytokine determinants of viral tropism. Nat Rev Immunol. 2009;9(9):645–55.
McFadden G. Poxvirus tropism. Nat Rev Microbiol. 2005;3(3):201–13.
Schneider-Schaulies J. Cellular receptors for viruses: links to tropism and pathogenesis. J Gen Virol. 2000;81(Pt 6):1413–29.
Borman AM, Le Mercier P, Girard M, et al. Comparison of picornaviral IRES-driven internal initiation of translation in cultured cells of different origins. Nucleic Acids Res. 1997;25(5):925–32.
Gromeier M, Alexander L, Wimmer E. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc Natl Acad Sci U S A. 1996;93(6):2370–5.
Ohka S, Nomoto A. The molecular basis of poliovirus neurovirulence. Dev Biol. 2001;105:51–8.
Yanagiya A, Ohka S, Hashida N, et al. Tissue-specific replicating capacity of a chimeric poliovirus that carries the internal ribosome entry site of hepatitis C virus in a new mouse model transgenic for the human poliovirus receptor. J Virol. 2003;77(19):10479–87.
Belsham GJ, Sonenberg N. Picornavirus RNA translation: roles for cellular proteins. Trends Microbiol. 2000;8(7):330–5.
Shiroki K, Ishii T, Aoki T, et al. Host range phenotype induced by mutations in the internal ribosomal entry site of poliovirus RNA. J Virol. 1997;71(1):1–8.
Macadam AJ, Pollard SR, Ferguson G, et al. Genetic basis of attenuation of the Sabin type 2 vaccine strain of poliovirus in primates. Virology. 1993;192(1):18–26.
Lu HH, Wimmer E. Poliovirus chimeras replicating under the translational control of genetic elements of hepatitis C virus reveal unusual properties of the internal ribosomal entry site of hepatitis C virus. Proc Natl Acad Sci U S A. 1996;93(4):1412–7.
Ida-Hosonuma M, Iwasaki T, Yoshikawa T, et al. The alpha/beta interferon response controls tissue tropism and pathogenicity of poliovirus. J Virol. 2005;79(7):4460–9.
García-Sastre A, Durbin RK, Zheng H, et al. The role of interferon in influenza virus tissue tropism. J Virol. 1998;72(11):8550–8.
Ryman KD, Klimstra WB, Nguyen KB, et al. Alpha/beta interferon protects adult mice from fatal Sindbis virus infection and is an important determinant of cell and tissue tropism. J Virol. 2000;74(7):3366–78.
Tompa DR, Immanuel A, Srikanth S, et al. Trends and strategies to combat viral infections: a review on FDA approved antiviral drugs. Int J Biol Macromol. 2021;172:524–41.
Kincaid O, Lipton HL. Viral myelitis: an update. Curr Neurol Neurosci Rep. 2006;6(6):469–74.
Tyler KL. Editorial commentary: failure of adjunctive valacyclovir to improve outcomes in herpes simplex encephalitis. Clin Infect Dis. 2015;61(5):692–4.
Van Rompay AR, Johansson M, Karlsson A. Phosphorylation of nucleosides and nucleoside analogs by mammalian nucleoside monophosphate kinases. Pharmacol Ther. 2000;87(2–3):189–98.
Gumina G, Choi Y, Chu CK. Chapter 1: Recent advances in antiviral nucleosides. In: Chu CK, editor. antiviral nucleosides. Amsterdam: Elsevier; 2003. p. 1–76.
Kausar S, Said Khan F, Ishaq Mujeeb Ur Rehman M, et al. A review: mechanism of action of antiviral drugs. Int J Immunopathol Pharmacol. 2021;35:20587384211002620.
Jordheim LP, Durantel D, Zoulim F, et al. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov. 2013;12(6):447–64.
Whitley RJ, Gnann JW. Viral encephalitis: familiar infections and emerging pathogens. Lancet. 2002;359(9305):507–13.
Ibrahim AI, Obeid MT, Jouma MJ, et al. Prevalence of herpes simplex virus (types 1 and 2), varicella-zoster virus, cytomegalovirus, and human herpesvirus 6 and 7 DNA in cerebrospinal fluid of Middle Eastern patients with encephalitis. J Clin Microbiol. 2005;43(8):4172–4.
Steiner I, Budka H, Chaudhuri A, et al. Viral meningoencephalitis: a review of diagnostic methods and guidelines for management. Eur J Neurol. 2010;17(8):999-e57.
Kimberlin DW, Lin CY, Jacobs RF, et al. Safety and efficacy of high-dose intravenous acyclovir in the management of neonatal herpes simplex virus infections. Pediatrics. 2001;108(2):230–8.
Whitley R, Arvin A, Prober C, et al. A controlled trial comparing vidarabine with acyclovir in neonatal herpes simplex virus infection. Infectious Diseases Collaborative Antiviral Study Group. N Engl J Med. 1991;324(7):444–9.
