
Abstract
Background
Cognitive impairment is a core feature of schizophrenia. While first- and second-generation antipsychotic drugs treat psychotic exacerbations, no treatment is approved for the cognitive dysfunction. We have identified ASP4345, a positive allosteric modulator of the dopamine type 1 (D1) receptor that selectively binds to, and enhances the activity of, D1 receptors. ASP4345 has the potential to be an effective and well-tolerated treatment option for cognitive impairment associated with schizophrenia.
Objective
The objective of this study was to determine the pharmacokinetics of ASP4345 in two phase I single ascending-dose and multiple ascending-dose studies.
Methods
Both phase I studies were randomized, double blind, and placebo controlled. The single dose-ascending study assessed pharmacokinetics of single oral doses of 3–900 mg of ASP4345 or placebo in the fasted state in healthy adult volunteers. This study also assessed cerebrospinal fluid pharmacokinetics, as well as the effects of food on pharmacokinetic parameters. The multiple ascending-dose study (NCT02720263) assessed the pharmacokinetics of multiple oral doses of 3–150 mg of ASP4345 in patients with schizophrenia or schizoaffective disorder receiving stable antipsychotic drug treatment. The pharmacokinetic data from both studies were summarized using descriptive statistics.
Results
The plasma concentration–time profile in both studies showed a rapid increase in concentrations of ASP4345. The median time to maximum concentration range was 1.00–2.26 h in the single ascending-dose study in the fasted state and 1.25–3.02 h in the multiple ascending-dose study at steady state. There were less than dose-proportional increases in maximum concentration and area under the curve in the single ascending-dose study, where doses had a range from 3 to 900 mg, and in the multiple ascending-dose study in patients with stabilized schizophrenia or schizoaffective disorder, where doses had a range from 3 to 150 mg. The mean terminal elimination half-life was dose independent and had a range from 9.12 to 14.3 h in the single ascending-dose study and from 11.1 to 26.8 h in the multiple ascending-dose study. Additionally, in the single ascending-dose study, absorption of 300 mg of ASP4345 was slightly delayed when administered in the fed state compared with the fasted state; median time to maximum concentration was 1.5 h under the fasting state and 4.0 h under fed states. All other pharmacokinetic parameters were comparable for both conditions. ASP4345 appeared in the cerebrospinal fluid with some delay; time to maximum concentration range was from 2.48 to 7.98 h in cerebrospinal fluid compared with 0.75 to 1.03 h in plasma (median cerebrospinal fluid/plasma = 0.188). The ratio of cerebrospinal fluid to total plasma for area under the curve from 0 to 24 h (0.157–0.573%) and maximum concentration (0.0899–0.311%) and the ratio of cerebrospinal fluid to unbound plasma for maximum concentration (25.0–86.4%) confirm the distribution of ASP4345 into the brain.
Conclusions
The pharmacokinetics of ASP4345 suggest that single daily dosing is appropriate for ASP4345. Furthermore, the concentration of ASP4345 in cerebrospinal fluid compared to free drug concentrations in plasma provides evidence of penetration of ASP4345 into the brain.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Cognition is often impaired in patients with schizophrenia but drugs currently used to treat schizophrenia do not improve cognition. We report two studies, one performed in healthy adults and the other in adults with schizophrenia, that determined a safe dose for further clinical study of a new oral drug, ASP4345. |
ASP4345 was found in blood circulation and in the cerebrospinal fluid, an important characteristic for an oral drug to be investigated in cognition and schizophrenia. We found that ASP4345 taken once-daily is appropriate for future clinical trials in patients to determine its effect on cognitive impairment associated with schizophrenia. |
1 Introduction
The first large-scale meta-analysis linking cognitive impairment and schizophrenia was published in 1998, based on results from 204 schizophrenia trials conducted between 1980 and 1997 [1]. Cognitive impairment associated with schizophrenia (CIAS) includes significant deficits across several cognitive performance domains including visual and auditory attention, verbal learning, episodic memory, executive function, and speed of processing [2, 3]. Studies have also confirmed that CIAS exists in patients who are drug naïve as well as in those taking antipsychotic medications [3]. A number of approaches enhance learning or memory in animal models; nonetheless, to date, no pharmacotherapy has demonstrated consistent and clinically meaningful efficacy in improving CIAS in clinical trials [4].
