
Abstract
Background and Objective
CYP2C19-mediated drug interactions of acid-reducing agents are clinically important given the high possibility of concomitant administration with CYP2C19 substrates. This study aimed to evaluate the effect of tegoprazan on the pharmacokinetics (PK) of a CYP2C19 substrate, proguanil, compared with vonoprazan or esomeprazole.
Methods
A two-part, randomized, open-label, two-sequence, three-period crossover study was conducted in 16 healthy CYP2C19 extensive metabolizers (eight subjects per part). In each period, a single oral dose of atovaquone/proguanil 250/100 mg was administered alone or co-administered with tegoprazan 50 mg, esomeprazole 40 mg (Part 1 only) or vonoprazan 20 mg (Part 2 only). The plasma and urine concentrations of proguanil and its metabolite, cycloguanil, were measured up to 48 h post-dose. PK parameters were calculated using a non-compartmental method and compared between administered alone and co-administered with tegoprazan, vonoprazan or esomeprazole.
Results
Co-administration of tegoprazan did not significantly affect the systemic exposure of proguanil and cycloguanil. In contrast, co-administration of vonoprazan or esomeprazole increased proguanil systemic exposure and decreased cycloguanil systemic exposure, and the magnitude of the corresponding change was greater with esomeprazole co-administration than vonoprazan co-administration.
Conclusion
Tegoprazan, unlike vonoprazan and esomeprazole, exhibited negligible CYP2C19-mediated PK interaction. It suggests that as an alternative to other acid-reducing agents, tegoprazan can be used concomitantly with CYP2C19 substrates in clinical settings.
Trial Registration
Clinicaltrials.gov identifier: NCT04568772 (Registered on September 29, 2020).
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Tegoprazan, a novel potassium-competitive acid blocker, is expected to exert negligible cytochrome P450 (CYP) 2C19 interaction based on in vitro studies. Considering the clinical importance of CYP2C19-mediated drug interactions of acid-reducing agents, this study evaluated the effect of tegoprazan on CYP2C19 activity, compared with vonoprazan or esomeprazole, using proguanil as a CYP2C19 substrate. |
Tegoprazan had no meaningful effect on proguanil pharmacokinetics, which indicates that tegoprazan is neither an inhibitor nor an inducer of CYP2C19. In contrast to tegoprazan, vonoprazan and esomeprazole both decreased the CYP2C19-mediated metabolism of proguanil. |
Tegoprazan exhibited insignificant CYP2C19-mediated pharmacokinetic interaction. Our findings suggest that as an alternative to other acid-reducing agents, tegoprazan can be used concomitantly with CYP2C19 substrates in clinical practice without any dose adjustment. |
1 Introduction
Acid-reducing agents, especially proton pump inhibitors (PPIs), are widely used to control gastric acid-related disorders and prevent drug-induced gastrointestinal complications [1, 2]. Because most PPIs are predominantly metabolized by cytochrome P450 (CYP) 2C19 and have CYP2C19 inhibitory effects, the intake of PPIs in combination with CYP2C19 substrates requires caution regarding pharmacokinetic (PK) drug interactions and, by extension, their clinical relevance [2, 3]. Notably, concomitant PPI administration is recommended as a gastroprotective agent in patients who receive antiplatelet therapy with clopidogrel, which is a well-known CYP2C19 substrate [1, 4]. Accordingly, the CYP2C19-mediated drug interactions of PPIs are clinically important issues given the high possibility of concomitant use with CYP2C19 substrates.
CYP2C19-mediated drug interactions of PPIs may result in unwarranted clinical outcomes. PPIs, including esomeprazole, exhibit PK interactions with CYP2C19 substrates, such as diazepam, phenytoin, warfarin, and clopidogrel, most of which are of clinical significance [5,6,7]. In particular, clopidogrel, which is often used in combination with PPIs in clinical practice, requires the formation of an active metabolite by CYP2C19 to exert its antiplatelet activity [4], and it was reported that concomitant PPI administration reduced the antiplatelet effect of clopidogrel and ultimately increased the cardiovascular events of patients [8, 9]. Therefore, alternative acid-reducing agents to PPIs that can be used concomitantly with CYP2C19 substrates without caution are needed.
Potassium-competitive acid blockers (P-CABs) are a novel class of acid-reducing agents that overcome several limitations of PPIs, such as nocturnal acid breakthrough [10]. It is noteworthy that the concomitant administration of P-CABs is likely not restricted by CYP2C19-mediated drug interactions considering their elimination pathways are mainly via CYP3A4, not CYP2C19, and because of their relatively low in vitro CYP2C19 inhibition potential [10, 11].
