
Abstract
Background and Objective
Imarikiren hydrochloride (TAK-272; SCO-272) is a novel direct renin inhibitor. The objective of this study was to determine the effects of renal impairment (RI) or hepatic impairment (HI) on the pharmacokinetics and safety of imarikiren.
Methods
This phase I, open-label, parallel-group comparative study evaluated the pharmacokinetics and safety of a single 40 mg oral dose of imarikiren in RI [mild, moderate, severe, or end-stage renal disease (ESRD), and on hemodialysis] or HI (mild or moderate) subjects compared with subjects with normal renal or hepatic function.
Results
Following administration of a single 40 mg oral imarikiren dose, the geometric mean imarikiren area under the plasma concentration–time curve from time zero to infinity (AUC∞) and maximum observed plasma concentration (Cmax) in subjects with mild, moderate, and severe RI (including non-hemodialysis and ESRD), and hemodialysis subjects compared with normal renal function subjects were approximately 0.5-, 1.2-, 2.7-, and 1.8-fold, respectively, for AUC∞; and approximately 0.6-, 0.8-, 2.1-, and 1.4-fold, respectively, for Cmax. The mean fraction of excretion of imarikiren in dialysate was ~ 3% during the 4 h dialysis period. The geometric mean imarikiren AUC∞ and Cmax in mild and moderate HI subjects compared with normal hepatic function subjects were approximately 1.0- and 1.4-fold, respectively, for AUC∞, and approximately 0.9- and 1.3-fold, respectively, for Cmax. No deaths or serious adverse events were observed; all adverse events were mild or moderate in intensity.
Conclusions
RI and HI are associated with limited changes in imarikiren pharmacokinetics. Imarikiren was safe and well-tolerated, regardless of the severity of RI or HI.
Clinical Trial Registration
ClinicalTrials.gov Identifier: NCT02367872.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Imarikiren was safe and well-tolerated, regardless of the severity of renal impairment (RI) or hepatic impairment (HI). |
RI or HI were associated with limited changes in the pharmacokinetics of imarikiren. |
1 Introduction
Direct inhibition of renin, the rate-limiting enzyme of the renin–angiotensin (R–A) system, is a promising treatment strategy for cardiovascular/renal disease [1], with one direct renin inhibitor (DRI) (aliskiren) approved for the treatment of hypertension. Because of their ability to inhibit the whole R–A system, DRIs may have more potent therapeutic effects than agents that do not inhibit the whole R–A system, such as angiotensin II receptor blockers (ARBs) and angiotensin-converting enzyme (ACE) inhibitors [1]. In clinical studies, treatment with aliskiren resulted in reduced blood pressure [2] and decreased urinary albumin compared with placebo [3]; however, aliskiren has relatively low bioavailability [4] and produces dose-related gastrointestinal adverse events (AEs) [5].
Imarikiren hydrochloride (TAK-272; SCO-272) is a novel oral DRI [4]. In non-clinical studies, imarikiren showed potent renin inhibitory activity equivalent to aliskiren, but with increased bioavailability in animals [4]. In an in vitro analysis of metabolism using hepatic microsomes, the major metabolite of imarikiren in humans was M-I and it was shown that cytochrome P450 (CYP) 3A4/5 was mainly involved in human metabolism of imarikiren [6]. In in vitro tests, imarikiren and M-I demonstrated strong inhibition of plasma renin activity in humans, with half-maximal inhibitor concentrations of 2.1 and 0.43 nM, respectively. In vitro plasma protein binding of imarikiren in mice, rats, monkeys, and humans decreased in proportion to plasma imarikiren concentration in all species [6]. An analysis of imarikiren using a variety of protein solutions indicated that imarikiren may mainly bind to α1-acid glycoprotein (AGP) in human plasma [7]. In an in vivo study, while imarikiren and M-I were detected in the urine and feces of rats and monkeys and in the bile of rats, the excretion of imarikiren as unchanged drug in urine and bile was low [6]. Thus, absorbed imarikiren was thought to be eliminated from the body by metabolism.