Dopamine neurotransmission plays a central role in the pathology of schizophrenia [5]. Blockade of dopamine type 2 receptors in the ventral tegmental area and the striatum appears most responsible for ameliorating or preventing psychotic exacerbation. In contrast, optimal dopamine levels, particularly via the stimulation of the dopamine type 1 (D1) receptors in the dorsolateral prefrontal cortex, are required for adequate cognitive functioning [6, 7]. The Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) program, which aimed to investigate a range of putative molecular targets for treating cognition, concluded that the D1 receptor is a promising target for improving cognition [8]. However, clinical development of D1 agonists has been associated with poor bioavailability [9] and safety concerns, particularly seizures [10] and orthostatic hypotension [11]. Because of the close structural and pharmacological similarities between many of the dopamine receptor families, therapies developed to target them are typically non-specific. More recently, two positive allosteric modulators of the D1 receptor that bind to sites on the receptor that are distinct from the orthosteric binding site have been identified [12]. By binding to the less conserved regions of the receptor and allosterically enhancing functional effects of endogenous dopamine, positive allosteric modulators minimize undesired activation of the D1 receptor in a non-physiological manner and provide reduced or no receptor desensitization compared with orthosteric D1 receptor agonists. Therefore, allosteric modulators may represent a treatment for disorders that otherwise are without treatment options. This approach may be akin to a subtle “dimmer switch” compared with agonist or antagonist activity that causes complete activation (regardless of ambient dopaminergic tone) or inhibition [13].
ASP4345, a novel small-molecule positive allosteric modulator, selectively binds to, and enhances the activity of, D1 receptors stimulated by endogenous dopamine. It may, therefore, represent a potential treatment option for CIAS. ASP4345 was recently assessed in two phase I studies: a single ascending-dose trial and a multiple ascending-dose trial. The objective of the single dose trial was to determine the safety, tolerability, and pharmacokinetics of ASP4345 in healthy male and female volunteers. The primary endpoints of the multiple-ascending dose trial were to determine the safety and tolerability of ASP435, and to evaluate the pharmacokinetics of multiple ascending oral doses of ASP4345 in patients with schizophrenia. The exploratory endpoint was to assess the effects of ASP4345 on cognitive function and neurophysiological biomarkers in patients with stabilized schizophrenia or schizoaffective disorder (reported elsewhere). Presented here are the pharmacokinetic data from these two phase I studies.
2 Methods
2.1 Overview and Design
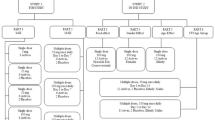
The two studies were randomized, double-blind, placebo-controlled phase I trials. The single ascending-dose study included two parts. In Part 1, the safety, tolerability, and pharmacokinetics of ASP4345 were assessed in healthy volunteers under fasted conditions, and the effect of food on the pharmacokinetics of ASP4345 was evaluated. Healthy volunteers were assigned to receive ASP4345 or placebo using a 3:1 randomization schedule (six cohorts of eight subjects each, with six subjects in each cohort receiving ASP4345 and two subjects receiving matching placebo under fasted conditions). Part 2 of this study aimed to evaluate the plasma and cerebrospinal fluid (CSF) pharmacokinetics of single oral doses of ASP4345 in healthy volunteers, and utilized a 4:1 randomization schedule. The multiple-dose study (NCT02720263) enrolled patients in up to four dose cohorts using a 3:1 randomization schedule. The study protocols and the ethical, scientific, and medical appropriateness of the study were reviewed by the institutional review board prior to study initiation, and all healthy volunteers and patients provided written informed consent. Studies were conducted in accordance with ethical principles based on the Declaration of Helsinki, Good Clinical Practice, International Council for Harmonization guidelines, and applicable laws and regulations.
