
Abstract
The synaptic vesicle glycoprotein SV2A belongs to the major facilitator superfamily (MFS) of transporters and is an integral constituent of synaptic vesicle membranes. SV2A has been demonstrated to be involved in vesicle trafficking and exocytosis, processes crucial for neurotransmission. The anti-seizure drug levetiracetam was the first ligand to target SV2A and displays a broad spectrum of anti-seizure activity in various preclinical models. Several lines of preclinical and clinical evidence, including genetics and protein expression changes, support an important role of SV2A in epilepsy pathophysiology. While the functional consequences of SV2A ligand binding are not fully elucidated, studies suggest that subsequent SV2A conformational changes may contribute to seizure protection. Conversely, the recently discovered negative SV2A modulators, such as UCB0255, counteract the anti-seizure effect of levetiracetam and display procognitive properties in preclinical models. More broadly, dysfunction of SV2A may also be involved in Alzheimer’s disease and other types of cognitive impairment, suggesting potential novel therapies for levetiracetam and its congeners. Furthermore, emerging data indicate that there may be important roles for two other SV2 isoforms (SV2B and SV2C) in the pathogenesis of epilepsy, as well as other neurodegenerative diseases. Utilization of recently developed SV2A positron emission tomography ligands will strengthen and reinforce the pharmacological evidence that SV2A is a druggable target, and will provide a better understanding of its role in epilepsy and other neurological diseases, aiding in further defining the full therapeutic potential of SV2A modulation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Synaptic vesicle glycoprotein SV2A is involved in vesicle trafficking and exocytosis, and appears to exert a role in epilepsy pathophysiology. |
Levetiracetam was the first anti-seizure drug to target SV2A, followed by brivaracetam, which selectively targets SV2A. |
SV2A and its isoforms, SV2B and SV2C, may also be involved in the pathogenesis of other neurodegenerative diseases, including Alzheimer’s disease. |
The recent identification of SV2A positron emission tomography tracer ligands and selective SV2A ligands that activate, antagonize and modulate the function of SV2A should facilitate a further understanding of the function and therapeutic potential of SV2A. |
1 Introduction
Epilepsy is the most common neurological condition affecting individuals of all ages and a cause of substantial morbidity and mortality [1, 2]. The mainstay of epilepsy therapy is the prophylactic use of anti-seizure drugs (ASDs; also commonly known as anticonvulsant or antiepileptic drugs [AEDs]), which provide symptomatic treatment of spontaneously recurrent seizures, the predominant symptom of underlying epilepsy [3]. Available ASDs thus target mechanisms associated with seizure generation and propagation (or ictogenesis) in the epileptic brain. Although the approximately 20 clinically used ASDs act by a variety of mechanisms (Fig. 1), they all act to reduce hyperexcitability by either decreasing excitatory or enhancing inhibitory neurotransmission. This involves modulation of voltage-gated ion channels, enhancement of γ-aminobutyric acid (GABA)-mediated inhibition, interactions with elements of the synaptic release machinery, or blockade of ionotropic glutamate receptors [4, 5]. However, approximately 30 % of patients with epilepsy are not controlled by current medications [3] and, despite a high prevalence of acquired epilepsy, no preventive treatment exists for patients at risk of developing epilepsy [3]. Thus, novel treatments for the pharmacoresistant patient and the prevention of epilepsy are major unmet clinical needs [3].
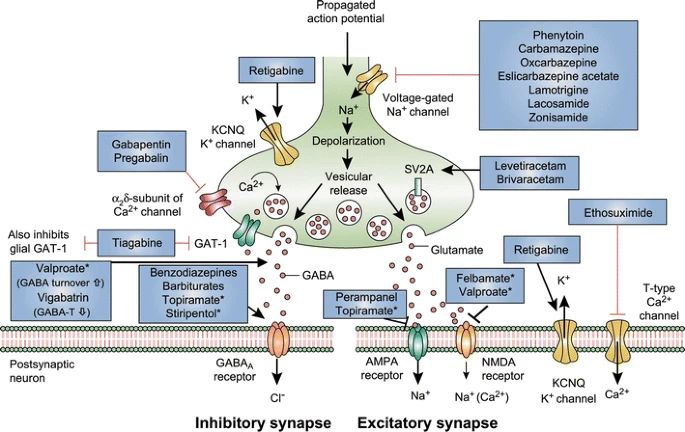
Mechanism of action of clinically approved anti-seizure drugs. Updated and modified from Löscher and Schmidt [151]. Drugs marked with asterisks indicate that these compounds act by multiple mechanims (not all mechanisms shown here). GABA-T GABA aminotransferase, GAT GABA transporter, SV2A synaptic vesicle protein 2A, GABA gamma-aminobutyric acid, NMDA N-methyl-D-aspartate, AMPA α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, KCNQ a family of voltage-gated potassium channels (also known as the Kv7 family)
The processes underlying epileptogenesis (the development of epilepsy following brain injury or other predisposing factors) and pharmacoresistance are complex and likely reflect multifactorial phenomena, involving alterations in networks rather than in single targets [3]. This emphasizes the therapeutic promise of druggable targets that regulate multiple molecular pathways (‘master regulators’), which may be driving these molecular and physiological alterations. This review focusses on the synaptic vesicle glycoprotein SV2A, which is thought to be involved in vesicle function and neurotransmitter release in normal and pathological brain conditions, and is altered during epileptogenesis and in pharmacoresistant epilepsy. In addition, there is increasing evidence that SV2A may constitute a master regulator in different disease processes, including epilepsy (see Sect. 3.4). Importantly, SV2A is the primary molecular target of the ASD levetiracetam [6, 7] and the selective SV2A ligand brivaracetam [8]. The latter was recently approved in Europe by the European Medicines Agency (EMA) and in the US by the Food and Drug Administration (FDA) as adjunctive treatment for drug refractory partial-onset seizures in patients with epilepsy aged 16 years and above [9]. These drugs have not only contributed a further understanding of the functions of SV2A but also constitute a new category of highly effective and well tolerated ASDs that have led to a paradigm shift in ASD discovery.
2 The Discovery of Levetiracetam and its Effect on SV2A
The discovery of the anti-seizure efficacy of levetiracetam is an example of a successful joint endeavor between academia and the industry. Levetiracetam, initially termed ucb L059, is the S-enantiomer of the ethyl analog of piracetam (Fig. 2), a nootropic drug that is indicated for cortical myoclonus, cerebrovascular insufficiencies, and age-associated cognitive impairment. Levetiracetam was discovered in 1977 at UCB Pharma in Belgium during research to identify a second-generation nootropic agent to replace piracetam [10]; however, levetiracetam failed to show significant clinical activity in patients with cognitive impairment. Instead, subsequent pharmacological testing with levetiracetam showed seizure suppression in the audiogenic seizure-susceptible DBA/2 mice, a genetic animal model of epilepsy [11]. These and other findings triggered UCB Pharma to commission Löscher, in 1989, to perform a further characterization of the anti-seizure efficacy of levetiracetam in the amygdala kindling model, a predictive rat model of temporal lobe epilepsy (TLE) [12]. For comparison, levetiracetam was also evaluated in the maximal electroshock seizure (MES) and subcutaneous pentylenetetrazole seizure tests [12], used as initial screens in many ASD discovery programs at that time, including the Anticonvulsant Screening Program (ASP) of the National Institute of Neurological Disorders and Stroke (NINDS) in the US [13]. Surprisingly, levetiracetam was ineffective in the standard MES and pentylenetetrazole tests in rats and mice, up to doses of 500 mg/kg intraperitoneally, but was highly effective in suppressing partial and secondarily generalized convulsive seizures in amygdala-kindled rats [12]. In contrast to the lack of effect in the standard MES and pentylenetetrazole tests, levetiracetam (27–108 mg/kg intraperitoneally) moderately increased the thresholds for MES (MEST) and myoclonic and clonic seizures induced by timed intravenous infusion of pentylenetetrazole with maximum effects of approximately 30 % at the highest dose tested. In kindled rats, levetiracetam increased the focal seizure threshold (after-discharge threshold [ADT]) by 150 % at 54 mg/kg intraperitoneally, demonstrating the particular efficacy of levetiracetam in this model [12]. Figure 3 illustrates the striking difference in levetiracetam’s efficacy to increase the seizure threshold in amygdala kindled rats versus MEST in non-kindled rats, suggesting that the chronic brain alterations associated with kindling render levetiracetam effective. At doses up to 1700 mg/kg intraperitoneally, levetiracetam was inactive in tests measuring ‘minimal neurological deficit’, i.e. the rotarod and chimney tests, indicating a high therapeutic index [12]. These findings revealed a unique profile that made levetiracetam the first known ASD to display efficacy in fully kindled rats, while showing absence of anti-seizure activity in the two major screening models (MES, pentylenetetrazole) used over several decades for identifying new ASD candidates (Table 1). This was in sharp contrast to the profile of numerous previously screened ASDs in these models [10], and unexpected because focal and secondarily generalized seizures in amygdala kindled rats are typically more difficult to suppress by ASDs than acute seizures in the MES test [14]. Based on these data, Löscher and Hönack [12] proposed that levetiracetam should be particularly effective against partial seizures, which was later confirmed by clinical trials, thus validating the data from the kindling model [10]. In subsequent studies in the early 1990s, published by Löscher et al. in 1998 [15], it was shown that treatment of rats with levetiracetam during amygdala kindling retards kindling acquisition and that this effect outlasted the period of drug treatment, indicating potential antiepileptogenic or disease-modifying activity, which was subsequently confirmed by other groups (see Sect. 6). This distinct profile of levetiracetam prompted UCB Pharma to pursue levetiracetam as an ASD candidate. Scientists at UCB continued to test levetiracetam in various animal models, including acute, chronic, and genetic models of seizures or epilepsy, and data from these experiments (Table 1) further confirming a unique profile of levetiracetam consisting of a broad spectrum of efficacy in animal models of acquired and genetic epilepsy, in contrast with a lack of anti-seizure activity in various models of acute seizure induction in normal animals [16, 17]. Following numerous clinical trials, levetiracetam was eventually approved by the FDA in November 1999 and the EMA in September 2000 as a novel ASD [10]. In subsequent years, several clinical studies confirmed the efficacy and safety of levetiracetam, and the drug became the most prescribed newer generation ASD for the treatment of epilepsy [18].

Chemical structures of the racetams piracetam, levetiracetam, brivaracetam and seletracetam

Effect of LEV on the a MEST in non-kindled rats, and b threshold for secondarily generalized seizures in amygdala-kindled rats. c, d Effect of BRV and SEL on the GST in amygdala-kindled rats. Data are shown as means ± SEM of a 20 non-kindled and b–d 9 fully kindled rats in percentage seizure threshold increase above vehicle control seizure thresholds. Significant differences to control thresholds are indicated by an asterisk (p < 0.05). Note the marked difference in anti-seizure efficacy of LEV in a non-kindled vs. b kindled rats. All drugs also significantly increased the threshold for focal seizures (ADT) in kindled rats. The minimum doses significantly increasing ADT were 1.25 mg/kg (LEV), 0.68 mg/kg (BRV), and 0.0074 mg/kg (SEL). Data on MEST in non-kindled rats are taken from Löscher and Hönack [12], and data on kindled rats are from Potschka and Löscher (unpublished observations). LEV levetiracetam, MEST maximal electroshock seizure threshold, GST generalized seizure threshold, BRV brivaracetam, SEL seletracetam, SEM standard error of the mean, ADT after-discharge threshold, i.p. intraperitoneally
The unusual preclinical profile of levetiracetam indicated that its antiepileptic properties likely involve a novel mechanism not shared by other ASDs. Indeed, a vast number of studies, conducted at therapeutically relevant concentrations, did not show any direct interaction of levetiracetam with mechanisms through which other ASDs act, including inhibition of sodium and low-voltage activated calcium (T-type) channels [17]. However, some cellular and molecular effects of levetiracetam have been reported at therapeutically relevant concentrations, including inhibition of high-voltage-gated (N-type) calcium and AMPA currents, inhibition of calcium release from intracellular stores, effects on the metabolism and turnover of GABA in discrete brain regions, reduction in the firing activity of GABAergic neurons in substantia nigra pars reticulata in vivo, and reversal of zinc-induced inhibition of GABAA receptors [5]. Although these effects may contribute to the unique pharmacological profile of levetiracetam, their modest magnitude suggested that they do not represent the primary mechanism of action.