De Broucker T, Mailles A, Chabrier S, et al. Acute varicella zoster encephalitis without evidence of primary vasculopathy in a case-series of 20 patients. Clin Microbiol Infect. 2012;18(8):808–19.
Herlin LK, Hansen KS, Bodilsen J, et al. Varicella zoster virus encephalitis in Denmark from 2015 to 2019: a nationwide prospective cohort study. Clin Infect Dis. 2021;72(7):1192–9.
Kohil A, Jemmieh S, Smatti MK, et al. Viral meningitis: an overview. Arch Virol. 2021;166(2):335–45.
Asundi A, Cervantes-Arslanian AM, Lin NH, et al. Infectious myelitis. Semin Neurol. 2019;39(4):472–81.
Pouplin T, Pouplin JN, Van Toi P, et al. Valacyclovir for herpes simplex encephalitis. Antimicrob Agents Chemother. 2011;55(7):3624–6.
Gnann JW, Sköldenberg B, Hart J, et al. Herpes simplex encephalitis: lack of clinical benefit of long-term valacyclovir therapy. Clin Infect Dis. 2015;61(5):683–91.
Momméja-Marin H, Lafaurie M, Scieux C, et al. Herpes simplex virus type 2 as a cause of severe meningitis in immunocompromised adults. Clin Infect Dis. 2003;37(11):1527–33.
Price RW, Spudich S. Antiretroviral therapy and central nervous system HIV type 1 infection. J Infect Dis. 2008;197(Suppl. 3):S294–306.
d'Arminio Monforte A, Cinque P, Mocroft A, et al. Changing incidence of central nervous system diseases in the EuroSIDA cohort. Ann Neurol. 2004;55(3):320–8.
Sacktor N, Skolasky RL, Seaberg E, et al. Prevalence of HIV-associated neurocognitive disorders in the Multicenter AIDS cohort study. Neurology. 2016;86(4):334–40.
Letendre S, Marquie-Beck J, Capparelli E, et al. Validation of the CNS penetration-effectiveness rank for quantifying antiretroviral penetration into the central nervous system. Arch Neurol. 2008;65(1):65–70.
Letendre SL, Ellis RJ, Ances BM, et al. Neurologic complications of HIV disease and their treatment. Top HIV Med. 2010;18(2):45–55.
Daliparty VM, Balasubramanya R. HIV encephalitis. Treasure Island: StatPearls Publishing LLC; 2021.
Avedissian SN, Dyavar SR, Fox HS, et al. Pharmacologic approaches to HIV-associated neurocognitive disorders. Curr Opin Pharmacol. 2020;54:102–8.
Yilmaz A, Price RW, Gisslén M. Antiretroviral drug treatment of CNS HIV-1 infection. J Antimicrob Chemother. 2011;67(2):299–311.
Patel K, Ming X, Williams PL, et al. Impact of HAART and CNS-penetrating antiretroviral regimens on HIV encephalopathy among perinatally infected children and adolescents. AIDS. 2009;23(14):1893–901.
Sadowski LA, Upadhyay R, Greeley ZW, et al. Current drugs to treat infections with herpes simplex viruses-1 and -2. Viruses. 2021;13(7).
Varga A, Lionne C, Roy B. Intracellular metabolism of nucleoside/nucleotide analogues: a bottleneck to reach active drugs on HIV reverse transcriptase. Curr Drug Metab. 2016;17(3):237–52.
Groaz E, De Jonghe S. Overview of biologically active nucleoside phosphonates. Front Chem. 2020;8: 616863.
Slusarczyk M, Serpi M, Pertusati F. Phosphoramidates and phosphonamidates (ProTides) with antiviral activity. Antiviral Chem Chemother. 2018;26:2040206618775243.
Thornton PJ, Kadri H, Miccoli A, et al. Nucleoside phosphate and phosphonate prodrug clinical candidates. J Med Chem. 2016;59(23):10400–10.
Robertson KR, Miyahara S, Lee A, et al. Neurocognition with maraviroc compared with tenofovir in HIV. AIDS. 2016;30(15):2315–21.
Robertson K, Jiang H, Kumwenda J, et al. Improved neuropsychological and neurological functioning across three antiretroviral regimens in diverse resource-limited settings: AIDS Clinical Trials Group study a5199, the International Neurological Study. Clin Infect Dis. 2012;55(6):868–76.
Ma Q, Ocque AJ, Morse GD, et al. Switching to tenofovir alafenamide in elvitegravir-based regimens: pharmacokinetics and antiviral activity in cerebrospinal fluid. Clin Infect Dis. 2020;71(4):982–8.
Nicol MR, Pastick KA, Taylor J, et al. Cerebrospinal fluid and brain tissue penetration of tenofovir, lamivudine, and efavirenz in postmortem tissues with cryptococcal meningitis. Clin Transl Sci. 2019;12(5):445–9.
Fox HS, Niu M, Morsey BM, et al. Morphine suppresses peripheral responses and transforms brain myeloid gene expression to favor neuropathogenesis in SIV infection. Front Immunol. 2022;13:1012884.