2.2 Healthy Volunteers and Patients
The single ascending-dose study included healthy men and women between 18 and 55 years of age, with a body mass index between 18.5 and 30 kg/m2. Additionally, the healthy volunteers enrolled in Part 1 of the study were required to consume a standardized high-fat meal (approximately 800–1000 cal: 50–60% from fat, 15% from protein, 25% from carbohydrates); these conditions were expected to impact gastrointestinal physiology such that systemic drug availability is maximally affected.
The multiple ascending-dose study included men and women between 18 and 60 years of age with a body mass index between 18.5 and 40 kg/m2 and a confirmed diagnosis of schizophrenia or schizoaffective disorder based on the Diagnostic and Statistical Manual of Mental Disorders, 5th edition criteria. All patients in the multiple ascending-dose study were required to be receiving stable doses of risperidone, quetiapine, olanzapine, ziprasidone, brexpiprazole, aripiprazole, paliperidone, or lurasidone for at least 2 months for oral formulations, or at least 3 months for depot formulations, prior to screening. It was expected that an additional month for depot formulations would increase the chance that stable peak-to-trough antipsychotic concentrations would be observed.
For both the single ascending-dose and multiple ascending-dose studies, men and women of childbearing potential were required to use at least two forms of highly effective birth control starting at screening, continuing throughout the study period, and for 90 days after study drug administration. Women who were pregnant within 6 months prior to the screening assessment or breastfeeding within 3 months prior to screening were not eligible for inclusion in either study. Participants were excluded from both studies if they had any known or suspected hypersensitivity to ASP4345, a history of suicide attempt or suicidal behavior, or suicidal ideation within the last 3 months; liver chemistry tests above the upper limit of normal (aspartate aminotransferase, alanine aminotransferase, alkaline phosphatase, gamma glutamyl transferase, total bilirubin); clinically significant history of allergic conditions; history or evidence of any clinically significant cardiovascular, gastrointestinal, endocrinologic, hematologic, hepatic, immunologic, metabolic, urologic, pulmonary, neurologic, dermatologic, psychiatric, renal, and/or other major disease or malignancy (as judged by the investigator); mean pulse, mean systolic blood pressure or mean diastolic blood pressure, and mean corrected QT interval using Fridericia’s formula above the prespecified range; and/or a history of smoking cigarettes (or equivalent amount of tobacco) or alcohol consumption above the prespecified limit. In addition, patients’ previous medications could not include mild, moderate, or strong inhibitors or inducers of cytochrome P450 (CYP) 3A4 or CYP2C19; these exclusion criteria were based on data (on file) that suggest that CYP3A4 and CYP2C19 are CYP enzymes that are involved in the metabolism of ASP4345.
2.3 Treatments and Study Design
In the single ascending-dose study, all eligible healthy volunteers received ASP4345 or matching placebo as a single oral dose following randomization. In Part 1, single oral doses of 3, 10, 30, 100, 300, or 900 mg of ASP4345 or placebo were administered to six separate cohorts under fasted conditions. In addition, Part 1 included a seventh open-label cohort, wherein participants received a single 300-mg oral dose of ASP4345 within 30 min after starting and 5 min after completion of a high-fat breakfast per the recommendations from the US Food and Drug Administration (Guidance for Industry for Food Effect Bioavailability Studies and Fed Bioequivalence Studies, December 2002) to evaluate the effect of food on the pharmacokinetics of ASP4345. In Part 2, all participants were included in a single cohort and received 300 mg of ASP4345 or matching placebo administered under fasted conditions.