In 1993, pioneering studies at UCB Pharma led to the identification of a specific binding site for [3H]-levetiracetam, which became known as the levetiracetam binding site (LBS), preferentially localized on synaptic plasma membranes with high prevalence in a number of brain structures, including the hippocampus, cortex, and cerebellum [19]. Levetiracetam was found to bind saturably, reversibly, and stereospecifically to LBS, unlike other tested ASDs or CNS drugs that lacked any significant affinity to LBS. Further studies also demonstrated a strong correlation between the affinity of a series of levetiracetam analogs to the LBS, both in vitro and ex vivo, and their anticonvulsant potency in the audiogenic seizure model [19]. In 2003, the molecular nature of LBS was discovered and identified as the synaptic vesicle glycoprotein 2A (SV2A) [6]. Subsequently, the correlation between SV2A binding affinity and anti-seizure potency was confirmed in the audiogenic seizure model and extended to other models of epilepsy (Fig. 4) [20]. Further supportive evidence for a role of SV2A in the anti-seizure activity of levetiracetam came from studies in heterozygous SV2A knockout (+/−) mice. In these animals harboring a 50 % reduction in SV2A proteins, levetiracetam displayed a reduced anti-seizure activity compared with wild-type animals, while valproate, not acting through SV2A, had equal efficacy in both strains [21]. These findings provided further support for SV2A being a valid and novel target for further ASD discovery research and its role in epilepsy [9].

Correlation between the anti-seizure activity of a series of LEV analogs in different epilepsy models and their SV2A in vitro binding affinities. a Genetically sound-susceptible mice; b corneally kindled mice; c rats with spontaneous absence-like EEG seizures from the GAERS strain. SV2A binding affinities pIC50 (−logIC50) were measured in rat brain membranes with the use of [3H]ucb 30889. Protective potencies, based on dose-response studies, are shown as pED50 (−logED50). All correlations were statistically significant (p < 0.01). From Kaminski et al. [20]. LEV levetiracetam, EEG electroencephalogram, GAERS genetical absence epilepsy rats from Strasbourg, SV2A synaptic vesicle protein 2A. ED 50 median effective dose, IC 50 half maximal inhibitory concentration, pED 50 logED50, pIC 50 logIC50
3 The Role of SV2A in Modulating Neuronal Excitability in the Brain
Synaptic transmission is initiated when an action potential triggers neurotransmitter release from presynaptic nerve terminals by synaptic vesicle exocytosis. Traditionally, synaptic vesicle exocytosis is divided into four sequential stages (Fig. 5). First, neurotransmitters are transported into synaptic vesicles, then vesicles dock at the active zone (docking), and prime at the plasma membrane (priming or preactivation). Finally, primed vesicles are triggered for exocytosis by Ca2+ [22–24]. Vesicle exocytosis is followed by vesicle endocytosis and recycling (Fig. 5). SV2, one of the first synaptic vesicle proteins identified, is a component of all vertebrate synaptic vesicles [25]. Three SV2 isoforms are known, two major (SV2A and SV2B) and one minor isoform (SV2C). These proteins constitute a distinct protein family containing 12 transmembrane regions (Fig. 6a). The expression of these SV2 isoforms has been mapped in the central nervous system [26]. SV2A is the most abundant isoform, ubiquitously present throughout all brain areas, with the exception of the trigeminal and facial nuclei [27]. SV2A is expressed in excitatory and inhibitory synapses, as observed in the hippocampus, cerebellum, and cortex [27], and is particularly abundant in subcortical areas, such as the thalamus and basal ganglia [28]. In contrast to SV2A, SV2B has a more restricted expression pattern; however, it is widely coexpressed with SV2A in many, but not all, synaptic vesicles of nerve terminals, including the hippocampal pyramidal cells [27, 28]. SV2C displays an even more restricted pattern of expression, which, under physiological conditions, is mainly limited to basal ganglia and the brain stem [29, 30]. SV2A is the only SV2 isoform expressed in many inhibitory, GABAergic neurons [27, 31]. The human SV2A gene is located in the q arm of chromosome 1 at locus 21.2; it is approximately 14.565 bp in size, and encodes a 4353 bp messenger RNA (mRNA) with 13 exons, which is translated to an 82.6 kDa protein composed of 742 amino acids (Fig. 6a) [26].

Schematic representation of the dynamics of SVs at the presynaptic terminal, illustrating detailed mechanism of NT release and synaptic vesicle recycling. SVs are specialized spheroidal membrane structures that traffic along the axon to the presynaptic terminals, where they internalize and store NTs (blue dots). The SV membrane harbors proteins, some of them highly glycosylated (synapsin, synaptotagmin, SV2, and synaptophysin), whose precise sorting is required for an efficient neurotransmission. A magnified view of an SV (top left) shows the main identified vesicular proteins (except for the proton pump). Some SV proteins, such as synapsins or SV2s, come in multiple isoforms. Once SVs are loaded by NTs, a number of processes lead to excocytosis. (1) NT-loaded SVs dock to the presynaptic membrane by interaction of Rab3 with the RIM protein. Docking displays the SVs in close contact with the SNARE ternary complex (syntaxin 1, SNAP25, and VAMP) and SNARE effectors (MUNCs). (2) Preactivation (priming). In response to increased presynaptic concentrations of Ca2+, sensed by synaptophysin, proteins of the complex change conformation, inducing SVs to fuse with the plasma membrane and release their contents into the synaptic cleft. (3) Fusion/release. Subsequently, empty SVs undergo coating with clathrin polymers, and dynamin-driven scission from plasma membrane. (4) Coating/scission allows SV membrane proteins to be recycled. SV2s are thought to be involved in several of these processes, including calcium-dependent exocytosis, NT loading/retention in synaptic vesicles, and synaptic vesicle priming, as well as transport of vesicle constituents. Updated and modified from Rossetto et al. [23]. SVs synaptic vesicles, NT neurotransmitter, SV2 synaptic vesicle protein-2, Syt synaptotagmin, VAMP vesicle integral membrane protein, SNARE soluble N-ethylmaleimide-sensitive factor attachment protein receptor, RIM proteins a family of active zone proteins, SNAP25 synaptosomal-associated protein 25, MUNC mammalian uncoordinated proteins, ADP adenosine diphosphate, ATP adenosine triphosphate

Schematic representation of SV2A. a SV2A is comprised of 2 TM domains (each of which is comprised of 6 TM helices) and three extramembranous domains, namely the amino domain (blue); a long intervening loop spanning region between TM helices 6 and 7 (orange); and an extracellular domain between TM helices 7 and 8 (green). 2D protein topology figure generated using Protter [152]. Models of b inward-open (based on GlpT template) and c outward-open (based on FucP) conformations of the SV2A protein [86] aligned using the numbering system for proteins of the major facilitator superfamily [153]. TM transmembrane, 2D 2-dimensional, SV2A synaptic vesicle protein-2A, GlpT glycerol-3-phosphate transporter, FucP L-fucose-proton symporter
Although it is known that SV2A is involved in normal synaptic vesicle function, its exact role is not fully understood [26]. Among the various functions proposed are roles in calcium-dependent exocytosis, neurotransmitter loading/retention in synaptic vesicles, and synaptic vesicle priming, as well as transport of vesicle constituents [5]. The latter is supported by homology with the major facilitator superfamily (MFS) of membrane transport proteins [32] and by the suggestion that SV2A is a galactose transporter [33]. Mice lacking SV2A generated by targeted gene disruption [34] appear normal at birth, but do not grow normally; they express seizures and die within 3 weeks, suggesting multiple neural alterations. Interestingly, hippocampal SV2A expression increases in wild-type mice between postnatal day (PND) 5 and 7, and remains stable from PND 7–10 [35]. The observed alterations of SV2A expression in hippocampus are consistent with the appearance of seizures in SV2A knockout animals at early postnatal age and the hypothesis that SV2A absence favors epileptic seizures. Electrophysiological studies of spontaneous inhibitory transmission in the hippocampal CA3 region of these animals revealed that loss of SV2A induces a decrease in action-potential-dependent GABA transmission, without changes in action-potential-independent transmission. Altered transmission in SV2A (−/−) animals was not attributable to changes in the number or morphology of synapses or SVs, indicating that SV2 proteins are not structural components of synapses nor are they required for SV biogenesis [34, 36]. These results support the hypothesis that SV2A is a key player in the control of exocytosis, and, consequently, has a key involvement in neurotransmission [34]. The strong epilepsy phenotype of animals with deletion of the SV2A gene, together with the fact that SV2A binding is involved in the mechanism of action of levetiracetam, has stimulated studies to further characterize the potential role of SV2A in the epileptogenic process.
3.1 SV2A in Animal Models of Epilepsy
Gorter et al. [37] were the first to describe that SV2A gene expression is transiently downregulated during epileptogenesis in the entorhinal cortex of rats in which epilepsy was induced by sustained electrical stimulation of the angular bundle. In a subsequent study, van Vliet et al. [38] reported that SV2A protein expression was decreased in the latent period of this model in the inner molecular layer and stratum lucidum of the hippocampus. In chronic epileptic rats, SV2A was decreased throughout the hippocampus, specifically in rats with a progressive form of epilepsy, resembling the hippocampal SV2A decrease observed in patients with TLE and hippocampal sclerosis [38].
In a systems level, functional genomics analysis of chronic epilepsy using the intrahippocampal kainate model of TLE in rats, Winden et al. [39] found that the network structure and connectivity of the SV2A gene showed significant changes between normal and epileptogenic dentate gyrus, becoming more highly connected in epileptic brain, possibly indicating that SV2A had gained a more important role in synaptic function in epilepsy than under normal conditions. The authors suggested that genes upregulated due to seizures may regulate neuronal excitability through effects on vesicular trafficking, and that SV2A represents a central (hub) gene in this respect. This hypothesis would contribute to an explanation of the unique pharmacological profile of levetiracetam, which shows consistent efficacy in animal models of acquired and genetic epilepsy but lacks an effect against acute seizure induction in normal animals.
In spontaneously epileptic rats (SERs), Hanaya et al. [40] found low SV2A expression, particularly in the frontal and entorhinal cortices and the inner molecular layer and dentate gyrus of the hippocampal formation. SERs are characterized by a double mutation (zi/zi, tm/tm) which causes tonic convulsions, absence seizures, and sclerosis-like changes in the hippocampus [40]. Levetiracetam inhibits both tonic convulsions and absence seizures in these rats, despite the low level of SV2A expression.
In apparent contrast to the reduced expression of SV2A found in the hippocampus and other brain regions of two rat models of TLE [38, 40], increased expression of SV2A has been reported for the kindling model of TLE [41–44]. A likely, at least partial, reason for this discrepancy is the massive neuronal loss in the hippocampus and parahippocampal areas, occurring in post-status epilepticus (SE) models of TLE (and many TLE patients), but not kindling. When rats were kindled by repeated short (0.8 s) unilateral stimulations of the amygdala, entorhinal cortex or septum, the expression of synaptosomal SV2s (SV2A/SV2B) in the ipsilateral hippocampus significantly increased, regardless of the stimulation site [41]. The asymmetric accumulation of SV2s that was observed 4 weeks after the last kindling stimulation was specific to the hippocampus; it was not seen at synaptically distant sites (occipital cortex and cerebellum) that are not thought to be directly involved in the neural network of kindling. In a subsequent study, Matveeva et al. [42] reported that the ipsilateral accumulation of SV2s in the hippocampus persists for at least 1 year and can be prevented by treating rats with levetiracetam during kindling acquisition. In contrast, levetiracetam had no effect on SV2 expression in non-kindled rats.
An increase in hippocampal SV2A expression was also observed in mice kindled by repeated administration of pentylenetetrazole [43, 44]. Twenty-four hours after the last pentylenetetrazole injection, intense SV2A expression was found in the hilus of the dentate gyrus, but not in other regions, including the stratum lucidum of CA3 and the synaptic layer surrounding CA1 and CA3 pyramidal cells [44]. In addition, pentylenetetrazole kindling selectively elevated SV2A expression in GABAergic interneurons located in the hilus of the DG, but not in mossy glutamatergic fibers [44]. The authors postulate that increased SV2A expression in the hippocampus of kindled rats is a compensatory mechanism that facilitates inhibitory GABAergic neurotransmission [43, 44].
SV2A-deficient mice exhibit increased kindling acquisition, both when kindled via repeated electrical stimulation of the amygdala or by corneal stimulation [21]. A proepileptic phenotype of SV2A (+/−) heterozygous mice was also observed in pilocarpine, kainate, pentylenetetrazol and 6 Hz models, but not in the MES test [21]. This profile matches the protective profile associated with the activity of levetiracetam in these models (Table 1).