The pharmacokinetics and safety of imarikiren have been assessed in two human studies to date. In a phase I single-dose study conducted in Japan, imarikiren at doses of 5, 10, 25, 50, 100, and 200 mg was well-tolerated in healthy, non-elderly adult male subjects [8]. In that study, imarikiren was rapidly absorbed after oral administration, and the area under the plasma concentration–time curve (AUC) from time zero to infinity (AUC∞) and maximum observed plasma concentration (Cmax) of imarikiren generally increased in proportion to the dose of imarikiren (mean AUC∞ range across doses: 211–12,060 ng·h/mL; mean Cmax range across doses: 25–2614 ng/mL) [8]. Imarikiren was detected in urine, and the mean cumulative fraction of urinary excretion of imarikiren as unchanged drug up to 72 h after treatment was 12.9–15.9%. In another phase I study conducted in Japan with healthy subjects, multiple oral administrations of imarikiren (80 mg for elderly; 80 or 160 mg for non-elderly subjects) for 7 days were safe and well-tolerated with no drug accumulation [9]. Apparent clearance ranged from 14.1 to 21.5 L/h. In addition, potent and sustained suppression of plasma renin activity was observed [9].
Based on the results of these non-clinical and clinical studies, imarikiren was thought to mainly be metabolized by the liver and partially excreted as unchanged drug through the kidneys. Therefore, the present phase I study was designed to evaluate the effect of renal impairment (RI) or hepatic impairment (HI) on the pharmacokinetics and safety of single doses of imarikiren.
2 Methods
2.1 Study Design and Participants
This was a phase I, open-label, parallel-group, comparative study to evaluate the effect of RI or HI on the pharmacokinetics and safety of imarikiren after a single oral administration. The study protocol was approved by the institutional review boards for each study site. All subjects provided written informed consent.
Subjects were eligible for the study if they were aged 20–85 years, weighed at least 45 kg for males and 40 kg for females, and had a body mass index < 35 kg/m2 at screening and Day 1. Key exclusion criteria included previous exposure to imarikiren, history of cancer, known hypersensitivity or allergies to any component of the imarikiren formulation or renin inhibitors, history of kidney or liver transplantation, onset of myocardial infarction or coronary revascularization within 6 months before screening, and clinically significant hyperkalemia.
Subjects with RI were grouped into one of four cohorts: mild RI [estimated glomerular filtration rate (eGFR) ≥ 60 and < 90 mL/min/1.73 m2], moderate RI (eGFR ≥ 30 and < 60 mL/min/1.73 m2), severe RI or end-stage renal disease (ESRD) but not on hemodialysis (eGFR < 30 mL/min/1.73 m2), and subjects on hemodialysis. The classification was based on guidelines of the Japanese Society of Nephrology [10, 11] as well as a US Food and Drug Administration (FDA) guideline [12]. Exclusion criteria for RI cohorts included alanine aminotransferase (ALT) or aspartate aminotransferase (AST) > 2.0 times the upper limit of normal (ULN) and sitting systolic blood pressure < 110 mmHg at screening. A cohort of subjects with normal renal function (eGFR ≥ 90 mL/min/1.73 m2) was matched to within ± 10 years of the mean age and ± 20% of the mean weight for all subjects with RI who were administered the study drug. Exclusion criteria for the normal renal function cohort included sitting systolic blood pressure < 80 mmHg at screening with repeated instances of findings (dizziness postural, facial pallor, cold sweats) suggestive of hypotension and ALT or AST > 2.0 times the ULN at screening. The planned number of subjects for each renal cohort was six.
Subjects with HI were grouped into one of two cohorts: mild HI (Child-Pugh class A) and moderate HI (Child-Pugh class B). These cohorts were included per FDA guidelines [13]. Exclusion criteria for the HI cohorts included sitting systolic blood pressure < 80 mmHg at screening with repeated instances of findings (dizziness postural, facial pallor, cold sweats) suggestive of hypotension and eGFR < 60 mL/min/1.73 m2 at screening. A cohort of subjects with normal hepatic function was matched within ± 10 years of the mean age and ± 20% of the mean weight for all subjects with HI who were administered the study drug. Exclusion criteria for this cohort were the same as those for the normal renal function cohort. The planned number of subjects for each hepatic cohort was six.
2.2 Treatment
The study consisted of a screening period (Day − 28 to Day − 2), a pretreatment examination (Day − 1), a treatment period (Day 1 to Day 6), and a follow-up period (Day 7 to Day 8). Each subject was hospitalized on Day − 1 after pretreatment examination, discharged on Day 6, and then visited the study site on Day 8. For subjects on hemodialysis, the study course was repeated: Course 1 [hemodialysis performed from 2 h after study drug administration (Day 1)] and Course 2 [hemodialysis not performed on the day of study drug administration (Day 1) and was to be started after an interval of at least 24 h after study drug administration]. Course 2 was performed within 56 days after Day 8 of Course 1 (within 28–56 days for subjects with a body weight of < 50 kg).