Götte M. Remdesivir for the treatment of COVID-19: the value of biochemical studies. Curr Opin Virol. 2021;49:81–5.
Nevalainen OPO, Horstia S, Laakkonen S, et al. Effect of remdesivir post hospitalization for COVID-19 infection from the randomized SOLIDARITY Finland trial. Nat Commun. 2022;13(1):6152.
Berlin DA, Gulick RM, Martinez FJ. Severe COVID-19. N Engl J Med. 2020;383(25):2451–60.
Métifiot M, Marchand C, Pommier Y. HIV integrase inhibitors: 20-year landmark and challenges. Adv Pharmacol. 2013;67:75–105.
Jóźwik IK, Passos DO, Lyumkis D. Structural biology of HIV integrase strand transfer inhibitors. Trends Pharmacol Sci. 2020;41(9):611–26.
Zhao AV, Crutchley RD, Guduru RC, et al. A clinical review of HIV integrase strand transfer inhibitors (INSTIs) for the prevention and treatment of HIV-1 infection. Retrovirology. 2022;19(1):22.
Gandhi RT, Bedimo R, Hoy JF, et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2022 recommendations of the International Antiviral Society-USA Panel. JAMA. 2023;329(1):63–84.
Gelé T, Furlan V, Taburet AM, et al. Dolutegravir cerebrospinal fluid diffusion in HIV-1-infected patients with central nervous system impairment. Open Forum Infect Dis. 2019;6(6):ofz174.
Letendre SL, Mills AM, Tashima KT, et al. ING116070: a study of the pharmacokinetics and antiviral activity of dolutegravir in cerebrospinal fluid in HIV-1-infected, antiretroviral therapy-naive subjects. Clin Infect Dis. 2014;59(7):1032–7.
Tiraboschi J, Imaz A, Khoo S, et al. Total and unbound bictegravir concentrations and viral suppression in cerebrospinal fluid of human immunodeficiency virus-infected patients (Spanish HIV/AIDS Research Network, PreEC/RIS 56). J Infect Dis. 2020;221(9):1425–8.
Jakimiuk A, Piechal A, Wiercińska-Drapało A, et al. Central nervous system disorders after use of dolutegravir: evidence from preclinical and clinical studies. Pharmacol Rep. 2023;75(5):1138–51.
Ma Q, Schifitto G, Venuto C, et al. Effect of dolutegravir and sertraline on the blood brain barrier (BBB). J Neuroimmune Pharmacol. 2020;15(1):7–9.
Huang C, Hoque T, Bendayan R. Antiretroviral drugs efavirenz, dolutegravir and bictegravir dysregulate blood–brain barrier integrity and function. Front Pharmacol. 2023;14:1118580.
Piret J, Boivin G. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother. 2011;55(2):459–72.
Azimi T, Tavakolian S, Goudarzi H, et al. Global estimate of phenotypic and genotypic ganciclovir resistance in cytomegalovirus infections among HIV and organ transplant patients; a systematic review and meta-analysis. Microb Pathog. 2020;141:104012.
Pardridge WM. Drug transport across the blood-brain barrier. J Cereb Blood Flow Metab. 2012;32(11):1959–72.
Banks WA. Characteristics of compounds that cross the blood-brain barrier. BMC Neurol. 2009;9(Suppl 1):S3.
Lycke J, Malmeström C, Ståhle L. Acyclovir levels in serum and cerebrospinal fluid after oral administration of valacyclovir. Antimicrob Agents Chemother. 2003;47(8):2438–41.
Whitley RJ, Blum MR, Barton N, et al. Pharmacokinetics of acyclovir in humans following intravenous administration: a model for the development of parenteral antivirals. Am J Med. 1982;73(1a):165–71.
Lahiri CD, Reed-Walker K, Sheth AN, et al. Cerebrospinal fluid concentrations of tenofovir and emtricitabine in the setting of HIV-1 protease inhibitor-based regimens. J Clin Pharmacol. 2016;56(4):492–6.
Garcia CR, Rad AT, Saeedinejad F, et al. Effect of drug-to-lipid ratio on nanodisc-based tenofovir drug delivery to the brain for HIV-1 infection. Nanomedicine. 2022;17(13):959–78.
Ashraf T, Kao A, Bendayan R. Functional expression of drug transporters in glial cells: potential role on drug delivery to the CNS. Adv Pharmacol. 2014;71:45–111.
Hosoyamada M, Sekine T, Kanai Y, et al. Molecular cloning and functional expression of a multispecific organic anion transporter from human kidney. Am J Physiol. 1999;276(1):F122-8.
Naud J, Laurin L-P, Michaud J, et al. Effects of chronic renal failure on brain drug transporters in rats. Drug Metab Dispos. 2012;40(1):39–46.
Mori S, Takanaga H, Ohtsuki S, et al. Rat organic anion transporter 3 (rOAT3) is responsible for brain-to-blood efflux of homovanillic acid at the abluminal membrane of brain capillary endothelial cells. J Cerebral Blood Flow Metab. 2003;23(4):432–40.