In the multiple ascending-dose study, ASP4345 or placebo was administered orally as a single daily dose for 14 days. The dose was administered under fasted conditions on days 1, 7, and 14 and under fed conditions on days 2, 3, 4, 5, 6, 8, and 13. The four cohorts initially received ASP4345 (3 mg, followed successively by 15 mg, 50 mg, and 150 mg) or placebo. The subsequent dose concentrations were decided by the sponsor and investigator/delegate based on the nature, frequency, and severity of the safety profile. Based on a food-effect analysis, it was shown that food had no effect on the pharmacokinetics of ASP4345. Nevertheless, to minimize any eventual impact that food might have on proof of pharmacology measures, cognitive testing, and electrophysiological measures (auditory steady-state response, mismatch negativity), doses were administered under fasted conditions on days 1, 7, and 14.
The ASP4345 capsules were supplied in doses of 0.5, 5, and 50 mg. The capsules were made of a hard gelatin with a yellow cap and Swedish orange body containing ASP4345-HCl, anhydrous silicic acid, and low-substituted hydroxypropyl cellulose. Placebo capsules had the same appearance and storage conditions as the ASP4345 capsules. Placebo capsules were made of a hard gelatin with a yellow cap and Swedish orange body containing low-substituted hydroxypropyl cellulose.
2.4 Study Outcomes
To assess the pharmacokinetic profile of ASP4345 and its metabolites, plasma and urine samples were collected in Part 1 of the single ascending-dose study and the multiple ascending-dose study; plasma samples and CSF samples were collected in Part 2 of the single ascending-dose study. The sampling time points for blood/plasma and urine, as well as the assessments of central nervous system effects, were adjusted by the investigators based on the emerging data. The total blood volume collected did not exceed 500 mL per participant. Both studies used Phoenix software version 6.3 (Certara, St. Louis, MO, USA) and a non-compartmental approach to calculate plasma pharmacokinetic parameters. The parameters assessed during these studies were maximum concentration (Cmax), time to maximum concentration (Tmax), area under the concentration vs time curve to the last measurable time (AUClast), area under the concentration vs time curve from the time of dosing extrapolated to infinity (AUCinf), area under the concentration–time curve for the dosing interval (AUC), terminal elimination half-life, apparent total systemic clearance after extravascular dosing (CL/F), apparent volume of distribution during the terminal elimination phase after a single extravascular dose, apparent volume of distribution during the terminal elimination phase after extra-vascular dosing (Vz/F), accumulation ratio calculated from AUCτ, ss, and AUCτ after single dosing (Rac AUC), and lag time.
ASP4345 pharmacokinetic samples were analyzed using a validated liquid chromatography-tandem mass spectrometry method. For plasma, the standard curve range was 50.0–50,000 pg/mL for ASP4345, using a sample volume of 50 µL. Concentrations are reported on the basis of the free form. Standards, quality controls, blanks, and samples were combined with the internal standard and were extracted using supported liquid extraction with methyl tert-butyl ether. The solvent methyl tert-butyl ether was evaporated, and the remaining residue was dissolved in reconstitution solution. Separation was achieved on a UPLC HSS T3 (50 × 2.1 mm, 1.8 µm) column with an isocratic mobile phase composed of 45:55:0.05 (v/v/v) H2O:ACN:formic acid. For CSF, the analyte and the internal standard were extracted from 125 μL of human CSF by supported liquid extraction. Internal standard solution (25 μL, 100 pg/mL) was added to thawed CSF samples (1250-μL aliquot) in a 96-well plate. The samples were mixed, and then subjected to supported liquid extraction using a 96-well plate (200 mg). The eluates resulting from extractions were evaporated to dryness under a stream of nitrogen (nominal 40 °C). The residues were re-dissolved in the reconstitution solvent and submitted to analysis by liquid chromatography-tandem mass spectrometry. The method had been qualified in terms of specificity, reproducibility, precision, and robustness. The lower limit of quantification had been set to 50 ng/mL.