Meanwhile, it was recently reported that vonoprazan, one of the P-CABs, acts as a clinically relevant CYP2C19 inhibitor at therapeutic doses although its CYP2C19 half-maximum inhibitory concentration (IC50) value (13 µM) was sufficiently higher than its unbound maximum plasma concentration (Cmax) (approximately 0.01 µM) at the therapeutic dose [12]. This result may partially explain the previously reported attenuation of the antiplatelet function of clopidogrel by vonoprazan [13]. These unexpected findings suggest the need for clinical evaluations of the CYP2C19-mediated PK interactions of other P-CABs for use in combination with CYP2C19 substrates in clinical settings.
Tegoprazan is a newly approved P-CAB for the treatment of acid-related diseases in several countries, including the Republic of Korea. Given its sufficient acid suppression, tegoprazan also has potential for combination therapy with various drugs including CYP2C19 substrates, as a gastroprotective agent in addition to an acid suppressant [14]. Even though tegoprazan is expected to exert negligible CYP2C19 interaction based on in vitro studies (IC50 >30 µM), the CYP2C19-mediated PK interaction of tegoprazan has not been clinically evaluated. Indeed, like vonoprazan, the unbound Cmax of tegoprazan (approximately 1.3 µM) at the therapeutic dose is much lower than the aforementioned IC50 value [15].
This study was aimed to evaluate the effect of tegoprazan on the PK of a CYP2C19 substrate, proguanil, compared with vonoprazan or esomeprazole. Proguanil was chosen as the CYP2C19 substrate in this study because it is primarily metabolized to cycloguanil by CYP2C19, and approximately 40–60% of the dose is renally excreted [16].
2 Methods
The Institutional Review Board of Seoul National University Hospital and the Korean Ministry of Food and Drug Safety approved the study protocol and informed consent form. This study was conducted in accordance with the Korean Good Clinical Practice guidelines and the principles of the Declaration of Helsinki. The study was registered in the public clinical trial registry on September 29, 2020 (ClinicalTrials.gov identifier: NCT04568772). Eligible subjects were recruited through a recruitment notice at the Seoul National University Hospital Clinical Trials Center, including the website (https://ctcr.snuh.org/). Prior to any study-related procedures, written informed consent was obtained from all individual subjects.
2.1 Study Population
Eligible subjects were healthy CYP2C19 extensive metabolizers carrying the CYP2C19 *1/*1 diplotype who were 19–50 years old with a body mass index (BMI) of 19.0–30.0 kg/m2. Subjects who had evidence or a history of gastrointestinal disorders likely to influence drug absorption and/or who ingested drug metabolizing inducers (e.g., barbiturates) or inhibitors (e.g., clarithromycin) within 4 weeks prior to the first administration and/or other medications within 2 weeks prior to the first administration were excluded from the study. The exclusion criteria also included aspartate aminotransferase or alanine aminotransferase values 1.5 times greater than the upper normal limit or Modification of Diet in Renal Disease (MDRD) glomerular filtration ratio (GFR) values < 80 mL/min. The enrolled subjects were prohibited from intake of any medication without the prior permission of the investigators and the consumption of grapefruit products, caffeine, or alcohol throughout the study. The sample size was determined empirically based on the exploratory and descriptive characteristics of the study without calculating study power for any statistical hypothesis.
2.2 Study Design
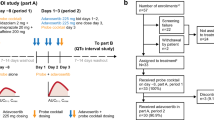
This study had a two-part, randomized, open-label, two-sequence, three-period crossover design (Fig. 1). Each sequence consisted of the following three treatments (i.e., one control treatment and two co-administration treatments): a single oral administration of atovaquone/proguanil 250/100 mg alone [control], 6-day pretreatment with once-daily oral doses of tegoprazan 50 mg, followed by a single oral administration of atovaquone/proguanil 250/100 mg concomitant with tegoprazan 50 mg [co-administration], and 6-day pretreatment with once-daily oral doses of esomeprazole 40 mg, followed by a single oral administration of atovaquone/proguanil 250/100 mg concomitant with esomeprazole 40 mg (Part 1) or 6-day pretreatment with once-daily oral doses of vonoprazan 20 mg, followed by a single oral administration of atovaquone/proguanil 250/100 mg concomitant with vonoprazan 20 mg (Part 2) [co-administration]. In accordance with the guideline for clinical drug interaction studies, the dose of each study drug was set to the therapeutic dose for a substrate, proguanil, and to the approved maximum dose for perpetrators, tegoprazan, vonoprazan, and esomeprazole, respectively [17]. The following drug products were used: atovaquone/proguanil 250/100 mg (Malarone® Tab., GlaxoSmithKline Inc., Seoul, Republic of Korea), tegoprazan 50 mg (K-CAB® Tab. 50 mg, HK inno.N Corp., Seoul, Republic of Korea), esomeprazole 40 mg (Nexium® Tab. 40 mg, AstraZeneca Pharmaceutical Co., Ltd., Seoul, Republic of Korea), or vonoprazan 20 mg (Takecab® Tab. 20 mg, Takeda Pharmaceutical Co., Ltd., Tokyo, Japan).