Cha SH, Sekine T, Fukushima JI, et al. Identification and characterization of human organic anion transporter 3 expressing predominantly in the kidney. Mol Pharmacol. 2001;59(5):1277–86.
Minuesa G, Volk C, Molina-Arcas M, et al. Transport of lamivudine [(-)-beta-L-2’,3’-dideoxy-3’-thiacytidine] and high-affinity interaction of nucleoside reverse transcriptase inhibitors with human organic cation transporters 1, 2, and 3. J Pharmacol Exp Ther. 2009;329(1):252–61.
Zhang X, Wang R, Piotrowski M, et al. Intracellular concentrations determine the cytotoxicity of adefovir, cidofovir and tenofovir. Toxicol In Vitro. 2015;29(1):251–8.
Ye J, Liu Q, Wang C, et al. Benzylpenicillin inhibits the renal excretion of acyclovir by OAT1 and OAT3. Pharmacol Rep. 2013;65(2):505–12.
Bahn A, Ljubojevic M, Lorenz H, et al. Murine renal organic anion transporters mOAT1 and mOAT3 facilitate the transport of neuroactive tryptophan metabolites. Am J Physiol Cell Physiol. 2005;289(5):C1075-84.
Oshima N, Onimaru H, Matsubara H, et al. Uric acid, indoxyl sulfate, and methylguanidine activate bulbospinal neurons in the RVLM via their specific transporters and by producing oxidative stress. Neuroscience. 2015;304:133–45.
Wada S, Tsuda M, Sekine T, et al. Rat multispecific organic anion transporter 1 (rOAT1) transports zidovudine, acyclovir, and other antiviral nucleoside analogs. J Pharmacol Exp Ther. 2000;294(3):844–9.
Hasegawa M, Kusuhara H, Endou H, et al. Contribution of organic anion transporters to the renal uptake of anionic compounds and nucleoside derivatives in rat. J Pharmacol Exp Ther. 2003;305(3):1087–97.
Aloy B, Tazi I, Bagnis CI, et al. Is tenofovir alafenamide safer than tenofovir disoproxil fumarate for the kidneys? AIDS Rev. 2016;18(4):184–92.
Takeda M, Khamdang S, Narikawa S, et al. Human organic anion transporters and human organic cation transporters mediate renal antiviral transport. J Pharmacol Exp Ther. 2002;300(3):918–24.
Gasser PJ, Hurley MM, Chan J, et al. Organic cation transporter 3 (OCT3) is localized to intracellular and surface membranes in select glial and neuronal cells within the basolateral amygdaloid complex of both rats and mice. Brain Struct Funct. 2017;222(4):1913–28.
Yoshikawa T, Naganuma F, Iida T, et al. Molecular mechanism of histamine clearance by primary human astrocytes. Glia. 2013;61(6):905–16.
Busch AE, Karbach U, Miska D, et al. Human neurons express the polyspecific cation transporter hOCT2, which translocates monoamine neurotransmitters, amantadine, and memantine. Mol Pharmacol. 1998;54(2):342–52.
Ronaldson PT, Davis TP. Transport mechanisms at the blood-brain barrier and in cellular compartments of the neurovascular unit: focus on CNS delivery of small molecule drugs. Pharmaceutics. 2022;14(7):1501.
Aronica E, Gorter JA, Redeker S, et al. Localization of breast cancer resistance protein (BCRP) in microvessel endothelium of human control and epileptic brain. Epilepsia. 2005;46(6):849–57.
Daood M, Tsai C, Ahdab-Barmada M, et al. ABC transporter (P-gp/ABCB1, MRP1/ABCC1, BCRP/ABCG2) expression in the developing human CNS. Neuropediatrics. 2008;39(4):211–8.
van Vliet EA, Redeker S, Aronica E, et al. Expression of multidrug transporters MRP1, MRP2, and BCRP shortly after status epilepticus, during the latent period, and in chronic epileptic rats. Epilepsia. 2005;46(10):1569–80.
Lee G, Babakhanian K, Ramaswamy M, et al. Expression of the ATP-binding cassette membrane transporter, ABCG2, in human and rodent brain microvessel endothelial and glial cell culture systems. Pharm Res. 2007;24(7):1262–74.
Dallas S, Zhu X, Baruchel S, et al. Functional expression of the multidrug resistance protein 1 in microglia. J Pharmacol Exp Ther. 2003;307(1):282–90.
Declèves X, Regina A, Laplanche JL, et al. Functional expression of P-glycoprotein and multidrug resistance-associated protein (Mrp1) in primary cultures of rat astrocytes. J Neurosci Res. 2000;60(5):594–601.
Hirrlinger J, König J, Dringen R. Expression of mRNAs of multidrug resistance proteins (Mrps) in cultured rat astrocytes, oligodendrocytes, microglial cells and neurones. J Neurochem. 2002;82(3):716–9.