Subjects received a single oral dose of imarikiren 40 mg on Day 1 of each course. All subjects received imarikiren with 150 mL of water, after having fasted for ≥ 10 h. Dietary products and drinks that contained alcohol or caffeine were prohibited from 72 h before treatment to the end of the follow-up examination. Subjects were given consistent instructions regarding diet and exercise therapies (e.g., instructions regarding calories and salt intake) throughout the study period and instructed to comply with them.
2.3 Pharmacokinetic Assessment
Blood sampling (3 mL per sample) for pharmacokinetic assessment was performed on Day 1 (1 h before and 0.5, 1, 1.5, 2, 3, 4, 5, 8, 10, 12, and 14 h after treatment administration), Day 2 (24 and 36 h after treatment administration), and on Days 3–6 (48, 72, 96, and 120 h after treatment administration). For in vitro measurement of imarikiren plasma protein binding ratio, a blood sample (30 mL) was collected during the pretreatment examination (Day – 1). In Course 1, plasma sampling for pharmacokinetics in hemodialysis subjects was collected from the arterial-side and venous-side blood circuits at 3, 4, and 5 h after treatment administration. Urine sampling for pharmacokinetic assessment was performed on Days 1–2 (pretreatment, 0–4, 4–8, 8–12, and 12–24 h after treatment), Days 2–3 (24–48 h after treatment), and Days 3–4 (48–72 h after treatment). Dialysis fluid samples for pharmacokinetic analyses were taken on Day 1 of Course 1 (pretreatment and from 2.5 h after treatment to the end of dialysis at an interval of 30 min).
2.4 Bioanalytical Assay
Concentrations of imarikiren in plasma, urine, and dialysate and the metabolite M-I in plasma and urine were measured at CMIC Pharma Science Co., Ltd. (formerly JCL Bioassay Corporation) by liquid chromatography with tandem mass spectrometry (LC–MS/MS). Internal standards (IS) were deuterated forms of imarikiren hydrochloride and M-I.
For the pharmacokinetic analyses, frozen plasma, urine, and dialysate samples were sent to the analytical laboratory and stored at − 70 °C or below until LC–MS/MS. For plasma sample preparation, 50 μL of plasma sample was mixed with 10 μL of methanol, 900 μL of ammonium acetate (10 mmol/L), and 20 μL of IS solution (200 ng/mL), and the resultant solution was purified. This solution was mixed with 500 μL of 10 mmol/L ammonium acetate/acetic acid (1000:1, v/v) and 300 μL of the mixture was filtered through 0.22 μm polyvinylidene fluoride (PVDF) Ultrafree®-MC centrifugal filters (Merck Millipore, Burlington, MA, U.S.A.). A total of 10 μL of the filtrate was injected to LC–MS/MS. For urine sample preparation, 20 μL of urine sample was mixed with 20 μL of methanol and 100 μL of IS solution (200 ng/nL) followed by further mixing and centrifugation. The supernatant (20 μL) was mixed with 400 μL of 10 mmol/L ammonium acetate/acetonitrile/acetic acid (65:35:0.1, v/v/v) and then filtered in a similar manner as for the plasma samples. The LC–MS/MS injection volume for the urine samples was 3 μL. For sample preparation of dialysate samples, 50 μL of dialysis solution was mixed with 50 μL of acetonitrile, 10 μL of methanol, and 20 μL of IS solution (200 ng/mL) and then mixed and centrifuged. This mixture was filtered as described previously for the plasma samples. The LC–MS/MS injection volume for the dialysate samples was 5 μL.
The LC–MS/MS system consisted of a Nexera ultra high-performance liquid chromatograph (Shimadzu Corporation, Kyoto, Japan) and either a Triple Quad™ 5500 (plasma and urine assays; AB Sciex, Redwood City, CA, USA) or API 4000™ (dialysate assay; AB Sciex) mass spectrometer. Chromatographic separation was performed on an XBridge™ C18 column (Waters Corporation, Milford, MA, USA) at a column temperature of 40 °C. The mass spectrometer was equipped with electrospray ionization and turbo spray; multiple reaction monitoring with the positive ion mode was used. Data analysis was performed using Analyst® version 1.4.2 or 1.5.2 (AB Sciex). The lower limit of quantification was 0.5 ng/mL for plasma and dialysate assays and 20 ng/mL for the urine assay. The percentage accuracy for concentration determination was 100 ± 15%.