3.2 Potential Role of SV2A in Models of Pharmacoresistant Epilepsy
In the early 1990s, Löscher and Rundfeldt developed the first animal model of pharmacoresistant seizures, i.e. the phenytoin-resistant amygdala-kindled rat [45]. This model was discovered by evaluating the anti-seizure effect of phenytoin in large groups of kindled rats, resulting in three subgroups—responders, non-responders, and variable responders (cf, [46]). In phenytoin non-responders, even large doses of phenytoin did not increase the threshold for focal seizures (ADT), whereas ADT was significantly and reproducibly increased in the responder subgroup. The lack of anti-seizure response in non-responders extended to various other ASDs, with the remarkable exception of the SV2A ligand levetiracetam [47], which suggests that SV2A could be involved in the response of phenytoin non-responders to levetiracetam. Wang et al. [48] selected phenytoin- and phenobarbital-resistant rats from a large group of amygdala-kindled rats and found that hippocampal SV2A mRNA and protein expression were significantly lower in resistant rats compared with ASD responders. Hippocampal low-frequency stimulation increased SV2A expression in the resistant rats and exerted anti-seizure effects [48]. The authors suggest that decreased levels of SV2A are involved in the mechanism underlying pharmacoresistant epilepsy. This suggestion is supported by the findings of decreased SV2A levels in different types of pharmacoresistant epilepsy in patients (see Sect. 3.3).
The high efficacy of levetiracetam in models of pharmacoresistant epilepsy was also supported by experiments with the 6-Hz mouse model of difficult-to-treat partial seizures [49]. Bankstahl et al. [50] applied this model in mice that had been rendered epileptic by a pilocarpine-induced SE, and observed that levetiracetam was much more effective in blocking 6-Hz seizures in epileptic mice rather than non-epileptic mice, which was substantiated by Leclercq and Kaminski [51]. In SV2A-deficient mice, anti-seizure efficacy of levetiracetam in the 6-Hz model was significantly reduced (approximately 50 %), consistent with reduced binding to SV2A in these mice [21]. In contrast, valproate produced the same anti-seizure effect in wild-type and SV2A-deficient mice. It is tempting to speculate that the striking increase in the anti-seizure effect of levetiracetam on 6-Hz seizures in epileptic mice is mediated by epilepsy-associated changes of its target, SV2A [50, 51]. Indeed, profound changes in synaptic vesicle release machinery were reported in post-SE models of TLE; however, SV2A density in individual synaptic boutons has not been directly measured in these studies [52, 53].
3.3 SV2A in Patients with Pharmacoresistant Epilepsy
SV2A expression has been analyzed in patients experiencing different types of intractable epilepsy, owing to the availability of brain tissue removed from these patients during epilepsy surgery [26]. As in post-SE rat models of TLE (see Sect. 3.1), SV2A expression was reduced by 30–50 % in the anterior temporal neocortex of adult patients with TLE [54]. Weak SV2A immunoreactivity (IR) was observed in the tissue from TLE patients, whereas strong IR was present in brain tissue from control patients [54]. SV2A expression, measured by Western blotting, was significantly decreased in the temporal neocortex of TLE patients compared with control patients [54]. Likewise, SV2A expression was decreased in the resected hippocampus of patients with TLE and hippocampal sclerosis [37]. A substantial loss of SV2A IR was observed in the hippocampal neuropil, except in the inner molecular layer of the dentate gyrus [37]. These observations were recently further substantiated with positron emission tomography (PET) studies, where decreased binding (approximately 50 %) of an SV2A tracer (11C-UCB-J) was observed ipsilaterally to the seizure focus in patients with TLE [55]. Malformations that cause intractable epilepsy, such as focal cortical dysplasia and cortical tubers, also show significantly reduced SV2A IR [56]. SV2A IR within the dysplastic cortex had less staining than the normal cortex. This decrease was also observed by Western blot analysis in both the focal cortical dysplasia and the tuberous sclerosis complex [56]. These results suggest that decreased SV2A expression may contribute to the instability of neuronal networks and therefore to the progression of epilepsy [54, 56]. Further work is needed to fully interpret those findings since immunohistological and PET assessments of SV2A expression could be confounded by neuronal loss often observed in TLE. It is essential to study SV2A at the level of individual synapses in patients with TLE since recent reports indicate significant increases in synaptic vesicle density within limbic structures of animals with chronic TLE [52, 53].
In addition, the expression and distribution of SV2A have been investigated in patients with brain tumors with or without epilepsy [57]. Strong and diffuse SV2A IR was observed throughout all layers of the control cortex. Similarly, strong SV2A IR (with the same diffuse distribution pattern) was observed in peritumoral cortical specimens from patients with and without epilepsy. Modest SV2A IR was observed within the tumor area [57]. Although the mechanisms of SV2A function are not fully understood, a reduction in SV2A expression appears to lead to abnormal neurotransmission and the development of seizures [34, 36]. However, as SV2A expression in the peritumoral tissue was similar in patients with or without epilepsy (although tumor cells of patients with and without epilepsy showed modest SV2A IR and lower SV2A protein expression than the control cortex), the role of SV2A in epileptogenesis in tumor patients remains questionable [57]. Alternatively, the reduced SV2A expression and subsequent loss of SV2A function may lead to dysfunction of SV exocytosis and to altered neurotransmission, contributing to epileptogenesis in patients with brain tumors [57]. In a subsequent prospective study, de Groot et al. [58] found that SV2A expression in tumor and peritumoral tissue correlated with the efficacy of levetiracetam in post-surgical patients, suggesting an association between SV2A expression and levetiracetam response, an association that has been previously observed [21].
Recently, the first evidence that an SV2A mutation can cause epilepsy in humans was reported [59]. These investigators found an homozygous mutation in the SV2A gene in a patient with intractable epilepsy. Exome sequencing identified a homozygous arginine to glutamine mutation in amino acid position 383 (R383Q) in exon 5 of the SV2A gene. Both parents were carriers for the R383Q variant, suggesting that R383Q is a recessive mutation. There were no other candidate alterations in the exome that could explain the phenotype in the patient.
3.4 SV2A in Cognitive Impairment
Elevated hippocampal excitability is observed in a number of conditions that confer risk for Alzheimer’s disease (AD), including amnestic mild cognitive impairment (aMCI) [60]. By using treatment with a low dose of levetiracetam (125 mg twice daily), hippocampal activation in patients with aMCI was reduced to a level that did not differ from the control group, and memory performance was significantly improved [60], substantiating previous experiments with levetiracetam in a rat model of age-related memory loss [61]. By using differential coexpression analysis (DCA) to disentangle causative events from secondary changes within transcriptome-wide gene expression data sets, SV2A has been identified as a causal ‘node’ or ‘master regulator’ in late-onset AD, involved in modulation of amyloid β/A4 precursor protein (APP) processing [62]. In in vitro experiments in cells cultured from carriers of apolipoprotein E (APOE)-4, which is the greatest genetic risk factor for late-onset AD, treatment with the SV2A ligand levetiracetam could correct APOE4-related alteration in APP processing [62]. Levetiracetam treatment of APP transgenic mice has similarly been reported to improve cognitive function, whereas other anti-seizure agents fail to do so [63]. In a cell model for aging and early late-onset AD, levetiracetam improved several aspects of mitochondrial dysfunction, which were abolished when SV2A was knocked down by small interfering RNA (siRNA), indicating that interfering with SV2A at the mitochondrial level, and thereby improving mitochondrial function, might represent an additional therapeutic effect of levetiracetam to improve symptoms of late-onset AD [64]. Taken together, these data support a potential role of SV2A in AD and have stimulated an interest in exploring the therapeutic potential of levetiracetam as a novel therapeutic approach for AD treatment.
In a prospective, randomized, case-control study of AD patients with seizures, treatment with either levetiracetam, phenobarbital, or lamotrigine exhibited the same efficacy to suppress seizures, but, in addition, treatment with levetiracetam was associated with improved cognitive performance, specifically improved attention, short-term memory, and oral fluency, while this was not seen with phenobarbital or lamotrigine [65]. Instead, patients receiving phenobarbital showed a significant worsening of cognitive performance and patients treated with lamotrigine showed a slight decline in cognitive function [65]. Currently, several phase II and III studies with levetiracetam in AD patients are being performed (see ClinicialTrials.gov).
Several high-affinity SV2A ligands were recently discovered that are devoid of anti-seizure activity and selectively counteract the anti-seizure effect of levetiracetam, but not that of other ASDs [66]. Due to their opposite action to levetiracetam, UCB scientists speculated that this finding may reflect similarity to other receptors (GABA and AMPA) and ion channel (KCNQ2/3) systems where pharmacological modulation opposite to the one providing anti-seizure effects elevates neuronal excitability and results in procognitive effects. This was confirmed when testing several SV2A ligands, devoid of anti-seizure properties, in the object recognition test in mice. Among these, UCB0255 was selected as a lead candidate. It displays high selectivity and affinity (pKi = 7.9) for SV2A and reveals a pharmacology for SV2A opposite that of levetiracetam by an absence of anti-seizure properties and an ability to counteract the seizure protection afforded by levetiracetam [66]. Further characterization of UCB0255 in various animal models of cognition substantiated the procognitive hypothesis of this class of SV2A ligands [66].
The finding that both an anticonvulsant SV2A ligand (levetiracetam) and a SV2A ligand devoid of anticonvulsant properties (UCB0255) exert procognitive activity indicates that these compounds interact differently, depending on the circumstances. The procognitive effect of levetiracetam may be observed in conditions where there is background hyperexcitability (e.g. MCI), while SV2A ligands devoid of anticonvulsant effects may exert their procognitive activity by enhancement of neuronal excitability in advanced AD associated with hypoexcitability (e.g. analogous to cholinergic drugs). Clearly, what the underlying cause of the cognitive deficit is, i.e. hyper- or hypoexcitability needs to be carefully assessed before treatment with either type of SV2A ligand is considered.
3.5 Role of SV2B and SV2C Isoforms
Little is known about physiological functions of SV2B and SV2C isoforms beyond observations indicating different expression patterns in the brain, or association with particular types of either excitatory or inhibitory neurons. It appears that SV2B levels are very low in the synaptic vesicles of GABAergic neurons compared with SV2A, and appear to have a preferential localization in glutamatergic neurons [67]. However, the functional consequence of this finding may be limited, at least under physiological conditions, since SV2B knockout mice lack any overt phenotype [68]. Furthermore, both the frequency and amplitude of spontaneous excitatory and inhibitory postsynaptic currents is not significantly altered in SV2B knockout mice [69]. The role of SV2B may become more apparent under disease conditions since SV2B knockout are protected against Aβ25–35-induced oxidative stress and decrease in choline acetyltransferase activity in the hippocampus [68]. Furthermore, SV2B knockout mice failed to show Aβ25–35-induced impairments in a range of cognitive domains [68]. The SV2B knockout mice, unlike SV2A-deficient mice, have only a limited phenotype in seizure models and show a slight decrease in vulnerability to 6 Hz-, pilocarpine- and kainate-induced seizures and reduced pentylenetetrazole seizure threshold (Leclercq and Kaminski, unpublished observations).
Similar to SV2B knockout mice, the SV2C-deficient animals also do not display any overt phenotype under physiological conditions, but changes in SV2C expression have been reported in tissue from disease models and patients. Crèvecoeur et al. [70] studied the expression pattern of all three isoforms of SV2 in the hippocampus of patients with TLE and hippocampal sclerosis. They found that the expression of SV2 isoforms is altered with a marked decrease of SV2A and SV2B paralleling synaptic loss and a selective increase of SV2C in sprouting mossy fibers. Similar findings were made in mice with TLE resulting from electrical stimulation of basolateral amygdala followed by self-sustained SE, where selective SV2C overexpression was seen in the CA4 and CA3 hippocampal (stratum lucidum) areas [71]. Interestingly, SV2C mRNA expression was also significantly increased in the striatum of animals challenged with 6-hydroxydopamine, a model of Parkinson’s disease [72]. Together, these data indicate that SV2C may be involved in presynaptic remodeling in response to neuronal hyperexcitability or cellular stress conditions.
3.6 Investigation of SV2A by Positron Emission Tomography Tracers
A highly selective SV2A radiotracer for brain imaging by PET could help to further understand the physiological role of SV2A and to reveal its fundamental implication in various neurological diseases, characterized by a synaptic dysfunction such as epilepsy or by a synaptic deconnection such as in AD. Recently, [18F]UCB-H, a high-affinity PET ligand for SV2A, was developed [73, 74]. This tracer shows a nanomolar affinity for SV2A in the brain, which has been demonstrated using µPET in a preclinical study by Warnock et al. [75] in the rat brain. After a single intravenous administration in rats, [18F]UCB-H showed rapid and high-level brain uptake, with a free brain-to-plasma ratio of greater than 1 at 5, 10, and 20 min after administration. The uptake of [18F]UCB-H in the brain corresponded to the reported distribution of SV2A, with widespread tracer uptake in the entire brain. The first human dosimetry study of [18F]UCB-H [76] showed that the highest number of disintegrations per organ occurred in the liver, followed by the bladder and the brain. Other PET tracers based on 11C-labeling have been also recently developed in preclinical species, e.g. [11C]UCB-A [77] and [11C]UCB-J [78]. As with UCB-H, UCB-J [(R)-1-((3-(methyl-11C)pyridin-4-yl)methyl)-4-(3,4,5-trifluorophenyl)pyrrolidin-2-one] is a heterocyclic non-acetamide compound [74]. Its whole body biodistribution was studied in non-human primates, demonstrating high uptake and fast kinetics in the brain [78]. Preblocking with levetiracetam 10 and 30 mg/kg resulted in approximately 60 and 90 % occupancy, respectively. The recent evaluation demonstrated that [11C]-UCB-J has exceptional imaging qualities for PET measurements of SV2A in the human brain, including high brain uptake, rapid kinetics appropriate for the 11C radiolabel, a high and reliably measurable free fraction in plasma, and suitable regional time–activity curves [78]. [11C]UCB-J displays rapid metabolism in human subjects and excellent brain uptake, which is highest in the striatum and the cortex, while being moderate in the cerebellum and the thalamus (Fig. 7a) [55]. Interestingly, evaluation of a patient with TLE indicated a significant reduction of [11C]UCB-J binding in the right mesial temporal lobe colocalized with mesial temporal lobe sclerosis seen on MRI (Fig. 7b) [55].