Nies AT, Jedlitschky G, König J, et al. Expression and immunolocalization of the multidrug resistance proteins, MRP1-MRP6 (ABCC1-ABCC6), in human brain. Neuroscience. 2004;129(2):349–60.
Bendayan R, Ronaldson PT, Gingras D,et al. In situ localization of P-glycoprotein (ABCB1) in human and rat brain. J Histochem Cytochem. 2006;54(10):1159–67.
Volk HA, Burkhardt K, Potschka H, et al. Neuronal expression of the drug efflux transporter P-glycoprotein in the rat hippocampus after limbic seizures. Neuroscience. 2004;123(3):751–9.
Bernstein HG, Hölzl G, Dobrowolny H, et al. Vascular and extravascular distribution of the ATP-binding cassette transporters ABCB1 and ABCC1 in aged human brain and pituitary. Mech Ageing Dev. 2014;141–142:12–21.
Adams JL, Greener BN, Kashuba ADM. Pharmacology of HIV integrase inhibitors. Curr Opin HIV AIDS. 2012;7(5):390–400.
Errasti-Murugarren E, Pastor-Anglada M. Drug transporter pharmacogenetics in nucleoside-based therapies. Pharmacogenomics. 2010;11(6):809–41.
Patel SH, Ismaiel OA, Mylott WR, et al. Cell-type specific differences in antiretroviral penetration and the effects of HIV-1 Tat and morphine among primary human brain endothelial cells, astrocytes, pericytes, and microglia. Neurosci Lett. 2019;712:134475.
McDonnell AM, Dang CH. Basic review of the cytochrome p450 system. J Adv Pract Oncol. 2013;4(4):263–8.
Mano ECC, Scott AL, Honorio KM. UDP-glucuronosyltransferases: structure, function and drug design studies. Curr Med Chem. 2018;25(27):3247–55.
Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther. 2013;138(1):103–41.
Ouzzine M, Gulberti S, Ramalanjaona N, et al. The UDP-glucuronosyltransferases of the blood-brain barrier: their role in drug metabolism and detoxication. Front Cell Neurosci. 2014;8:349.
Voirol P, Jonzier-Perey M, Porchet F, et al. Cytochrome P-450 activities in human and rat brain microsomes. Brain Res. 2000;855(2):235–43.
Gilham DE, Cairns W, Paine MJ, et al. Metabolism of MPTP by cytochrome P4502D6 and the demonstration of 2D6 mRNA in human foetal and adult brain by in situ hybridization. Xenobiotica. 1997;27(1):111–25.
Ghosh C, Marchi N, Desai NK, et al. Cellular localization and functional significance of CYP3A4 in the human epileptic brain. Epilepsia. 2011;52(3):562–71.
Wahlström A, Winblad B, Bixo M, et al. Human brain metabolism of morphine and naloxone. Pain. 1988;35(2):121–7.
Castellino S, Moss L, Wagner D, et al. Metabolism, excretion, and mass balance of the HIV-1 integrase inhibitor dolutegravir in humans. Antimicrob Agents Chemother. 2013;57(8):3536–46.
Gambaro SE, Robert MC, Tiribelli C, et al. Role of brain cytochrome P450 mono-oxygenases in bilirubin oxidation-specific induction and activity. Arch Toxicol. 2016;90(2):279–90.
Di L, Balesano A, Jordan S, et al. The role of alcohol dehydrogenase in drug metabolism: beyond ethanol oxidation. AAPS J. 2021;23(1):20.
Martínez SE, Vaglenova J, Sabrià J, et al. Distribution of alcohol dehydrogenase mRNA in the rat central nervous system: consequences for brain ethanol and retinoid metabolism. Eur J Biochem. 2001;268(19):5045–56.
Raskin NH, Sokoloff L. Brain alcohol dehydrogenase. Science. 1968;162(3849):131–2.
Strasfeld L, Chou S. Antiviral drug resistance: mechanisms and clinical implications. Infect Dis Clin N Am. 2010;24(2):413–37.
Zdanowicz MM. The pharmacology of HIV drug resistance. Am J Pharm Educ. 2006;70(5):100.
Bacon TH, Levin MJ, Leary JJ, et al. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin Microbiol Rev. 2003;16(1):114–28.
Lurain NS, Chou S. Antiviral drug resistance of human cytomegalovirus. Clin Microbiol Rev. 2010;23(4):689–712.
Chen Y, Scieux C, Garrait V, et al. Resistant herpes simplex virus type 1 infection: an emerging concern after allogeneic stem cell transplantation. Clin Infect Dis. 2000;31(4):927–35.
Wyles DL, Patel A, Madinger N, et al. Development of herpes simplex virus disease in patients who are receiving cidofovir. Clin Infect Dis. 2005;41(5):676–80.
Jabs DA, Enger C, Forman M, et al. Incidence of foscarnet resistance and cidofovir resistance in patients treated for cytomegalovirus retinitis: the Cytomegalovirus Retinitis and Viral Resistance Study Group. Antimicrob Agents Chemother. 1998;42(9):2240–4.