Study design
The enrolled subjects were randomly assigned to one of two sequences in each part and received their respective treatment with 150 mL of water after at least 10 h of overnight fasting. The respective treatment was separated by a 7-day washout period, which was set based on the turnover half-life (t½) of CYP2C19 and the t½ of the study drugs [14, 16, 18,19,20].
For PK analyses of proguanil and cycloguanil, blood samples were obtained at pre-dose and 1, 2, 3, 4, 6, 8, 10, 24, and 48 h after atovaquone/proguanil dosing, and urine samples were collected at pre-dose and 0–8, 8–24, and 24–48 h after atovaquone/proguanil dosing. At each blood sampling point, approximately 8 mL of blood was collected in a sodium heparin tube and then centrifuged at 4 °C and 3000 rpm for 10 min, and the supernatant was separated in Eppendorf tubes® and stored at −70 °C until analysis. At each urine collection interval, the collected urine was gently mixed, and separated and stored in the same manner as the blood samples.
2.3 CYP2C19 Genotyping
To identify CYP2C19 extensive metabolizers, DNA was extracted from whole blood using a Maxwell® CSC Blood DNA Kit and Maxwell® CSC Instrument (Promega, Madison, WI, USA), and TaqMan allelic discrimination assays were performed in a real-time polymerase chain reaction (PCR) System (Applied Biosystems, Foster City, CA, USA). A 10-µL PCR mixture was prepared with 5 µL of 2× TaqMan Universal Master Mix II, 0.5 µL of 20× Drug Metabolism Genotyping Assay Mix, 3.5 µL of DNase-free water, and 1 µL of DNA. Genotyping for the CYP2C19*2 allele (rs4244285, assay ID: C__25986767_70) and CYP2C19*3 allele (rs4986893, assay ID: C__27861809_10) was performed using validated TaqMan Genotyping Assays. The PCRs were carried out in the order of initial denaturation at 95 °C for 10 min followed by 40 cycles of denaturation at 95 °C for 15 seconds and annealing/extension at 60 °C for 1 minute. The allelic discrimination results were determined using 7500 Real-Time PCR System software version 2.0.6 (Applied Biosystems, Foster City, CA, USA). Based on the genotyping results, only CYP2C19 extensive metabolizers carrying the CYP2C19 *1/*1 diplotype were included in the study.
2.4 Determination of Plasma and Urine Proguanil and Cycloguanil Concentrations
The plasma and urine concentrations of proguanil and cycloguanil were determined using validated high-performance liquid chromatography (HPLC; Agilent 1260/1290 Infinity system, Agilent Technologies) coupled with tandem mass spectrometry (MS/MS; API4000, AB Sciex for plasma samples and Agilent 6460, Agilent Technologies for urine samples). For plasma specimens, 50 μL of plasma was mixed with 10 μL of internal standard solution (0.2 μg/L proguanil-d4 in 50% acetonitrile and 0.1 μg/L cycloguanil-d4 in 50% acetonitrile, respectively), followed by 1 mL of 100% acetonitrile. Mixed solutions were centrifuged at 4 °C and 14,000 rpm for 5 min. The supernatant was dried at 40 °C and dissolved with 100 μL of 30% acetonitrile, followed by centrifugation at 4 °C and 14,000 rpm for 5 min. Then, 5 μL of the supernatant was subjected to HPLC-MS/MS analysis. For urine specimens, 50 μL of urine was mixed with 10 μL of internal standard solution (proguanil-d4 12.5 μg/L in 50% acetonitrile and cycloguanil-d4 10 μg/L in 50% acetonitrile, respectively), followed by 930 μL of 100% acetonitrile. Mixed solutions were centrifuged at 4 °C and 14,000 rpm for 5 min. The supernatant was dried at 40 °C and dissolved with 500 μL of 0.1% formic acid in 20% acetonitrile, followed by centrifugation at 4 °C and 14,000 rpm for 5 min. Then, 5 μL of the supernatant was subjected to HPLC-MS/MS analysis.