In vitro plasma protein binding of imarikiren was measured at Tsukuba Laboratories, Nemoto Science Co., Ltd (Ibariki, Japan) using the ultracentrifugation method. In brief, a test substance solution consisting of a 2.20 mg aliquot of [14C] imarikiren hydrochloride dissolved in 20.5 of acetonitrile was prepared. Spiked plasma samples at a concentration of 0.5 μg/mL of imarikiren were prepared by combining a 3.2 mL aliquot of human plasma sample and a 16 μL aliquot of the test substance solution under ambient conditions. A 100 μL aliquot of the spiked plasma sample was mixed with 10 mL of liquid scintillator Hionic-Fluor™ (PerkinElmer Inc., Waltham, MA, USA). The radioactivity of this sample was measured with a liquid scintillation counter. Another 3 mL aliquot of the spiked plasma sample was centrifuged at 274,000 or 316,000×g for 14 h at 4 °C. The radioactivity of a 500 μL aliquot of the supernatant just under the lipid layer was measured as described previously. The plasma protein binding ratio of radioactivity in plasma was calculated as the difference in concentrations of radioactivity in the supernatant and spiked plasma sample divided by the concentration of radioactivity in the spiked plasma sample.
2.5 Endpoints
The primary endpoints were pharmacokinetics, including plasma concentrations of imarikiren and M-I, the ratio of urinary excretion of imarikiren as unchanged drug and M-I (or ratio of dialysate excretion of imarikiren for subjects on hemodialysis), and the imarikiren plasma protein binding ratio. AEs, vital signs, weight, 12-lead electrocardiogram (ECG) findings, and laboratory results served as safety endpoints.
2.6 Statistical Analysis
Enrollment of six subjects per cohort was planned. Pharmacokinetic endpoints were analyzed in the set of subjects who received study drug and could be evaluated for plasma, urine, or dialysate pharmacokinetics.
The following pharmacokinetic parameters were calculated by non-compartmental analysis using WinNonlin® version 6.3 (Pharsight, Mountain View, CA, USA) based on plasma concentrations of imarikiren and M-I up to 120 h after treatment: AUC∞, Cmax, time to first occurrence of Cmax (tmax), and terminal disposition phase half-life. Actual sampling times were used for calculation of these pharmacokinetic parameters. Summary statistics for plasma concentrations and pharmacokinetic parameters were calculated.
The unbound pharmacokinetic parameters (e.g., AUC∞,u and Cmax,u) were calculated from pharmacokinetic parameters of total fractions of imarikiren and the protein binding ratio.
The cumulative fraction of urinary excretion [percentage of dose administered (as imarikiren)] for imarikiren and M-I up to each urine collection period was calculated for each subject based on urinary concentrations of imarikiren and M-I and urine output during each urine collection period. Summary statistics were calculated for each cohort. The fraction of excretion in dialysis fluid was calculated for each subject based on the concentration in dialysis fluid of imarikiren and dialysis fluid output during each dialysis fluid collection period. Summary statistics for the fraction of excretion in dialysis fluid were also calculated.
For the AUC∞ and Cmax of imarikiren and M-I, an analysis of variance was applied to the natural log-transformed values to calculate the difference between subjects with RI and healthy adult subjects and the two-sided confidence interval (confidence coefficients 90 and 95%). A similar analysis was performed to evaluate the effects of HI on the pharmacokinetics of imarikiren.
Safety was assessed for subjects treated with at least one dose of study drug. AEs were coded using the Medical Dictionary for Regulatory Activities (MedRA®) version 19.0 and summarized using preferred terms and system organ classes.
3 Results
3.1 Subject Disposition
A total of 69 subjects from four study sites in Japan signed the informed consent form to participate in this study. Of these, 48 entered the treatment period, received study drug, and completed all planned study visits. The safety and plasma pharmacokinetic analysis sets included all 48 subjects (six per cohort). Among 42 subjects who were not on hemodialysis, 41 subjects were included in the urine pharmacokinetic analysis set; one subject was excluded because of a missed pharmacokinetic endpoint for urine concentration. All six subjects on hemodialysis were included in the dialysate pharmacokinetic analysis set.
3.2 Baseline Subject Characteristics
Across RI cohorts, the mean subject age ranged from 59.7 to 71.2 years (Table 1). The mean baseline eGFR was consistent with baseline RI status. The mean baseline AGP levels in the severe RI or ESRD cohort and the hemodialysis cohort were higher than those in the other RI cohorts. There were no other apparent differences in baseline characteristics among RI cohorts.
Across HI cohorts, the mean age of subjects ranged from 61.0 to 61.5 years. No apparent differences in baseline characteristics were observed among the HI cohorts.