Characterization of a novel SV2A PET radiotracer [11C]UCB-J in humans. a Axial, coronal and sagittal brain MRI images in a healthy volunteer subject (top row); regional volume of distribution maps (VT) during test (middle row) and retest (bottom row) PET scanning with [11C]UCB-J in the same subject. b Axial and coronal brain MRI images in a patient with temporal lobe epilepsy (top row); standardized uptake value maps (SUV) of [11C]UCB-J in the same subject (bottom row). Arrows show unilateral mesial temporal sclerosis (MTS) seen on MRI (top row) and reduced radioligand binding in the corresponding areas of the right mesial temporal lobe of the same subject (bottom row). From Finnema et al. [55]. SV2A synaptic vesicle protein-2A, PET positron emission tomography, MRI magnetic resonance imaging, SUV standardized uptake value, MTS mesial temporal sclerosis, V T volume of distribution
In addition to their potential value in the study of epilepsy, SV2A PET tracers could be of interest as useful tools to measure synaptic density in humans [55, 75]. SV2A does not appear to affect vesicle formation as synaptic morphology and vesicle density were not altered in SV2A knockout animals [34, 36]. These data suggested that SV2A could influence SV cycling. As this cycling is a continuous process within the presynaptic terminal, it could be postulated that reduced uptake of an SV2A PET tracer is a consequence of synaptic loss [75], which clearly appears to be the case in patients with mesial TLE [55]. Such a non-invasive measure of synaptic density could be of great value, not only for the diagnosis of epilepsy but also other neurodegenerative disorders, including AD. In addition to UCB-H and UCB-J, levetiracetam itself has been 11C-labeled and used as a PET tracer [79], but no imaging data with this radiotracer have been reported to date.
4 SV2A as a Target for Anti-Seizure Drugs: Levetiracetam, Brivaracetam and Beyond
As discussed above, there are different lines of evidence to suggest that modulation of SV2A constitutes the primary mechanism of action of levetiracetam [7, 80], although other effects may contribute to its pharmacodynamic activities. In a critical review on the role of SV2A for the effects of levetiracetam, Surges et al. [81] concluded in 2008 that “although levetiracetam shares some targets with other ASDs, levetiracetam exhibits unique mechanisms of action and a novel and unique binding site”. However, the latter authors also noted that “unfortunately to date, there are no available reports on the specific binding site for levetiracetam on the SV2A molecule”. Respective studies are complicated by the fact that SV2A has not been structurally characterized with X-ray crystallography or nuclear magnetic resonance. By sequence homology, SV2A has been described as a member of the Major Facilitator Superfamily (MFS) of transporters [32, 82, 83] and is considered to adopt the architecture found in the X-ray crystallography of the MFS transporter family. Structural evidence for this eventually came from protein tomography experiments [84]. Using this technique, two major conformations could be inferred. Although the sequence identity between SV2A and other MFS proteins is very low, typically <20 %, homology models have been constructed using available MFS x-ray structures as templates. In an early study Shi et al. [85] selected three residues—F658, G659 and V661—based on their alignment with functionally relevant residues in LacY for site-directed mutagenesis (SDM) studies with levetiracetam and structurally-related analogs such as ucb 30889, collectively referred to as racetams, to elucidate residues that are important for racetam binding. Furthermore, Shi et al. [85] constructed an SV2A homology model using the LacY crystal structure as a template, and in conjunction with racetam docking studies they proposed additional residues for SDM studies. In total, through binding affinity experiments, 14 mutants were found to be implicated in racetam binding (F277A, W300A, W300F, Y462A, K694A, G303A, F658A, V661A, I663A, W666A, N667A, S294A, M301A, G659A) and were proposed to encompass the SV2A racetam binding site. This work was extended in the study of Lee et al. [86], wherein they considered two conformational states based on two additional templates: L-fucose-proton symporter as a template for the outward-open state, and Glycerol-3-phosphate transporter as a template for the inward-open state (Figs. 6b, c). On the basis of the homology modeling, docking and subsequent molecular dynamics simulations, these researchers observed that two additional residues—W454 and D670—may contribute to racetam binding. These predictions were subsequently borne out by SDM performed on W454A and D670A in conjunction with binding affinity assays [86]. Taken together with the studies of Shi et al. [85], the outward-open conformation of SV2A (i.e. with the putative binding site exposed to the interior of the synaptic vesicle) presents a levetiracetam-binding pocket that is in greater agreement with the SDM experiments.
In the former studies, only the transmembrane regions of SV2A were modeled, which was a consequence of the fact that a complete MFS protein structure has yet to be solved. Recently, Correa-Basurto et al. [87] built a complete model of the SV2A protein with levetiracetam, along with other racetams (brivaracetam and seletracetam) docked into it. The subsequent complexes were then simulated using molecular dynamics in an attempt to further characterize SV2A–racetam binding site interactions. Despite the short timescale simulations and the challenge and caveats of modeling the extramembranous domains, the studies support the putative binding pocket observed by Shi et al. [85] and Lee et al. [86], and, furthermore, identified additional hydrophobic and hydrogen bond interactions with T456, S665 and L689, which may be important for ligand recognition within the putative racetam binding site.
Studies with an SV2A positive allosteric modulator (UCB1244283) indicated that the SV2A protein contains multiple interacting binding sites that are linked via the conformation of the protein [88]. One of the functional consequences of levetiracetam binding to the SV2A protein in brain slices was found to be a reduction in exocytosis [89]. In hippocampal slices from non-epileptic rats, treatment with levetiracetam reduces neurotransmission in response to fast stimulus trains [89], consistent with the drug blocking the effects of SV2 on vesicle priming. Further studies showed that levetiracetam decreased vesicular release in a dose-, time-, and stimulation-dependent manner, suggesting that repetitive stimulation is required to allow levetiracetam entry into recycling synaptic vesicles and intravesicular levetiracetam binding to SV2A [90]. Most interestingly, the latency of drug action is much shorter when neurons are stimulated [91], suggesting that levetiracetam preferentially targets hyperactive synapses. Based on the experiments of Meehan et al. [91] in hippocampal slices, Mendoza-Torreblanca et al. [26] suggested that levetiracetam enters synaptic vesicles during recycling and endocytosis (see Fig. 5) and escapes during synaptic vesicle fusion, and that the site of binding between levetiracetam and SV2A is an intravesicular portion of the protein or a protein portion that can be accessed from the intravesicular side. However, given the pharmacokinetic properties of levetiracetam (and brivaracetam) [see Sect. 5], one would expect these compounds to reach their target through diffusion of membranes. The lag time observed for the compounds to act might be related to diffusion rate into tissues or slices, but additional studies are needed to address the location and relevance of the position of the SV2A binding site(s).
By studying the effects of overexpressing SV2A in hippocampal neurons cultured to form autaptic synapses, Nowack et al. [92] found that elevated expression of SV2 resulted in a neurotransmission phenotype that resembled that seen in neurons from SV2 knockout mice, suggesting that too much SV2 is as detrimental to neuronal function as too little. Levetiracetam reversed the effects of excess SV2A, suggesting that levetiracetam may modulate SV2 protein interactions. Furthermore, levetiracetam was shown to accelerate the induction of supply rate depression at excitatory synapses during incipient epileptic activity, an effect that was absent in SV2A-deficient mice [93]. While the underlying mechanistic effect of ligand binding is not fully elucidated, one hypothesis could be that ligand binding to SV2A would stabilize a certain conformation of the protein, resulting in potentiation or optimization of its function, and thereby providing seizure protection [88].
Brivaracetam displays a 15- to 30-fold higher affinity to SV2A than levetiracetam and is selective for SV2A [8, 9]. Rat hippocampal slice studies comparing brivaracetam and levetiracetam using high-frequency neuronal stimulation, indicated that brivaracetam augments synaptic depression and thereby decreases synaptic transmission at 100-fold lower concentrations than levetiracetam [94]. In studies with dye-loaded vesicles, brivaracetam slowed stimulation-induced destaining (i.e. slowed vesicle release) significantly more than levetiracetam [94], indicating that brivaracetam reduces vesicle mobilization more effectively than levetiracetam, which may explain their distinct pharmacodynamic properties (see Sect. 5).
Additionally, different binding properties for levetiracetam and brivaracetam to the SV2A protein might also contribute to different pharmacological efficacies. These were revealed by further studies with the SV2A allosteric modulator UCB1244283 on human recombinant SV2A expressed in HEK293 cells, and in human cortex [88, 95]. In cells expressing human SV2A, [3H]brivaracetam labeled twice as many sites than [3H]levetiracetam. Moreover, co-incubation of [3H]levetiracetam with the allosteric modulator produced a modest increase in affinity for SV2A, but a doubling in maximum binding capacity. In contrast, co-incubation with the allosteric modulator increased the affinity of [3H]brivaracetam to a much greater degree (tenfold), while the increase observed in maximum binding capacity was more modest (1.3-fold). Taken together, these data suggest that levetiracetam and brivaracetam recognize, induce, or stabilize different conformations of the SV2A protein, which may provide the molecular rationale for their distinct pharmacodynamic properties.
The accumulated experience from drug discovery efforts for new anti-seizure SV2A ligands has revealed a diversity in anti-seizure profiles that not only appears to correlate with differences in affinity but also with different binding properties for SV2A. Taken together with the recent identification of high affinity SV2A ligands, devoid of anti-seizure properties, and an allosteric modulator, this provides a diverse armamentarium of SV2A ligands able to both activate, antagonize, and modulate the function of SV2A. Hopefully, this will facilitate further understanding of the function and therapeutic potential of SV2A as a target for CNS drugs.
5 Preclinical and Clinical Profile of Levetiracetam and its Congeners
Based on the unique preclinical pharmacology and mechanism of action of levetiracetam as a novel ASD, a major drug discovery program was triggered at UCB with the purpose of optimizing the therapeutic benefit of discovery of the SV2A mechanism. The goal was to identify selective, high-affinity SV2A ligands possessing anti-seizure properties superior to levetiracetam [96]. Approximately 12,000 compounds were screened in vitro for SV2A binding affinity; 1200 were further screened in vivo for seizure protection in audiogenic seizure-susceptible mice, and approximately 30 compounds were selected and broadly characterized in a variety of animal models of seizures and epilepsy [9]. This led to the identification of two distinct anti-seizure racetam families, both with high affinity for SV2A but with different pharmacological properties, which were named after their lead compounds—brivaracetam and seletracetam (Fig. 2). In vitro models of epilepsy showed that, while both brivaracetam and seletracetam inhibited epileptiform hypersynchronization, brivaracetam demonstrated a more pronounced ability to inhibit neuronal hyperexcitability than seletracetam [9]. This likely explains brivaracetam’s seizure protection in the MES and subcutaneous pentylenetetrazole tests (Table 1), albeit at relatively high doses, and its significant protection against the partial seizure phase in animal models of partial epilepsy [97]. Brivaracetam also differs from levetiracetam by its selective, high affinity and differential interaction with SV2A [9]. This likely explains its superior ability to inhibit neurotransmitter release and more potent and complete seizure suppression than levetiracetam in animal models of partial, generalized, and drug-resistant seizures (Table 1), a profile that triggered the decision to focus on brivaracetam for further clinical development. Interestingly, initial preclinical studies have also revealed brivaracetam to possess a more significant antiepileptogenic potential than levetiracetam (see Sect. 6).