2.5 Statistical Analyses
In both the single ascending-dose and multiple ascending-dose studies, the pharmacokinetic analysis set (PKAS) were used for the analysis of all pharmacokinetic data. The PKAS included all randomized participants who took at least one dose of ASP4345 or matching placebo, had sufficient data available to derive at least one pharmacokinetic parameter, and for whom the time of dosing on the day of sampling was known. The plasma, urine, and CSF concentrations and pharmacokinetic parameters of ASP4345 were summarized using descriptive statistics, including mean, standard deviation, coefficient of variation, median, minimum, maximum, and geometric mean, where applicable.
An analysis of variance model was used to determine the effect of food on the pharmacokinetics of ASP4345 in the open-label cohort of Part 1 of the single ascending-dose study. Specifically, to assess the effect of food on the pharmacokinetics of ASP4345 in study Part 1, between the food effect cohort and the cohort with a corresponding dose level under fasted conditions, an analysis of variance model with fixed effects for treatment (fed or fasted) was fitted on natural logarithm transformed Cmax and AUC. Within the analysis of variance, the least-squares (LS) mean differences between fed and fasted, along with the 90% CI on the differences, were estimated. The LS means for Cmax and AUC were back-transformed to produce the geometric LS means. The geometric LS mean ratios and their corresponding 90% CI for each pharmacokinetic parameter were presented by back-transforming and were expressed as percentages.
3 Results
3.1 Participant Disposition
A total of 58 healthy volunteers were randomized in Part 1 of the single ascending-dose study: 46 to the ASP4345 treatment groups and 12 to matching placebo. Of the 46 healthy volunteers, 44 (95.7%) were included in the PKAS; two volunteers discontinued for reasons other than treatment-emergent adverse events. Five healthy volunteers were randomized in Part 2 of the single ascending-dose study: four to the ASP4345 300-mg treatment group and one to matching placebo. All five healthy volunteers were included in the PKAS.
A total of 48 patients were randomized in the multiple ascending-dose study: 36 patients to the ASP4345 treatment groups and 12 patients to matching placebo. All 36 (100%) randomized patients who received ASP4345 were included in the PKAS.
3.2 Baseline Demographics
The demographic and baseline characteristics across treatments in each study are summarized in Table 1. In the single ascending-dose study, healthy volunteers ranged in age from 21 to 55 years, most (91.1%) were male, and overall median body mass index (BMI) was in the normal-to-overweight range. In the multiple ascending-dose study, patients ranged in age from 27 to 60 years, most (75%) were male, primarily African American (83.3%), and overall median BMI was in the normal-to-obese range.
3.3 Single-Dose Pharmacokinetics Under Fasting Conditions
The AUC of ASP4345 in the single ascending-dose study demonstrated a rapid increase in plasma concentration (Fig. 1), and the median Tmax range was 1.00–1.50 h for the 3- to 300-mg ASP4345 doses (Table 2). Absorption of ASP4345 for the 900-mg dose was slower, with a median Tmax of 2.26 h. Both Cmax and AUCinf increased in a less than dose-proportional manner in the ASP4345 3- to 900-mg dose range as the regression line fitting the data deviated from the reference line (Fig. 2). The percentage coefficient of variance for Cmax and AUCinf was higher for the ASP4345 900-mg dose compared with doses in the 3- to 300-mg range. The average apparent elimination half-life range was 9.12–14.3 h and was dose independent. The apparent oral clearance and volume of distribution showed a general trend of increase with increasing doses in the 3- to 900-mg dose range (mean CL/F was in the range of 4.14–19.7 L/h and mean VZ/F was in the range of 66.5–289 L).