3.3 Pharmacokinetics in Renal Impairment Cohorts
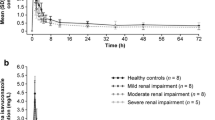
The mean plasma concentration–time curves for imarikiren following oral administration of a single 40 mg dose of imarikiren to subjects with RI or a matched cohort of subjects with normal renal function are shown in Fig. 1a; those of Course 1 (with hemodialysis) and Course 2 (without hemodialysis) for subjects on hemodialysis are shown in Fig. 1b.

Mean imarikiren plasma concentration–time curves following administration of a single 40 mg dose of imarikiren to subjects in cohorts classified according to a renal function status; b with or without hemodialysis period (hemodialysis subjects); and c hepatic function status (inset: semi-log plot of mean plasma concentration–time curves). Data are mean + standard deviation. HI hepatic impairment, RI renal impairment
Pharmacokinetic parameters of imarikiren for the renal cohorts are shown in Table 2. The geometric mean AUC∞ values ranged from 1527 to 8106 ng∙h/mL among cohorts. Compared with subjects with normal renal function, the geometric mean imarikiren AUC∞ was approximately 49% lower in the mild RI cohort, 18% higher in the moderate RI cohort, 173% higher in the severe RI or ESRD cohort, and 84% higher in the hemodialysis cohort (Course 2: without hemodialysis) (Table 2). The geometric mean imarikiren Cmax was approximately 43% lower in the mild RI cohort, 23% lower in the moderate RI cohort, 106% higher in the severe RI or ESRD cohort, and 43% higher in the hemodialysis cohort (Course 2: without hemodialysis) than in the normal renal function cohort (Table 2). There was no consistent trend in the mean plasma concentrations of imarikiren with severity of RI, while the mean plasma concentrations of imarikiren were generally similar with or without hemodialysis for subjects on hemodialysis.
The mean [standard deviation (SD)] unbound fraction of imarikiren in plasma was 5.30% (3.13%) in subjects with normal renal function, 5.60% (0.88%) in subjects with mild RI, 3.97% (0.88%) in subjects with moderate RI, 1.95% (0.36%) in subjects with severe RI, and 3.60% (0.60%) in subjects on hemodialysis. The unbound pharmacokinetic parameters of imarikiren in subjects with RI and the effect of RI on them are shown in Electronic Supplementary Material Table S1.
The mean plasma concentrations of M-I for each timepoint of each cohort were much lower than those of the parent drug. Compared with the normal renal function cohort, the geometric mean M-I AUC∞ was approximately 40% lower in the mild RI cohort, 3% lower in the moderate RI cohort, 160% higher in the severe RI or ESRD cohort, and 220% higher in the hemodialysis cohort (Course 2: without hemodialysis); the geometric mean M-I Cmax was approximately 30% lower in the mild RI cohort, 45% lower in the moderate RI cohort, 25% higher in the severe RI or ESRD cohort, and 4% higher in the hemodialysis cohort (Course 2: without hemodialysis).
The mean (SD) cumulative fraction of urinary excretion (percentage of dose) of imarikiren as unchanged drug at 72 h following imarikiren administration was 13.23% (1.33%) in the normal renal function cohort, 10.71% (3.22%) in the mild RI cohort, 12.82% (2.66%) in the moderate RI cohort, and 4.95% (3.32%) in the severe RI or ESRD cohort. Renal clearance values are shown in Table 2. For total compounds (imarikiren plus M-I), the mean (SD) cumulative fraction of urinary excretion (percentage of dose) at 72 h after imarikiren administration was 13.79% (1.28%) in the normal renal function cohort, 11.19% (3.37%) in the mild RI cohort, 13.10% (2.66%) in the moderate RI cohort, and 5.02% (3.34%) in the severe RI or ESRD cohort.
The mean imarikiren excretion fraction in dialysate was low (Table 2). The mean Course 1 (with hemodialysis) to Course 2 (without hemodialysis) plasma pharmacokinetic parameter ratios were 0.85 for the AUC from time zero to time of last quantifiable concentration and 1.00 for Cmax.
3.4 Pharmacokinetics in Hepatic Impairment Cohorts
The mean plasma concentration–time curves for imarikiren following oral administration of a single 40 mg dose of imarikiren to subjects with HI or a matched cohort of subjects with normal hepatic function are shown in Fig. 1c.