Brivaracetam displays a faster onset of anti-seizure action in preclinical models than levetiracetam [9, 98], which is likely due to levetiracetam having a relatively low lipophilicity (LogD −0.64) contrasting a higher lipophilicity of brivaracetam (LogD 1.04), leading to a higher brain–blood barrier permeability of brivaracetam [98]. As a consequence, displacement experiments in rhesus monkey with the SV2A PET tracer [11C]UCB-J following single intravenous administration of brivaracetam (5 mg/kg) and levetiracetam (30 mg/kg) showed that plasma levels comparable to those following intravenous administration of brivaracetam 100 mg and levetiracetam 1500 mg in epilepsy patients provided a faster displacement of [11C]UCB-J by brivaracetam than levetiracetam [98]. A sevenfold difference was observed in brain entry speed, with brivaracetam having a predicted drug entry half-time of only 3 min, whereas this figure was 23 min for levetiracetam [98]. Together with high solubility and tolerability, and low risk of drug–drug interactions, the faster SV2A occupancy by brivaracetam than levetiracetam supports a promising potential of brivaracetam for the treatment of acute seizures, including SE. The latter is also reinforced by significant activity of brivaracetam in animal models of SE [98].
An initial report suggested that inhibition of voltage-gated sodium channels at low micromolar concentrations may contribute to the anti-seizure activity of brivaracetam [99]. However, several subsequent studies suggested that the anti-seizure mechanism of brivaracetam is unrelated to inhibition of voltage-gated sodium channels [9]. Further studies on other potential ASD mechanisms, including voltage-gated potassium channels, high- and low-voltage-gated calcium channels, and GABA and glutamate currents, have shown no effect of brivaracetam at therapeutically relevant concentrations, suggesting that brivaracetam represents the first, selective SV2A ligand for epilepsy treatment [9].
The increased affinity for SV2A of brivaracetam over levetiracetam shown in vitro correlates well with observations from various in vivo models of seizures and epilepsy (Table 1) that show brivaracetam to have a markedly higher potency over levetiracetam for protection of both generalized and partial seizures [8, 80, 100]. An example for dose–effect comparisons between levetiracetam and brivaracetam in amygdala-kindled rats is shown in Fig. 3. Furthermore, in contrast to levetiracetam, brivaracetam is also effective against seizures induced in the classical MES and pentylenetetrazole seizure tests, albeit at relatively high doses (Table 1). A particularly remarkable difference in potency is seen in the 6-Hz model of pharmacoresistant partial seizures. In this model, use of a high stimulation current (44 mA) results in resistance of induced seizures to many ASDs [49]. Anti-seizure ED50 of levetiracetam is 1089 mg/kg, whereas brivaracetam suppresses seizures with an ED50 of 4.4 mg/kg (Table 1), indicating a 250-fold higher anti-seizure potency in this model [101].
Pharmacologic studies have shown that brivaracetam, in contrast to levetiracetam, exhibits significant activity in animal models beyond epilepsy, which likely reflects the improved ability of its selective SV2A mechanism to inhibit synaptic transmission and vesicle release. These preclinical findings suggest potential activity of brivaracetam in neuropathic pain, migraine, and tremor [9]. The latter seems of particular interest as case reports [102] and open-label observations [103] have suggested potential anti-tremor activity of levetiracetam in some tremor types, although this has not been confirmed by double-blind, placebo controlled trials [104–107]. A determination of the ratio between doses inducing adverse effects on motor function in the rotarod test and seizure suppression, i.e. the therapeutic index, which may be lower in epileptic than non-epileptic individuals [3], showed brivaracetam to have a lower therapeutic index in corneal-kindled mice than levetiracetam (46 vs. 148, respectively); however, it was markedly higher than that observed for classical and other newer ASDs (therapeutic indexes 2–21) [9]. In contrast, rotarod testing in amygdala-kindled rats showed brivaracetam to have a higher therapeutic index than levetiracetam (4 vs. 2, respectively) [108].
Clinical studies have shown that levetiracetam is generally well tolerated [109]. The most common adverse effects in epilepsy patients are somnolence, asthenia, and dizziness, which usually appear early after initiation of therapy and generally resolve without medication withdrawal. The most serious adverse effects of levetiracetam are behavioral in nature and are more common in children and in adult epilepsy patients with a prior history of behavioral problems and psychiatric disease [110]. The more selective SV2A mechanism of brivaracetam versus levetiracetam triggered UCB to perform a small, open-label study to assess if brivaracetam may be associated with fewer behavioral adverse effects [111]. This study showed that epilepsy patients experiencing behavioral adverse effects associated with levetiracetam benefited from switching to brivaracetam. The good tolerability of brivaracetam was also seen in several larger clinical epilepsy trials [112–116].
Several randomized, double-blind, placebo-controlled studies in idiopathic generalized epilepsies have shown a significant response of myoclonic seizures to levetiracetam treatment, and the drug is approved by the EMA and FDA as add-on treatment of juvenile myoclonic epilepsy [117, 118]. Levetiracetam is also frequently proposed as a treatment opportunity for various types of myoclonus [119], and open-label trial data even suggested potential antimyoclonic effects in patients with Unverricht–Lundborg disease [120, 121]. However, two phase III studies evaluating the efficacy and safety of adjunctive therapy with brivaracetam in these patients did not demonstrate a statistically significant symptomatic relief of action myoclonus [122].
Brivaracetam has been clinically evaluated as adjunctive therapy for adults with refractory partial-onset seizures. It has demonstrated significant efficacy and good tolerability in both phase II and III clinical studies [123, 124] and has recently been approved by the EMA and FDA for this indication. While brivaracetam and levetiracetam demonstrated similar clinical efficacy, indirect comparisons using available trials remain problematic due to the lack of homogeneity between patient populations and other confounders (e.g. increasing refractoriness, increasing placebo response rates over time) [125]. Interestingly, a post hoc analysis of trial data from a recent major clinical study with brivaracetam [116] on the impact of levetiracetam exposure suggests seizure reduction in patients never exposed to levetiracetam as well as those previously exposed to levetiracetam, although the effect appeared to be greater in the levetiracetam-naïve population. The results of the exploratory post hoc subgroup analysis by levetiracetam exposure showed that statistically significant (p < 0.05 vs. placebo) seizure reduction occurred in both previously levetiracetam-exposed and levetiracetam-naive patients. Percentage reductions in seizure frequency over placebo per 28 days and the ≥50 % responder rates were higher across treatment groups for levetiracetam-naive patients than for levetiracetam-exposed patients. However, it is important to note that there was a response in patients with previous levetiracetam exposure. This finding suggests the possibility that brivaracetam may be an effective ASD in some patients who have previously not responded to levetiracetam, although these post hoc findings would need to be confirmed in further clinical trials. In additional post hoc analyses, greater efficacy was seen in the levetiracetam-naive group than in the prior levetiracetam group, and this tendency was also seen with other ASDs (e.g. carbamazepine-, lamotrigine-, and topiramate-naive groups compared with their prior-exposed counterparts) [126]. These findings support previous reports that suggest patients who did not respond to previous ASDs are more likely to demonstrate pharmacoresistance to additional ASD treatment regimens [127, 128].
A review of the pharmacokinetic properties of brivaracetam in humans [101] has shown it to be well absorbed from the gastrointestinal tract and to show linear pharmacokinetics following single oral doses of up to 600 mg. The compound is weakly bound to plasma proteins (≤20 %) and its volume of distribution is close to that of total body water (0.6 L/kg). The terminal half- life of brivaracetam is approximately 8 h and does not vary with dose [101]. Similar to brivaracetam, a review of levetiracetam [129] has shown linear and time-independent pharmacokinetics, with low intra- and intersubject variability. The drug is rapidly and almost completely absorbed after oral administration, and its bioavailability is not affected by food. Levetiracetam has low plasma protein binding and its volume of distribution is close to the volume of total body water. Its plasma half-life across studies is approximately 6–8 h but the clinical duration of action can last up to 30 h after a single dose, an observation that led to use of a twice-daily dosing regimen in clinical trials [129].
Seletracetam is the lead compound of the second anti-seizure family, identified during the discovery program at UCB focused on identifying selective and high affinity SV2A ligands with anti-seizure properties superior to levetiracetam (Fig. 2). Seletracetam was chosen from a large group of SV2A ligands based not only on its higher affinity for this target (tenfold greater affinity for SV2A than levetiracetam) but also on its potency and efficacy, as observed in in vivo and in vitro testing [130]. As shown in Table 1 and Fig. 3, seletracetam is much more potent than levetiracetam or brivaracetam in the amygdala kindling model, but, in contrast to brivaracetam, ineffective in the MES and pentylenetetrazole tests, thus demonstrating that the two families of anti-seizure SV2A ligands markedly differ in their pharmacology. Seletracetam shows very potent seizure suppression in models of acquired or genetic epilepsy, as well as high CNS tolerability in various animal models [130, 131]. As with brivaracetam, seletracetam is more selective for SV2A than levetiracetam [130]. In clinical phase I studies, seletracetam was rapidly absorbed, volume of distribution was approximately 0.6 L/kg, and plasma terminal half-life was approximately 8 h in young healthy male subjects [131].
6 SV2A Ligands for the Prevention of Epilepsy?
A major goal of contemporary epilepsy research is the identification of therapies to prevent the development of recurrent seizures in individuals at risk, including those with brain injuries, infections, tumors, febrile and afebrile SE, cortical dysplasias, or genetic epilepsy susceptibility [132]. The first indication that levetiracetam may exert not only anti-seizure activity but also antiepileptogenic or disease-modifying activity came from the study by Löscher et al. [15] in the amygdala-kindling model. When rats were treated with levetiracetam during kindling acquisition at daily intraperitoneal doses of 13, 27, or 54 mg/kg, the drug dose-dependently suppressed the increase in seizure severity and duration induced by repeated amygdala stimulation. After termination of daily treatment with 54 mg/kg, duration of behavioral seizures and after-discharges recorded from the amygdala remained significantly shorter compared with vehicle controls, although amygdala stimulations were continued in the absence of drug. Therefore, these data indicated that levetiracetam did not simply mask the expression of kindled seizures through an anti-seizure effect but exerted a true disease-modifying effect [15]. The only other ASD, for which a similar effect had been described previously, was high-dose treatment with valproate [133], whereas other ASDs just retarded kindling by their suppressive effect on kindled seizures [134]. The initial observation of Löscher et al. with levetiracetam in the amygdala-kindling model [15] was subsequently confirmed by Stratton et al. [135], who found that levetiracetam, but not lamotrigine (which was used as a comparator), delayed or prevented the expected increase in after-discharge duration during further kindling after drug washout. Furthermore, levetiracetam, but not valproate, was found to suppress pentylenetetrazole kindling at doses that did not suppress pentylenetetrazole-induced seizures [136].
After the initial observation of an antiepileptogenic or disease-modifying potential of levetiracetam in the kindling model [15], a gene expression analysis was performed to identify the mechanisms responsible for these effects [137]. Previously described epilepsy-related genes, such as neuropeptide Y (NPY), thyrotropin-releasing hormone (TRH) and glial fibrillary acidic protein (GFAP), were upregulated by kindling, and partially normalized by levetiracetam. Similarly, Husum et al. [138] reported that the process of amygdala kindling is associated with an upregulation of hippocampal NPY and brain-derived neurotropic factor (BDNF) which is prevented by levetiracetam. In a subsequent study by Matveeva et al. [42], it was shown that levetiracetam also inhibits the kindling-induced increase of the synaptic vesicle protein SV2A.
However, several studies showed that levetiracetam did not prevent epilepsy when administered after an electrically- or chemically-induced SE that induces epileptogenesis and, after a latent period, epilepsy with spontaneous recurrent seizures [139]. This negative outcome of levetiracetam studies in post-SE models was unexpected and gave rise to critical arguments on study design used for drug testing in the SE models [140]. Indeed, SE induced by systemic administration of chemoconvulsants such as pilocarpine or kainate, or by sustained electrical stimulation of amygdala or hippocampus, may induce too severe and widespread brain injury for allowing any antiepileptogenic or disease-modifying effects of drugs administered after such SE [139]. Interestingly, Margineanu et al. [141] reported that, although prophylactic treatment with levetiracetam after a pilocarpine-induced SE did not seem to prevent development of spontaneous seizures, it completely prevented the development of hippocampal hyperexcitability, i.e. increased amplitude of population spikes recorded in the dentate gyrus and reduced paired-pulse inhibition in the CA1 area. More recently, Sugaya et al. [142] reported that the treatment of rats with levetiracetam over 25 days following SE, induced by intracerebroventricular administration of kainate, reduced the frequency and duration of electrographic seizures recorded 32 days after the termination of treatment [142], which is the first proof that levetiracetam does exert disease-modifying activity in a post-SE model of TLE.
In contrast to the conflicting results from post-SE studies, studies in genetic animal models of epilepsy have consistently shown antiepileptogenic properties of levetiracetam when administered before the developmental expression of seizure activity [132]. Thus, levetiracetam was found to exert antiepileptogenic or disease-modifying effects in SERs [143, 144], indicating that studies on kindling acquisition may be more predictive for such effects than data from SE models, and that post-SE studies may lead to false negative data on antiepileptogenic drug potential [139].