The lower limits of quantification for proguanil and cycloguanil were 1 μg/L and 0.5 μg/L, respectively, in plasma and 200 μg/L and 100 μg/L, respectively, in urine. The accuracies for proguanil and cycloguanil were 92.87–100.19% and 96.10–98.68%, respectively, in plasma samples and 96.65–99.75% and 99.28–100.95%, respectively, in urine samples. The precision coefficients of variation for proguanil and cycloguanil were ≤ 5.65% and ≤ 6.49%, respectively, in plasma samples and ≤ 2.32% and ≤ 2.69%, respectively, in urine samples.
2.5 Pharmacokinetic Analysis
The PK parameters of proguanil and cycloguanil were derived using a non-compartmental method in Phoenix® WinNonlin® version 8.2 (Certara, St. Louis, MO, USA). The area under the concentration–time curve (AUC) from 0 to last measurable time point (AUClast) was calculated using the linear-up and log-down trapezoidal rule and extrapolated to infinity (AUCinf) using the terminal elimination rate constant (λz). Cmax and the time to reach Cmax (Tmax) were determined directly from the observed plasma concentration–time profiles. The elimination t½ was calculated as ln(2) divided by λz, and the apparent clearance (CL/F) was calculated as the dose divided by AUCinf. The fraction excreted unchanged in the urine (fe) was calculated as the cumulative amount of proguanil in urine up to 48 h post-dose divided by the dose. The metabolic ratio was calculated as the AUCinf ratio of cycloguanil to proguanil, and the apparent formation clearance (CLF/F) was calculated as the cumulative amount of cycloguanil in urine up to 48 h post-dose divided by AUClast of proguanil.
2.6 Safety Evaluation
Safety was evaluated based on adverse event (AE) monitoring, clinical laboratory tests, vital signs, physical examination, and 12-lead electrocardiogram (ECG) throughout the study. Each finding from the safety evaluation was assessed regarding its clinical significance and relationship with the treatment by the investigators.
2.7 Statistical Analysis
Statistical analyses were performed using SAS® version 9.4 (SAS Institute Inc., Cary, NC, USA). The demographic characteristics were compared between parts and sequences in each part using Wilcoxon's rank sum test. For control and tegoprazan co-administration treatments, the PK data from Part 1 and Part 2 were pooled. The PK parameters of proguanil and cycloguanil were summarized by treatment and compared between administered alone and co-administered with tegoprazan, vonoprazan, or esomeprazole. Analysis of variance was performed for log-transformed AUC and Cmax, and the effects on the PK of proguanil and cycloguanil were compared by estimating the geometric mean ratios of each co-administration to control and the corresponding confidence intervals. Additionally, CYP2C19 metabolism-related PK parameters, the metabolic ratio and CLF/F of cycloguanil were evaluated using Dunnett’s t test.
3 Results
3.1 Study Population
A total of 19 healthy Korean male subjects, eight in Part 1 and 11 in Part 2, were enrolled in this study. Of those, three subjects involved in Part 2 discontinued the study before the second administration due to the practical issues of the site, and in consequence, 16 subjects, eight in each part, completed the study. All of the enrolled subjects were CYP2C19 extensive metabolizers. PK was analyzed in 16 subjects who had completed the study as planned, and safety was evaluated in 19 subjects who had taken the treatment at least once.
The arithmetic mean (± standard deviation) values of age, height, weight, and BMI of the enrolled subjects were 32.63 (± 7.70) years, 172.30 (± 6.29) cm, 73.57 (± 9.59) kg, and 24.79 (± 3.04) kg/m2, respectively. The demographic characteristics of the enrolled subjects were not significantly different between parts or sequences in each part. Furthermore, the enrolled subjects did not take any medications other than the study drugs throughout the study.
3.2 Effect of Tegoprazan on the Pharmacokinetics of Proguanil and Cycloguanil
Co-administration of tegoprazan did not significantly affect the PK of proguanil and cycloguanil. When co-administered with tegoprazan, the plasma concentrations and the degree of urinary excretion of proguanil and cycloguanil were marginally higher or comparable to when administered alone (Figs 2 and 3). The systemic exposure of proguanil and cycloguanil was not significantly changed by concomitant tegoprazan administration, showing comparable AUClast and Cmax values (Fig. 4, Table 1). In addition, the metabolic ratio and CLF/F of cycloguanil were similar regardless of tegoprazan co-administration (Fig. 5, Table 1), which indicated that tegoprazan had a negligible effect on the CYP2C19-mediated metabolism of proguanil to cycloguanil.