Pharmacokinetic parameters for imarikiren by HI cohort are shown in Table 3. The geometric mean AUC∞ values ranged from 2289 to 3126 ng∙h/mL among cohorts. Compared with the normal HI cohort, the geometric mean imarikiren AUC∞ was approximately 2% higher in the mild HI cohort and 37% higher in the moderate HI cohort. The geometric mean Cmax values ranged from 446.0 to 688.1 ng/mL among cohorts; the geometric mean imarikiren Cmax was approximately 15% lower in the mild HI cohort and 32% higher in the moderate HI cohort than in the normal hepatic function cohort (Table 3). The mean plasma concentrations of imarikiren over time were similar in subjects with normal hepatic function or mild HI, but slightly higher in subjects with moderate HI.
The mean (SD) unbound fractions of imarikiren in plasma were 4.80% (0.77%) in the normal HI cohort, 7.18% (4.05%) in the mild HI cohort, and 12.82% (6.55%) in the moderate HI cohort. The unbound pharmacokinetic parameters of imarikiren in subjects with HI and the effect of HI on them are shown in Electronic Supplementary Material Table S2.
The mean plasma concentrations of M-I for each timepoint of each cohort were much lower than those of the parent drug. The geometric mean M-I AUC∞ was approximately 30% higher in the mild HI cohort and 93% higher in the moderate HI cohort than in the normal hepatic function cohort; the geometric mean M-I Cmax was approximately 8% lower in the mild HI cohort and 39% higher in the moderate HI cohort than in the normal hepatic function cohort.
For imarikiren, the mean (SD) cumulative fraction of urinary excretion (percentage of dose) as unchanged drug at 72 h after imarikiren administration was 13.98% (5.04%) in the normal hepatic function cohort, 15.21% (2.62%) in the mild HI cohort, and 21.22% (4.94%) in the moderate HI cohort. The mean imarikiren renal clearance values are shown in Table 3. The mean (SD) cumulative fraction of urinary excretion (percentage of dose) at 72 h after imarikiren administration for total compounds (imarikiren plus M-I) was 14.43% (5.07%), 15.76% (2.71%), and 22.01% (5.10%), respectively.
3.5 Safety
Thirteen subjects experienced a total of 16 treatment-emergent AEs (TEAEs; Table 4). TEAEs reported in more than one subject were decreased blood pressure (n = 3), increased ALT (n = 2), and orthostatic hypotension (n = 2). All subjects reported TEAEs that were mild in intensity except for one subject in the severe RI or ESRD cohort who experienced moderate hemorrhagic anemia and gastroduodenal ulcer events, both of which were considered by the investigator as unrelated to study drug.
The TEAEs that were considered to be related to study drug by the investigator were decreased blood pressure [n = 3; one in the severe RI or ESRD cohort, one in the hemodialysis cohort (Course 1: with hemodialysis), and one in the moderate HI cohort], increased ALT (one subject in the mild RI cohort), and headache (one subject in the moderate HI cohort).
There were no deaths, serious AEs, or TEAEs leading to study drug discontinuation. No clinically relevant mean changes from baseline were observed in clinical laboratory evaluations (after taking subjects’ background into consideration), vital signs, and weight except for the cases of decreased blood pressure noted previously. There were no clinically relevant mean changes from baseline 12-lead ECG parameters, and no 12-lead ECG findings were considered ‘abnormal, clinically significant’ at any timepoint by the investigator.
4 Discussion
In general, chronic kidney disease detrimentally affects glomerular filtration, tubular secretion, and tubular reabsorption, resulting in impaired renal excretion for many drugs [14]. Hepatic disease may also impair metabolic pathways and biliary excretion, resulting in reduced drug elimination [13]. To date, the effect of RI or HI on the pharmacokinetics of DRIs has been studied during the course of the development of aliskiren. In pharmacokinetic studies of aliskiren, RI resulted in up to 2.3-fold increases in the exposure of aliskiren compared with that in matched healthy subjects [14], and HI did not significantly change the pharmacokinetic profile of aliskiren following administration of a single aliskiren dose [15].
This is the first clinical study to evaluate the effect of RI and HI on the pharmacokinetics and safety of imarikiren. We found that RI was associated with altered exposure (AUC) and Cmax of imarikiren and that HI was associated with altered exposure (AUC) and Cmax of unbound imarikiren. Following administration of a single 40 mg oral dose of imarikiren, the geometric mean imarikiren AUC∞ and Cmax in subjects with mild, moderate, and severe RI (including non-hemodialysis and ESRD), and hemodialysis subjects compared with subjects with normal renal function were approximately 0.5-, 1.2-, 2.7-, and 1.8-fold, respectively, for AUC∞ and approximately 0.6-, 0.8-, 2.1-, and 1.4-fold, respectively, for Cmax. The mean fraction of urinary excretion of imarikiren in dialysate was approximately 3% during the 4 h dialysis period and the exposures of hemodialysis subjects during Courses 1 and 2 were similar, indicating that hemodialysis did not remove imarikiren in a meaningful manner from the systemic circulation and imarikiren could be administered regardless of dialysis timing, as might have been expected based on the high protein binding of imarikiren. The geometric mean imarikiren AUC∞ and Cmax in subjects with mild and moderate HI compared with subjects with normal hepatic function were approximately 1.0- and 1.4-fold, respectively, for AUC∞ and approximately 0.9- and 1.3-fold, respectively, for Cmax. The geometric mean AUC∞ and Cmax of the metabolite M-I in subjects changed similarly to those of imarikiren; however, this increased concentration of M-I (an active metabolite) is unlikely to be clinically relevant because the mean plasma concentrations of M-I were much lower than those of imarikiren in both RI and HI cohorts.