Clinical pilot trials directed by Dr. Pavel Klein (Mid-Atlantic Epilepsy and Sleep Center, Bethesda, MD, USA) determined the safety and feasibility of using levetiracetam to decrease the risk of post-traumatic epilepsy, resulting in promising data [145–147]. In one of these studies [145], an open-label, non-randomized study in 126 adults and children, assessing the safety, tolerability, and pharmacokinetics of levetiracetam, treatment in patients with traumatic brain injury (TBI) for 1 month, initiated within 8 h of the injury, showed a non-significant trend toward fewer patients receiving levetiracetam treatment (11 %) developing epilepsy after 2 years than those who were untreated (20 %). A large, controlled epilepsy prevention trial in patients with TBI is planned. An uncontrolled, retrospective analysis of patients undergoing craniotomy and treated postoperatively with either levetiracetam or phenytoin showed that fewer patients who received levetiracetam (26 %) developed epilepsy after 1 year than those treated with phenytoin (36 %), although the difference did not reach statistical significance [148]. Interestingly, a recent retrospective analysis of seizure outcome implications of ASD use in patients who underwent temporal lobectomy demonstrated perioperative use of levetiracetam to predict a more favorable outcome [149]. While all ASDs were equally effective to suppress early (acute symptomatic) seizures after surgery, at 5 years, patients taking levetiracetam exhibited a substantial ‘seizure freedom advantage’ over other drugs, including carbamazepine, lamotrigine, oxcarbazepine, phenytoin, and topiramate. The authors speculated that this might reflect an antiepileptogenic action of levetiracetam under the assumption that the late recurrence (>6 months post-surgery) of epilepsy reflects a new epileptogenic process following removal of the original epileptic focus. Overall, the available preclinical and clinical data indicate that continued clinical evaluation of the antiepileptogenic potential of levetiracetam is worthwhile.
In this context, it is noteworthy that the selective, high-affinity SV2A ligand brivaracetam shows more pronounced antiepileptogenic effects than levetiracetam in kindling models in mice [108]. In a rat model of post-SE TLE, in which SE is induced by intermittent stimulation of the perforant path, treatment with brivaracetam or a combination of brivaracetam and diazepam 10 min after establishment of self-sustained SE reduced the number of chronic spontaneous recurrent seizures occurring both 6 weeks and 1 year after treatment, indicating a disease-modifying effect, although the contribution of initial insult modification cannot be excluded [150]. The favorable evidence from these initial studies supports the continued evaluation of the antiepileptogenic potential of brivaracetam.
7 Conclusions
SV2A represents an integral constituent of synaptic vesicle membranes in presynaptic terminals and is involved in vesicle trafficking and exocytosis, which is crucial for neurotransmission [36]. The SV2A protein contains multiple interacting binding sites that are linked via the conformation of the protein [88]. Levetiracetam was the first ASD to target SV2A, and thereby revealed that SV2A is involved in the underlying pathology of certain kinds of epilepsy, which is supported by various preclinical and clinical studies showing altered expression of SV2A in epilepsy. Furthermore, dysfunction of SV2A may potentially be involved in AD and other types of cognitive impairment, thus providing novel therapeutic indications to consider for levetiracetam and its congeners. One of the functional consequences of levetiracetam binding to the SV2A protein in brain slices was found to be a reduction in exocytosis [89]; however, the underlying mechanistic effect of ligand binding is not fully elucidated. One hypothesis would be that ligand binding to SV2A would promote/stabilize a conformation that enables the protein to fulfill a role in providing seizure protection [88]. The recent development of negative SV2A modulators, such as UCB0255, which counteracts the anti-seizure effect of levetiracetam [66], will help to further clarify how levetiracetam specifically interacts with SV2A. There is evidence that levetiracetam may alter short-term synaptic plasticity [89, 91] known to arise from simultaneous activation of multiple positive (enhancement) and negative (depression) mechanisms within presynaptic terminals [93]. Increasing the impact of depression at excitatory synapses might have potential for treating epilepsy because more synaptic depression would dampen excitatory feedback in neural circuitry during incipient seizures, thus switching the balance between excitatory and inhibitory synaptic drive to favor inhibition [93]. The effect of levetiracetam on activity-dependent synaptic plasticity would also provide an explanation for the disease-modifying or antiepileptogenic effects of this drug in both genetic and acquired forms of experimental epilepsy. The recent development of PET ligands for SV2A allows imaging of this protein in the brain in vivo. This will hopefully facilitate a further understanding of its role in epilepsy and other diseases, and enable new and innovative ways to define the full therapeutic potential of modulating SV2A function.
Finally, many questions remain to be explored, including why SV2A is highly expressed in some brain areas but absent in others; the differential regulation of SV2A expression in different models of epilepsy and other pathologies; and the exact mechanism of action of levetiracetam and its congeners on SV2A. Emerging data, albeit not as broad as that reported for SV2A, seem to suggest a potential role of SV2B and SV2C in epilepsy and other neurodegenerative diseases, and these additional SV2 isoforms could also become novel drug targets. As shown in this review, the recent availability of SV2A PET ligands and brivaracetam will enable research to further address the physiology and pathophysiology of SV2A, as well as the pharmacology and therapeutic utility of SV2A ligands. This represents a rapidly evolving field of research that has the capability to address many of these relevant and impactful questions in the near future.
References
Chang BS, Lowenstein DH. Epilepsy. N Engl J Med. 2003;349:1257–66.
Banerjee PN, Filippi D, Allen HW. The descriptive epidemiology of epilepsy: a review. Epilepsy Res. 2009;85:31–45.
Löscher W, Klitgaard H, Twyman RE, et al. New avenues for antiepileptic drug discovery and development. Nat Rev Drug Discov. 2013;12:757–76.
Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5:553–64.
Rogawski MA, Löscher W, Rho JM. Mechanisms of action of antiseizure drugs and the ketogenic diet. Cold Spring Harb Perspect Med. 2016;6:a022780.
Lynch BA, Lambeng N, Nocka K, et al. The synaptic vesicle protein SV2A is the binding site for the antiepileptic drug levetiracetam. Proc Natl Acad Sci USA. 2004;101:9861–6.
Gillard M, Chatelain P, Fuks B. Binding characteristics of levetiracetam to synaptic vesicle protein 2A (SV2A) in human brain and in CHO cells expressing the human recombinant protein. Eur J Pharmacol. 2006;536:102–8.
Gillard M, Fuks B, Leclercq K, et al. Binding characteristics of brivaracetam, a selective, high affinity SV2A ligand in rat, mouse and human brain: relationship to anti-convulsant properties. Eur J Pharmacol. 2011;664:36–44.
Klitgaard H, Matagne A, Nicolas JM, et al. Brivaracetam: rationale for discovery and preclinical profile of a selective SV2A ligand for epilepsy treatment. Epilepsia. 2016;57(4):538–48.
Klitgaard H, Verdru P. Levetiracetam: the first SV2A ligand for the treatment of epilepsy. Expert Opin Drug Discov. 2007;2:1537–45.
Gower AJ, Noyer M, Verloes R, et al. UCB L059, a novel anti-convulsant drug: pharmacological profile in animals. Eur J Pharmacol. 1992;222:193–203.
Löscher W, Hönack D. Profile of UCB L059, a novel anticonvulsant drug, in models of partial and generalized epilepsy in mice and rats. Eur J Pharmacol. 1993;232:147–58.
Kupferberg HJ. Antiepileptic drug development program: a cooperative effort of government and industry. Epilepsia. 1989;30(Suppl. 1):S51–6.
Löscher W, Jäckel R, Czuczwar SJ. Is amygdala kindling in rats a model for drug-resistant partial epilepsy? Exp Neurol. 1986;93:211–26.
Löscher W, Hönack D, Rundfeldt C. Antiepileptogenic effects of the novel anticonvulsant levetiracetam (ucb L059) in the kindling model of temporal lobe epilepsy. J Pharmacol Exp Ther. 1998;284:474–9.
Klitgaard H, Matagne A, Gobert J, et al. Evidence for a unique profile of levetiracetam in rodent models of seizures and epilepsy. Eur J Pharmacol. 1998;353:191–206.
Klitgaard H. Levetiracetam: the preclinical profile of a new class of antiepileptic drugs? Epilepsia. 2001;42(Suppl 4):13–8.
Brodie MJ, French JA. Role of levetiracetam in the treatment of epilepsy. Epileptic Disord. 2003;5(Suppl 1):S65–72.
Noyer M, Gillard M, Matagne A, et al. The novel antiepileptic drug levetiracetam (ucb L059) appears to act via a specific binding site in CNS membranes. Eur J Pharmacol. 1995;286:137–46.
Kaminski RM, Matagne A, Leclercq K, et al. SV2A protein is a broad-spectrum anticonvulsant target: functional correlation between protein binding and seizure protection in models of both partial and generalized epilepsy. Neuropharmacology. 2008;54:715–20.
Kaminski RM, Gillard M, Leclercq K, et al. Proepileptic phenotype of SV2A-deficient mice is associated with reduced anticonvulsant efficacy of levetiracetam. Epilepsia. 2009;50:1729–40.
Südhof TC, Rizo J. Synaptic vesicle exocytosis. Cold Spring Harb Perspect Biol. 2011;3:a005637.
Rossetto O, Pirazzini M, Montecucco C. Botulinum neurotoxins: genetic, structural and mechanistic insights. Nat Rev Microbiol. 2014;12:535–49.
Abad-Rodriguez J, Diez-Revuelta N. Axon glycoprotein routing in nerve polarity, function, and repair. Trends Biochem Sci. 2015;40:385–96.
Buckley K, Kelly RB. Identification of a transmembrane glycoprotein specific for secretory vesicles of neural and endocrine cells. J Cell Biol. 1985;100:1284–94.
Mendoza-Torreblanca JG, Vanoye-Carlo A, Phillips-Farfan BV, et al. Synaptic vesicle protein 2A: basic facts and role in synaptic function. Eur J Neurosci. 2013;38:3529–39.
Bajjalieh SM, Frantz GD, Weimann JM, et al. Differential expression of synaptic vesicle protein 2 (SV2) isoforms. J Neurosci. 1994;14:5223–35.
Bajjalieh SM, Peterson K, Linial M, et al. Brain contains two forms of synaptic vesicle protein 2. Proc Natl Acad Sci USA. 1993;90:2150–4.
Janz R, Südhof TC. SV2C is a synaptic vesicle protein with an unusually restricted localization: anatomy of a synaptic vesicle protein family. Neuroscience. 1999;94:1279–90.
Dardou D, Dassesse D, Cuvelier L, et al. Distribution of SV2C mRNA and protein expression in the mouse brain with a particular emphasis on the basal ganglia system. Brain Res. 2011;1367:130–45.
Gronborg M, Pavlos NJ, Brunk I, et al. Quantitative comparison of glutamatergic and GABAergic synaptic vesicles unveils selectivity for few proteins including MAL2, a novel synaptic vesicle protein. J Neurosci. 2010;30:2–12.
Bajjalieh SM, Peterson K, Shinghal R, et al. SV2, a brain synaptic vesicle protein homologous to bacterial transporters. Science. 1992;257:1271–3.
Madeo M, Kovacs AD, Pearce DA. The human synaptic vesicle protein, SV2A, functions as a galactose transporter in Saccharomyces cerevisiae. J Biol Chem. 2014;289:33066–71.
Crowder KM, Gunther JM, Jones TA, et al. Abnormal neurotransmission in mice lacking synaptic vesicle protein 2A (SV2A). Proc Natl Acad Sci USA. 1999;96:15268–73.
Crevecoeur J, Foerch P, Doupagne M, et al. Expression of SV2 isoforms during rodent brain development. BMC Neurosci. 2013;14:87.
Janz R, Goda Y, Geppert M, et al. SV2A and SV2B function as redundant Ca2+ regulators in neurotransmitter release. Neuron. 1999;24:1003–16.
Gorter JA, van Vliet EA, Aronica E, et al. Potential new antiepileptogenic targets indicated by microarray analysis in a rat model for temporal lobe epilepsy. J Neurosci. 2006;26:11083–110.
van Vliet EA, Aronica E, Redeker S, et al. Decreased expression of synaptic vesicle protein 2A, the binding site for levetiracetam, during epileptogenesis and chronic epilepsy. Epilepsia. 2009;50:422–33.
Winden KD, Karsten SL, Bragin A, et al. A systems level, functional genomics analysis of chronic epilepsy. PLoS One. 2011;6:e20763.
Hanaya R, Hosoyama H, Sugata S, et al. Low distribution of synaptic vesicle protein 2A and synaptotagimin-1 in the cerebral cortex and hippocampus of spontaneously epileptic rats exhibiting both tonic convulsion and absence seizure. Neuroscience. 2012;221:12–20.