Mean plasma concentration vs time profile of ASP4345 by treatment group in Part 1 of the single ascending-dose study (semi-log scale)

Assessment of dose proportionality on the area under the concentration–time curve from the time of dosing extrapolated to infinity (AUCinf) in Part 1 (log–log scale)
3.4 Single-Dose Pharmacokinetics Under Fed Conditions
Under fed conditions in the single ascending-dose study, Cmax decreased by 3.95% compared with fasted conditions (Table 3). In contrast, AUClast and AUCinf increased by 4.04% and 2.97%, respectively, under fed conditions compared with fasted conditions. Median Tmax was higher under fed conditions than under fasted conditions (4.0 h vs 1.5 h). All other pharmacokinetic parameters were comparable between the fed and fasted conditions.
3.5 Single-Dose Pharmacokinetics in Cerebrospinal Fluid
In Part 2 of the single ascending-dose study, CSF concentrations were quantifiable from 30 min up to 24 h post-dose in all participants. ASP4345 was rapidly absorbed in plasma with Tmax in the range of 0.75–1.03 h, but appeared in the CSF with a slight delay; median Tmax was 4.73 h with a range of 2.48–7.98 h. The distribution of ASP4345 into the brain was confirmed based on the ratio of CSF to total plasma for AUC24 of 0.157–0.573%, Cmax of 0.0899–0.311%, and ratio of CSF to unbound plasma for Cmax of 25.0–86.4%. The low ratio of CSF to total plasma (median CSF/plasma = 0.188) is likely due to high plasma protein binding as ASP4345 is highly bound (> 99%) to human plasma proteins in vitro.
3.6 Multiple-Dose Pharmacokinetics
The concentration–time profile in the multiple ascending-dose study showed an increase in plasma concentrations of ASP4345 with increasing dose (Fig. 3); mean steady-state conditions were observed by day 5 based on visual inspection of the trough values over time. However, we noted that one or two patients in each of the treatment groups had more variability prior to achieving steady state. The coefficient for variation for the Cmax range was 20.2–48.8% and for AUCtau was 39.4–49.9% across treatment groups. Rac (AUC) values were approximately one- to two-fold at steady state, with no notable difference between treatment groups (Table 4). The mean elimination half-life range was 11.1–26.8 h and median Tmax range was 1.25–3.02 h. No dose dependency was observed for rate of absorption, lag time, or elimination half-life, and no evidence of time variant pharmacokinetics was detected throughout the study.

Mean plasma concentration vs time profile of ASP4345 by treatment group at day 14 in the multiple ascending-dose study (semi-log scale)
4 Discussion
These two phase I studies of the pharmacokinetics of ASP4345 revealed that single daily dosing is appropriate for ASP4345. The single ascending-dose study recorded rapid absorption of ASP4345 (for all doses 3–900 mg) under fasted conditions (median Tmax range 1.00–2.28 h) and low-to-moderate between-patient variability based on plasma Cmax (11.4–79.8%) and AUCinf (31.0–76.6%). The plasma concentration–time profile was biphasic with a slow decline during the alpha phase, which suggests slow distribution from plasma to tissue and is consistent with the delay in Tmax in CSF compared with plasma Tmax. The elimination half-life was approximately 9–14 h after single administration, and the degree of accumulation in AUCtau was one- to two-fold after repeated doses, which supports once-daily dosing of ASP4345.
This study also determined that food did not alter the pharmacokinetics of ASP4345. Additionally, ASP4345 was shown to reach the brain based on the average CSF to total plasma ratios of Cmax (0.167%) and AUC24 (0.277%), and the average CSF to unbound plasma ratio of Cmax (46.5%). The pharmacokinetic data from the multiple ascending-dose study (ASP4345 in the dose range of 3–150 mg) were consistent with the data from the single ascending-dose study. There were less than dose-proportional increases in Cmax and AUC in the single ascending-dose study, where the dose range was 3–900 mg, and in the multiple ascending-dose study in patients with stabilized schizophrenia or schizoaffective disorder, where doses had a range of 3–150 mg. The favorable pharmacokinetic data from the two phase I studies support further clinical development of ASP4345 as a single daily dose.