Consistent with the mechanisms by which RI may impair drug elimination, imarikiren exposure was inversely correlated with eGFR and renal clearance, and the mean cumulative excretion ratio for total compounds was lower in subjects with RI than in those with normal function.
Another possible mechanism for the effect of RI or HI on the pharmacokinetics of imarikiren is variation in plasma AGP levels with RI or HI. AGP (also known as orosomucoid) is produced mainly in hepatocytes [16]. It has a normal plasma concentration between 0.6 and 1.2 mg/mL, i.e., 60–120 mg/dL, and is an important plasma protein involved in the binding and transport of drugs in addition to albumin secreted by hepatocytes [16]. Serum concentrations of AGP are known to increase with the presence of inflammation. Non-clinical data from rats suggest that plasma AGP levels can affect the pharmacokinetics of imarikiren. When imarikiren was administered to rats whose AGP levels were 10 and 27 times higher than control rats, the AUC and Cmax of total imarikiren increased to 11 and 35 times and 12 and 23 times higher than those in the control rats, respectively [7]. Consistent with these observations, in the current study, AGP levels and exposure of total imarikiren increased with RI, whereas the unbound fraction of imarikiren decreased. In addition, plasma AGP levels slightly decreased with HI, which may have been a factor in the lack of considerable effect of mild or moderate HI on imarikiren exposure (based on total plasma concentrations of imarikiren). The effect of severe HI was not evaluated in this study; subjects with severe HI are expected to be outside the target population of imarikiren.
Although single oral administration of imarikiren 40 mg was found to be safe and well-tolerated in all renal and hepatic cohorts, further consideration is needed regarding the appropriate dosing regimen based on the severity of RI or HI.
A limitation of this study is that it was only a single-dose study with limited patient numbers, and the effect of RI or HI on the steady-state pharmacokinetics of imarikiren was not evaluated. In addition, the pharmacodynamics of imarikiren were not evaluated in this study. Furthermore, the results of this study are limited to guidance only and cannot be applied directly to patients with hypertension and concomitant RI and/or HI. Given that the use of aliskiren in combination with an ARB or an ACE inhibitor is restricted in subjects with diabetes mellitus or RI based on findings showing these therapies may cause more harm than benefit [3], the pharmacokinetics and safety of DRI-based therapy merit additional study in these populations.
5 Conclusion
RI and HI were associated with some changes in the pharmacokinetics of imarikiren (total or unbound) following a single dose (40 mg) of imarikiren. Despite increased exposure in some cohorts, imarikiren was safe and well-tolerated, regardless of the severity of RI or HI.
References
Azizi M, Menard J. Renin inhibitors and cardiovascular and renal protection: an endless quest? Cardiovasc Drugs Ther. 2013;27:145–53.
Triller DM, Evang SD, Tadrous M, Yoo BK. First renin inhibitor, aliskiren, for the treatment of hypertension. Pharm World Sci. 2008;30:741–9.
Parving HH, Persson F, Lewis JB, Lewis EJ, Hollenberg NK, Investigators AS. Aliskiren combined with losartan in type 2 diabetes and nephropathy. N Engl J Med. 2008;358:2433–46.
Imaeda Y, Tokuhara H, Fukase Y, et al. Discovery of TAK-272: A novel, potent, and orally active renin inhibitor. ACS Med Chem Lett. 2016;7:933–8.
Noden Pharma. Tekturna prescribing information. 2017. http://www.accessdata.fda.gov/drugsatfda_docs/label/2017/210709s000lbl.pdf. Accessed August 24, 2018
Ebihara T, Nishihara M, Takahashi J, Jinno F, Tagawa Y. Differences in nonclinical pharmacokinetics between species and prediction of human pharmacokinetics of TAK-272 (SCO-272), a novel orally active renin inhibitor. Biopharm Drug Dispos. 2018;39:175–83.