Mean plasma concentration–time profiles of a proguanil and b cycloguanil following a single oral administration of atovaquone/proguanil 250/100 mg alone or co-administered with tegoprazan 50 mg, vonoprazan 20 mg or esomeprazole 40 mg in linear scale. The error bars represent the standard deviations. aThe data from Part 1 and Part 2 were pooled

Mean cumulative urinary excretion–time profiles of a proguanil and b cycloguanil following a single oral administration of atovaquone/proguanil 250/100 mg alone or co-administered with tegoprazan 50 mg, vonoprazan 20 mg or esomeprazole 40 mg in linear scale. The error bars represent the standard deviations. aThe data from Part 1 and Part 2 were pooled

Effect of tegoprazan, vonoprazan or esomeprazole on the pharmacokinetics of a proguanil and b cycloguanil. The symbols and error bars represent the geometric mean ratios and 90% confidence intervals, respectively. The shadowed region represents the conventional bioequivalence range (0.80–1.25). aThe data from Part 1 and Part 2 were pooled. AUClast area under the plasma concentration–time curve from 0 to last measurable time point, Cmax maximum plasma concentration, Esomeprazole a single oral administration of atovaquone/proguanil 250/100 mg co-administered with esomeprazole 40 mg, Proguanil alone a single oral administration of atovaquone/proguanil 250/100 mg alone, Tegoprazan a single oral administration of atovaquone/proguanil 250/100 mg co-administered with tegoprazan 50 mg, Vonoprazan a single oral administration of atovaquone/proguanil 250/100 mg co-administered with vonoprazan 20 mg

Comparison of a metabolic ratio and b CLF/F of cycloguanil following a single oral administration of atovaquone/proguanil 250/100 mg alone and co-administered with tegoprazan 50 mg, vonoprazan 20 mg or esomeprazole 40 mg. The horizontal lines, boxes, vertical lines and symbols represent the median, interquartile range, minimum to maximum and individual values, respectively. The significance was evaluated by Dunnett’s t-test. aThe data from Part 1 and Part 2 were pooled. bCumulative amount of cycloguanil in urine up to 48 h post-dose divided by AUClast of proguanil. AUClast area under the plasma concentration–time curve from 0 to last measurable time point, CLF/F apparent formation clearance
3.3 Effect of Vonoprazan or Esomeprazole on the Pharmacokinetics of Proguanil and Cycloguanil
Co-administration of vonoprazan or esomeprazole considerably changed the PK of proguanil and cycloguanil. When co-administered with vonoprazan or esomeprazole compared with when administered alone, the plasma concentrations and the degree of urinary excretion of proguanil were higher, and the corresponding profiles of cycloguanil were lower (Figs. 2 and 3). With each concomitant vonoprazan and esomeprazole administration, the systemic exposure of proguanil increased 18% and 32% in AUClast, respectively, and the systemic exposure of cycloguanil decreased 25% and 51% in AUClast, respectively, showing greater changes in esomeprazole co-administration than in vonoprazan co-administration (Fig. 4, Table 1). Correspondingly, the metabolic ratio and CLF/F of cycloguanil were reduced with the co-administration of vonoprazan or esomeprazole (Fig. 5, Table 1), which indicated that vonoprazan and esomeprazole had meaningful effects on the CYP2C19-mediated metabolism of proguanil to cycloguanil.
3.4 Safety
A total of three treatment-emergent AEs, two in Part 1 and one in Part 2, were reported in two subjects throughout the study. All cases occurred after the administration of control treatment and were mild in intensity, and no serious AEs occurred. Moreover, no clinically significant changes or issues in clinical laboratory tests, vital signs, physical examination, and 12-lead ECG were observed.
4 Discussion
Considering the clinical importance of CYP2C19-mediated drug interactions of acid-reducing agents, the current study investigated the effect of tegoprazan on CYP2C19 activity, compared with vonoprazan or esomeprazole, using proguanil as a CYP2C19 substrate. It is worth noting that we evaluated the potential of acid-reducing agents for not only CYP2C19 inhibition but also CYP2C19 induction by administering the corresponding drugs for 7 days [21, 22].
In the present study, tegoprazan had no meaningful effect on the PK of the CYP2C19 substrate, indicating that tegoprazan is neither an inhibitor nor an inducer of CYP2C19. Our findings align with the results of in vitro studies using human liver microsomes, in which tegoprazan showed limited inhibition of CYP2C19 (IC50 >30 µM). In contrast to tegoprazan, vonoprazan and esomeprazole both decreased the CYP2C19-mediated metabolism of the CYP2C19 substrate, indicating that vonoprazan and esomeprazole are CYP2C19 inhibitors, and esomeprazole exhibited greater CYP2C19 inhibition than vonoprazan. Taken together, these results suggest favorable properties of tegoprazan in comparison with other anti-reducing agents when used concomitantly with CYP2C19 substrates in clinical settings.