The maximum tolerated dose could not be determined in the single ascending-dose study. We also noted an increasing trend of CL/F and VZ/F with increasing doses. This trend suggests a decrease in the bioavailability of the drug with increasing dose, and provides an explanation for the less than dose-proportional increase in Cmax and AUCinf for the 3- to 900-mg ASP4345 dose range. In addition, the percentage of CV for Cmax and AUCinf for the ASP4345 900-mg dose compared with doses in the 3- to 300-mg range may be a result of the greater number of capsules administered at the 900-mg dose.
Further clinical development of ASP4345 as a potential treatment for cognitive impairment is warranted because CIAS is an important symptom domain for patients with schizophrenia and there is no approved medication for CIAS treatment [14, 15]. Over the past few decades, a number of compounds have been tested in clinical trials based on targets identified from basic scientific research conducted to understand the mechanisms involved in CIAS [16]. Unfortunately, none have demonstrated adequate efficacy in large clinical trials. The CIAS clinical trials are challenging owing to concerns associated with patient recruitment and the length of time required to identify response to treatment [15].
Two D1 receptor agonists, DHX and ABT-431, have shown promising efficacy results [17,18,19]; however, clinical use of these agonists is limited by low bioavailability [9] and safety concerns, particularly seizures [10] and hypotension [11]. The results thus far support the development of D1 receptor agonists that are more target specific [9].
In the current study, a few limitations should be noted. First, future studies would benefit from evaluating populations that are more balanced in terms of sex relative to the current study, which was 91.1% male. Furthermore, the BMI varied between groups. While we did not notice any BMI-related trends in our data, future studies might benefit from evaluating more homogenous groups, as it pertains to weight/BMI. Last, while the selected number of subjects was considered sufficient to achieve the study objectives, future studies may benefit from larger group/sample sizes.
5 Conclusions
These two phase I studies showed that ASP4345 is rapidly absorbed, has a long half-life, and reaches the brain, and its pharmacokinetics are not affected by food. The results of these phase I studies support the feasibility of further investigation of the efficacy and safety of ASP4345 in CIAS. A phase IIa, randomized, double-blind, placebo-controlled, parallel-group study to assess the safety and efficacy of ASP4345 (NCT03557931) has been completed and results are forthcoming.
Data availability
Researchers may request access to anonymized participant-level data, trial-level data, and protocols from Astellas-sponsored clinical trials at https://www.clinicalstudydatarequest.com. For the Astellas criteria on data sharing, see https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx.
References
Heinrichs RW, Zakzanis KK. Neurocognitive deficit in schizophrenia: a quantitative review of the evidence. Neuropsychology. 1998;12(3):426–45.
Schaefer J, Giangrande E, Weinberger DR, Dickinson D. The global cognitive impairment in schizophrenia: consistent over decades and around the world. Schizophr Res. 2013;150(1):42–50.
Fatouros-Bergman H, Cervenka S, Flyckt L, Edman G, Farde L. Meta-analysis of cognitive performance in drug-naive patients with schizophrenia. Schizophr Res. 2014;158(1–3):156–62.
Keefe RS, Buchanan RW, Marder SR, et al. Clinical trials of potential cognitive-enhancing drugs in schizophrenia: what have we learned so far? Schizophr Bull. 2013;39(2):417–35.
Davis KL, Kahn RS, Ko G, Davidson M. Dopamine in schizophrenia: a review and reconceptualization. Am J Psychiatry. 1991;148(11):1474–86.
Goldberg TE, Bigelow LB, Weinberger DR, Daniel DG, Kleinman JE. Cognitive and behavioral effects of the coadministration of dextroamphetamine and haloperidol in schizophrenia. Am J Psychiatry. 1991;148(1):78–84.
Goldman-Rakic PS. The cortical dopamine system: role in memory and cognition. Adv Pharmacol. 1998;42:707–11.