Bertagnolio S, Hermans L, Jordan MR, et al. Clinical impact of pretreatment human immunodeficiency virus drug resistance in people initiating nonnucleoside reverse transcriptase inhibitor-containing antiretroviral therapy: a systematic review and meta-analysis. J Infect Dis. 2021;224(3):377–88.
Chimukangara B, Lessells RJ, Rhee SY, et al. Trends in pretreatment HIV-1 drug resistance in antiretroviral therapy-naive adults in South Africa, 2000–2016: a pooled sequence analysis. EClinicalMedicine. 2019;9:26–34.
Neuhann F, de Forest A, Heger E, et al. Pretreatment resistance mutations and treatment outcomes in adults living with HIV-1: a cohort study in urban Malawi. AIDS Res Ther. 2020;17(1):22.
Julin JE, van Burik JH, Krivit W, et al. Ganciclovir-resistant cytomegalovirus encephalitis in a bone marrow transplant recipient. Transpl Infect Dis. 2002;4(4):201–6.
Bergmann M, Beer R, Kofler M, et al. Acyclovir resistance in herpes simplex virus type I encephalitis: a case report. J Neurovirol. 2017;23(2):335–7.
Hlebowicz M, Parczewski M, Jakubowski P. HIV encephalitis in a patient on antiretroviral therapy: a case report. Int J STD AIDS. 2019;30(6):617–9.
Tamarit Mdel P, Quereda C, Gonzalez-Rozas M, et al. HIV type 1 viral encephalitis after development of viral resistance to plasma suppressive antiretroviral therapy. AIDS Res Hum Retroviruses. 2012;28(1):83–6.
Smit TK, Brew BJ, Tourtellotte W, et al. Independent evolution of human immunodeficiency virus (HIV) drug resistance mutations in diverse areas of the brain in HIV-infected patients, with and without dementia, on antiretroviral treatment. J Virol. 2004;78(18):10133–48.
Saleh MAA, Loo CF, Elassaiss-Schaap J, et al. Lumbar cerebrospinal fluid-to-brain extracellular fluid surrogacy is context-specific: insights from LeiCNS-PK3.0 simulations. J Pharmacokinet Pharmacodyn. 2021;48(5):725–41.
Märtson A-G, Edwina AE, Kim HY, et al. Therapeutic drug monitoring of ganciclovir: where are we? Ther Drug Monitor. 2022;44(1):138–47.
Calcagno A, Trunfio M, D'Avolio A, et al. The impact of age on antiretroviral drug pharmacokinetics in the treatment of adults living with HIV. Expert Opin Drug Metabol Toxicol. 2021;17(6):665–76.
Wang L, Soon GH, Seng K-Y, et al. Pharmacokinetic modeling of plasma and intracellular concentrations of raltegravir in healthy volunteers. Antimicrob Agents Chemother. 2011;55(9):4090–5.
Burns RN, Hendrix CW, Chaturvedula A. Population pharmacokinetics of tenofovir and tenofovir-diphosphate in healthy women. J Clin Pharmacol. 2015;55(6):629–38.
Habtewold A, Aklillu E, Makonnen E, et al. Population pharmacokinetic model linking plasma and peripheral blood mononuclear cell concentrations of efavirenz and its metabolite, 8-hydroxy-efavirenz. HIV patients Antimicrobial Agents Chemother. 2017;61(8):10–1128.
Letendre SL, Capparelli EV, Ellis RJ, et al. Indinavir population pharmacokinetics in plasma and cerebrospinal fluid. The HIV Neurobehavioral Research Center Group. Antimicrob Agents Chemother. 2000;44(8):2173–5.
Sy SKB, Malmberg R, Matsushima A, et al. Effect of reducing the paediatric stavudine dose by half: a physiologically-based pharmacokinetic model. Int J Antimicrob Agents. 2015;45(4):413–9.
Gallo JM. Hybrid physiologically-based pharmacokinetic model for remdesivir: application to SARS-CoV-2. Clin Transl Sci. 2021;14(3):1082–91.
Ito M, Kusuhara H, Ose A, et al. Pharmacokinetic modeling and Monte Carlo simulation to predict interindividual variability in human exposure to oseltamivir and its active metabolite, Ro 64–0802. AAPS J. 2017;19(1):286–97.
Badhan RK, Chenel M, Penny JI. Development of a physiologically-based pharmacokinetic model of the rat central nervous system. Pharmaceutics. 2014;6(1):97–136.
Johnson M, Kozielska M, Pilla Reddy V, et al. Translational modeling in schizophrenia: predicting human dopamine D2 receptor occupancy. Pharm Res. 2016;33(4):1003–17.
Gaohua L, Neuhoff S, Johnson TN, et al. Development of a permeability-limited model of the human brain and cerebrospinal fluid (CSF) to integrate known physiological and biological knowledge: estimating time varying CSF drug concentrations and their variability using in vitro data. Drug Metab Pharmacokinet. 2016;31(3):224–33.