Compared with a previous vonoprazan–proguanil study, the current study showed a consistent tendency in the effect of both vonoprazan and esomeprazole on proguanil PK, but the cycloguanil-to-proguanil ratio was lower overall in all treatments (e.g., metabolic ratio for ‘proguanil alone’: 0.35 in this study vs 0.84 in the previous study) [12]. A recent study found that hepatic uptake by OCT1 played a rate-limiting role in the hepatic metabolism of proguanil to cycloguanil [23]. Therefore, the observed phenomenon may be attributed to the resulting OCT1 deficiency in the enrolled subjects given the high frequency of OCT1-deficient alleles in Koreans [24]. However, it does not appear to have affected the interaction results observed in this study because we used a crossover design.
CYP2C19 is highly genetically polymorphic with resulting differences in drug metabolic capacity between phenotypes, and the frequency of CYP2C19 genetic polymorphism is particularly higher in Asian populations than in other races [25,26,27]. A previous study reported that the metabolic degree of proguanil to cycloguanil varied significantly between CYP2C19 phenotypes [28]. Accordingly, only CYP2C19 extensive metabolizers were recruited in the present study to exclude gene-related confounding factors. Our findings represent the metabolism-specific interaction by tegoprazan.
The US Food and Drug Administration recommends lansoprazole and omeprazole as CYP2C19-sensitive index substrates for clinical drug interaction studies [29]. However, these drugs increase gastric pH by themselves and this increase in gastric pH likely affects their absorption due to their physicochemical properties, classified as Biopharmaceutical Classification System (BCS) class II [30, 31]. Therefore, lansoprazole and omeprazole are considered inappropriate for use in drug metabolism-mediated interaction studies of acid-reducing agents. Based on these points, the present drug interaction study utilized proguanil, which belongs to BCS class III [32], as a CYP2C19 substrate to exclude the possibility of absorption-related drug interaction owing to gastric pH elevation by acid-reducing agents.
This study used the drug product of a fixed-dose combination of atovaquone/proguanil 250/100 mg, which is the only formulation commercially available in the Republic of Korea. Atovaquone, an antiprotozoal agent, is excreted mostly unchanged in the feces (≥94%) and is rarely metabolized by CYP2C19 as well as not affecting CYP2C19 activity [16, 33, 34], implying no CYP2C19-mediated PK interaction with proguanil. Indeed, the concomitant administration of atovaquone did not have a meaningful effect on proguanil PK [35]. Although atovaquone has in vitro CYP3A4 inhibition potential (IC50 of 4.7 µM), it does not appear to influence in vivo CYP3A4 activity at clinically relevant concentrations due to its high protein binding [16, 34]. Thus, it seems that atovaquone did not influence the achievement of steady states of the acid-reducing agents used in this study though the PK of these drugs was not evaluated.
Overall, our well controlled study provided robust results on CYP2C19-mediated PK interaction by tegoprazan in comparison with other acid-reducing agents, which may be generalized to other CYP2C19 substrates.
5 Conclusion
Tegoprazan, unlike vonoprazan and esomeprazole, exhibited negligible CYP2C19-mediated PK interaction. Our findings suggest that as an alternative to other acid-reducing agents, tegoprazan can be used concomitantly with CYP2C19 substrates in clinical settings without any dose adjustment.
References
Targownik LE, Fisher DA, Saini SD. AGA clinical practice update on de-prescribing of proton pump inhibitors: expert review. Gastroenterology. 2022;162(4):1334–42.
Ogawa R, Echizen H. Drug-drug interaction profiles of proton pump inhibitors. Clin Pharmacokinet. 2010;49(8):509–33.
Wedemeyer R-S, Blume H. Pharmacokinetic drug interaction profiles of proton pump inhibitors: an update. Drug Saf. 2014;37(4):201–11.
Jiang X-L, Samant S, Lesko LJ, Schmidt S. Clinical pharmacokinetics and pharmacodynamics of clopidogrel. Clin Pharmacokinet. 2015;54(2):147–66.
Andersson T, Hassan-Alin M, Hasselgren GR, Röhss K. Drug interaction studies with esomeprazole, the (S)-isomer of omeprazole. Clin Pharmacokinet. 2001;40(7):523–37.
Frelinger AL 3rd, Lee RD, Mulford DJ, Wu J, Nudurupati S, Nigam A, et al. A randomized, 2-period, crossover design study to assess the effects of dexlansoprazole, lansoprazole, esomeprazole, and omeprazole on the steady-state pharmacokinetics and pharmacodynamics of clopidogrel in healthy volunteers. J Am Coll Cardiol. 2012;59(14):1304–11.