Ebihara T, Shimizu H, Yamamoto M, Higuchi T, Jinno F, Tagawa Y. The effect of elevated α1-acid glycoprotein on the pharmacokinetics of TAK-272 (SCO-272), an orally active renin inhibitor, in rats. Xenobiotica. 2018. https://doi.org/10.1080/00498254.2018.1480817 (Epub 2018 Jun 25).
Matsuno K, Kuroda S, Tanaka S, Nakamichi H, Kagawa T, Komura E. Pharmacokinetics, pharmacodynamics and safety of a single dose of imarikiren, a novel renin inhibitor, in healthy male subjects. Basic Clin Pharmacol Toxicol. 2018. https://doi.org/10.1111/bcpt.13050 (Epub 2018 May 30).
Matsuno K, Tanaka S, Hashimoto T, Nakamichi H, Kagawa T, Koumura E. A randomized, single-center, double-blind, placebo-controlled multiple-dose phase 1 study to evaluate the safety, pharmacokinetics, and pharmacodynamics of imarikiren in healthy adult nonelderly and elderly male subjects. J Clin Pharmacol. 2018. https://doi.org/10.1002/jcph.1142 (Epub 2018 May 7).
Japanese Society of Nephrology. Clinical practice guidebook for diagnosis and treatment of chronic kidney disease (in Japanese). Tokyo: Tokyo-Igakusha; 2012.
Japanese Society of Nephrology. Evidence-based clinical practice guidelines for CKD. Clin Exp Nephrol. 2009;13:537–66.
US Department of Health and Human Services, Food and Drug Administration, Center for Drug evaluation and research. Guidance for industry: Pharmacokinetics in patients with impaired renal function - Study Design, Data Analysis, and Impact on Dosing and Labeling; 2010. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM204959.pdf. Accessed August 24, 2018
Food and Drug Administration. Guidance for industry: pharmacokinetics in patients with impaired hepatic function: study design, data analysis, and impact on dosing and labeling. Rockville: Food and Drug Administration; 2003.
Vaidyanathan S, Bigler H, Yeh C, et al. Pharmacokinetics of the oral direct renin inhibitor aliskiren alone and in combination with irbesartan in renal impairment. Clin Pharmacokinet. 2007;46:661–75.
Vaidyanathan S, Warren V, Yeh C, Bizot MN, Dieterich HA, Dole WP. Pharmacokinetics, safety, and tolerability of the oral renin inhibitor aliskiren in patients with hepatic impairment. J Clin Pharmacol. 2007;47:192–200.
Allen PC, Hill EA, Stokes AM. Plasma proteins: analytical and preparative techniques. 1st ed. Oxford: Blackwell; 1977.
Acknowledgements
Medical writing assistance was provided by BlueMomentum, an Ashfield company, and supported by Takeda.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by Takeda Pharmaceutical Company Ltd.
Conflict of Interest
Yukio Shimasaki, TK, Takuya Saiki, Kumi Matsuno, Yuhei Sano, Shingo Kuroda, and Emiko Koumura report current or past employment with Takeda Pharmaceutical Company Ltd. EK reports stock/stock options with Takeda. Yukio Shimasaki, Masashi Sakaki, Minoru Itou, Tokurou Kobayashi, Masako Aso, Tomoya Kagawa, Takuya Saiki, Kumi Matsuno, Yuhei Sano, Kohei Shimizu, Shingo Kuroda, and Emiko Koumura report that medical writing assistance was provided by BlueMomentum, an Ashfield company, and supported by Takeda.
Ethical Standards
All procedures performed in studies involving human participants were in accordance with Japanese regulatory requirements and the ethical standards of the World Medical Association’s Declaration of Helsinki. The study protocol was approved by the institutional review board of each study site: Kurume Clinical Pharmacology Clinic (Institutional Review Board of Kurume Clinical Pharmacology Clinic), Moriya Keiyu Hospital (Review Board of Human Rights and Ethics for Clinical Studies Institutional Review Board), Kitasato University Hospital (Kitasato University Hospital Institutional Review Board), and Showa University Hospital (Institutional Review Board of Showa University Hospital).
Informed Consent
Informed consent was obtained from all individual participants included in the study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Shimasaki, Y., Sakaki, M., Itou, M. et al. Pharmacokinetics and Safety After a Single Dose of Imarikiren in Subjects with Renal or Hepatic Impairment. Clin Drug Investig 38, 1041–1051 (2018). https://doi.org/10.1007/s40261-018-0695-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-018-0695-4