Matveeva EA, Vanaman TC, Whiteheart SW, et al. Asymmetric accumulation of hippocampal 7S SNARE complexes occurs regardless of kindling paradigm. Epilepsy Res. 2007;73:266–74.
Matveeva EA, Vanaman TC, Whiteheart SW, et al. Levetiracetam prevents kindling-induced asymmetric accumulation of hippocampal 7S SNARE complexes. Epilepsia. 2008;49:1749–58.
Ohno Y, Ishihara S, Terada R, et al. Preferential increase in the hippocampal synaptic vesicle protein 2A (SV2A) by pentylenetetrazole kindling. Biochem Biophys Res Commun. 2009;390:415–20.
Ohno Y, Okumura T, Terada R, et al. Kindling-associated SV2A expression in hilar GABAergic interneurons of the mouse dentate gyrus. Neurosci Lett. 2012;510:93–8.
Löscher W, Rundfeldt C. Kindling as a model of drug-resistant partial epilepsy: selection of phenytoin-resistant and nonresistant rats. J Pharmacol Exp Ther. 1991;258:483–9.
Löscher W. Animal models of intractable epilepsy. Prog Neurobiol. 1997;53:239–58.
Löscher W, Reissmüller E, Ebert U. Anticonvulsant efficacy of gabapentin and levetiracetam in phenytoin-resistant kindled rats. Epilepsy Res. 2000;40:63–77.
Wang L, Shi J, Wu G, et al. Hippocampal low-frequency stimulation increased SV2A expression and inhibited the seizure degree in pharmacoresistant amygdala-kindling epileptic rats. Epilepsy Res. 2014;108:1483–91.
Barton ME, Klein BD, Wolf HH, et al. Pharmacological characterization of the 6 Hz psychomotor seizure model of partial epilepsy. Epilepsy Res. 2001;47:217–28.
Bankstahl M, Bankstahl JP, Löscher W. Pilocarpine-induced epilepsy in mice alters seizure thresholds and the efficacy of antiepileptic drugs in the 6-Hertz psychomotor seizure model. Epilepsy Res. 2013;107:205–16.
Leclercq K, Kaminski RM. Status epilepticus induction has prolonged effects on the efficacy of antiepileptic drugs in the 6-Hz seizure model. Epilepsy Behav. 2015;49:55–60.
Buckmaster PS, Yamawaki R, Thind K. More docked vesicles and larger active zones at basket cell-to-granule cell synapses in a rat model of temporal lobe epilepsy. J Neurosci. 2016;36:3295–308.
Upreti C, Otero R, Partida C, et al. Altered neurotransmitter release, vesicle recycling and presynaptic structure in the pilocarpine model of temporal lobe epilepsy. Brain. 2012;135:869–85.
Feng G, Xiao F, Lu Y, et al. Down-regulation synaptic vesicle protein 2A in the anterior temporal neocortex of patients with intractable epilepsy. J Mol Neurosci. 2009;39:354–9.
Finnema SJ, Nabulsi NB, Eid T, et al. Imaging synaptic density in the living human brain using positron emission tomography. Sci Transl Med. 2016;8:348ra96.
Toering ST, Boer K, de Groot M, et al. Expression patterns of synaptic vesicle protein 2A in focal cortical dysplasia and TSC-cortical tubers. Epilepsia. 2009;50:1409–18.
de Groot M, Toering ST, Boer K, et al. Expression of synaptic vesicle protein 2A in epilepsy-associated brain tumors and in the peritumoral cortex. Neuro Oncol. 2010;12:265–73.
de Groot M, Aronica E, Heimans JJ, et al. Synaptic vesicle protein 2A predicts response to levetiracetam in patients with glioma. Neurology. 2011;77:532–9.
Serajee FJ, Huq AM. Homozygous mutation in synaptic vesicle glycoprotein 2a gene results in intractable epilepsy, involuntary movements, microcephaly, and developmental and growth retardation. Pediatr Neurol. 2015;52:642–6.
Bakker A, Krauss GL, Albert MS, et al. Reduction of hippocampal hyperactivity improves cognition in amnestic mild cognitive impairment. Neuron. 2012;74:467–74.
Koh MT, Haberman RP, Foti S, et al. Treatment strategies targeting excess hippocampal activity benefit aged rats with cognitive impairment. Neuropsychopharmacology. 2010;35:1016–25.
Rhinn H, Fujita R, Qiang L, et al. Integrative genomics identifies APOE epsilon4 effectors in Alzheimer’s disease. Nature. 2013;500:45–50.
Sanchez PE, Zhu L, Verret L, et al. Levetiracetam suppresses neuronal network dysfunction and reverses synaptic and cognitive deficits in an Alzheimer’s disease model. Proc Natl Acad Sci USA. 2012;109:E2895–903.
Stockburger C, Miano D, Baeumlisberger M, et al. A mitochondrial role of SV2a protein in aging and Alzheimer’s disease: studies with levetiracetam. J Alzheimers Dis. 2015;50:201–15.
Cumbo E, Ligori LD. Levetiracetam, lamotrigine, and phenobarbital in patients with epileptic seizures and Alzheimer’s disease. Epilepsy Behav. 2010;17:461–6.
Detrait E, Laruelle M, Lamberty Y, et al. 5:00 pm negative modulation of the synaptic vesicle protein (SV2A): a new pharmacological target for cognitive deficit associated with schizophrenia. Schizophr Res. 2014;153(Suppl 1):S66
Bragina L, Fattorini G, Giovedi S, et al. Analysis of synaptotagmin, SV2, and Rab3 expression in cortical glutamatergic and GABAergic axon terminals. Front Cell Neurosci. 2011;5:32.
Detrait E, Maurice T, Hanon E, et al. Lack of synaptic vesicle protein SV2B protects against amyloid-beta 25–35-induced oxidative stress, cholinergic deficit and cognitive impairment in mice. Behav Brain Res. 2014;271:277–85.
Venkatesan K, Alix P, Marquet A, et al. Altered balance between excitatory and inhibitory inputs onto CA1 pyramidal neurons from SV2A-deficient but not SV2B-deficient mice. J Neurosci Res. 2012;90:2317–27.
Crèvecoeur J, Kaminski RM, Rogister B, et al. Expression pattern of synaptic vesicle protein 2 (SV2) isoforms in patients with temporal lobe epilepsy and hippocampal sclerosis. Neuropathol Appl Neurobiol. 2014;40:191–204.
Crèvecoeur J, Kaminski R, Rogister B, et al. Expression of synaptic vesicle protein 2C (SV2C) is selectively increased in hippocampal sclerosis with mossy fiber sprouting. Eur J Neurol. 2011;18(Suppl. 2):175.
Dardou D, Monlezun S, Foerch P, et al. A role for Sv2c in basal ganglia functions. Brain Res. 2013;1507:61–73.
Bretin F, Warnock G, Bahri MA, et al. Preclinical radiation dosimetry for the novel SV2A radiotracer [18F]UCB-H. EJNMMI Res. 2013;3:35.
Mercier J, Archen L, Bollu V, et al. Discovery of heterocyclic nonacetamide synaptic vesicle protein 2A (SV2A) ligands with single-digit nanomolar potency: opening avenues towards the first SV2A positron emission tomography (PET) ligands. Chem Med Chem. 2014;9:693–8.
Warnock GI, Aerts J, Bahri MA, et al. Evaluation of 18F-UCB-H as a novel PET tracer for synaptic vesicle protein 2A in the brain. J Nucl Med. 2014;55:1336–41.
Bretin F, Bahri MA, Bernard C, et al. Biodistribution and radiation dosimetry for the novel SV2A radiotracer [(18)F]UCB-H: first-in-human study. Mol Imaging Biol. 2015;17:557–64.
Estrada S, Lubberink M, Thibblin A, et al. [(11)C]UCB-A, a novel PET tracer for synaptic vesicle protein 2A. Nucl Med Biol. 2016;43:325–32.
Nabulsi N, Mercier J, Holden D, et al. Synthesis and preclinical evaluation of 11C-UCB-J as a PET tracer for imaging the synaptic vesicle glycoprotein 2A in the brain. J Nucl Med. 2016;57(5):777–84.
Cai H, Mangner TJ, Muzik O, et al. Radiosynthesis of (11)C-levetiracetam: a potential marker for PET imaging of SV2A expression. ACS Med Chem Lett. 2014;5:1152–5.
Kaminski RM, Gillard M, Klitgaard H. Targeting SV2A for discovery of antiepileptic drugs. In: Noebels JL, Avoli M, Rogawski MA, Olsen RW, Delgado-Escueta AV, editors. Jasper’s basic mechanisms of the epilepsies. 4th ed. New York: Oxford; 2012. p. 974–83.
Surges R, Volynski KE, Walker MC. Is levetiracetam different from other antiepileptic drugs? Levetiracetam and its cellular mechanism of action in epilepsy revisited. Ther Adv Neurol Disord. 2008;1:13–24.
Feany MB, Lee S, Edwards RH, et al. The synaptic vesicle protein SV2 is a novel type of transmembrane transporter. Cell. 1992;70:861–7.
Gingrich JA, Andersen PH, Tiberi M, et al. Identification, characterization, and molecular cloning of a novel transporter-like protein localized to the central nervous system. FEBS Lett. 1992;312:115–22.
Lynch BA, Matagne A, Brannstrom A, et al. Visualization of SV2A conformations in situ by the use of Protein Tomography. Biochem Biophys Res Commun. 2008;375:491–5.
Shi J, Anderson D, Lynch BA, et al. Combining modelling and mutagenesis studies of synaptic vesicle protein 2A to identify a series of residues involved in racetam binding. Biochem Soc Trans. 2011;39:1341–7.
Lee J, Daniels V, Sands ZA, et al. Exploring the interaction of SV2A with racetams using homology modelling, molecular dynamics and site-directed mutagenesis. PLoS One. 2015;10:e0116589.
Correa-Basurto J, Cuevas-Hernandez RI, Phillips-Farfan BV, et al. Identification of the antiepileptic racetam binding site in the synaptic vesicle protein 2A by molecular dynamics and docking simulations. Front Cell Neurosci. 2015;9:125.
Daniels V, Wood M, Leclercq K, et al. Modulation of the conformational state of the SV2A protein by an allosteric mechanism as evidenced by ligand binding assays. Br J Pharmacol. 2013;169:1091–101.
Yang XF, Weisenfeld A, Rothman SM. Prolonged exposure to levetiracetam reveals a presynaptic effect on neurotransmission. Epilepsia. 2007;48:1861–9.
Yang XF, Rothman SM. Levetiracetam has a time- and stimulation-dependent effect on synaptic transmission. Seizure. 2009;18:615–9.
Meehan AL, Yang X, McAdams BD, et al. A new mechanism for antiepileptic drug action: vesicular entry may mediate the effects of levetiracetam. J Neurophysiol. 2011;106:1227–39.
Nowack A, Malarkey EB, Yao J, et al. Levetiracetam reverses synaptic deficits produced by overexpression of SV2A. PLoS One. 2011;6:e29560.
Garcia-Perez E, Mahfooz K, Covita J, et al. Levetiracetam accelerates the onset of supply rate depression in synaptic vesicle trafficking. Epilepsia. 2015;56:535–45.
Yang X, Bognar J Jr, He T, et al. Brivaracetam augments short-term depression and slows vesicle recycling. Epilepsia. 2015;56:1899–909.
Wood M, Urbain D, Gillard M. Evidence for a differential interaction of brivaracetam and levetiracetam with the SV2A protein. Epilepsia. 2015;56(Suppl. 1):215.
Kenda BM, Matagne AC, Talaga PE, et al. Discovery of 4-substituted pyrrolidone butanamides as new agents with significant antiepileptic activity. J Med Chem. 2004;47:530–49.
Klitgaard H, Matagne A. Mechanisms of action of levetiracetam and newer SV2A ligands. In: Shorvon S, Pedley TA, editors. The epilepsies. 3rd ed. Philadelphia: Butterworth Heineman Elsevier; 2008. p. 28–38.
Nicolas JM, Hannestad J, Holden D, et al. Brivaracetam, a selective high-affinity synaptic vesicle protein 2A (SV2A) ligand with preclinical evidence of high brain permeability and fast onset of action. Epilepsia. 2016;57:201–9.
Zona C, Pieri M, Carunchio I, et al. Brivaracetam (ucb 34714) inhibits Na(+) current in rat cortical neurons in culture. Epilepsy Res. 2010;88:46–54.
Mumoli L, Palleria C, Gasparini S, et al. Brivaracetam: review of its pharmacology and potential use as adjunctive therapy in patients with partial onset seizures. Drug Design Develop Ther. 2015;9:5719–25.