Zakaria Z, Badhan R. Development of a region-specific physiologically based pharmacokinetic brain model to assess hippocampus and frontal cortex pharmacokinetics. Pharmaceutics. 2018;10(1):1–14.
Saleh MAA, de Lange ECM. Impact of CNS diseases on drug delivery to brain extracellular and intracellular target sites in human: a “WHAT-IF” simulation study. Pharmaceutics. 2021;13(1):95.
Saleh MAA, Hirasawa M, Sun M, et al. The PBPK LeiCNS-PK3.0 framework predicts nirmatrelvir (but not remdesivir or molnupiravir) to achieve effective concentrations against SARS-CoV-2 in human brain cells. Eur J Pharm Sci. 2023;181:106345.
Heitman AM, Bies RR, Schwartz SL. A physiologically-based pharmacokinetic model of the brain considering regional lipid variance. J Pharmacol Exp Ther. 2022;383(3):217–26.
Soldin OP, Mattison DR. Sex differences in pharmacokinetics and pharmacodynamics. Clin Pharmacokinet. 2009;48(3):143–57.
Courchesne M, Manrique G, Bernier L, et al. Gender differences in pharmacokinetics: a perspective on contrast agents. ACS Pharmacol Transl Sci. 2024;7(1):8–17.
Clewell HJ, Teeguarden J, McDonald T, et al. Review and evaluation of the potential impact of age- and gender-specific pharmacokinetic differences on tissue dosimetry. Crit Rev Toxicol. 2002;32(5):329–89.
Cosgrove KP, Mazure CM, Staley JK. Evolving knowledge of sex differences in brain structure, function, and chemistry. Biol Psychiatry. 2007;62(8):847–55.
Zhang M, Rottschäfer V, de Lange ECM. The potential impact of CYP and UGT drug-metabolizing enzymes on brain target site drug exposure. Drug Metab Rev. 2023;56:1–30.
Muenchhoff M, Goulder PJ. Sex differences in pediatric infectious diseases. J Infect Dis. 2014;209(Suppl. 3):S120–6.
Sundermann EE, Heaton RK, Pasipanodya E, et al. Sex differences in HIV-associated cognitive impairment. AIDS. 2018;32(18):2719–26.
Liguori C, Pierantozzi M, Spanetta M, et al. Subjective neurological symptoms frequently occur in patients with SARS-CoV2 infection. Brain Behav Immun. 2020;88:11–6.
Scully EP, Haverfield J, Ursin RL, et al. Considering how biological sex impacts immune responses and COVID-19 outcomes. Nat Rev Immuno. 2020;20(7):442–7.
Fish KN, Soderberg-Naucler C, Mills LK, et al. Human cytomegalovirus persistently infects aortic endothelial cells. J Virol. 1998;72(7):5661–8.
Racaniello VR. One hundred years of poliovirus pathogenesis. Virology. 2006;344(1):9–16.
Lafay F, Coulon P, Astic L, et al. Spread of the CVS strain of rabies virus and of the avirulent mutant AvO1 along the olfactory pathways of the mouse after intranasal inoculation. Virology. 1991;183(1):320–30.
Fletcher NF, Wilson GK, Murray J, et al. Hepatitis C virus infects the endothelial cells of the blood-brain barrier. Gastroenterology. 2012;142(3):634–43.
Colpitts TM, Conway MJ, Montgomery RR, et al. West Nile virus: biology, transmission, and human infection. Clin Microbiol Rev. 2012;25(4):635–48.
Lim SM, Koraka P, Osterhaus ADME, et al. West Nile virus: immunity and pathogenesis. Viruses. 2011;3(6):811–28.
Malygin AA, Bondarenko EI, Ivanisenko VA, et al. C-terminal fragment of human laminin-binding protein contains a receptor domain for venezuelan equine encephalitis and tick-borne encephalitis viruses. Biochemistry. 2009;74(12):1328–36.
Zaitsev BN, Benedetti F, Mikhaylov AG, et al. Force-induced globule-coil transition in laminin binding protein and its role for viral-cell membrane fusion. J Mol Recognit. 2014;27(12):727–38.
Palus M, Vancova M, Sirmarova J, et al. Tick-borne encephalitis virus infects human brain microvascular endothelial cells without compromising blood-brain barrier integrity. Virology. 2017;507:110–22.
Elphick GF, Querbes W, Jordan JA, et al. The human polyomavirus, JCV, uses serotonin receptors to infect cells. Science. 2004;306(5700):1380–3.
Boothpur R, Brennan DC. Human polyoma viruses and disease with emphasis on clinical BK and JC. J Clin Virol. 2010;47(4):306–12.
Chapagain ML, Verma S, Mercier F, et al. Polyomavirus JC infects human brain microvascular endothelial cells independent of serotonin receptor 2A. Virology. 2007;364(1):55–63.