Blume H, Donath F, Warnke A, Schug BS. Pharmacokinetic drug interaction profiles of proton pump inhibitors. Drug Saf. 2006;29(9):769–84.
Przespolewski ER, Westphal ES, Rainka M, Smith NM, Bates V, Gengo FM. Evaluating the effect of six proton pump inhibitors on the antiplatelet effects of clopidogrel. J Stroke Cerebrovasc Dis. 2018;27(6):1582–9.
Serbin MA, Guzauskas GF, Veenstra DL. Clopidogrel-proton pump inhibitor drug-drug interaction and risk of adverse clinical outcomes among PCI-treated ACS patients: a meta-analysis. J Manag Care Spec Pharm. 2016;22(8):939–47.
Oshima T, Miwa H. Potent potassium-competitive acid blockers: a new era for the treatment of acid-related diseases. J Neurogastroenterol Motil. 2018;24(3):334–44.
Nishihara M, Yamasaki H, Czerniak R, Jenkins H. In vitro assessment of potential for CYP-inhibition-based drug-drug interaction between vonoprazan and clopidogrel. Eur J Drug Metab Pharmacokinet. 2019;44(2):217–27.
Funakoshi R, Tomoda Y, Kudo T, Furihata K, Kusuhara H, Ito K. Effects of proton pump inhibitors, esomeprazole and vonoprazan, on the disposition of proguanil, a CYP2C19 substrate, healthy volunteers. Br J Clin Pharmacol. 2019;85(7):1454–63.
Kagami T, Yamade M, Suzuki T, Uotani T, Hamaya Y, Iwaizumi M, et al. Comparative study of effects of vonoprazan and esomeprazole on antiplatelet function of clopidogrel or prasugrel in relation to CYP2C19 genotype. Clin Pharmacol Ther. 2018;103(5):906–13.
Han S, Choi HY, Kim YH, Nam JY, Kim B, Song GS, et al. Randomised clinical trial: safety, tolerability, pharmacokinetics, and pharmacodynamics of single and multiple oral doses of tegoprazan (CJ-12420), a novel potassium-competitive acid blocker, healthy male subjects. Aliment Pharmacol Ther. 2019;50(7):751–9.
Moon SJ, Shin N, Kang M, Kim B, Kim MG. Pharmacokinetic interactions between tegoprazan and naproxen, aceclofenac, and celecoxib in healthy Korean male subjects. Clin Ther. 2022;44(7):930-44.e1.
HIGHLIGHTS OF PRESCRIBING INFORMATION: MALARONE (atovaquone and proguanil hydrochloride) tablets. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/021078s023lbl.pdf. Accessed 29 Oct 2022.
Guidance for Industry: Clinical Drug Interaction Studies—Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions. 2020. U.S. Department of Health and Human Services. Food and Drug Administration. Center for Drug Evaluation and Research (CDER) https://www.fda.gov/media/134581/download. Accessed 13 Feb 2023.
Hassan-Alin M, Andersonn T, Bredberg E, Röhss K. Pharmacokinetics of esomeprazole after oral and intravenous administration of single and repeated doses to healthy subjects. Eur J Clin Pharmacol. 2000;56(9–10):665–70.
Sakurai Y, Nishimura A, Kennedy G, Hibberd M, Jenkins R, Okamoto H, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of single rising TAK-438 (Vonoprazan) doses in healthy male Japanese/non-Japanese subjects. Clin Transl Gastroenterol. 2015;6(6): e94.
Yang J, Liao M, Shou M, Jamei M, Yeo KR, Tucker GT, et al. Cytochrome P450 Turnover: Regulation of Synthesis and Degradation, Methods for Determining Rates, and Implications for the Prediction of Drug Interactions. Curr Drug Metab. 2008;9(5):384–94.
Inui N, Akamatsu T, Uchida S, Tanaka S, Namiki N, Karayama M, et al. Chronological effects of rifampicin discontinuation on cytochrome P450 activity in Healthy Japanese volunteers, using the cocktail method. Clin Pharmacol Ther. 2013;94(6):702–8.
Hakkola J, Hukkanen J, Turpeinen M, Pelkonen O. Inhibition and induction of CYP enzymes in humans: an update. Arch Toxicol. 2020;94(11):3671–722.
Matthaei J, Seitz T, Jensen O, Tann A, Prukop T, Tadjerpisheh S, et al. OCT1 deficiency affects hepatocellular concentrations and pharmacokinetics of cycloguanil, the active metabolite of the antimalarial drug proguanil. Clin Pharmacol Ther. 2019;105(1):190–200.