Bialer M, Johannessen SI, Levy RH, et al. Progress report on new antiepileptic drugs: a summary of the Ninth Eilat Conference (EILAT IX). Epilepsy Res. 2009;83:1–43.
Ferlazzo E, Morgante F, Rizzo V, et al. Successful tratment of Holmes tremor by levetiracetam. Mov Disord. 2008;23:2001–3.
Fasano A, Deuschl G. Therapeutic advances in tremor. Mov Disord. 2015;30:1557–65.
Feys P, D’hooghe MB, Nagels G, et al. The effect of levetiracetam on tremor severity and functionality in patients with multiple sclerosis. Mult Scler. 2009;15:371–8.
Chitsaz A, Mehrbod N, Etemadifar M, et al. Does levetiracetam decrease of the rubral tremor in patients with multiple sclerosis. J Res Med Sci. 2013;18:S78–80.
Elble RJ, Lyons KE, Pahwa R. Levetiracetam is not effective for essential tremor. Clin Neuropharmacol. 2007;30:350–6.
Hellriegel H, Raethjen J, Deuschl G, et al. Levetiracetam in primary orthostatic tremor: a double-blind placebo-controlled crossover study. Mov Disord. 2011;26:2431–4.
Matagne A, Margineanu DG, Kenda B, et al. Anti-convulsive and anti-epileptic properties of brivaracetam (ucb 34714), a high-affinity ligand for the synaptic vesicle protein, SV2A. Br J Pharmacol. 2008;154:1662–71.
French JA, Gazzola DM. New generation antiepileptic drugs: what do they offer in terms of improved tolerability and safety? Ther Adv Drug Saf. 2011;2:141–58.
Sirsi D, Safdieh JE. The safety of levetiracetam. Expert Opin Drug Saf. 2007;6:241–50.
Yates SL, Fakhoury T, Liang W, et al. An open-label, prospective, exploratory study of patients with epilepsy switching from levetiracetam to brivaracetam. Epilepsy Behav. 2015;52:165–8.
French JA, Costantini C, Brodsky A, et al. Adjunctive brivaracetam for refractory partial-onset seizures: a randomized, controlled trial. Neurology. 2010;75:519–25.
Biton V, Berkovic SF, Abou-Khalil B, et al. Brivaracetam as adjunctive treatment for uncontrolled partial epilepsy in adults: a phase III randomized, double-blind, placebo-controlled trial. Epilepsia. 2014;55:57–66.
Ryvlin P, Werhahn KJ, Blaszczyk B, et al. Adjunctive brivaracetam in adults with uncontrolled focal epilepsy: results from a double-blind, randomized, placebo-controlled trial. Epilepsia. 2014;55:47–56.
Ferlazzo E, Russo E, Mumoli L, et al. Profile of brivaracetam and its potential in the treatment of epilepsy. Neuropsychiatr Dis Treat. 2015;11:2967–73.
Klein P, Schiemann J, Sperling MR, et al. A randomized, double-blind, placebo-controlled, multicenter, parallel-group study to evaluate the efficacy and safety of adjunctive brivaracetam in adult patients with uncontrolled partial-onset seizures. Epilepsia. 2015;56:1890–8.
Noachtar S, Andermann E, Meyvisch P, et al. Levetiracetam for the treatment of idiopathic generalized epilepsy with myoclonic seizures. Neurology. 2008;70:607–16.
Crespel A, Gelisse P, Reed RC, et al. Management of juvenile myoclonic epilepsy. Epilepsy Behav. 2013;28:S81–6.
Levy A, Chen R. Myoclonus: pathophysiology and treatment options. Curr Treat Options Neurol. 2016;18:21.
Magaudda A, Gelisse P, Genton P. Antimyoclonic effects of levetiracetam in 13 patients with Unverricht–Lundborg disease: clinical observations. Epilepsia. 2004;45:678–81.
Genton P, Gelisse P. Antimyoclonic effect of levetiracetam. Epileptic Disord. 2000;2:209–12.
Kälviäinen R, Genton P, Andermann E, et al. Brivaracetam in Unverricht–Lundborg disease (EPMI): results from two randomized, double-blind, placebo-controlled studies. Epilepsia. 2016;57:210–21.
Klein P, Tyrlikova I, Brazdil M, et al. Brivaracetam for the treatment of epilepsy. Expert Opin Pharmacother. 2016;17:283–95.
Lattanzi S, Cagnetti C, Foschi N, et al. Brivaracetam add-on for refractory focal epilepsy: a systematic review and meta-analysis. Neurology. 2016;86:1344–52.
Swallow E, Fang A, Signorovitch J, Plumb J, Borghs S. Can indirect comparison methods mitigate evolving trial populations in adjunctive antiepileptic drug trials? A propensity-score matched indirect comparison of brivaracetam and levetiracetam. 21st ISPOR Annual International Meeting, 21–25 May 2016, Washington DC.
Chung S, Klein P, Sperling M, et al. Efficacy of brivaracetam (BRV) as adjunctive therapy in partial-onset (focal) seizures among patients with prior levetiracetam (LEV), carbamazepine (CBZ), lamotrigine (LTG), or topiramate (TPM) exposure. Neurology. 2016;86(16 Suppl):P2.027.
Kwan P, Brodie MJ. Early identification of refractory epilepsy. N Engl J Med. 2000;342:314–9.
Mohanraj R, Brodie MJ. Diagnosing refractory epilepsy: response to sequential treatment schedules. Eur J Neurol. 2006;13:277–82.
Bialer M, Johannessen SI, Kupferberg HJ, et al. Progress report on new antiepileptic drugs: a summary of the Fifth Eilat Conference (EILAT V). Epilepsy Res. 2001;43:11–58.
Bennett B, Matagne A, Michel P, et al. Seletracetam (UCB 44212). Neurotherapeutics. 2007;4:117–22.
Matagne A, Margineanu DG, Potschka H, et al. Profile of the new pyrrolidone derivative seletracetam (ucb 44212) in animal models of epilepsy. Eur J Pharmacol. 2009;614:30–7.
Kaminski RM, Rogawski MA, Klitgaard H. The potential of antiseizure drugs and agents that act on novel molecular targets as antiepileptogenic treatments. Neurotherapeutics. 2014;11:385–400.
Silver JM, Shin C, McNamara JO. Antiepileptogenic effects of conventional anticonvulsants in the kindling model of epilepsy. Ann Neurol. 1991;29:356–63.
Dudek FE. Commentary: a skeptical view of experimental gene therapy to block epileptogenesis. Neurotherapeutics. 2009;6:319–22.
Stratton SC, Large CH, Cox B, et al. Antiepileptogenic-like effects of lamotrigine in a rat amygdala kindling model. Epilepsy Res. 2003;53:95–106.
Ohno Y, Ishihara S, Terada R, et al. Antiepileptogenic and anticonvulsive actions of levetiracetam in a pentylenetetrazole kindling model. Epilepsy Res. 2010;89:360–4.
Gu J, Lynch BA, Anderson D, et al. The antiepileptic drug levetiracetam selectively modifies kindling-induced alterations in gene expression in the temporal lobe of rats. Eur J Neurosci. 2004;19:334–45.
Husum H, Bolwig TG, Sanchez C, et al. Levetiracetam prevents changes in levels of brain-derived neurotrophic factor and neuropeptide Y mRNA and of Y1- and Y5-like receptors in the hippocampus of rats undergoing amygdala kindling: implications for antiepileptogenic and mood-stabilizing properties. Epilepsy Behav. 2004;5:204–15.
Löscher W, Brandt C. Prevention or modification of epileptogenesis after brain insults: experimental approaches and translational research. Pharmacol Rev. 2010;62:668–700.
Dudek FE, Bertram EH, Staley KJ. Antiepileptogenesis therapy with levetiracetam: data from kindling versus status epilepticus models. Epilepsy Curr. 2008;8:28–30.
Margineanu DG, Matagne A, Kaminski RM, et al. Effects of chronic treatment with levetiracetam on hippocampal field responses after pilocarpine-induced status epilepticus in rats. Brain Res Bull. 2008;77:282–5.
Sugaya Y, Maru E, Kudo K, et al. Levetiracetam suppresses development of spontaneous EEG seizures and aberrant neurogenesis following kainate-induced status epilepticus. Brain Res. 2010;1352:187–99.
Yan HD, Ji-qun C, Ishihara K, et al. Separation of antiepileptogenic and antiseizure effects of levetiracetam in the spontaneously epileptic rat (SER). Epilepsia. 2005;46:1170–7.
Russo E, Citraro R, Scicchitano F, et al. Comparison of the antiepileptogenic effects of an early long-term treatment with ethosuximide or levetiracetam in a genetic animal model of absence epilepsy. Epilepsia. 2010;51:1560–9.
Klein P, Herr D, Pearl PL, et al. Results of phase 2 safety and feasibility study of treatment with levetiracetam for prevention of posttraumatic epilepsy. Arch Neurol. 2012;69:1290–5.
Klein P, Herr D, Pearl PL, et al. Results of phase II pharmacokinetic study of levetiracetam for prevention of post-traumatic epilepsy. Epilepsy Behav. 2012;24:457–61.
Pearl PL, McCarter R, McGavin CL, et al. Results of phase II levetiracetam trial following acute head injury in children at risk for posttraumatic epilepsy. Epilepsia. 2013;54:e135–7.
Milligan TA, Hurwitz S, Bromfield EB. Efficacy and tolerability of levetiracetam versus phenytoin after supratentorial neurosurgery. Neurology. 2008;71:665–9.
Jehi LE, Irwin AI, Kayyali H, et al. Levetiracetam may favorably affect seizure outcome after temporal lobectomy. Epilepsia. 2012;53:979–86.
Wasterlain C, Suchomelova L, Matagne A, et al. Short-term and long-term effects of brivaracetam in an animal model of status epilepticus. Epilepsia. 2009;50(Suppl. 10):13.
Löscher W, Schmidt D. Epilepsy: perampanel—new promise for refractory epilepsy? Nat Rev Neurol. 2012;8:661–2.
Omasits U, Ahrens CH, Muller S, et al. Protter: interactive protein feature visualization and integration with experimental proteomic data. Bioinformatics. 2014;30:884–6.
Lee J, Sands ZA, Biggin PC. A numbering system for MFS transporter proteins. Front Mol Biosci. 2016;3:21.
Marescaux C, Vergnes M. Genetic absence epilepsy in rats from strasbourg (GAERS). Ital J Neurol Sci. 1995;16:113–8.
Ebert U, Reissmüller E, Löscher W. The new antiepileptic drugs lamotrigine and felbamate are effective in phenytoin-resistant kindled rats. Neuropharmacology. 2000;39:1893–903.
Hönack D, Löscher W. Kindling increases the sensitivity of rats to adverse effects of certain antiepileptic drugs. Epilepsia. 1995;36:763–71.
Bialer M, Twyman RE, White HS. Correlation analysis between anticonvulsant ED50 values of antiepileptic drugs in mice and rats and their therapeutic doses and plasma levels. Epilepsy Behav. 2004;5:866–72.
Matagne A, Klitgaard H. Validation of corneally kindled mice: a sensitive screening model for partial epilepsy in man. Epilepsy Res. 1998;31:59–71.
Löscher W. Fit for purpose application of currently existing animal models in the discovery of novel epilepsy therapies. Epilepsy Res. 2016;126:157–84.
Löscher W, Nau H, Marescaux C, Vergnes M. Comparative evaluation of anticonvulsant and toxic potencies of valproic acid and 2-en-valproic acid in different animal models of epilepsy. Eur J Pharmacol 1984;99(2–3):211–8.
Acknowledgments
Karine Leclercq is greatly acknowledged for sharing the seizure model data generated with SV2B knockout mice. The help of Dr. Laurent Provins with preparation of Fig. 4 is greatly appreciated. We would also like to thank Dr. Joanna Lee and Prof. Philip Biggin for providing the inward-open and outward-open models of SV2A. We are grateful to Dr. Sjoerd Finnema for sharing PET imaging data obtained with [11C]UCB-J radiotracer and preparation of Fig. 7. We thank Dr. Barbara Bennett, UCB, for her review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Wolfgang Löscher’s initial animal studies on levetiracetam were supported by grants from UCB Pharma. No funding was obtained for preparing this current review. The open access payment for this article was funded by UCB Pharma.
Conflict of interest
Michel Gillard, Zara Sands, Rafal M. Kaminski, and Henrik Klitgaard are employees of UCB Pharma (Braine-l’Alleud, Belgium). Wolfgang Löscher has no conflicts of interest to declare.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Löscher, W., Gillard, M., Sands, Z.A. et al. Synaptic Vesicle Glycoprotein 2A Ligands in the Treatment of Epilepsy and Beyond. CNS Drugs 30, 1055–1077 (2016). https://doi.org/10.1007/s40263-016-0384-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-016-0384-x