O'Hara BA, Morris-Love J, Gee GV, et al. JC virus infected choroid plexus epithelial cells produce extracellular vesicles that infect glial cells independently of the virus attachment receptor. PLoS Pathog. 2020;16(3):e1008371.
Alexaki A, Wigdahl B. HIV-1 infection of bone marrow hematopoietic progenitor cells and their role in trafficking and viral dissemination. PLoS Pathog. 2008;4(12):e1000215.
Jha NK, Ojha S, Jha SK, et al. Evidence of coronavirus (CoV) pathogenesis and emerging pathogen SARS-CoV-2 in the nervous system: a review on neurological impairments and manifestations. J Mol Neurosci. 2021;71(11):2192–209.
Wang X, Huang DY, Huong S-M, et al. Integrin alphavbeta3 is a coreceptor for human cytomegalovirus. Nat Med. 2005;11(5):515–21.
Wang D, Yu Q-C, Schröer J, et al. Human cytomegalovirus uses two distinct pathways to enter retinal pigmented epithelial cells. Proc Natl Acad Sci U S A. 2007;104(50):20037–42.
Albright AV, Shieh JT, Itoh T, et al. Microglia express CCR5, CXCR4, and CCR3, but of these, CCR5 is the principal coreceptor for human immunodeficiency virus type 1 dementia isolates. J Virol. 1999;73(1):205–13.
Lünemann A, Lünemann JD, Roberts S, et al. Human NK cells kill resting but not activated microglia via NKG2D- and NKp46-mediated recognition. J Immunol. 2008;181(9):6170–7.
Maherally Z, Smith JR, An Q, et al. Receptors for hyaluronic acid and poliovirus: a combinatorial role in glioma invasion? PLoS ONE. 2012;7(2):e30691.
Gromeier M, Solecki D, Patel DD, et al. Expression of the human poliovirus receptor/CD155 gene during development of the central nervous system: implications for the pathogenesis of poliomyelitis. Virology. 2000;273(2):248–57.
Xia H, Lazartigues E. Angiotensin-converting enzyme 2 in the brain: properties and future directions. J Neurochem. 2008;107(6):1482–94.
Palmberger TF, Hombach J, Bernkop-Schnürch A. Thiolated chitosan: development and in vitro evaluation of an oral delivery system for acyclovir. Int J Pharm. 2008;348(1–2):54–60.
Stahl JP, Azouvi P, Bruneel F, et al. Guidelines on the management of infectious encephalitis in adults. Med Mal Infect. 2017;47(3):179–94.
Britton PN, Eastwood K, Paterson B, et al. Consensus guidelines for the investigation and management of encephalitis in adults and children in Australia and New Zealand. Intern Med J. 2015;45(5):563–76.
Li M, Si L, Pan H, et al. Excipients enhance intestinal absorption of ganciclovir by P-gp inhibition: assessed in vitro by everted gut sac and in situ by improved intestinal perfusion. Int J Pharm. 2011;403(1–2):37–45.
Tamai I, Sai Y, Ono A, et al. Immunohistochemical and functional characterization of pH-dependent intestinal absorption of weak organic acids by the monocarboxylic acid transporter MCT1. J Pharm Pharmacol. 1999;51(10):1113–21.
World Health Organization. WHO guidelines approved by the Guidelines Review Committee, in consolidated guidelines on HIV prevention, testing, treatment, service delivery and monitoring: recommendations for a public health approach. Geneva: World Health Organization; 2021.
Reese MJ, Savina PM, Generaux GT, et al. In vitro investigations into the roles of drug transporters and metabolizing enzymes in the disposition and drug interactions of dolutegravir, a HIV integrase inhibitor. Drug Metab Dispos. 2013;41(2):353–61.
Reznicek J, Ceckova M, Cerveny L, et al. Emtricitabine is a substrate of MATE1 but not of OCT1, OCT2, P-gp, BCRP or MRP2 transporters. Xenobiotica. 2017;47(1):77–85.
Ghodke Y, Anderson PL, Sangkuhl K, et al. PharmGKB summary: zidovudine pathway. Pharmacogenet Genom. 2012;22(12):891–4.
Yang K. What do we know about remdesivir drug interactions? Clin Transl Sci. 2020;13(5):842–4.
Acknowledgments
Ming Sun thanks the China Scholarship Council for the funding (No. 202106370020).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the preparation of this article.
Conflict of Interest
Ming Sun, Martijn L. Manson, Tingjie Guo, and Elizabeth C. M. de Lange have no conflicts of interest that are directly relevant to the content of this article.
Ethical Approval
Not applicable.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Not applicable.
Code Availability
Not applicable.
Author Contributions
Wrote or contributed to the writing of the manuscript: MS, MLM, TG, and ECMdL.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Sun, M., Manson, M.L., Guo, T. et al. CNS Viral Infections—What to Consider for Improving Drug Treatment: A Plea for Using Mathematical Modeling Approaches. CNS Drugs 38, 349–373 (2024). https://doi.org/10.1007/s40263-024-01082-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-024-01082-3