Kang H-J, Song I-S, Shin HJ, Kim W-Y, Lee C-H, Shim J-C, et al. Identification and functional characterization of genetic variants of human organic cation transporters in a Korean population. Drug Metab Dispos. 2007;35(4):667–75.
Sienkiewicz-Oleszkiewicz B, Wiela-Hojeńska A. CYP2C19 polymorphism in relation to the pharmacotherapy optimization of commonly used drugs. Pharmazie. 2018;73(11):619–24.
Lee SS, Lee SJ, Gwak J, Jung HJ, Thi-Le H, Song IS, et al. Comparisons of CYP2C19 genetic polymorphisms between korean and vietnamese populations. Ther Drug Monit. 2007;29(4):455–9.
Dehbozorgi M, Kamalidehghan B, Hosseini I, Dehghanfard Z, Sangtarash M, Firoozi M, et al. Prevalence of the CYP2C19*2 (681 G>A), *3 (636 G>A) and *17 (-806 C>T) alleles among an Iranian population of different ethnicities. Mol Med Report. 2018;17(3):4195–202.
Watkins WM, Mberu EK, Nevill CG, Ward SA, Breckenridge AM, Koech DK. Variability in the metabolism of proguanil to the active metabolite cycloguanil in healthy Kenyan adults. Trans R Soc Trop Med Hyg. 1990;84(4):492–5.
Drug Development and Drug Interactions | Table of Substrates, Inhibitors and Inducers. 2022. https://www.fda.gov/drugs/drug-interactions-labeling/drug-development-and-drug-interactions-table-substrates-inhibitors-and-inducers. Accessed 29 Oct 2022.
Geng L, Han L, Huang L, Wu Z, Wu Z. High anti-acid omeprazole lightweight capsule for gastro-enteric system acid-related disorders treatment. J Clin Gastroenterol Treat. 2019;5:068.
Wu C, Sun L, Sun J, Yang Y, Ren C, Ai X, et al. Profiling biopharmaceutical deciding properties of absorption of lansoprazole enteric-coated tablets using gastrointestinal simulation technology. Int J Pharm. 2013;453(2):300–6.
Plöger GF, Abrahamsson B, Cristofoletti R, Groot DW, Langguth P, Mehta MU, et al. Biowaiver monographs for immediate release solid oral dosage forms: proguanil hydrochloride. J Pharm Sci. 2018;107(7):1761–72.
Rolan PE, Mercer AJ, Tate E, Benjamin I, Posner J. Disposition of atovaquone in humans. Antimicrob Agents Chemother. 1997;41(6):1319–21.
Thapar MM, Ashton M, Lindegårdh N, Bergqvist Y, Nivelius S, Johansson I, et al. Time-dependent pharmacokinetics and drug metabolism of atovaquone plus proguanil (Malarone) when taken as chemoprophylaxis. Eur J Clin Pharmacol. 2002;58(1):19–27.
Gillotin C, Mamet JP, Veronese L. Lack of a pharmacokinetic interaction between atovaquone and proguanil. Eur J Clin Pharmacol. 1999;55(4):311–5.
Acknowledgements
We thank all subjects for their participation in this study. In addition, we gratefully acknowledge the investigators, coordinators and site personnel involved in this study. This study was funded in full by HK inno.N Corp., Seoul, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was funded by HK inno.N Corp., Seoul, Republic of Korea.
Conflict of Interest
The authors declare no conflicts of interest in this work.
Ethics Approval
The protocol and informed consent form of this study were reviewed and approved by the Ministry of Food and Drug Safety of Korea and by the Institutional Review Board at Seoul National University Hospital. This study was performed in accordance with the Korean Good Clinical Practice guidelines and the Declaration of Helsinki.
Consent to Participate
Written consent was obtained from each subject prior to any study-related procedures.
Consent for Publication
Not applicable.
Availability of Data and Material
The individual de-identified participant data supporting published results are available with approval from the corresponding author on reasonable request, at any time after publication.
Code Availability
Not applicable.
Author Contributions
YE and LS wrote the manuscript. YE and LS designed the research. YE, SCJ, I-JJ, and LS performed the research. YE and LS analyzed the data. All the authors reviewed and revised the paper. All the authors approved the final version of the manuscript.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Yang, E., Ji, S.C., Jang, IJ. et al. Evaluation of CYP2C19-Mediated Pharmacokinetic Drug Interaction of Tegoprazan, Compared with Vonoprazan or Esomeprazole. Clin Pharmacokinet 62, 599–608 (2023). https://doi.org/10.1007/s40262-023-01228-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01228-4