Tamminga CA. The neurobiology of cognition in schizophrenia. J Clin Psychiatry. 2006;67 Suppl. 9:9–13 (discussion 36–42).
Arnsten AF, Girgis RR, Gray DL, Mailman RB. Novel dopamine therapeutics for cognitive deficits in schizophrenia. Biol Psychiatry. 2017;81(1):67–77.
Starr MS, Starr BS. Seizure promotion by D1 agonists does not correlate with other dopaminergic properties. J Neural Transm Park Dis Dement Sect. 1993;6(1):27–34.
Blanchet PJ, Fang J, Gillespie M, et al. Effects of the full dopamine D1 receptor agonist dihydrexidine in Parkinson's disease. Clin Neuropharmacol. 1998;21(6):339–43.
Luderman KD, Conroy JL, Free RB, et al. Identification of positive allosteric modulators of the D1 dopamine receptor that act at diverse binding sites. Mol Pharmacol. 2018;94(4):1197–209.
Abdel-Magid AF. Allosteric modulators: an emerging concept in drug discovery. ACS Med Chem Lett. 2015;6(2):104–7.
Jablensky A. The 100-year epidemiology of schizophrenia. Schizophr Res. 1997;28(2–3):111–25.
Keefe RSE. Why are there no approved treatments for cognitive impairment in schizophrenia? World Psychiatry. 2019;18(2):167–8.
Goff DC, Hill M, Barch D. The treatment of cognitive impairment in schizophrenia. Pharmacol Biochem Behav. 2011;99(2):245–53.
Rosell DR, Zaluda LC, McClure MM, et al. Effects of the D1 dopamine receptor agonist dihydrexidine (DAR-0100A) on working memory in schizotypal personality disorder. Neuropsychopharmacology. 2015;40(2):446–53.
George MS, Molnar CE, Grenesko EL, et al. A single 20 mg dose of dihydrexidine (DAR-0100), a full dopamine D1 agonist, is safe and tolerated in patients with schizophrenia. Schizophr Res. 2007;93(1–3):42–50.
Mu Q, Johnson K, Morgan PS, et al. A single 20 mg dose of the full D1 dopamine agonist dihydrexidine (DAR-0100) increases prefrontal perfusion in schizophrenia. Schizophr Res. 2007;94(1–3):332–41.
Acknowledgements
We acknowledge all patients and their families for their participation in this study. In addition, we also acknowledge the investigators, coordinators, and site personnel involved in this study. This research was designed and sponsored by Astellas Pharma Global Development, Inc. (Northbrook, IL, USA). Writing and editorial assistance under the authors’ guidance was provided by Shilpa Lalchandani, PhD, Laura McCormick, PhD, and Elizabeth Hermans, PhD, of OPEN Health Medical Communications (Chicago, IL, USA).
Funding
Financial support for the development of this article was provided by the study sponsor, Astellas Pharma, Inc., Northbrook, IL, USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Amit Desai, Lauren Benner, Ruishan Wu, Tolga Uz, Gerard J. Marek, and Tong Zhu are employees of Astellas Pharma Global Development, Inc. Lev Gertsik is an employee of California Clinical Trials Medical Group.
Ethics approval
Studies were conducted in accordance with ethical principles based on the Declaration of Helsinki, Good Clinical Practice, International Council for Harmonization guidelines, and applicable laws and regulations.
Consent to participate
All healthy volunteers and patients provided written informed consent.
Consent for publication
All healthy volunteers and patients provided written informed consent.
Code availability
Not applicable.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Desai, A., Benner, L., Wu, R. et al. Pharmacokinetics of ASP4345 from Single Ascending-Dose and Multiple Ascending-Dose Phase I Studies. Clin Pharmacokinet 60, 79–88 (2021). https://doi.org/10.1007/s40262-020-00911-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-020-00911-0